

Abstract
Purpose
To evaluate the safety and efficacy of topical TOP1630, a novel nonsystemic kinase inhibitor, in dry eye disease (DED).
Patients and methods
A randomized, double-masked, parallel-group trial of 0.1% TOP1630 ophthalmic solution TID or placebo (vehicle without active drug) was conducted in DED subjects (n=61). Key eligibility criteria consistent with enrolling a moderate to severe DED population included >6 months DED history; OSDI© score ≥18; Schirmer’s test score ≤10 and ≥1 mm/5 minutes; tear film break-up time >1 and <7 seconds; and dry eye exacerbation in corneal staining and ocular discomfort in a Controlled Adverse Environment (CAE®). After a 7-day run-in period with placebo TID, eligible subjects were randomized to TOP1630 or placebo for 28 days. No supplemental artificial tears or rescue medication were allowed.
Results
TOP1630 was safe, well-tolerated, and efficacious in treating DED symptoms and signs. No serious adverse events (AEs) or withdrawals due to treatment emergent AEs occurred. Drop comfort scores showed TOP1630 to be comfortable and comparable with placebo. Significant symptom improvements were seen for TOP1630 vs placebo for ocular discomfort (P=0.02 post-CAE), grittiness/foreign body sensation (on four independent assessment scales, each P<0.05), worst DED symptom (diary, P=0.06), and ocular pain (VAS, P=0.03). Sign improvements were seen for total ocular surface (all regions), corneal sum, and conjunctival sum staining with TOP1630 compared with placebo (each P<0.05).
Conclusion
TOP1630 had placebo-like tolerability and produced improvements in multiple symptom and sign endpoints in both environmental and challenge settings. The emergent TOP1630 benefit–risk profile for DED treatment is highly favorable and supports further development.
Keywords: dry eye, DED, TOP1630, ocular inflammation
Introduction
Dry eye disease (DED) is a chronic, multifactorial inflammatory disorder of the lacrimal functional unit characterized by ocular discomfort, pain, and visual disturbances.1,2 This disorder is associated with aging, contact lens wear, refractive surgery, and immune diseases, and it affects 15%–30% of the over-50s, depending on ethnicity.3,4 DED negatively impacts visual, social, and physical functioning and quality of life particularly when moderate–severe.5 Current therapies for treating DED have significant limitations.6 Cyclosporine (Restasis®) has limited efficacy, tolerability issues, a slow onset of action, and is approved only for treating a single sign of the disease (indicated in patients whose tear production is presumed to be suppressed due to ocular inflammation associated with keratoconjunctivitis sicca).7–9 Corticosteroids are commonly prescribed off-label and are effective on both signs and symptoms of DED, but are restricted to short-term use as a consequence of serious ocular adverse events (AEs).10 Lifitegrast (Xiidra®) has recently been approved, and clinical studies have shown that after 12 weeks of treatment, there can be an improvement in selected signs and symptoms of DED, albeit with some commonly occurring drug tolerance issues.11,12 Consequently, a substantial unmet medical need still exists for a fast-acting, effective, safe, and well-tolerated immunomodulatory therapy to address both the signs and the symptoms of DED.13
Inflammation has an important role in DED pathophysiology.6 Nonsystemic kinase inhibitors (NSKIs) represent a novel class of pharmacological agents that selectively target key kinases fundamental to inflammatory cell signaling in innate and adaptive immune responses.14 In DED, the NSKI targets p38α, Src family kinases (Src and Lck), and Syk are upregulated at the gene level in patients compared with healthy volunteers.14 NSKIs have broad, potent anti-inflammatory effects in vitro and in vivo, exhibiting potent inhibition of cytokine release in cellular assays mimicking both innate and adaptive immune systems, as well as in vivo models.15 These potent anti-inflammatory agents are small molecules designed for topical administration and demonstrate an exemplary safety profile in preclinical and clinical studies with very low systemic exposure.15 Recent investigations have highlighted the potential of NSKIs in alleviating inflammatory conditions such as ulcerative colitis (UC), COPD, and rheumatoid arthritis.15–18 In this study, we investigated the safety and efficacy of the topical ocular NSKI TOP1630 in DED.
Patients and methods
The study was conducted in accordance with the tenets of the Declaration of Helsinki and the provisions of the International Conference on Harmonization Harmonized Guideline on Good Clinical Practice E6. All subjects provided written informed consent after explanation of the nature and possible consequences of the study. The research was approved by Alpha IRB (San Clemente, CA, USA; Office for Human Research Protections [OHRP]/Food and Drug Administration [FDA] registration number IRB00006205). The clinical trial was registered on www.ClinicalTrials.gov (NCT03088605).
TOP1630 investigational medicinal product
TOP1630 was manufactured by Onyx Scientific Limited (Sunderland, United Kingdom) in compliance with current good manufacturing practices using a published route and was formulated as a 0.1% (1 mg/mL) ophthalmic solution.19 Matched placebo comprised vehicle solution (sterile water containing potassium phosphate, mannitol, polyoxyl 40 stearate, and pH modifiers) with no TOP1630. TOP1630 and placebo were prepared as preservative-free, sterile, clear, colorless solutions presented in single-use 1 mL “natural” low-density polyethylene eye dropper bottles.
The investigational medicinal product was manufactured by Bio-Concept Laboratories, Inc. (Salem, NH, USA). Labeling, final packaging, and release of the clinical trial material were performed by Ora, Inc. (Andover, MA, USA).
Study design and conduct
This study was a single-center, randomized, double-masked, placebo-controlled trial to evaluate the safety and efficacy of topical TOP1630 0.1% ophthalmic solution in patients with DED. This investigational dose was well tolerated in a small pilot investigation assessing drop comfort, conducted under the same protocol with eight DED subjects sequentially receiving TOP1630 0.01% TID to 0.1% TID or placebo in a randomized double-masked schema (single day ascending dosing with a further period of 4 days’ dosing at the highest dose 0.1% TID, with interim rest days between dose steps).
Subjects who met eligibility criteria (see below for main inclusion and exclusion criteria) were randomized in a 1:1 ratio to receive treatment with either TOP1630 or matched placebo following a 7-day run-in period with placebo TID in both eyes (OU). The run-in period was followed by a 28-day treatment period with randomized treatment TID OU. No supplemental artificial tears or rescue medication were allowed during the study period. There was a total of four scheduled study visits: two during screening (visit 1 at day −7 and visit 2 at day 1) and two during treatment (visit 3 at day 15 and visit 4 at day 29 on completion of which subjects exited the study). Dry eye signs and symptoms and safety measures were assessed at each clinic visit (see Table 1 for assessment schedule). The study was conducted at Andover Eye Associates, Andover, MA, USA.
Table 1.
Schedule of main events and safety and efficacy assessments
| Procedure | Visit 1b (day −7±1) | Visit 2b (day 1) | Visit 3b (day 15±1) | Visit 4b (day 29±2) | ||||
|---|---|---|---|---|---|---|---|---|
| Pre-CAE | Post-CAE | Pre-CAE | Post-CAE | Pre-CAE | Post-CAE | Pre-CAE | Post-CAE | |
| Informed consent/HIPAA | X | |||||||
| Medical/medication history and demographics | X | |||||||
| Ocular discomfort (Ora Calibra/dry eye symptoms) | X | X | X | X | X | X | X | X |
| OSDI© Questionnaire | X | X | X | X | ||||
| Visual acuity | X | X | X | X | ||||
| Review of inclusion/exclusion criteria | X | X | X | X | ||||
| Slit-lamp biomicroscopy | X | X | X | X | X | X | X | X |
| VAS symptom assessment | X | X | X | X | ||||
| Conjunctival redness (Ora Calibra scale) | X | X | X | X | X | X | X | X |
| Lid margin redness (Ora Calibra scale) | X | X | X | X | ||||
| Posterior lid edge evaluation (Ora Calibra scale) | X | X | X | X | ||||
| TFBUT | X | X | X | X | X | X | X | X |
| Fluorescein staining (Ora Calibra and NEI) | X | X | X | X | X | X | X | X |
| Lissamine green staining (Ora Calibra and NEI) | X | X | X | X | X | X | X | X |
| Corneal sensitivity (Cochet–Bonnet) | X | X | ||||||
| OPI 2.0 | X | X | ||||||
| Unanesthetized Schirmer’s test | X | X | X | X | ||||
| Diary card dispense (D)/collect (C) | D | C | D | C | D | C | ||
| CAE® Exposure | X | X | X | X | ||||
| CAE discomfort (Ora Calibra ocular discomfort) | X | X | X | X | ||||
| Intraocular pressure (noncontact) | X | X | ||||||
| Undilated fundus examination | X | X | ||||||
| Randomization | X | |||||||
| Adverse event query | X | X | X | X | X | X | X | X |
| Exit subject from study | X | |||||||
Abbreviations: CAE, controlled adverse environment; HIPAA, Health Insurance Portability and Accountability Act; NEI, National Eye Institute; OPI, Ocular Protection Index; OSDI, Ocular Surface Disease Index; TFBUT, tear film break-up time.
Subjects
Main criteria for inclusion were as follows: Adult subjects (≥18 years old) who had a reported history of DED in OU for at least 6 months prior to visit 1 and an associated history of use or desire to use eye drops for dry eye symptoms for at least 6 months. Subjects must have had, at visits 1 and 2, an Ocular Surface Disease Index© (OSDI©; Allergan Inc) score ≥18; a score of ≥2 in at least one symptom of the Ora Calibra Ocular Discomfort and 4-Symptom Questionnaire before Controlled Adverse Environment (CAE®) exposure; and in at least one eye (the same eye) before CAE exposure Schirmer’s test score of ≤10 and ≥1 mm, a conjunctival redness score of ≥1 (Ora Calibra scale), a tear film breakup time (TFBUT) >1 and <7 seconds, a corneal fluorescein staining score of ≥2 (Ora Calibra scale) in at least one region (eg, inferior, superior, or central), and a total lissamine green score of ≥2 (Ora Calibra scale) based on the sum of the temporal and nasal regions of the conjunctiva. Subjects had to show an exacerbation of corneal staining and ocular discomfort in the CAE at both visits 1 and 2.
Main exclusion criteria were as follows: Any clinically significant ocular conditions including active blepharitis, meibomian gland dysfunction, lid margin inflammation, active ocular allergies; contact lens use within 14 days of visit 1 or anticipated use during the study; previous laser-assisted in situ keratomileusis surgery within the last 12 months or any other ocular surgeries within the last 6 months; ocular cyclosporine or lifitegrast use within 90 days of visit 1; topical ocular steroid use within 30 days of visit 1; any planned ocular and/or lid surgeries over the study period; taking any topical ophthalmic prescription (including medications for glaucoma) or over-the-counter solutions, artificial tears, gels or scrubs, and could not discontinue those medications for the duration of the trial or had used any of those medications in the 24 hours prior to visit 1; have had corrected visual acuity greater than or equal to the logarithm of the minimum angle of resolution (logMAR) +0.7 as assessed by Early Treatment of Diabetic Retinopathy Study (ETDRS) scale in OU at visit 1.
Treatment protocol
Subjects meeting all eligibility criteria at visits 1 and 2 were randomized (following 7-day run-in period on placebo) to receive treatment with TOP1630 or placebo.
Subjects were instructed to dose by instilling one drop in each eye TID in approximately 6-hour intervals (morning, afternoon, and evening before bed). Subjects were instructed to not use study drug on the day of visits 2, 3, and 4, prior to the visit.
The sponsor, investigator, study staff, and subject were masked to the subject’s treatment arm during the randomization process and throughout the study.
Outcome measures
The primary objective of the study was to compare the safety and tolerability of TOP1630 ophthalmic solution with placebo in subjects with DED.
The secondary objectives were to compare the efficacy of TOP1630 ophthalmic solution with placebo for the treatment of the signs and symptoms of DED.
A summary of scheduled assessments and study visits is shown in Table 1.
Efficacy measures
Efficacy assessments included environmental and CAE change in DED symptoms and ocular surface staining.20
Dry eye symptoms
Ocular Discomfort Scale
At each visit, ocular discomfort scores were subjectively graded by subjects rating each eye separately according to a 5-point (0–4) Ocular Discomfort Scale (Ora Calibra), where 0= none and 4= severe.
Ocular Discomfort and 4-Symptom Questionnaire
Each day, subjects graded the severity of their DED symptoms in their diary in the morning, afternoon, and evening before instilling the study drug.
Subjects rated the severity of each of the following symptoms, with regard to how both their eyes felt: overall ocular discomfort, burning, dryness, grittiness, and stinging according to a 6-point (0–5) scale (Ora Calibra Ocular Discomfort and 4-Symptom Questionnaire), where 0= none and 5= worst.
Symptom VAS
At each visit, subjects were asked questions regarding ocular discomfort (unrelated to study drug instillation) over the previous week. The VAS comprised a 7-item 100-point scale: 0= no discomfort, 100= maximal discomfort for assessment of burning/stinging, itching, foreign body sensation, blurred vision, eye dryness, photophobia, and pain.
Ocular Surface and Disease Index (OSDI) Questionnaire
Subjects were asked to recall symptom-related parameters experienced in the previous week.21
Dry eye signs
Lissamine green staining
Staining was graded in defined regions of the ocular surface with the Ora Calibra Corneal and Conjunctival Staining Scale (5-point scale, 0= none to 4= confluent).
Staining was also graded in the conjunctiva using the National Eye Institute (NEI)/Industry Workshop scale (4-point scale, 0= no staining present to 3= severe staining).
Fluorescein staining
Staining was graded with the Ora Calibra Corneal and Conjunctival Staining Scale and NEI scale (each scale as detailed above, with NEI grading of five areas of the cornea).
Other measures
Unanesthetized Schirmer’s test (mm/5 minutes), TFBUT (average of two to three measurements), and Ora Calibra Ocular Protection Index 2.0,22 each after instillation of 5 µL of 2% preservative-free sodium fluorescein solution were also assessed.
Conjunctival redness and lid margin redness were each graded according to a 5-point (0–4) scale (Ora Calibra scale), where 0= normal and 4= severe. Posterior lid edge evaluation was graded according to a 4-point (0–3) scale (Ora Calibra scale), where 0= normal and 3= severe.
Safety measures
Safety assessments and AE recording were assessed at each visit and included slit-lamp biomicroscopy, intraocular pressure, visual acuity, undiluted fundoscopy, and corneal sensitivity.
Drop comfort assessment
Drop comfort was assessed on day 1 (visit 2) and day 15 (visit 3) following the first dose each day of study drug administered in the clinic. The Ora Calibra Drop Comfort Scale (0–10 scale, with a score of 0 indicating very comfortable and 10 indicating very uncomfortable) was used to assess drop comfort for each eye separately upon instillation and to 2 minutes postinstillation. The worst eye from each subject was used for this analysis.
Statistical analysis
The primary objectives of the study were to evaluate safety and tolerability, and exploratory hypothesis testing was performed for efficacy endpoints. With a planned sample size of 60 subjects, the study had 79% probability of detecting AEs occurring at a rate of 5% or more in either treatment arm.
The analysis populations included the following:
Safety population: Safety population included all subjects who received at least one dose of the investigational medicinal product.
Modified intent-to-treat: The modified intent-to-treat (mITT) population included all randomized subjects with one postbaseline efficacy assessment. Efficacy analysis was performed on the mITT population.
All statistical analyses of the Safety and Efficacy Assessment were performed (by SDC Phoenix, AZ, USA) after the study was completed and the database had been locked and released for unmasking. Statistical programming and analyses were performed using SAS® (SAS Institute, Middleton, MA, USA) version 9.4. The Statistical Analysis Plan was finalized and signed before unmasking of treatment assignments. All efficacy analyses were conducted two-sided at a significance level of 0.1.
The treatment effect was estimated from the difference between TOP1630 and placebo groups in the mean within-group change as a percentage of respective TOP1630 and placebo baseline mean values.
Safety endpoints were analyzed for OU. For efficacy endpoints, eyes were eligible for analysis if they met all of the inclusion criteria, but the unit of analysis was the “worst eye.” In the case that OU were eligible for analysis, the study eye was the eye with worse (higher) increase in total corneal staining change from pre-CAE to post-CAE at visit 2 on the Calibra scale. If the increase in total corneal staining was the same in OU, then the study eye was the eye with the largest increase in ocular discomfort (Calibra scale) from pre-CAE to post-CAE. If the ocular discomfort symptom increase was the same in OU, then the right eye was selected as the worst eye.
Safety: AEs were coded using the MedDRA dictionary (version 20.0). Frequencies and percentages of subjects with treatment-emergent adverse events (TEAEs), serious TEAEs, and TEAEs causing premature discontinuation were determined by treatment group.
Tolerability data: Drop comfort was summarized using quantitative summary statistics.
Secondary and exploratory endpoints: The continuous and ordinal secondary efficacy variables collected at each visit were summarized descriptively. All visit-based data were analyzed at each visit as well as change from baseline (visit 2) matched relative to the CAE. Sign and symptom endpoints were analyzed by visit and time point (where applicable pre-CAE and post-CAE) using two-sample t-tests and Wilcoxon rank sum tests. Changes from baseline were also analyzed using two-sample t-tests and analysis of covariance (ANCOVA) models adjusting for baseline values. Symptoms recorded on the daily diary were analyzed using repeated measures ANCOVA models, where baseline scores were calculated as the average of the scores in the placebo run-in period and postbaseline scores were calculated as weekly morning, afternoon, evening, and daily averages. The efficacy analyses were performed using observed data only. As an exploratory study, there was no adjustment for multiplicity.
Results
One hundred sixty subjects were screened and 61 DED subjects enrolled at visit 2, of which 31 were randomized to TOP1630 and 30 to placebo. One subject in the TOP1630 group discontinued prior to study completion due to subject’s choice (decision not to continue) and did not provide data at day 15 and day 29. The remaining 60 subjects completed the study. A summary of subject disposition is shown in Table 2. Baseline demographics were comparable with no notable differences between TOP1630 and placebo groups. Baseline OSDI total score was also similar in the two groups. DED was reported in the ocular medical history for all subjects, with cataract nuclear (32.8%), cataract operation (21.3%), and vitreous detachment (11.5%) being the next most commonly reported conditions and procedures (all others occurred in <5% of subjects). A summary of demographic data and baseline characteristics is presented in Table 3. No ocular concomitant medications were reported during the study. Mean (SD) treatment compliance with study drug dosing was 98.9% (2.03%) in the TOP1630 group and 97.7% (6.05%) in the placebo group.
Table 2.
Summary of subject disposition
| TOP1630 (N=31) n (%) | Placebo (N=30) n (%) | All subjects (N=61) n (%) | |
|---|---|---|---|
| Analysis populationsa | |||
| mITT | 31 (100.0) | 30 (100.0) | 61 (100.0) |
| Per-protocol | 29 (93.5) | 28 (93.3) | 57 (93.4) |
| Safety | 31 (100.0) | 30 (100.0) | 61 (100.0) |
| Study completion | |||
| Completed | 30 (96.8) | 30 (100.0) | 60 (98.4) |
| Discontinued | 1 (3.2) | 0 | 1 (1.6) |
| Reason for discontinuation | |||
| Adverse event | 0 | 0 | 0 |
| Protocol violations | 0 | 0 | 0 |
| Administrative reasons | 0 | 0 | 0 |
| Sponsor termination of study | 0 | 0 | 0 |
| Subject choice | 1 | 0 | 1 |
Notes:
The mITT population was analyzed as randomized. For the treatment assignments, the safety and per-protocol populations were analyzed as treated.
Abbreviation: mITT, modified intent-to-treat.
Table 3.
Demographic and baseline information by treatment group
| TOP1630 (N=31) | Placebo (N=30) | |
|---|---|---|
| Age (years) | ||
| Mean (SD) | 62.5 (11.4) | 65.3 (11.9) |
| Median | 63.0 | 63.0 |
| Min, max | 33, 81 | 37, 89 |
| <65 years | 17 (54.8%) | 16 (53.3%) |
| ≥65 years | 14 (45.2%) | 14 (46.7%) |
| Sex | ||
| Male | 11 (35.5%) | 14 (46.7%) |
| Female | 20 (64.5%) | 16 (53.3%) |
| Duration of DED (days) | ||
| Median (min, max) | 3,770 (835, 17,999) | 4,309 (378, 24,575) |
| OSDI© total score | ||
| Mean (SD) | 41.2 (14.4) | 38.0 (13.7) |
Notes: N in the headers represents the total number of subjects in each respective treatment group within the modified intention-to-treat population. Percentages are based on the total number of subjects in each treatment group.
Abbreviations: DED, dry eye disease; OSDI, Ocular Surface Disease Index.
Safety
Twelve of the 61 subjects (19.7%) reported 16 TEAEs. Six subjects (19.4%) in the TOP1630 group reported seven TEAEs, and six subjects (20.0%) in the placebo group reported nine TEAEs. All TEAEs were mild to moderate in intensity. Ten ocular TEAEs were reported (Table 4); of these, seven were considered related to study treatment. None of the ocular TEAEs were severe and none resulted in subject withdrawal or required modification in study treatment. The majority (two) on TOP1630 were mild in severity (one moderate in severity).
Table 4.
Ocular treatment emergent adverse events
| System organ class Preferred term | TOP1630 (N=31) | Placebo (N=30) | All subjects (N=61) | |||
|---|---|---|---|---|---|---|
| Events | Subjects n (%) | Events | Subjects n (%) | Events | Subjects n (%) | |
| Total | 5 | 4 (12.9) | 5 | 4 (13.3) | 10 | 8 (13.1) |
| Eye disorders | 3 | 2 (6.5) | 2 | 2 (6.7) | 5 | 4 (6.6) |
| Visual acuity reduced | 1 | 1 (3.2) | 1 | 1 (3.3) | 2 | 2 (3.3) |
| Eye discharge | 0 | 0 | 1 | 1 (3.3) | 1 | 1 (1.6) |
| Vision blurred | 1 | 1 (3.2) | 0 | 0 | 1 | 1 (1.6) |
| Vitreous floaters | 1 | 1 (3.2) | 0 | 0 | 1 | 1 (1.6) |
| General disorders and administration site conditions | 1 | 1 (3.2) | 3 | 3 (10.0) | 4 | 4 (6.6) |
| Instillation site pain | 1 | 1 (3.2) | 2 | 2 (6.7) | 3 | 3 (4.9) |
| Instillation site discomfort | 0 | 0 | 1 | 1 (3.3) | 1 | 1 (1.6) |
| Injury, poisoning, and procedural complications | 1 | 1 (3.2) | 0 | 0 | 1 | 1 (1.6) |
| Procedural pain | 1 | 1 (3.2) | 0 | 0 | 1 | 1 (1.6) |
Notes: SOCs are listed in the order of descending frequency for all subjects. PTs are listed in the order of descending frequency within each SOC for all subjects. N in the headers represents the number of subjects in each respective treatment group within the safety population. Subjects experiencing more than one TEAE within a given SOC or PT are counted once within that SOC or PT. Percentages are based on the total number of subjects in each treatment group. TEAEs were coded using MedDRA version 20.0.
Abbreviations: MedDRA, Medical Dictionary for Regulatory Activities; PT, preferred term; SOC, system organ class; TEAE, treatment-emergent adverse event.
Six nonocular TEAEs were reported and all were considered unrelated to study drug.
No deaths or treatment-emergent serious AEs occurred. There were no withdrawals from study drug due to any TEAE.
Drop comfort assessments
A difference favoring the drop comfort in the TOP1630 group was observed upon instillation at visit 3 (day 15) TOP1630 1.9 (SD 2.18), placebo 2.9 (2.23), P=0.0496. At 2 minutes postinstillation, drop comfort scores were: TOP1630 2.4 (1.98), placebo 2.7 (1.86), P=0.5564. Comparisons of in-office drop comfort scores at the remaining time points at visit 2 (day 1) and visit 3 provided no evidence of a difference between TOP1630 and placebo.
Efficacy endpoints
TOP1630 demonstrated statistically significant treatment effects vs placebo in a range of DED symptoms and signs, and an overall summary for key efficacy endpoints is shown in Figure 1.
Figure 1.

Key efficacy endpoints showing statistical significance (P<0.1) of TOP1630 compared with placebo for DED (mean change from baseline to day 29, modified intention-to-treat population).
Note: See Results and Figures for data and further details.
Abbreviations: CAE, Controlled Adverse Environment; DED, dry eye disease.
Symptoms
Ocular Discomfort Score
Baseline ocular discomfort scores were closely matched between the groups both pre- and post-CAE challenge. Pre-CAE, TOP1630 mean 2.4 (SD 0.96), placebo 2.3 (0.83); post-CAE challenge, TOP1630 3.7 (0.53), placebo 3.7 (0.52).
By day 29, a statistically significant difference favoring TOP1630 was noted for post-CAE discomfort (Figure 2), with a percentage treatment effect of 11%.
Figure 2.
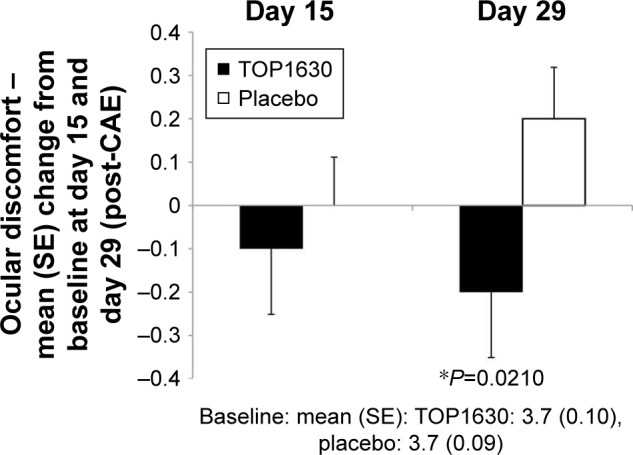
CAE-associated Ocular Discomfort score (Ora Calibra scale).
Note: Results showing mean change from baseline to day 15 and day 29 analysis (*analysis of covariance test P<0.1) in modified intention-to-treat population.
Abbreviation: CAE, controlled adverse environment.
Ocular Discomfort and 4-Symptom Questionnaire
Baseline scores were comparable between treatment groups. Statistically significant differences were noted at day 15 and day 29 in favor of TOP1630 for change from baseline post-CAE grittiness. At day 29, significant differences were also noted in favor of TOP1630 for pre-CAE grittiness and change from baseline in pre-CAE grittiness (Figure 3). The percentage treatment difference was 25% pre-CAE and 42% post-CAE in favor of TOP1630 at day 29.
Figure 3.

Grittiness score (Ora Calibra scale).
Note: Results showing mean change from baseline to day 15 and day 29 analysis (*analysis of covariance test P<0.1) in modified intention-to-treat population.
Abbreviation: CAE, controlled adverse environment.
A difference favoring the TOP1630 group was noted for change from baseline in post-CAE dryness at day 29: baseline post-CAE TOP1630 4.0 (0.93), placebo 4.0 (0.67), day 29 change from baseline TOP1630 −0.5 (0.73), placebo −0.1 (0.94), mean Δ −0.4, P=0.0741. The percentage treatment difference was 14% in favor of TOP1630 at day 29.
No statistically significant differences were observed on this assessment scale for ocular discomfort, burning, or stinging.
Ocular Surface Disease Index
For grittiness, baseline scores were comparable between groups: TOP1630 1.7 (1.24), placebo 1.9 (1.08). Statistically significant differences were noted at day 15 and day 29 in favor of TOP1630 for response score (data not shown) and for change from baseline in response score: TOP1630 −0.2 (0.86), placebo +0.1 (0.73) at day 15; and TOP1630 −0.2 (0.68), placebo +0.1 (0.78) at day 29; mean Δ −0.4 at both day 15 and day 29, P=0.0173 and P=0.0150, respectively. The percentage treatment difference was 17% in favor of TOP1630 at day 29.
No statistically significant differences between TOP1630 and placebo groups were noted for “painful or sore eyes,” “blurred vision,” or “poor vision.”
There were some statistically significant differences in change from baseline at day 15 that were not statistically significant by day 29 for the following: “eyes that are sensitive to light,” “reading,” “working with computer or bank machine (ATM),” “watching TV,” and “driving at night” (each in favor of placebo) and eyes feeling uncomfortable in “areas that are air conditioned” (in favor of TOP1630). There was no difference between the groups for the remaining symptom trigger assessments.
Ocular symptoms of DED – VAS
Baseline scores in the individual items were comparable between treatment groups, with the exception of ocular pain, which was more severe in the TOP1630 group, TOP1630 32.1 (29.71), placebo 19.1 (23.22), P=0.0618.
Differences were noted at day 15 and day 29 in favor of TOP1630 for change from baseline in burning/stinging: baseline TOP1630 40.7 (32.04), placebo 29.6 (23.84), day 15 TOP1630 −4.8 (13.62), placebo +5.8 (21.94); and day 29 TOP1630 −7.9 (15.76), placebo −0.6 (17.24); mean Δ −10.6 at day 15 and Δ −7.3 at day 29, P=0.064 and P=0.09, respectively. The percentage treatment difference was 21% in favor of TOP1630 at day 29.
Differences were noted at day 15 and day 29 in favor of TOP1630 for foreign body sensation and for change from baseline (day 29 only): baseline TOP1630 33.4 (29.10), placebo 37.9 (26.24), day 15 TOP1630 +1.7 (20.05), placebo +9.0 (18.97); and day 29 TOP1630 −3.0 (24.24) placebo +8.0 (16.45); mean Δ 7.2 at day 15 and Δ 11.0 at day 29, P=0.1023 and P=0.0213, respectively. The percentage treatment difference was 30% in favor of TOP1630 at day 29.
A difference was noted at day 29 in favor of TOP1630 for change from baseline in eye pain: baseline (see above), day 29 TOP1630 −11.0 (23.40), placebo +0.3 (14.77); mean Δ −11.3 at day 29, P=0.0292. The percentage treatment difference was 36% in favor of TOP1630 at day 29.
Differences were noted at day 15 and day 29 in favor of placebo for photophobia and for change from baseline in photophobia: baseline TOP1630 54.5 (32.26), placebo 47.9 (32.43), day 15 TOP1630 +5.1 (23.91), placebo −5.5 (19.28); and day 29 TOP1630 +1.8 (20.79), placebo −6.4 (18.89); mean Δ 10.6 at day 15 and Δ 8.2 at day 29, P=0.0284 and P=0.0606, respectively. No statistically significant differences between the TOP1630 group and the placebo group were noted on the VAS for the symptoms of itching, blurred vision, and dryness.
Daily diary symptomatology
Symptom results for grittiness and worst symptom are presented (baseline and day 15 to day 29) in Tables 5 and 6.
Table 5.
Diary card grittiness
| Period | Statistical measurement(s) | Time point | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Morning | Afternoon | Evening | Daily average | ||||||
| TOP1630 (N=31) | Placebo (N=30) | TOP1630 (N=31) | Placebo (N=30) | TOP1630 (N=31) | Placebo (N=30) | TOP1630 (N=31) | Placebo (N=30) | ||
| Baseline | n | 31 | 30 | 31 | 30 | 31 | 30 | 31 | 30 |
| Mean (SD) | 1.75 (1.16) | 2.07 (1.15) | 1.48 (1.01) | 1.55 (0.10) | 1.51 (1.06) | 1.56 (0.92) | 1.58 (1.03) | 1.74 (0.97) | |
| Mean Δa | −0.32 | – | −0.07 | – | −0.05 | – | −0.16 | – | |
| Day 15 to day 29 | n | 30 | 30 | 30 | 30 | 30 | 30 | 30 | 30 |
| Mean (SD) | 1.38 (1.06) | 1.95 (1.00) | 1.14 (0.87) | 1.61 (1.01) | 1.13 (0.87) | 1.61 (1.07) | 1.23 (0.89) | 1.74 (0.99) | |
| Mean Δa | −0.57 | – | −0.47 | – | −0.48 | – | −0.51 | – | |
| P-value (two-sample t-test) | 0.0354** | – | 0.0579* | – | 0.0651* | – | 0.0406** | – | |
| P-value (Wilcoxon rank sum test) | 0.0419** | – | 0.0870* | – | 0.0808* | – | 0.0717* | – | |
| Mean (SD) change from baseline | −0.37 (0.62) | −0.12 (0.53) | −0.34 (0.64) | 0.06 (0.36) | −0.38 (0.71) | 0.05 (0.48) | −0.32 (0.61) | 0.00 (0.38) | |
| Mean Δa | −0.21 | – | −0.37 | – | −0.39 | – | −0.31 | – | |
| P-value (two-sample t-test) | 0.1680 | – | 0.0071** | – | 0.0164** | – | 0.0186** | – | |
| P-value (ANCOVA model) | 0.0374** | – | 0.0028** | – | 0.0088** | – | 0.0062** | – | |
Notes: N in the headers represents the total number of subjects in each respective treatment group within the mITT population. Symptom score was assessed on a 0–5 scale; 0.5 increments were not allowed. Lower score indicates less symptomatology. Baseline was defined as the average of all days during the run-in period. Daily scores were obtained by averaging the morning, afternoon, and evening scores for that day.
TOP1630 value minus placebo value.
P<0.1;
P<0.05.
Abbreviations: ANCOVA, analysis of covariance; ∆, difference; mITT, modified intent-to-treat.
Table 6.
Diary card worst symptom (the symptom for each patient with the highest mean severity score during the run-in period)
| Period | Statistical measurement(s) | Time point | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Morning | Afternoon | Evening | Daily average | ||||||
| TOP1630 (N=31) | Placebo (N=30) | TOP1630 (N=31) | Placebo (N=30) | TOP1630 (N=31) | Placebo (N=30) | TOP1630 (N=31) | Placebo (N=30) | ||
| Baseline | n | 31 | 30 | 31 | 30 | 31 | 30 | 31 | 30 |
| Mean (SD) | 2.79 (1.00) | 3.08 (0.79) | 2.69 (0.90) | 2.69 (0.74) | 2.73 (0.82) | 2.72 (0.76) | 2.75 (0.86) | 2.85 (0.67) | |
| Mean Δa | −0.29 | – | 0.00 | – | 0.01 | – | −0.10 | – | |
| Day 15 to day 29 | n | 30 | 30 | 30 | 30 | 30 | 30 | 30 | 30 |
| Mean (SD) | 2.39 (0.99) | 2.90 (0.74) | 2.23 (0.87) | 2.56 (0.68) | 2.36 (0.77) | 2.56 (0.70) | 2.33 (0.83) | 2.69 (0.65) | |
| Mean Δa | −0.51 | – | −0.33 | – | −0.20 | – | −0.36 | – | |
| P-value (two-sample t-test) | 0.0270** | – | 0.1092 | – | 0.2826 | – | 0.0707* | – | |
| P-value (Wilcoxon rank sum test) | 0.0546* | – | 0.1205 | – | 0.3292 | – | 0.0986* | – | |
| Mean (SD) change from baseline | −0.40 (0.82) | −0.18 (0.50) | −0.46 (0.70) | −0.13 (0.55) | −0.37 (0.74) | −0.16 (0.58) | −0.42 (0.71) | −0.16 (0.46) | |
| Mean Δa | −0.22 | – | −0.30 | – | −0.21 | – | −0.26 | – | |
| P-value (two-sample t-test) | 0.2941 | – | 0.0644* | – | 0.3071 | – | 0.1480 | – | |
| P-value (ANCOVA model) | 0.0825* | – | 0.0360** | – | 0.2160 | – | 0.0628* | – | |
Notes: N in the headers represents the total number of subjects in each respective treatment group within the mITT population. Symptom score was assessed on a 0–5 scale; 0.5 increments were not allowed. Lower score indicates less symptomatology. Baseline was defined as the average of all days during the run-in period. Daily scores were obtained by averaging the morning, afternoon, and evening scores for that day. The symptom with the highest daily average score during the run-in period was selected as the worst dry eye symptom for the subject.
TOP1630 value minus placebo value.
P<0.1;
P<0.05.
Abbreviations: ANCOVA, analysis of covariance; ∆, difference; mITT, modified intent-to-treat.
Statistically significant differences were noted in favor of TOP1630 for grittiness and for change from baseline in grittiness for the morning, afternoon, and evening assessments, as well as for the daily average during the 14-day visit 3 (day 15) to visit 4 (day 29) diary period (Table 5). The percentage treatment difference in daily grittiness was 20% in favor of TOP1630 in the period to day 29.
Statistically significant differences were noted in favor of TOP1630 for ocular discomfort and for dryness for the morning assessment during the day 15 to day 29 diary period.
Statistically significant differences were noted in favor of TOP1630 for worst symptom (the symptom for each patient with the highest mean severity score during the run-in period) for the morning assessment, afternoon assessment (change from baseline only), and daily average during the day 15 to day 29 diary period (Table 6). The percentage treatment difference for daily worst symptom was 9% in favor of TOP1630 in the period to day 29. No statistically significant differences between the TOP1630 group and the placebo group were noted for the symptoms of burning or stinging.
Signs
Statistically significant differences consistently favored the TOP1630 group in total (sum of all regions) lissamine green staining (Calibra scale) for the change from baseline in pre-CAE and post-CAE scores at day 15 and day 29 (Figure 4A). The percentage treatment difference was 24% pre-CAE and 13% post-CAE in favor of TOP1630 at day 29. This outcome was also seen with corneal sum lissamine green staining, with statistically significant differences consistently favoring the TOP1630 group for each of these endpoints at day 15 and day 29 (Figure 4B). The percentage treatment difference in corneal staining was 29% pre-CAE and 21% post-CAE in favor of TOP1630 at day 29. For conjunctival sum lissamine green staining, statistically significant differences consistently favored the TOP1630 group for the change from baseline in pre-CAE total lissamine green staining at day 15 and day 29 (Figure 4C). The percentage treatment difference in conjunctival staining was 20% pre-CAE and 7% post-CAE in favor of TOP1630 at day 29.
Figure 4.

Lissamine green staining score (Ora Calibra scale).
Notes: Results showing mean change from baseline to day 15 and day 29 analysis (*analysis of covariance test P<0.1; †two-sample t-test P<0.1) in modified intention-to-treat population. (A) Total (cornea and conjunctiva) staining score; (B) corneal staining score; (C) conjunctival staining score.
Abbreviations: CAE, controlled adverse environment; SE, standard error.
Similarly, on the NEI scale, a difference favoring TOP1630 for the change from baseline in pre-CAE total lissamine green staining at day 29 was observed: baseline TOP1630 7.8 (4.46), placebo 6.7 (3.83), day 29 change from baseline TOP1630 −0.7 (3.99), placebo +1.1 (3.28); mean Δ −1.8, P=0.0613. The percentage treatment difference in NEI total staining was 25% in favor of TOP1630 at day 29.
Other signs
Corneal fluorescein staining, TFBUT, Ocular Protection Index, conjunctival redness, lid margin redness, posterior lid evaluation, and Schirmer’s test yielded some individual statistically significant differences between the treatment groups at various time points, but no discernable or consistent trends emerged for these endpoints.
Discussion
TOP1630 is a novel agent being developed for the treatment of DED with a mechanism of action that selectively targets key kinases that play a pivotal role in signaling in numerous immune cell types from both innate and adaptive pathways. The promising preclinical profiling of NSKIs14 and mechanism of action could translate into potent and broad efficacy in patients with DED, with effects at least as pronounced as established treatments but without the associated unwanted side effects.
In this first in human study TOP1630 demonstrated placebo-like safety and tolerability.
The observed TOP1630 profile appears highly promising when considered in reference to the only marketed DED immunomodulating products: lifitegrast (Xiidra®) and cyclosporine (Restasis, Cequa®), which are associated with considerable ocular AE and toleration burden.9,23,24 In contrast, the ocular comfort scores observed with TOP1630 (and placebo) in this study are similar to those observed with artificial tears.25
As is well recognized, demonstrating positive effects on signs and symptoms in DED using an intention-to-treat (ITT) analysis set and without interventions is difficult. To address this issue, a challenge environment was used in the form of CAE. Using this approach, TOP1630 demonstrated substantial treatment effects compared with placebo across multiple symptom and sign endpoints. Of note, positive effects were also observed outside the challenge environment, suggesting efficacy in an environmental setting. Moreover, there was an early onset of treatment effect, with positive effects with TOP1630 apparent by day 15, the first assessment time point.
Positive assessments consistently favored TOP1630 across a broad range of symptom endpoints, including relief of ocular dryness, pain/ocular discomfort, foreign body discomfort, and grittiness. The results for grittiness are particularly noteworthy, as a high degree of consistency was demonstrated across all assessment scales where this parameter was measured, across time and both inside and outside the CAE. Grittiness is a clinically relevant primary symptom of DED noted by up to about 50% of subjects4,26 and is a commonly assessed endpoint that is evaluated by established symptom scales used to diagnose and evaluate DED.27,28
A pronounced TOP1630 treatment effect was also seen for the relief of patients’ worst DED symptom (the symptom for each patient with the highest mean severity score during the run-in period). The worst symptom is a highly relevant, patient centric, measure of disease experience, and other studies have failed to show a treatment effect on this endpoint.29
Consistent with the promising efficacy profile on symptoms, positive effects on lissamine green staining for total, corneal, and conjunctival region sum scores were also seen. This finding, not observed in other studies, is indicative of a benefit on the total ocular surface and this endpoint may represent the most relevant staining measure in DED compared with individual segmental scores. Improvements favoring TOP1630 were achieved in all measurements and consistency of effect was similar to that seen with the symptom data.
The remaining sign endpoints of fluorescein staining, TFBUT, conjunctival redness, lid margin redness, posterior lid evaluation, and unanesthetized Schirmer’s test yielded some statistically significant differences between the treatment groups at various time points, but no discernable or consistent trends emerged. It is not, however, uncommon in DED trials to see an absence of a treatment effect on many of these signs due to the lack of correlation between these markers and the heterogeneous nature of the population.29 Subgroup analyses were not conducted to evaluate population subsets due to the limited size of study population.
Lissamine green is a gold standard assessment of ocular surface integrity alongside fluorescein.30,31 There is however little correlation between the staining results with fluorescein and lissamine green.31 Hence, the absence of effects with fluorescein staining on the cornea or conjunctiva in this study, despite positive effects being seen with lissamine green, is not unexpected and could be explained by the different staining properties on eye structures with each of these stains.30
Lissamine green staining may have greater disease relevance than fluorescein in DED for several reasons: 1) lissamine green is unique among the vital dyes, in that it is the only one not to stain healthy ocular cells;30,31 2) in patients with DED, cells that are more compromised are detected by lissamine green and not fluorescein;31 3) there is evidence to suggest that lissamine green staining of the conjunctiva correlates with the expression of inflammatory markers and immune cell infiltration in DED patients and mucus production.32 It is plausible that there is a differential protective effect of TOP1630 on cells that are more compromised and closer to apoptosis, which is detected by lissamine green and not fluorescein.
Despite the small sample size, TOP1630 showed statistically significant improvements compared with placebo on multiple symptom and sign endpoints. Although analysis of endpoints was not adjusted for multiplicity due to the exploratory nature of the endpoint assessments, high consistency of response was demonstrated for many of these improvements. These results were in the ITT study population, and no subgroup or post hoc analyses were required to achieve a positive overall picture of statistical significance on signs and/or symptoms, which is unusual in comparison with other published studies in this area where post hoc subgroup analyses have been required to establish populations that show a positive treatment effect.
Conclusion
In summary, topical NSKI TOP1630 is a novel anti-inflammatory drug with a unique mechanism of action that has demonstrated a highly promising benefit–risk profile for the treatment of DED and supports advancement to later stage development.
Data sharing statement
TopiVert Pharma Limited will consider data requests and may provide access to individual deidentified participant data from TopiVert Pharma Limited sponsored interventional clinical studies 1) for indications approved in the USA and/or EU or 2) in terminated programs (ie, development for all indications has been discontinued). TopiVert Pharma Limited will also consider requests for study-related documentation, namely the protocol and clinical study report. Data may be requested from TopiVert Pharma Limited trials 36 months after study completion. The deidentified participant data will be made available to researchers whose proposals meet generally accepted research criteria and other conditions, via a secure portal. For data access, data requestors must enter into a data access agreement with TopiVert Pharma Limited.
Acknowledgments
TopiVert Pharma Limited, London, UK, sponsored the study. Part of the material in this manuscript has been previously presented at the Association for Research in Vision and Ophthalmology Annual Meeting, May 2018.
Footnotes
Author contributions
GT was the principal investigator for the study. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
MT, CW, MCTF, AR, and SW are employees of TopiVert Pharma Limited. GO, JDS, and AD are consultants of TopiVert Pharma Limited. GO is an employee of Ora Inc. GT reports grants from TopiVert Pharma Limited, during the conduct of the study; grants from Oculeve, Allergan, ReGentree, Hanall, Mimetogen, AB2Bio, Aldeyra, Axerovision, BRIM, Diagnostear, Novaliq, Oyster point and grants, personal fees from Ora, Inc., outside the submitted work. MCTF reports personal fees from TopiVert Pharma Limited, outside the submitted work. MCTF has a patent US9499486 issued. JDS reports grants from TopiVert Pharma Limited, during the conduct of the study; grants from Allergan, Shire, Bausch & Lomb, Sun Pharma and Novaliq, outside the submitted work. The authors report no other conflicts of interest in this work.
References
- 1.Schein OD, Tielsch JM, Munõz B, Bandeen-Roche K, West S. Relation between signs and symptoms of dry eye in the elderly. A population-based perspective. Ophthalmology. 1997;104(9):1395–1401. doi: 10.1016/s0161-6420(97)30125-0. [DOI] [PubMed] [Google Scholar]
- 2.Begley CG, Chalmers RL, Abetz L, et al. The relationship between habitual patient-reported symptoms and clinical signs among patients with dry eye of varying severity. Invest Ophthalmol Vis Sci. 2003;44(11):4753–4761. doi: 10.1167/iovs.03-0270. [DOI] [PubMed] [Google Scholar]
- 3.Tomlinson A, Lemp MA, Asbell PA. Dry Eye Disease. New York: Thieme; 2007. pp. 1–15. [Google Scholar]
- 4.Farrand KF, Fridman M, Stillman IÖ, Schaumberg DA. Prevalence of diagnosed dry eye disease in the United States among adults aged 18 years and older. Am J Ophthalmol. 2017;182:90–98. doi: 10.1016/j.ajo.2017.06.033. [DOI] [PubMed] [Google Scholar]
- 5.Mcdonald M, Patel DA, Keith MS, Snedecor SJ. Economic and humanistic burden of dry eye disease in Europe, North America, and Asia: a systematic literature review. Ocul Surf. 2016;14(2):144–167. doi: 10.1016/j.jtos.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Chiaradia PA, Bardeci LA, Dankert S. Hot topics in dry eye disease. Curr Pharmaceut Des. 2017;23:608–623. doi: 10.2174/1381612822666161208094841. [DOI] [PubMed] [Google Scholar]
- 7.Schultz C. Safety and efficacy of cyclosporine in the treatment of chronic dry eye. Ophthalmol Eye Dis. 2014;6:OED.S16067–42. doi: 10.4137/OED.S16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ames P, Galor A. Cyclosporine ophthalmic emulsions for the treatment of dry eye: a review of the clinical evidence. Clin Investig. 2015;5(3):267–285. doi: 10.4155/cli.14.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Restasis® (cyclosporine) ophthalmic label [package insert] Irvine, CA: Allergan, Inc; 2012. [Google Scholar]
- 10.Colligris B, Crooke A, Huete-Toral F, Pintor J. An update on dry eye disease molecular treatment: advances in drug pipelines. Expert Opin Pharmacother. 2014;15(10):1371–1390. doi: 10.1517/14656566.2014.914492. [DOI] [PubMed] [Google Scholar]
- 11.Keating GM. Lifitegrast ophthalmic solution 5%: a review in dry eye disease. Drugs. 2017;77(2):201–208. doi: 10.1007/s40265-016-0681-1. [DOI] [PubMed] [Google Scholar]
- 12.Donnenfeld ED, Karpecki PM, Majmudar PA, et al. Safety of lifitegrast ophthalmic solution 5.0% in patients with dry eye disease: a 1-year, multicenter, randomized, placebo-controlled study. Cornea. 2016;35(6):741–748. doi: 10.1097/ICO.0000000000000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chao W, Belmonte C, Benitez del Castillo JM, et al. Report of the inaugural meeting of the TFOS i(2) = initiating innovation series: targeting the unmet need for dry eye treatment. Ocul Surf. 2016;14(2):264–316. doi: 10.1016/j.jtos.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Hagan S, Fyfe MCT, Ofori-Frimpong B, et al. Narrow spectrum kinase inhibitors demonstrate promise for the treatment of dry eye disease and other ocular inflammatory disorders. Invest Ophthalmol Vis Sci. 2018;59(3):1443–1453. doi: 10.1167/iovs.17-23479. [DOI] [PubMed] [Google Scholar]
- 15.Biancheri P, Foster MR, Fyfe MC, et al. Effect of narrow spectrum versus selective kinase inhibitors on the intestinal proinflammatory immune response in ulcerative colitis. Inflamm Bowel Dis. 2016;22(6):1306–1315. doi: 10.1097/MIB.0000000000000759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rowley A, Taylor M, Duggal A, et al. Sa1775 – a novel phase 1 trial design to evaluate safety, tolerability, pharmacokinetics and pharmacodynamics of TOP1288, a narrow spectrum kinase inhibitor, delivered topically to the colon via oral administration. Gastroenterology. 2018;154(6):S-390. [Google Scholar]
- 17.Onions ST, Ito K, Charron CE, et al. Discovery of narrow spectrum kinase inhibitors: new therapeutic agents for the treatment of COPD and steroid-resistant asthma. J Med Chem. 2016;59(5):1727–1746. doi: 10.1021/acs.jmedchem.5b01029. [DOI] [PubMed] [Google Scholar]
- 18.To WS, Aungier SR, Cartwright AJ, Ito K, Midwood KS. Potent anti-inflammatory effects of the narrow spectrum kinase inhibitor RV1088 on rheumatoid arthritis synovial membrane cells. Br J Pharmacol. 2015;172(15):3805–3816. doi: 10.1111/bph.13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker TM, Fyfe MCT, Jones G, et al. Kinase inhibitors. 9,751,837. United States patent. 2017
- 20.Ousler GW, Rimmer D, Smith LM, Abelson MB. Use of the controlled adverse environment (CAE) in clinical research: a review. Ophthalmol Ther. 2017;6(2):263–276. doi: 10.1007/s40123-017-0110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walt JG, Rowe MM, Stern KL. Evaluating the functional impact of dry eye: the Ocular Surface Disease Index [abstract] Drug Inf J. 1997;31:1436. [Google Scholar]
- 22.Abelson R, Lane KJ, Rodriguez J, et al. Validation and verification of the OPI 2.0 System. Clin Ophthalmol. 2012;6:613–622. doi: 10.2147/OPTH.S29431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiidra® (lifitegrast) prescribing information [package insert] Lexington, MA: Shire; 2016. [Google Scholar]
- 24.Cequa® (cyclosporine) ophthalmic label [package insert] Sun Pharma Global; 2018. (No location is given on the FDA label). [Google Scholar]
- 25.Torkildsen G, Brujic M, Cooper M, et al. Evaluation of a new artificial tear formulation for the management of tear film stability and visual function in patients with dry eye. Clin Opthalmol. 2017;11:1883–1889. doi: 10.2147/OPTH.S144369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olaniyan SI, Fasina O, Bekibele CO, Ogundipe AO. Dry eye disease in an adult population in South-West Nigeria. Cont Lens Anterior Eye. 2016;39(5):359–364. doi: 10.1016/j.clae.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 27.Mcmonnies CW. Key questions in a dry eye history. J Am Optom Assoc. 1986;57(7):512–517. [PubMed] [Google Scholar]
- 28.Schiffman RM, Christianson MD, Jacobsen G, Hirsch JD, Reis BL. Reliability and validity of the ocular surface disease index. Arch Ophthalmol. 2000;118(5):615–621. doi: 10.1001/archopht.118.5.615. [DOI] [PubMed] [Google Scholar]
- 29.Meerovitch K, Torkildsen G, Lonsdale J, et al. Safety and efficacy of MIM-D3 ophthalmic solutions in a randomized, placebo-controlled phase 2 clinical trial in patients with dry eye. Clin Ophthalmol. 2013;7:1275–1285. doi: 10.2147/OPTH.S44688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Efron N. Putting vital stains in context. Clin Exp Optom. 2013;96(4):400–421. doi: 10.1111/j.1444-0938.2012.00802.x. [DOI] [PubMed] [Google Scholar]
- 31.Korb DR, Herman JP, Finnemore VM, Exford JM, Blackie CA. An evaluation of the efficacy of fluorescein, rose bengal, lissamine green, and a new dye mixture for ocular surface staining. Eye Contact Lens. 2008;34(1):61–64. doi: 10.1097/ICL.0b013e31811ead93. [DOI] [PubMed] [Google Scholar]
- 32.Pflugfelder SC, de Paiva CS, Moore QL, et al. Aqueous tear deficiency increases conjunctival interferon-γ (IFN-γ) expression and goblet cell loss. Invest Opthalmol Vis Sci. 2015;56(12):7545–7550. doi: 10.1167/iovs.15-17627. [DOI] [PMC free article] [PubMed] [Google Scholar]