

Abstract
Revolutionary proteomics strategies have enabled rapid profiling of the cellular targets of electrophilic small molecules. However, precise means to directly interrogate how these individual electrophilic modifications at low occupancy functionally reshape signaling networks have until recently been largely limited. Here we highlight new methods that transcend proteomics platforms to forge a quantitative link between protein target-selective engagement and downstream signaling. We focus on recent progress in the study of non-enzyme assisted signaling mechanisms and crosstalk choreographed by native reactive electrophiles. Using this as a model, we offer a long-term vision on how these toolsets along with the fundamental biochemical knowledge of precision electrophile signaling may be harnessed to assist covalent ligand–target matching and ultimately amend a disease-specific signaling dysfunction.
Precision signaling capabilities of RES-modified protein cysteines
Many enzymes harness the specific properties of cysteine to promote stability and regulate function. For instance, cysteine is a commonly-used nucleophile in proteases[1, 2], and phosphatases[3]; and for the structural integrity of numerous proteins—such as antibodies and insulin—via specific disulphide linkages[4–6]. Many dedicated enzymes functionalize specific surface cysteines on client proteins, such as prenyltransferases that assist peripheral membrane localization[7]. Although much of the chemistry undertaken in these transformations can occur spontaneously under physiological conditions, enzymatic assistance is typically employed to ensure selectivity. However, it is becoming clear that there are numerous post-translational modifications (PTMs) that occur without enzyme assistance, yet apparently these non-enzyme-assisted (nE)-PTMs usher meaningful and precise biological responses[8–12] (Fig. 1). Among the most prevalent small-molecule modifiers orchestrating such nE-PTMs processes are native lipid-derived reactive electrophilic species (RES) that directly interact with specific nucleophilic proteins, and in some cases, specific cysteines[13, 14].
Figure 1. A representative pathway featuring multiple enzyme-/non-enzyme-assisted post-translational modifications (PTMs) that control cell-decision making.

[here, antioxidant response (AR) regulated by Nrf2 transcription factor]. Under non-stressed conditions, Keap1 (depicted as a monomer for simplicity), the negative regulator of Nrf2, recruits CuI3 and Rbx1 to ubiquitinate Nrf2 leading to Nrf2-proteasomal degradation[36]. Keap1 is subject to non-enzyme-mediated (nE)-PTM regulation wherein HNEylated Keap1 impedes Nrf2 binding, allowing Nrf2 accumulation, thereby upregulating AR. (Note: the precise chemical-state of HNE bound to Keap1 is unclear; the cyclized hemiacetal form is depicted here). Nrf2 is also subject to regulation by multiple phosphorylation (two shown for example). Identity of phospho-sites is thought to influence Nrf2 differential subcellular localization[37].
There are numerous subsets of native RES. We will primarily discuss α,β-unsaturated-carbonyl-based RES (α,β-RES). α,β-RES can be generated by either enzymatic or non-enzymatic oxidation of membrane lipids, specifically, poly-unsaturated fatty acids (PUFAs)[9]. Either mechanism can lead to localized production/elevation in α,β-RES. α,β-RES react with proteins largely irreversibly[13, 15] (or give long-lived adducts; e.g., t1/2 >4h in cells[16]). It is now widely accepted that α,β-RES are bioactive and can trigger biological responses[9, 10, 17–22]. Notably, α,β-RES with similar structures elicit different biological signaling outputs[23–25] and different enantiomers of the same α,β-RES also have differing biological effects[26–28]. Such behavior implies that there is(are) specific recognition processes occurring in cells. However, it is also unknown to what extent differences in metabolism/permeation/processing of individual RES contribute to these differing outputs. Indeed, a large number of contrasting—and often confounding—phenotypes have been ascribed to individual α,β-RES. For instance, the commonly-studied α,β-RES, 4-hydroxynonenal (HNE), can behave as either a tumor-suppressor or -promoter, depending on context, cell type, etc.[29].
Kinetic privilege in RES sensing
Given the muddied responses typically observed following bolus α,β-RES exposure of cells/organisms, decades of research have focused on α,β-RES-sensitive protein ID. This strategy has been productive, even though numerous mechanisms promote (semi-)selective labeling of sensor proteins, including RES localization, dynamic flux, and possibly differential metabolism of RES. Indeed, aside from contextual nuances, as most, if not all, cellular compartments are highly protein rich and protein diverse, HNE sensing must likely occur through elevation of the second-order rate of HNE adduction to specific proteins[17, 29, 30]. We have dubbed such proteins “kinetically-privileged sensors (KPSs)”[31]. Consistent with the “privileged” concept, KPSs typically (although not always) contain a single reactive cysteine (and often many by-stander cysteines)[17, 19, 20, 30, 32, 33]. It is currently difficult to define a kinetic threshold for KPSs with α,β-RES due to the limited kinetic characterization of RES responders in literature (Outstanding question 1)[34, 35], but by comparing alkylation efficiencies of KPSs with previously-characterized nucleophilic proteins, recent data highlight the capability of KPSs to undergo significant rate-enhancement over small-molecule thiols, (that react with α,β-RES at ca. 1 M−1s−1)[30].
Stoichiometric RES-modifications engender dominant signaling outputs
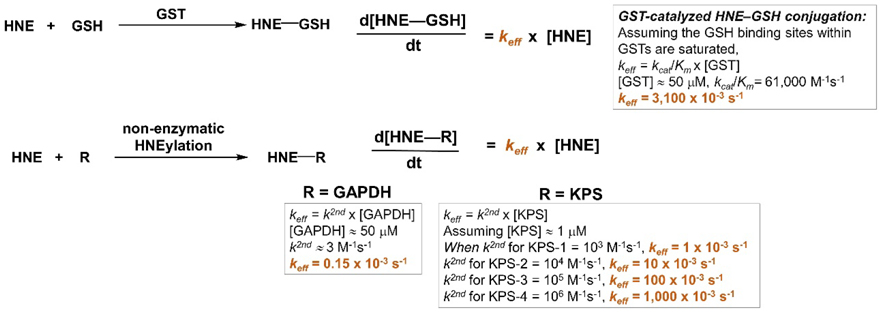
A common conception in the field points to cysteine pKa as a key factor in determining privileged sensing behavior[29]. However, cysteine thiolate is only 20-fold more reactive than the equilibrium mixture of cysteine and cysteine thiolate at physiological pH[38]. Although a marked improvement in reactivity, there is a high concentration of glutathione and protein-cysteines (1–10 mM each) in cells[39], and the rate of HNE-metabolizing enzymes (103–106 M−1s−1[40]) including various GST isoforms such as GST-A4–4 (principle enzyme to catalyze GSH–HNE conjugation in liver), alcohol dehydrogenase (NADH-dependent reduction), aldehyde dehydrogenase (NAD+-dependent oxidation) and aldo-keto reductase[40, 41] is substantial. Thus a 20-fold rate-enhancement alone is not likely able to endow sufficient reactivity to give meaningful signaling output (Box 1). Hence it remains an active area of investigation as to how kinetic privilege comes about.
BOX 1.
G-REX offers electrophile-limited conditions for KPS ID
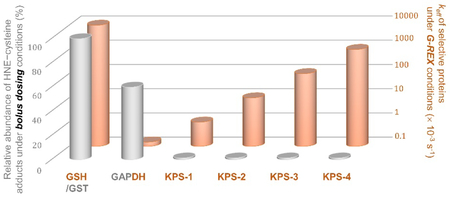
Under bolus dosing conditions, it is assumed that all KPSs and privileged responders to HNE, i.e., GAPDH and GST, will reach full HNE–cysteine occupancy. Therefore, this value [shown in grey with arbitrary unit (%)] is a function of the total amount of labeled POI cysteines. GST (at ~50 μM, possessing four cysteine residues on average) is set as the maximum responding protein here (with total 200 μM cysteine being labeled and set as 100%). In contrast, most KPSs are of low rank here [assuming a typical protein concentration (1 μM) with one reactive cysteine, which leads to total 1 μM KPS cysteine being labeled].
For G-REX proteomics profiling, the following equations are used to predict the effective rate constant (keff) of either GST-catalyzed GSH conjugation with HNE or non-enzymatic HNEylation of a given POI (orange bars).
Note: (1) We are assuming protein/GSH concentration remains at the steady state during the time course of G-REX study. (2) R is the POI containing (a) RES-sensing cysteine(s)
Data from proteome-target profiling as a function of specific α,β-RES reveal a positive correlation between labeling occupancy and respective RES dosage[22], suggesting specific RES binding sites may exist to optimize cellular responsiveness to stress in various contexts. On the other hand, non-covalent pre-association between RES and the protein target would render an effective increase in concentration of up to 108 M [42], thereby accelerating subsequent (intramolecular) covalent adduction. We have therefore hypothesized that in at least some instances, rapid rates of association prior to covalent modification could be achieved by some KPSs having an affinity to some(a) specific α,β-RES within the protein scaffold proximal to the nucleophilic cysteine. Interestingly, aside from improving labeling kinetics, such interactions could drive allosteric regulation, allowing (some) KPSs to undergo conformational changes upon α,β-RES labeling. For example, covalent modification on C117 of HSPB7 by HNE involves a fast association kinetics–the most potent sensing action thus far to our knowledge–and promotes β-sheet formation, indicating that HNE promotes folding, similar to a non-covalent ligand[30]. This sort of response could arise by the HNEylated-cysteine filling a vacant “hole (i.e., HNE-binding site)” in the non-alkylated, folded protein structure[13]. In this way, kinetics of labeling are enhanced, and signaling outputs are also modulated as a consequence of binding due to stabilizing a folded structure. For oligomeric proteins, such conformational changes could regulate reactivity of several protein partners, giving dominant phenotypic outputs. Alternatively, we have also predicted that α,β-RES-modification of protein-cysteines could chemically complement loss of function due to mutation of bulky amino acid residues, such as tryptophan[13]. Thus, α,β-RES modification could lead to direct gain of function. By contrast, one could propose that HNEylation disfavors (all) ground-state structure(s) due to steric clashes. However, this implies that HNEylated-cysteine is not congruent with pre-existing structure(s). Such a scenario, as one may predict, would lead to reduced kinetics of labeling, since an unexpected conformation of HNE-bound-protein that is unstable must be obtained in the transition state.
From this line of reasoning, one could predict that low RES-occupancy of some KPSs will trigger allosteric modulation to display high levels of signaling outputs [13, 31]. For instance, we have observed that ~12% of HNE occupancy of Akt3 kinase affords 20–30% loss of kinase activity in vivo[19]. We termed these proteins “privileged first responders” (PFRs) [17, 19], a subset of KPSs. The emerging data document that PFRs react rapidly with native RES HNE and elicit dominant responses. Other examples include Keap1[18, 20, 32, 43–45], Ube2V2[17] and PTEN[33, 43]. These data indicate to us that α,β-RES-sensing must be viewed holistically, in terms of both modification and signaling outputs. Indeed, it is absolutely critical to evaluate the signaling outputs modified upon α,β-RES modification of specific PFR/KPSs, and to understand, for each responder, to what extent modification at a specific occupancy affords meaningful biological outputs under physiological conditions[13]. Clearly, forging a quantitative link between specific target engagement and a specific phenotypic behavior/cellular output is interesting in its own right. But such quantitative relationships are also critical for drug design and optimizations [31].
Spoilt for choice or little by little? Interrogating signaling architectures shaped by precision RES modifications
There are two main approaches to study signaling pathways orchestrated by specific RES. These are bulk treatment with the RES in question (Fig. 2a), and T-REX, a relatively new development involving a non-invasive transgene [i.e., fusion protein of interest (POI)] harboring a photocaged RES that upon light-illumination liberates a specific RES of interest in the vicinity of the sensor-POI in the amount stoichiometric to the transgene-POI (Fig. 2b).
Figure 2.

(a) Studying RES signaling following bulk exposure of RES from outside of cells. Bolus RES dosing leads to modification/modulation of multiple signaling targets/pathways simultaneously. Correlating the resulting phenotypic output to on-target modification is often challenging following bulk RES exposure. (b) Interrogating precision RES signaling by T-REX. A functional Halo-POI fusion construct is expressed in live cells, worms, or fish. Treatment with a bio-inert cell/worm/fish-permeable photocaged-RES (i.e., T-REX probe that can deliver a specific RES) results in stoichiometric covalent binding of the photocaged-RES to Halo. After washing away the excess T-REX probe, photouncaging liberates a stoichiometric amount of RES within the microenvironment of Halo-POI protein. Provided the POI is a KPS to the RES in question, some level of substoichiometric RES-modification of the POI results. If the POI is a PFR, the resultant modified POI is sufficient to elicit (a) defined dominant response(s). The measured phenotypic responses can be directly related to the functional modification/target occupancy of POI by the RES. Validations using split construct and/or hypomorphic mutant(s) (see text and references cited therein) as negative controls, in addition to built-in T-REX controls (namely, light alone, T-REX-probe alone, vehicle alone) must be done in parallel to rule out any off-target responses.
A package deal: bolus dosing can ID many RES sensors.
Bolus dosing of RES is a conceptually-simple experiment: cells/organs/organisms are externally exposed to excess of a RES of choice for a given time, then a change in (a) specific pathway(s) is(are) readout (Fig. 2a). Since RES can affect membrane integrity, upregulate reactive oxygen species, are actively metabolized, and modify many proteins/macromolecules covalently in a time-dependent manner, numerous variables need to be controlled/considered during such regimens. Furthermore, as no single protein can be targeted selectively under bolus conditions, phenotypes are often not rescued by expression of hypomorphic sensing mutants. Nevertheless, a huge number of pathways have been implicated as α,β-RES sensitive following bolus RES treatment. This shows the versatility of bolus dosing and yet also highlights its complexities. In many ways, having such a simple protocol to study downstream signaling outputs is ideal. However, since a plethora of targets and pathways is triggered under bolus dosing and many outputs show hormesis, doubts have rightly been raised about the validity of bolus dosing. The relevance of the protein hits identified may be of limited scope because α,β-RES-sensing outputs identified by bolus dosing in vitro often shows that putative sensors have very low second-order reaction kinetics compared to typical KPSs identified under RES-limited conditions[30]. Under bulk treatment, fractional RES-occupancy on a target, modification of which alone is sufficient to trigger signaling, is challenging to determine, rendering direct correlation of RES-modification to phenotypic output difficult. Nevertheless, bolus dosing remains the go-to experiment most-commonly performed in the field, and provided conditions are carefully controlled and health of cells is maintained, useful information can be derived.
Step by step: T-REX documents that RES signals are sufficient to elicit downstream signaling.
T-REX[19, 33, 45] on the other hand delivers a specific RES to the microenvironment of a specific protein of interest (POI). Because the method currently requires genetic fusion of the POI with a protein-tag such as HaloTag[46] and custom design of small-molecule photocaged RES[32], this approach is less simple than direct treatment with any available RES. Furthermore, there is concern that forced complexation between liberated RES and POI due to proximity[47] and high effective RES-concentrations within the vicinity of POI may drive a reaction that would otherwise not occur. Out of many POI’s tested thus far (several of which were previously identified under bolus α,β-RES-exposure), only a few POIs are modified under T-REX-assisted α,β-RES-delivery conditions[17, 19, 33]. Importantly, each modified POI—at their intrinsic occupancy—can be directly evaluated for signaling capability (Fig. 2b). We stress that T-REX requires ectopic protein expression that can raise concerns for observing phenotypes of non-physiological relevance. Fortunately, the expression levels in various models (cells, fish, and worms) where successful T-REX has been executed, vary from close-to-endogenous to highly elevated, assuaging most of these concerns [17, 19, 44, 45]. Furthermore, in cultured cells and fish, we have used hypomorphic mutants (lacking HNE-sensor-cysteine but otherwise functional) [17, 19, 30]; and in cells, we have tested ‘Halo and POI split’ constructs, where HNE neither labels the POI nor elicits pathway response upon replicating T-REX using these constructs [20, 32, 33, 45]. We thus conclude that signaling arises from on-target modification. Finally, most of the phenotypes thus far measured for T-REX are dominant loss-of-function responses, which constitute a phenotype that is typically suppressed by protein-target overexpression.
Indeed, POIs that pass this T-REX signaling sufficiency test are PFRs. Typical delivery efficiencies of RES to sensor POIs (i.e., amount of RES that modifies the POI over that liberated) are ~5–30%. Only a small fraction among several POIs tested undergo RES-labeling during T-REX. Notably, unlike proteins identified by bolus dosing, proteins that are RES sensitive in T-REX-based screens (e.g., HSPB7[30], and Keap1[33]) tend to be hyper-reactive in in vitro assays. Obviously, given the relatively low-targeting efficiency of T-REX, significant amounts (sub-micromolar) of HNE are released to the cell. However, several controls show that adventitious HNE does not perturb cells (or off-target signaling pathways)[17, 19, 33]. Reiterating some of the points from above, dominant phenotypic outputs have been observed in a target- and specific cysteine-dependent manner[17, 19, 33]. T-REX-assisted Keap1-specific HNEylation stabilizes Nrf2 and stimulates antioxidant response (AR) to the same extent as bulk treatment with low-micromolar HNE[20, 32]. The ‘split construct’ does not upregulate AR upon T-REX[32]. PTEN is also deactivated by HNE under T-REX conditions using two orthogonal readouts−immunofluorescence (IF) of endogenous PIP3 levels in fixed cells and FRET-based ‘InPAkt’ reporter assay in living cells[33]. Using a similar set of experiments, downregulation of Akt3[19] has been shown. Although T-REX is currently unable to interrogate firing of multiple pathways simultaneously, the method can shed light on the complex outputs recorded under bolus dosing. For instance, Akt3 (antagonist of the FOXO tumor suppressor-signaling pathway) is inhibited by α,β-RES[19]; PTEN a positive regulator of the FOXO-pathway is also inhibited by α,β-RES[33]. It is likely the interplay of relative rates of modification and expression levels/activities/localizations of individual PFRs dictate dose- and time-dependent responses to α,β-RES under bolus dosing.
T-REX thus allows individual pathway analysis, similar to the use of specific inhibitors/stimulators to study kinase and ubiquitin pathways. However, given the paucity of bona fide α,β-RES sensors known, it is likely that we only have an incomplete picture of PFRs. To build up an understanding of precision electrophile signaling, one would ideally know which proteins sense RES/specific RES-chemotype, and under what context they perform their sensing functions. Thus we need be able to rapidly ID more potential PFRs and simultaneously profile their signaling activities.
Latest methods to identify PFRs
We propose the following criteria to evaluate methods to screen protein sensors. In our opinion, the ideal method can:
a. Detect low-occupancy modifications.
The trait of fractional occupancy is essential for identifying KPSs (thereby, PFRs), as these proteins are uniquely sensitive to stressed environments and may function through context-dependent (locale/redox environment, etc.) sensing and gain-of-function or dominant-loss-of-function signaling.
b. Readout modification directly.
The use of proxies to detect functionalization has grown in recent years, and such powerful methods can improve accuracy over a small number of cysteines. Limitations however exist as they cannot prove a direct interaction. Increased coverage over a larger portion of the cysteome (presently ~5%[31]) will also be desirable.
c. Control dose, timing, locale and duration of α,β-RES availability.
Since α,β-RES-modification is a time-dependent covalent labeling process, it is critical that the concentration of α,β-RES released as well as the time and location be known accurately. Spatiotemproally-controllable release sidesteps metabolism, permeation, distribution, etc., of these reactive species. In many instances, lifetime of α,β-RES may be modeled mathematically (Fig. 3-inset).
Figure 3. G-REX: Genome-wide ID of KPSs under electrophile-limited conditions.

General setup of G-REX: Cells ectopically expressing HaloTag are treated with the photocaged-RES (i.e., T-REX probe) [in this case, Ht-PreHNE (i.e., HaloTag-targetable precursor to HNE)]. HaloTag specifically binds the hexyl chloride linker of the probe with rapid second-order kinetics[46]. Unbound probe is washed out. Upon photouncaging, HNE(alkyne), in sub-stoichiometric amount to Halo concentration, is liberated rapidly inside cells (t1/2 < 1–2 min[32]) within the microenvironment of Halo, enabling low-occupancy covalent labeling of native KPSs by HNE. Cell lysis and Click coupling with biotin-azide allows enrichment of HNEylated KPS(s) by streptavidin pull-down and target-ID enabled by digest LC-MS/MS. Top enriched targets can be validated using T-REX and they must pass the same level of stringent negative-control tests (see Fig. 2b legend). Inset: The lifespan of HNE liberated in G-REX conditions is modeled (Kinetiscope, version 1.1.743.x64). krelease is the first-order rate constant of photouncaging. Assuming overall HNE deactivation/degradation follows a first-order kinetics, kdeact is the corresponding rate constant.
d. Assay α,β-RES-modification-driven signaling under conditions where most cellular processes are not grossly perturbed.
Redox/electrophile-sensitive proteins are likely to be most affected by heightened stress. Thus α,β-RES-sensing/signaling assays must be performed under conditions that mimic the “normal” cell, not cells that are already hyper-stressed.
e. Readout modification across the whole proteome.
Protein-cysteine modification/signaling is essentially a moonlighting function; in principle, any protein could be responsive to α,β-RES providing a substantial amount of time or reactive molecule. Thus, it is critical that methods have no hidden biases, such as ensuring protein target spectra of a specific RES remain the same regardless of proxy electrophile (e.g., iodoacetamide vs. other variants[48, 49]) used in indirect profiling, and good MS-sample-processing conditions including presence (and judicious choice) of a reducing agent to preserve aggregation-prone sensor proteins and labile RES-modifications.
f. Be applied to numerous types of specific α,β-RES and ideally other electrophiles/oxidants.
Many reactive small-signaling molecules are known and some of these may have differential signaling properties. For instance H2O2 (ROS) labels Akt2 [50] and HNE (RES) labels Akt3[19, 31]. The ability to parse signaling codes for specific reactive chemical signals is thus crucial to understanding PFRs.
g. Be applied to numerous different systems beyond cell culture/lysates.
Context is critical for RES/ROS sensing/signaling.
Altogether, the relevance of each criterion depends on the purpose of the experiment and the resolution required. However, an ideal method would score high in all these categories.
Screening for PFRs with T-REX
Commercial availability of plasmids encoding the human and mouse ORFeome fused to HaloTag, “Halo-ORFeome library” has enabled a medium-throughput screen for PFRs responding to α,β-RES using T-REX[19, 33]. In some respects, T-REX is ideal for this task, because T-REX can detect direct α,β-RES–POI labeling down to ~5% delivery efficiency. As alluded to above, specific novel sensor proteins (e.g., Akt3[19], RNR-α[33], and HSPB7[30, 33]) have been identified using this approach, and in some cases downstream signaling has been analyzed [17–20, 32, 33]. However, the method itself is not high-throughput. Typical screens have been conducted using panels of 10–20 proteins that are biased to reflect known or proposed α,β-RES (or ROS) sensors based on literature precedent. T-REX is compatible with a number of different RES[32, 33, 44], although the scope is currently limited to α,β-RES. Although T-REX works in cells[33], fish[17, 19], and worms[44, 45], ectopic expression of the POI is required and this may perturb “normal” physiological processes. Furthermore, a brief low-power UV-illumination (typically, 1–5 min, 365 nm, <5.0 mW/cm2) is used for photouncaging, and there may be some limitations that have not yet been encountered (Fig. 2b).
ω-Alkynyl linoleic acid incorporation
Cellular α,β-RES can be produced through the peroxidation of linoleic acids[9]. Much of this oxidation occurs in the mitochondria and plasma membranes that are rich in PUFAs. Building on this premise, a thoughtful means to generate “endogenous” clickable α,β-RES has recently surfaced[51]. Cells fed with terminal alkyne-labeled linoleic acid are activated with lipopolysaccharide-mimetics to produce (various forms of) terminal alkyne-labeled α,β-RES. Proteins labeled under these conditions are functionalized using biotin-Click coupling, and identified by streptavidin-agarose beads-based enrichment and LC-MS/MS (Fig. 4a). As it is an enrichment method, this approach can directly detect low-level occupancy modifications. Furthermore, proteins identified were mostly membrane-localized, indicating that this method mimics endogenous RES-sensing and that it is locale specific. However, the duration of stimulation was significant (24 h), relative to the t1/2 of HNE-labeled proteins (~4h)[16]. As cells were stimulated, “normal cellular processes were perturbed”; but, as the method intends to study activated RES-signaling, the experimental setting adequately meets the intended purpose, regardless of any limitations for overall generalizable utility. Finally, a defined α,β-RES is not generated under the reaction conditions, and further work is needed to understand what specific α,β-RES-chemotype(s) elicit(s) the observed labeling.
Figure 4.

(a) ω-Alkynyl linoleic acid incorporation for SILAC proteomics-based ID of RES-sensors [51]. Cells fed with terminal alkyne-labeled linoleic acid are activated with Kdo2-lipidA to produce terminal alkyne-labeled RES through lipid peroxidation. Proteins labeled under these conditions are identified through SILAC proteomics analysis. Plasma and mitochondrial membrane proteins are the most prevalent HNEylated targets identified under these conditions. (b) Competitive isoTOP-ABPP of RES-modified proteins[22]. Parallel sets of intact cells/lysates are treated with RES or DMSO. After cell lysis (where applicable), both sets of lysates are incubated with a reporter electrophile [e.g., iodoacetamide (IA)] which non-specifically labels (a subset of) cysteines. Using biotin-azide tags (housing differential isotope-labeled linker bearing TEV-protease cleavage site in between azide and biotin), Click coupling biotinylates the IA-labeled cysteines. Steptavidin pull-down followed by trypsin digest and TEV cleavage allow for LC-MS/MS analysis that indirectly identifies RES-modified cysteines based on loss of IA-labeled cysteine signals. Care must be exercised to exclude potential false negatives/positives. For instance, proteins (in dark green) subject to allosteric regulation, disulfide modification, and/or down-regulation in response to RES-bulk exposure, may be falsely identified as direct HNE-interactors. Proteins, [e.g., Keap1 (in light green)] with reactive cysteines with preference to undergo conjugate addition reaction to sp2-derived RES such as HNE, may not react extensively with sp3–centered electrophilic reporter probes (such as IA). Some proteins especially those unstable to oxidation-induced aggregation/precipitation may not be stable to sample processing conditions prior to MS analysis. (c) Venn diagram showing overlap of HNE-targeted proteins detected by G-REX (top 300)[17] and ABPP (all probe-labeled proteins)[22]. Note: the data originate from HEK293T and MDA-MB 231 cells in G-REX and ABPP, respectively.
Activity-based protein profiling
Activity-based protein profiling (ABPP) is an extremely versatile method applicable to almost any RES in any cell/organism (Fig. 4b). Unsurprisingly, of all current methods, ABPP has provided so far the most real-life relevant information. This is because no modification/derivatization of RES of interest is required, as the detection is based on secondary reactivity to a proxy electrophile such as iodoacetamide. Thus, target—RES engagement can be directly extrapolated from competitive profiling analysis[22, 48]. Although the precise correlation between occupancy and rate is not known, by varying RES dosage, the most reactive proteins can be inferred. It is worth noting that loss of target labeling by a proxy probe is what measured in the MS analysis as opposed to direct RES modifications, hence often limiting reliable assignment of low-occupancy RES-modifications. As loss of labeling by a secondary electrophile is measured, the MS data alone is not a definitive proof of RES-modification. Nonetheless, ABPP has been widely used to analyze drug targets/ligands[52, 53], lipid-derived electrophilic stress[22] and electrophilic metabolite toxicity[54], etc., in an impressive array of model systems. The key concept of ABPP is that cysteine reactive probes can evaluate availability of specific cysteines. This insight endows ABPP with the ability to rank protein-cysteine reactivity for around 1000–3000 specific cysteines within the cysteome. However, this level of accuracy comes at a price: given the large 200,000 cysteines in the cysteome a fraction of potential nucleophiles is being covered[31]. It is noteworthy that in ABPP, the electrophile is typically administered through bolus dosing. Such methods likely model drug dosing quite well, but as discussed above are not ideal for studying α,β-RES signaling. Encouraging strides have been made to address some of these limitations[55, 56], however, they have not been applied nearly as broadly as canonical ABPP.
G-REX
We recently disclosed a new high-throughput method to ID KPS(s) in cells. In this method, G-REX, HaloTag (unfused to a POI) is expressed in cells (Fig. 3). Under powerful promoters (e.g., CMV) the concentration of Halo protein was calculated to be ~9 µM in HEK293T cells, rendering the concentration of HNE released in cells from photocaged-HNE/HaloTag complex to be ~5 µM (assuming ~60% photouncaging efficiency)[17]. Treatment of Halo-expressing cells with HaloTag-targetable photocaged-HNE (Ht-PreHNE), followed by washout, then light illumination releases HNE within 1–2 min[32, 33]. After 5 min, cells were harvested and lysed. Interestingly, Click-coupling with fluorophore-conjugated azide showed no appreciable labeling of the cellular proteome when cells were exposed for 5 min to media containing 5 µM HNE(alkyne-functionalized). By contrast, a significant amount of proteins were labeled under G-REX. For direct target ID, HNE-labeled proteins following cell lysis can be tagged with biotin, then enriched by streptavidin-agarose beads and identified by LC-MS/MS. Ube2V2, and a similar protein Ube2V1, were identified as novel PFRs to HNE. Subsequent downstream assays using T-REX (employing functional Halo-Ube2V1/2 constructs) showed that substoichiometric HNEylation of Ube2V2 or Ube2V1 triggered specific downstream signaling activities. Critically, Ube2V2 and Ube2V1 are not enzymatically active. Instead, they allosterically regulate the activity of an E2-conjugating enzyme, Ubc13. These findings open new avenues to target PFRs that are not enzymes but allosteric proteins bearing functional PFR cysteines, modification of which regulate their (in this case, enzymatic) binding partner.
G-REX and ABPP data sets show little overlap
Given the above discussion, we compared G-REX data set profiling KPSs for HNE, with an ABPP data set profiling cysteines hyperactive to HNE. Published data enabled comparison of data stemming from different cell lines only (HEK293T and MDA-MB 231); however, as both data sets were taken from administration of cells to HNE (or photocaged-HNE followed by photouncaging), we assumed these may be the closest comparisons based on what is available. Taking the top 300 hits from G-REX (proteins with molecular weight between 15 and 25 kDa) and the whole ABPP data set (around 600 proteins) we found only 56 proteins overlap. Indeed, only 2 of the top 15 proteins from G-REX were in the ABPP data set, and neither of these two proteins was scored as HNE sensitive by ABPP. There are of course multiple possibilities for these divergent outcomes. Some explanations include: (1) Redox response of different cells/different passages may be different (there has been little systematic investigation of these phenomena in the literature); (2) Bolus HNE (treatment for ABPP) and intracellularly-generated HNE (under RES-limited G-REX conditions) affect the cell differently; (3) The proxy probe used in ABPP does not always label HNE-targeted cysteines and there is not always allosteric regulation among different cysteines. More systematic analysis of the different profiling methods and their limitations/assumptions will help us understand these nuances.
Relevance of α,β-RES sensors to drug mechanism and design
Covalent targeting of nucleophilic residue(s) proximal to a small-molecule-ligand/drug-binding site is a simple but effective strategy to afford selective and persistent target engagement. It has been successfully demonstrated in combating the occurrence of drug-resistant cancer mutations[57, 58]. This trait has spurred a growing interest in identifying targets of covalent drugs[31, 59–61]. In this regard ABPP has proven to be highly effective. For instance, important insights into the mechanism/targets of the blockbuster multiple sclerosis drug, Tecfidera, were derived using ABPP[53]. In this way, the authors implicated two cysteines within a single tryptic digest site in protein kinase C theta (PKCθ) as (a) target(s) of Tecfidera from a pool of 2500 cysteines. In spite of this significant success, ABPP was only able to paint an incomplete picture of Tecfidera’s targets: cells expressing a PKC mutant that is inert to Tecfidera were still sensitive to Tecfidera. Furthermore, Keap1, a protein labeled by Tecfidera[62, 63], was not detectable by ABPP. Thus, it was correctly concluded that there is(are) other Tecfidera target(s)—not covered by ABPP—yet to be discovered[53].
Although strides have been made in terms of target ID, design of covalent drugs is still not well developed and HT methods for covalent drug design are sorely lacking. The Cravatt laboratory has modified the ABPP protocol to develop a tool to identify “ligandable” cysteines[49]. These cysteines appear to be juxtaposed to a binding site, allowing for rapid labeling to occur through a templated reaction (possibly similar to how PFRs sense RES). In some instances, these reactive fragments were able to inhibit specific enzymes identified in the profiling method[22, 48]. Interestingly, many covalent drugs are targeted to the same electrophile-sensitive cysteine. For instance, Afatinib, Neratinib and Osimertinib are all approved drugs that can covalently bind to the C797 within eGFR[31, 59, 60]. In many other kinase—ligand/drug pairs, such as BTK (targeted by Ibrutinib and Acalabrutinib), ERBB2/HER2 (targeted by CP-724714 in Phase I trial for breast cancers), JAK3 (targeted by PF-06651600 in Phase II trial for Crohn’s disease and rheumatoid arthritis) and PI3Kα (targeted by PX-866 in Phase 2 trial for prostate cancer), cysteine residues engage with the respective drugs through covalent chemistry[58, 64]. Many of these drugs are derived from appending a moderately electrophilic motif to a previously known non-covalent inhibitor of a specific enzyme. Specifically, eGFR C797 and BTK C481 have been targeted by several successive generations of inhibitors through this approach. This trend clearly underscores the utility of “ligandable cysteines”. However, this progression also highlights limitations in current drug design: that is, it is hugely “enzyme centric” and restricted to a small number of known “druggable” proteins. We have noted the chemical similarities between endogenous α,β-RES and common drug covalent pharmocophores. We have thus proposed that endogenous α,β-RES sensing proteins are ideal drug targets[31]. Critically, our data from G-REX indicates α,β-RES sensors, such as Ube2v2 and Ube2V1, are not necessarily enzymes[17]. Thus, HT methods to ID α,β-RES-sensitive proteins are potential routes to develop covalent drugs and discover new covalent drug targets, possibly with novel mechanisms. It will be interesting to see how PFRs identified from T-REX, G-REX and labeled linoleic acid fare as covalent drug targets. In this regard, following results of HT-screens, the use of T-REX mechanistic interrogation on single-proteins/pathways could allow triaging of proteins for drug design.
Synopsis and Outlook
First considered to be a toxic metabolic byproduct, then slowly implicated as a means of cellular communication, α,β-RES have grown in importance as a signaling currency in spite of their simple structures[9]. Based on data from a host of labs, it is now highly likely that these molecules are involved in prompting cellular decision-making. Time can only tell if these molecules can help us develop better drugs, but it is certainly true that understanding more about α,β-RES will help understand several aspects of health and disease. We hope the new HT methods to ID α,β-RES sensors will stimulate drug discovery.
Outstanding questions.
How fast is kinetically fast? Although likely context dependent, it is important to set limits on how fast a kinetically privileged sensor has to be, to achieve high occupancy against the rest of the proteome.
Is there correlation between kinetics and privilege? We must understand how kinetics of labeling interplay with privilege of occupancy; for instance, are fast kinetics a consequence of privileged first responding? Can low kinetics be adequately compensated by strongly dominant outputs?
What are the evolutionary/structural requirements for privileged sensing? The study of enzymology has helped us enormously to predict protein structure and function. Such an understanding of privileged sensing is currently not available. Studying evolutionary conservation of cysteine sensors and the structural requirements that allow sensing to occur is a must for us to be able to analyze HT-data sets quickly and to predict which proteins could be sensors computationally.
What is the overlap between different profiling methods? There are significant technical differences between the profiling methods we discuss, but, one would nevertheless expect there to be significant overlap between hits found by these protocols. This is not the case. Rigorous comparison of the methods would help to give researchers outside the field confidence in the data produced. Hits should be backed up with careful single-protein functional analyzes in vivo (T-REX and accompanying controls) and in vitro.
How can these sensors be harvested for drug design? The behavior of privileged sensors seems like an ideal match for drug design. Privileged sensors are modified rapidly, exert dominant outputs, and they interact selectively and irreversibly with their targets. Unlike many drug–target interactions, modulation of a privileged sensor can occur on proteins with no enzymatic function, and it can also relay stimulatory or inhibitory signals. The pharmaceutical communities have been developing multiple generations of covalent kinase inhibitors around limited druggable/ligandable cysteines. Thus, each novel, and potentially druggable, privileged cysteine is of huge value to the community. It is clear that the various proteomics profiling platforms that enable distinct sets of cysteines to be investigated in specific cellular/disease contexts will offer improved insights into rationale design of personalized covalent inhibitors.
Highlights.
T-REX enables a quantitative link between RES-occupancy and the magnitude of on-target signaling output(s).
T-REX is ideal for studying substiochiometric RES-signaling events. The KPSs identified by T-REX display significantly enhanced kinetics of RES-adduction than previously-identified sensors.
PFRs—a subset of KPS—are “sensitized” for RES-signaling to elicit dominant signaling outputs as a consequence of low-occupancy RES-modifications.
Innovative proteomics profiling methods have expanded our knowledge of RES-signaling targets/events in varied biological contexts.
ABPP is a versatile method that can be used in RES-signaling and druggable ligand/target ID.
A new HT-method (G-REX) enables profiling RES-sensor interactomes at defined endogenous level/space/time of RES-signaling. G-REX enables a direct read-out of low-occupancy sensors modified by a specific RES-chemotype.
BOX 2.
Evaluation of proteomics profiling platforms based on the discussed selection criteria for screening and identifying PFRs
| Method╲Criteria | a | b | c | d | e | f | g |
|---|---|---|---|---|---|---|---|
| T-REX | high | high | high | high | low | low | medium |
| ω-Alkynvl linoleic acid incorporation |
high | high | low | low | medium | low | currently low |
| ABPP | lowi | low | medium | low | low | high | high |
| G-REX | high | high | medium | low | high | low | unknown |
Acknowledgements and Disclosure
NSF CAREER (CHE-1351400), Beckman Young Investigator, NIH-New-Innovator (1DP2GM114850), Office of Naval Research (ONR) Young Investigator (N00014–17-1–2529), the Sloan fellowship (FG-2016–6379), and Pershing Square Sohn Cancer Research Alliance programs (to Y.A.) are acknowledged for personnel, shared supplies, and equipment supporting electrophile signaling research program in the Aye lab. zebrafish husbandry and microinjection/imaging facility (NIHR01 NS026539, PI: J. Fetcho), Cornell NMR facility (NSF MRI: CHE-1531632, PI: Y.A.), Cornell Imaging Center (NIH 1S10RR025502, PI: R.M. Williams), Cornell proteomics facility (NIH SIG grant 1S10RR025449–01) for resources and instrumentation. A full patent application relating to the use of REX technologies and a provisional patent involving RES-derived covalent inhibitors have been filed by Cornell University.
References
- 1.Otto H-H and Schirmeister T (1997) Cysteine proteases and their inhibitors. Chem Rev 97 (1), 133–172. [DOI] [PubMed] [Google Scholar]
- 2.Sanman LE and Bogyo M (2014) Activity-based profiling of proteases. Annu Rev Biochem 83, 249–73. [DOI] [PubMed] [Google Scholar]
- 3.Tonks NK (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol 7 (11), 833–46. [DOI] [PubMed] [Google Scholar]
- 4.Sevier CS and Kaiser CA (2002) Formation and transfer of disulphide bonds in living cells. Nat Rev Mol Cell Biol 3 (11), 836–47. [DOI] [PubMed] [Google Scholar]
- 5.Bechtel TJ and Weerapana E (2017) From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteomics (doi: 10.1002/pmic.201600391) 17 (6), 1600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herrmann JM et al. (2015) Highlight: dynamics of thiol-based redox switches. Biol Chem 396 (5), 385–7. [DOI] [PubMed] [Google Scholar]
- 7.Hang HC and Linder ME (2011) Exploring protein lipidation with chemical biology. Chem Rev 111 (10), 6341–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holmstrom KM and Finkel T (2014) Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 15 (6), 411–21. [DOI] [PubMed] [Google Scholar]
- 9.Schopfer FJ et al. (2011) Formation and signaling actions of electrophilic lipids. Chem Rev 111 (10), 5997–6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobs AT and Marnett LJ (2010) Systems Analysis of Protein Modification and Cellular Responses Induced by Electrophile Stress. Acc Chem Res 43 (5), 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winterbourn CC (2008) Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol 4 (5), 278–86. [DOI] [PubMed] [Google Scholar]
- 12.Brewer TF et al. (2015) Chemical approaches to discovery and study of sources and targets of hydrogen peroxide redox signaling through NADPH oxidase proteins. Annu Rev Biochem 84, 765–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poganik JR et al. (2018) Getting the message? Native reactive electrophiles pass two out of three thresholds to be bona fide signaling mediators. Bioessays, e1700240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Long MJ and Aye Y (2016) The Die Is Cast: Precision electrophilic modifications contribute to cellular decision making. Chem Res Toxicol 29 (10), 1575–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long MJ et al. (2017) Subcellular redox targeting: Bridging in vitro and in vivo chemical biology. ACS Chem Biol 12 (3), 586–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J et al. (2015) Quantitative chemoproteomics for site-specific analysis of protein alkylation by 4-hydroxy-2-nonenal in cells. Anal Chem 87 (5), 2535–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Y et al. (2018) Ube2V2 Is a Rosetta Stone bridging redox and ubiquitin codes, coordinating DNA damage responses. ACS Cent Sci 4 (2), 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Long MJ et al. (2017) beta-TrCP1 Is a Vacillatory Regulator of Wnt Signaling. Cell Chem Biol 24 (8), 944–957 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long MJC et al. (2017) Akt3 is a privileged first responder in isozyme-specific electrophile response. Nat Chem Biol 13 (3), 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parvez S et al. (2015) Substoichiometric hydroxynonenylation of a single protein recapitulates whole-cell-stimulated antioxidant response. J Am Chem Soc 137 (1), 10–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cubillos-Ruiz JR et al. (2015) ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 161 (7), 1527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang C et al. (2014) A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Methods 11, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chipuk JE et al. (2012) Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148 (5), 988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galligan JJ et al. (2014) Stable histone adduction by 4-oxo-2-nonenal: a potential link between oxidative stress and epigenetics. J Am Chem Soc 136 (34), 11864–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mills EL et al. (2018) Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556 (7699), 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dabrowski MJ et al. (2010) Stereoselective effects of 4-hydroxynonenal in cultured mouse hepatocytes. Chem Res Toxicol 23 (10), 1601–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wakita C et al. (2009) Stereochemical configuration of 4-hydroxy-2-nonenal-cysteine adducts and their stereoselective formation in a redox-regulated protein. J Biol Chem 284 (42), 28810–28822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.West JD et al. (2004) Induction of apoptosis in colorectal carcinoma cells treated with 4-hydroxy-2-nonenal and structurally related aldehydic products of lipid peroxidation. Chem Res Toxicol 17 (4), 453–462. [DOI] [PubMed] [Google Scholar]
- 29.Parvez S et al. (2018) Biological specificity from chemical promiscuity: functional signaling choreographed by reactive electrophilic and oxidative species. Chem. Rev (invited contribution) - revised and resubmitted. [Google Scholar]
- 30.Surya SL et al. (2018) Cardiovascular small heat shock protein HSPB7 Is a kinetically privileged reactive electrophilic species (RES) sensor. ACS Chem Biol ( 10.1021/acschembio.7b00925) 13 (7), 1824–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long MJC and Aye Y (2017) Privileged electrophile sensors: A resource for covalent drug development. Cell Chem Biol 24 (7), 787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin HY et al. (2015) A generalizable platform for interrogating target- and signal-specific consequences of electrophilic modifications in redox-dependent cell signaling. J Am Chem Soc 137 (19), 6232–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parvez S et al. (2016) T-REX on-demand redox targeting in live cells. Nat Protoc 11 (12), 2328–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hiratsuka A et al. (2000) 4-Hydroxy-2(E)-nonenal enantiomers: (S)-selective inactivation of glyceraldehyde-3-phosphate dehydrogenase and detoxification by rat glutathione S-transferase A4–4. Biochem J 349 (3), 729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doorn JA and Petersen DR (2003) Covalent adduction of nucleophilic amino acids by 4-hydroxynonenal and 4-oxononenal. Chem. Biol. Interact. 143–144, 93–100. [DOI] [PubMed] [Google Scholar]
- 36.Hayes JD and Dinkova-Kostova AT (2014) The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39 (4), 199–218. [DOI] [PubMed] [Google Scholar]
- 37.Huang HC et al. (2002) Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem 277 (45), 42769–74. [DOI] [PubMed] [Google Scholar]
- 38.LoPachin RM et al. (2009) Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: Nucleophilic targets and adduct formation. Chem. Res. Toxicol. 22, 1499–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eds. et al. (2007) Redox Biochemistry, John Wiley & Sons, Inc. . [Google Scholar]
- 40.Hiratsuka A et al. (2001) (S)-preferential detoxification of 4-hydroxy-2(E)-nonenal enantiomers by hepatic glutathione S-transferase isoforms in guinea-pigs and rats. Biochem J 355 (Pt 1), 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siems W and Grune T (2003) Intracellular metabolism of 4-hydroxynonenal. Mol. Aspects Med 24 (4), 167–175. [DOI] [PubMed] [Google Scholar]
- 42.Page MI and Jencks WP (1971) Entropic Contributions to Rate Accelerations in Enzymic and Intramolecular Reactions and the Chelate Effect. Proc Natl Acad Sci U S A 68, 1678–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fang X et al. (2013) Temporally controlled targeting of 4-hydroxynonenal to specific proteins in living cells. J Am Chem Soc 135 (39), 14496–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Long MJC et al. (2018) Precision Electrophile Tagging in Caenorhabditis elegans. Biochemistry 57 (2), 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hall-Beauvais AV et al. (2018) Single-protein-specific redox targeting in live mammalian cells and C. elegans. Curr Protoc Mol Biol (Accepted). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Los GV et al. (2008) HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3 (6), 373–382. [DOI] [PubMed] [Google Scholar]
- 47.Long MJ et al. (2016) On-Demand Targeting: Investigating Biology with Proximity-Directed Chemistry. J Am Chem Soc 138 (11), 3610–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weerapana E et al. (2008) Disparate proteome reactivity profiles of carbon electrophiles. Nat Chem Biol 4 (7), 405–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Backus KM et al. (2016) Proteome-wide covalent ligand discovery in native biological systems. Nature 534 (7608), 570–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wani R et al. (2011) Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc Nat Acad Sci 108 (26), 10550–10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beavers WN et al. (2017) Protein modification by endogenously generated lipid electrophiles: Mitochondria as the source and target. ACS Chem Biol 12 (8), 2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parker CG et al. (2017) Ligand and target discovery by fragment-based screening in human cells. Cell 168 (3), 527–541 e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blewett MM et al. (2016) Chemical proteomic map of dimethyl fumarate–sensitive cysteines in primary human T cells. Sci Signal 9 (445), rs10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whitby LR et al. (2017) Quantitative chemical proteomic profiling of the in vivo targets of reactive drug metabolites. ACS Chem Biol 12 (8), 2040–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bak DW et al. (2017) Identifying functional cysteine residues in the mitochondria. ACS Chem Biol 12 (4), 947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Y et al. (2018) Quantitative profiling of protein carbonylations in ferroptosis by an aniline-derived probe. J Am Chem Soc 140 (13), 4712–4720. [DOI] [PubMed] [Google Scholar]
- 57.Singh J et al. (2011) The resurgence of covalent drugs. Nat Rev Drug Discov 10 (4), 307–17. [DOI] [PubMed] [Google Scholar]
- 58.Zhao Z and Bourne PE (2018) Progress with covalent small-molecule kinase inhibitors. Drug Discov Today 23 (3), 727–735. [DOI] [PubMed] [Google Scholar]
- 59.Chaikuad A et al. (2018) The cysteinome of protein kinases as a target in drug development. Angew Chem Int Ed Engl 57 (16), 4372–4385. [DOI] [PubMed] [Google Scholar]
- 60.Ferguson FM and Gray NS (2018) Kinase inhibitors: the road ahead. Nat Rev Drug Discov 17, 353–377. [DOI] [PubMed] [Google Scholar]
- 61.Lanning BR et al. (2014) A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat Chem Biol 10 (9), 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Linker RA et al. (2011) Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 134 (Pt 3), 678–92. [DOI] [PubMed] [Google Scholar]
- 63.Brennan MS et al. (2015) Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro. PLoS One 10 (3), e0120254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Information obtained from http://www.guidetopharmacology.org & https://clinicaltrials.gov/.