

Abstract
G protein-coupled receptors (GPCRs) physically connect extracellular information with intracellular signal propagation. Membrane trafficking plays a supportive role by “bookending” signaling events: movement through the secretory pathway delivers GPCRs to the cell surface where receptors can sample the extracellular environment, while endocytosis and endolysosomal membrane trafficking provide a versatile system to titrate cellular signaling potential and maintain homeostatic control. Recent evidence suggests that, in addition to these important effects, GPCR trafficking actively shapes the cellular signaling response by altering the location and timing of specific receptor-mediated signaling reactions. Here, we review key experimental evidence underlying this expanding view, focused on GPCR signaling mediated through activation of heterotrimeric G proteins located in the cytoplasm. We then discuss lingering and emerging questions regarding the interface between GPCR signaling and trafficking.
Keywords: endosome, GPCR, G protein, signaling, trafficking
1 |. INTRODUCTION
G protein-coupled receptors (GPCRs) comprise the largest family of signaling receptors and integral membrane proteins encoded by mammalian genomes, and it is estimated that about one-third of all drugs presently used in the clinic target GPCRs.1–4 GPCRs are seven-transmembrane proteins that are cotranslationally inserted into the endoplasmic reticulum membrane and glycosylated in the endoplasmic reticulum and Golgi apparatus. GPCRs are so-named because many of their signaling effects are mediated through ligand-dependent allosteric coupling to heterotrimeric GTP-binding proteins (G proteins) that associate with cytoplasmic membrane leaflets.5,6
The beta-adrenergic receptor as a model of GPCR signaling-trafficking relationships.
Early evidence that membrane trafficking is relevant to GPCR signaling emerged through the course of studying desensitization of the beta-adrenergic receptor-coupled adenylyl cyclase activity transduced by the trimeric G protein, Gs.7 Short-term agonist exposure (on the order of minutes) resulted in a reversible reduction in cyclic AMP (cAMP) production without changing the total number of receptors detected in the cell, while more prolonged exposure (on the order of several hours or more) produced a downregulation of receptor number8 that required new protein synthesis to reverse.9 A nondestructive process of receptor “sequestration,” detected by subcellular fractionation and ligand accessibility assays, occurred over an intermediate time scale10–13 and was later shown to represent the iterative cycling of receptors between plasma membrane and endosomes in the presence of agonist.14 Agonists were found to stimulate this cycle primarily by promoting ligand-dependent accumulation of receptors in clathrin-coated pits (CCPs) that subsequently internalize irrespective of continued agonist binding,14,15 and the slower process of proteolytic downregulation was shown to be mediated by receptor sorting to lysosomes from a shared endosome intermediate.16–18 Ligand-dependent accumulation of beta-adrenergic receptors in CCPs—the key event initiating GPCR entry to the endocytic network—was found to be promoted through receptor phosphorylation mediated by a family of GPCR kinases (GRKs) followed by interaction with arrestin (or beta-arrestin) proteins that act as endocytic adaptors by binding also to the clathrin lattice structure and PIP2.19–21 GPCR phosphorylation and binding to arrestins were shown previously to attenuate G protein activation,22–25 and acidification of the endosome lumen was generally recognized to destabilize ligand binding to receptors.26
We focused above on beta-adrenergic receptors as an early and well-studied model of the GPCR signaling-trafficking interface, but considerable diversity can be observed even among closely related GPCR paralogues and splice variants.27,28 Nevertheless, there are now many examples of GPCRs that undergo regulated endocytosis as described above. General takeaways from studies of such GPCRs are that (a) ligand-induced activation of GPCRs and G proteins is initiated at the plasma membrane, and (b) GPCR endocytosis is associated with events that reduce or terminate G protein activation. These takeaways support a traditional model of the signaling-trafficking interface in which G protein activation is restricted to the plasma membrane and receptors are inactive after endocytosis (Figure 1, left side). It now appears that some GPCRs can retain or recover the ability to activate G proteins after they internalize, and some GPCRs can activate heterotrimeric G proteins from the biosynthetic pathway (Figure 1, right side). Receptor signaling reactions organized in discrete membrane compartments, and dynamically connected through membrane trafficking, is a widespread theme in cell biology.29 Moreover, G protein activation on endomembranes has been recognized since early studies of phototransduction initiated by the light-activated GPCR rhodopsin.30 Ligand-activated GPCRs, however, were long thought to operate exclusively at the plasma membrane or to use endomembranes only to initiate G protein-independent signaling cascades.31,32 The present review focuses on evidence regarding the hypothesis that ligand-activated GPCRs can activate cytoplasmic G proteins from endosomes and membrane compartments in the biosynthetic pathway, as well as the plasma membrane. We point the reader to other reviews that discuss additional aspects of this concept and mechanisms of G protein-independent signal activation from endosomes.33–36
FIGURE 1.
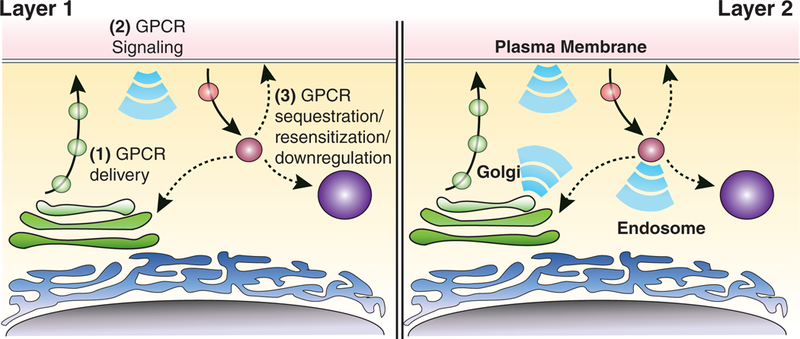
“Layers” in the GPCR trafficking/signaling relationship. Layer 1 (left side): Membrane trafficking processes “bookend” receptor signaling by trimeric G proteins from the plasma membrane (step 2), using biosynthetic (step 1) and endocytic (step 3) transport to modulate the number of functional surface receptors. Layer 2 (right side): Additional signaling can be initiated by receptor coupling to cytoplasmic G proteins from intracellular membranes. We propose that these layers function together to determine the integrated cellular response
2 |. G PROTEIN SIGNALING FROM ENDOSOMES: A CONTINUATION OR NEW BEGINNING?
As noted above, the traditional view of cellular GPCR signaling mediated by activation of heterotrimeric G proteins did not require any biochemical activity of receptors in endosomes. Indeed, early efforts to assess the potential of adrenergic receptors to initiate signaling from endosomes detected receptors and adenylyl cyclase activity in the same fraction but failed to detect functional coupling between them.13,37 Over the last decade, however, the hypothesis that GPCRs can initiate G protein-coupled signaling from endosomes as well as the plasma membrane has gained considerable experimental support.38–55
Evidence for GPCR-G protein signaling from endosomes.
Broadly considered, four experimental approaches have produced evidence supporting endosomal GPCR-G protein signaling. In the first, agonist application followed by washout showed a persistent component of the cellular response after agonist removal from the extracellular medium.39,40,42,45,48,54 In a second approach, the ability of membrane-permeant relative to membrane-impermeant antagonists to reverse the GPCR signaling was assessed; incomplete reversal by the membrane-impermeant antagonist was found.48,54 In a third approach, GPCR endocytosis was inhibited using genetic or chemical manipulations; endocytic blockade was found to reduce the strength and/or duration of downstream cellular responses.39,40,42,45,46,49,50 In a fourth approach, biosensors derived from single-domain antibodies (nanobodies) were used to localize active-conformation GPCRs as well as detect conformational activation of G protein; these studies reported a second phase of GPCR and G protein activation in endosomes, with a brief (seconds to about a minute) refractory period separating the arrival of receptors in endosomes from the second activation phase.46,53,54,56,57
Limitations of the present evidence.
While there is now reasonable evidence that some GPCRs initiate G protein signaling after endocytosis, endomembrane signaling by G proteins is not proven and the present evidence supporting it has limitations and caveats. A potential caveat of agonist washout experiments is that the ligand of interest may not be fully removed. Depending on the ligand and system, complete agonist washout is not trivial to achieve or verify.58–60 A caveat of genetic manipulations to inhibit endocytosis is that effects develop over a period of days, exceeding the time required for extensive remodeling of the plasma membrane and of the cellular proteome more broadly61–65; accordingly, genetic manipulations of endocytosis may have more widespread effects on cellular signaling than those resulting directly from blocking endocytosis of a particular GPCR. Chemical inhibitors of endocytosis act more rapidly but have demonstrated potential to produce additional off-target effects that complicate experimental interpretation.66–68 Studies using conformational biosensors to assess the activation state of GPCRs or G proteins are useful in that they can produce a “direct” location-specific readout, but a limitation of existing conformational biosensors is that they do not report functional signaling. Another caveat is that, depending on experimental conditions, such tools may significantly perturb the conformational landscape of the target that they are intended to sense or block critical signaling interactions.
The field is still grappling with how to deal with these problems. One approach is to combine ligand washout with endocytic inhibitor approaches, so that off-target effects of chemical/genetic inhibitors of endocytosis—as well as possible inefficiencies in ligand washout—can be internally controlled.39,45,49,54 Another is to deliberately exploit the potential of conformationally selective nanobodies to block GPCR signaling reactions, specifically localizing them to a defined target membrane using chemical recruitment tools and assessing effect on the cellular response.69 A third approach, which may also have therapeutic potential with further development, is to generate antagonist ligands that accumulate in endosomes to inhibit activation in this compartment selectively.49,50
Consequences of endosomal GPCR-G protein signaling.
One reported effect of GPCR-G protein signal initiation from endosomes is a prolonged or sustained cellular response, and another is an increased overall (or integrated) response magnitude.39,40 Ligand trapping in the endosome lumen is thought to prolong or enhance GPCR responses by favoring ligand rebinding after dissociation from the receptor,70 and G protein activation at endosomes may differ inherently from that at the plasma membrane because of location-specific differences in activation or regulatory machineries engaged (see below). GPCR-G protein activation in endosomes (or the trans-Golgi network after endocytic delivery) can also preferentially promote a subset of downstream effects such as GPCR-dependent transcriptional responses.39,40,49,56,57,71,72 Studies of GPCR-dependent transcriptional induction identified an additional function of endosomal G protein activation by GPCRs in enhancing cellular discrimination between similar ligands and increasing reliability of the cellular response.71,73
3 |. UNANSWERED QUESTIONS REGARDING ENDOSOMAL G PROTEIN SIGNALING
Is the mechanism of G protein activation at endosomes different from that at the plasma membrane?
A number of GPCRs that produce a persistent endosome-initiated response also mediate ligand-dependent recruitment of arrestins to this compartment.33 Whereas arrestin recruitment to the plasma membrane is associated with rapid termination of G protein activation, it has been proposed that arrestin recruitment to endosomes sustains it42,48 through formation of a complex in which the G protein and arrestin are simultaneously bound to the GPCR.48 Not all GPCRs that produce sustained responses from endosomes (such as the delta opioid and luteinizing hormone receptors45,54) recruit arrestin to endosomes, however, and other GPCRs that activate G proteins at endosomes (such as beta-adrenergic and D1 dopamine receptors 46,47) produce a transient rather than sustained cellular response. These findings raise the additional question of whether there might exist more than one mechanism of endosomal G protein activation by GPCRs, depending on the particular GPCR activated and/or the time scale of the endosome-initiated response.
What are other players in the signaling cascade and where are they working?
If GPCRs activate G proteins at the endosome, a next important question is whether relevant effectors and cofactors are present in sufficiently close physical proximity to mediate downstream signaling. There is evidence indicating that some G protein-dependent effector proteins are indeed present in the endosome limiting membrane but much remains unknown, and there is also evidence that heterotrimeric G proteins have the potential to traffic or translocate between membranes separately from GPCRs.74,75 Moreover, considering that plasma membrane and endosome membranes differ broadly in protein and lipid compositions, it is conceivable that there exist numerous mediators or regulators of compartment-specific GPCR signaling and trafficking that are presently unknown. Traditionally, the discovery of GPCR interaction partners has relied on genetic screens or biochemical fractionation. Newer technologies, such as proximity labeling combined with quantitative proteomics, have the potential to enable unbiased interrogation of GPCR-linked interaction networks in intact cells, while achieving both temporal and spatial resolution.76,77
How can location matter for signaling via diffusible second messengers?
As noted above, endosomal activation of G proteins has been reported to confer selectivity on GPCR-mediated transcriptional control.49,57,71 It is not clear how this is possible, particularly for cAMP-dependent transcriptional control, because this second messenger chemical is thought to diffuse very rapidly in the cytoplasm. Local signaling by cAMP is known to be enhanced by enforced proximity based on scaffolding, as well as by buffering and local hydrolysis of second messenger.78–80 It remains unknown if these strategies are sufficient to explain the high degree of signaling selectivity observed in compact cells where diffusion, based on theoretical considerations, would be expected to overwhelm them.79,81 Further work will be required to more fully delineate how spatial precision of second messenger signaling from the cytoplasm to the nucleus is achieved, particularly in compact cells lacking morphological elaborations capable of restricting diffusional access.
How is endosomal signaling regulated and terminated?
Multiple GPCRs have now been reported to mediate sustained signaling after agonist is removed from the extracellular environment.39,40,42,45,48–50,54 These findings raise a critical question: once an endosome is loaded with agonist and activated GPCR, how is activation terminated? Multiple mechanisms for terminating GPCR-G protein signaling from endosomes have been proposed and remain under investigation. A well-studied mechanism for regulating GPCR coupling to G proteins, as discussed above, is through phosphorylation and dephosphorylation of the receptor. This raises the question of the phosphorylation state(s) of GPCRs in endosomes. Beta-adrenergic receptor phosphorylation was found using metabolic labeling techniques to be rapidly induced in response to receptor activation but transient, with receptors isolated from endosomes in a net dephosphorylated state relative to receptors at the plasma membrane.82 However, studies carried out using phosphorylation site-specific antibodies reported persistent phosphorylation of GRK sites in the receptor tail and accumulation of GRK-phosphorylated receptor species in endosomes.83 Additional mechanisms that are proposed to terminate GPCR-G protein signaling from endosomes include ligand dissociation or destruction by endosomal acidification or proteolysis of the ligand,26 sequestration of GPCRs from the limiting membrane through transfer to intra-luminal vesicles (ILVs) mediated controlled by ubiquitin-dependent and -independent mechanisms,84–89 and binding of GPCRs to arrestin-like proteins. GPCR binding to arrestin-like proteins may directly inhibit receptor interaction with G proteins or control signaling indirectly by changing the residence time of receptors in the endosome limiting membrane before tubular exit.41,44,90–93 GPCR residence time in the endosome limiting membrane (or in subdomains thereof ) is known to vary considerably between different receptors and to produce effects both on the strength and duration of endosome-initiated signaling.57,94–97
4 |. G PROTEIN SIGNALING FROM THE BIOSYNTHETIC PATHWAY
While much attention has been focused on the potential of GPCRs to initiate G protein signaling from endosomes, the endosome membrane is not the only internal site at which GPCR-G protein activation has been detected or inferred. There is also evidence that some GPCRs have the potential to signal from the Golgi apparatus as well as the endoplasmic reticulum in some cases. Activation is thought to occur in two basic ways: In the first, the Golgi (or trans-Golgi network (TGN))-associated receptor pool arrives by cotrafficking with ligand from the plasma membrane.56 In the second, the relevant pool of GPCRs is derived by retention or retrieval during biosynthetic trafficking; in this case, ligand access is thought to be achieved through transmembrane transporters69,98 or, for sufficiently hydrophobic ligands, direct permeation through the membrane bilayer.54,69
A glutamate-activated GPCR, mGluR5, localizes at steady state primarily to the endoplasmic reticulum and outer nuclear membrane of neurons.99,100 Evidence that the intracellular receptor pool produces a functional response via cytoplasmic G proteins emerged by comparing effects of membrane-permeant and -impermeant ligands on calcium signaling; membrane-impermeant antagonists only partially blocked the cellular calcium response induced by glutamate, and a full response to glutamate required organic anion transporter and/or exchanger activities that enable this ligand to cross membranes.98 The beta-1 adrenergic receptor was found to be activated by its physiological agonist, epinephrine, at the Golgi apparatus of cardiac muscle cells, and ligand access was found to require an organic cation transporter activity.69 Sufficiently hydrophobic ligands, such as the clinically relevant drug dobutamine, were found to selectively activate the Golgi-associated pool of adrenergic receptors when transporter was inhibited. A Golgi-localized pool of opioid receptors was found to be activated only by small-molecule drugs, apparently by passive partitioning through membranes, while a panel of peptide ligands (including several natively produced opioid agonists) produced activation at the plasma membrane and in endosomes but not at the Golgi apparatus.54 Such observations further expand the concept of endomembrane GPCR signaling and suggest new opportunities for achieving selectivity in drug action based on the subcellular location of GPCR activation.
5 |. WHERE ARE WE NOW, AND WHERE DO WE GO FROM HERE?
There is presently considerable experimental support for the hypothesis that GPCRs can activate signaling via cytoplasmic G proteins from internal membrane compartments as well as from the plasma membrane. However, a broad caveat is that much of this knowledge is based on data derived from simplified cultured cell models. Such models are unlikely to fully mimic signaling reactions or regulation that occur in native cell types and tissues, and such models are also notoriously susceptible to distortion by protein overexpression. Improvements in technologies for achieving endogenous gene editing have potential to address the latter point, but this technology carries additional caveats such as clonal bias or adaptation effects that require careful control.101 Such (valid) concerns notwithstanding, simplified cell model systems offer significant advantages for mechanistic study, and we note that many aspects of GPCR signaling and trafficking that were first elucidated using such models have been subsequently validated in native systems. Nevertheless, a critical challenge moving forward is to develop improved approaches for investigating GPCR function under native conditions, while still enabling rigorous control and measurement. Recent efforts to combine genetic mouse models with advanced fluorescence imaging to examine GPCR ligand availability, activation and signaling in intact tissues45,102 suggest one promising direction for future investigation. Another interesting future direction is toward developing ligands that are able to selectively stimulate or inhibit GPCR activation at one subcellular location relative to another,49 both to experimentally manipulate location-based signaling and possibly as a basis to develop new drugs for the clinic with improved therapeutic specificity or efficacy.
ACKNOWLEDGMENTS
The authors thank Roshanak Irannejad, Nina Tsvetanova, Miriam Stoeber and Grace Peng for critical discussions and suggestions. We also thank other current and past members of the von Zastrow lab as well as many other colleagues, at University of California, San Francisco (UCSF) and elsewhere, for additional advice and comments. We regret being able to cite only a limited subset of relevant publications in this rapidly growing field because of space constraints. B.T.L. acknowledges NIH / NIDA K99 DA043607, and M.v.Z. acknowledges NIH / NIDA R01 DA010711 and DA012864 for supporting studies on GPCR signaling and trafficking related to the present review.
Footnotes
EDITORIAL PROCESS FILE
The Editorial Process File is available in the online version of this article.
REFERENCES
- 1.Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol 2013;53:531–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov 2017;16(12):829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weis WI, Kobilka BK. The molecular basis of G protein-coupled receptor activation. Annu Rev Biochem 2018;87:897–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol Med 2011; 17(3):126–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hepler JR, Gilman AG. G proteins. Trends Biochem Sci 1992;17(10): 383–387. [DOI] [PubMed] [Google Scholar]
- 6.Hilger D, Masureel M, Kobilka BK. Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol 2018;25(1):4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukherjee C, Caron MG, Lefkowitz RJ. Regulation of adenylate cyclase coupled beta-adrenergic receptors by beta-adrenergic catecholamines. Endocrinology 1976;99(2):347–357. [DOI] [PubMed] [Google Scholar]
- 8.Su YF, Harden TK, Perkins JP. Isoproterenol-induced desensitization of adenylate cyclase in human astrocytoma cells. Relation of loss of hormonal responsiveness and decrement in beta-adrenergic receptors. J Biol Chem 1979;254(1):38–41. [PubMed] [Google Scholar]
- 9.Doss RC, Perkins JP, Harden TK. Recovery of beta-adrenergic receptors following long term exposure of astrocytoma cells to catecholamine. Role of protein synthesis. J Biol Chem 1981;256(23): 12281–12286. [PubMed] [Google Scholar]
- 10.Chuang DM, Costa E. Beta-adrenergic receptors of frog erythrocytes. Biochemical sequelae following stimulation with isoproterenol. Neurochem Res 1979;4(6):777–793. [DOI] [PubMed] [Google Scholar]
- 11.Chuang DM, Costa E. Evidence for internalization of the recognition site of beta-adrenergic receptors during receptor subsensitivity induced by (−)-isoproterenol. Proc Natl Acad Sci U S A 1979;76(6): 3024–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Staehelin M, Simons P. Rapid and reversible disappearance of beta-adrenergic cell surface receptors. EMBO J 1982;1(2):187–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waldo GL, Northup JK, Perkins JP, Harden TK. Characterization of an altered membrane form of the beta-adrenergic receptor produced during agonist-induced desensitization. J Biol Chem 1983;258(22): 13900–13908. [PubMed] [Google Scholar]
- 14.von Zastrow M, Kobilka BK. Ligand-regulated internalization and recycling of human beta 2-adrenergic receptors between the plasma membrane and endosomes containing transferrin receptors. J Biol Chem 1992;267(5):3530–3538. [PubMed] [Google Scholar]
- 15.von Zastrow M, Kobilka BK. Antagonist-dependent and-independent steps in the mechanism of adrenergic receptor internalization. J Biol Chem 1994;269(28):18448–18452. [PubMed] [Google Scholar]
- 16.Gagnon AW, Kallal L, Benovic JL. Role of clathrin-mediated endocytosis in agonist-induced down-regulation of the beta2-adrenergic receptor. J Biol Chem 1998;273(12):6976–6981. [DOI] [PubMed] [Google Scholar]
- 17.Tsao PI, von Zastrow M. Type-specific sorting of G protein-coupled receptors after endocytosis. J Biol Chem 2000;275(15):11130–11140. [DOI] [PubMed] [Google Scholar]
- 18.Cao TT, Deacon HW, Reczek D, Bretscher A, von Zastrow M. A kinase-regulated PDZ-domain interaction controls endocytic sorting of the beta2-adrenergic receptor. Nature 1999;401(6750):286–290. [DOI] [PubMed] [Google Scholar]
- 19.Goodman OB Jr, Krupnick JG, Santini F, et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature 1996;383(6599):447–450. [DOI] [PubMed] [Google Scholar]
- 20.Ferguson SS, Downey WE 3rd, Colapietro AM, Barak LS, Ménard L, Caron MG. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 1996;271(5247): 363–366. [DOI] [PubMed] [Google Scholar]
- 21.Gaidarov I, Krupnick JG, Falck JR, Benovic JL, Keen JH. Arrestin function in G protein-coupled receptor endocytosis requires phosphoinositide binding. EMBO J 1999;18(4):871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. Beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science 1990;248(4962):1547–1550. [DOI] [PubMed] [Google Scholar]
- 23.Benovic JL, DeBlasi A, Stone WC, Caron MG, Lefkowitz RJ. Beta-adrenergic receptor kinase: primary structure delineates a multigene family. Science 1989;246(4927):235–240. [DOI] [PubMed] [Google Scholar]
- 24.Kühn H, Wilden U. Deactivation of photoactivated rhodopsin by rhodopsin-kinase and arrestin. J Recept Res 1987;7(1–4):283–298. [DOI] [PubMed] [Google Scholar]
- 25.Sibley DR, Strasser RH, Caron MG, Lefkowitz RJ. Homologous desensitization of adenylate cyclase is associated with phosphorylation of the beta-adrenergic receptor. J Biol Chem 1985;260(7):3883–3886. [PubMed] [Google Scholar]
- 26.Murphy JE, Padilla BE, Hasdemir B, Cottrell GS, Bunnett NW. Endosomes: a legitimate platform for the signaling train. Proc Natl Acad Sci U S A 2009;106(42):17615–17622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanowitz M, Hislop JN, von Zastrow M. Alternative splicing determines the post-endocytic sorting fate of G-protein-coupled receptors. J Biol Chem 2008;283(51):35614–35621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eichel K, Jullié D, von Zastrow M. β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat Cell Biol 2016;18(3):303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sigismund S, Confalonieri S, Ciliberto A, Polo S, Scita G, Di Fiore PP. Endocytosis and signaling: cell logistics shape the eukaryotic cell plan. Physiol Rev 2012;92(1):273–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stryer L Cyclic GMP cascade of vision. Annu Rev Neurosci 1986;9: 87–119. [DOI] [PubMed] [Google Scholar]
- 31.DeFea KA, Zalevsky J, Thoma MS, Déry O, Mullins RD, Bunnett NW. Beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol 2000;148(6):1267–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luttrell LM, Roudabush FL, Choy EW, et al. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci U S A 2001;98(5):2449–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomsen ARB, Jensen DD, Hicks GA, Bunnett NW. Therapeutic targeting of Endosomal G-protein-coupled receptors. Trends Pharmacol Sci 2018;39(10):879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jong Y-JI, Harmon SK, O’Malley KL. GPCR signalling from within the cell. Br J Pharmacol 2018;175(21):4026–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sposini S, Hanyaloglu AC. Evolving view of membrane trafficking and signaling systems for G protein-coupled receptors. Prog Mol Subcell Biol 2018;57:273–299. [DOI] [PubMed] [Google Scholar]
- 36.Shenoy SK, Lefkowitz RJ. β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci 2011;32(9):521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chuang DM, Dillon-Carter O, Spain JW, Laskowski MB, Roth BL, Coscia CJ. Detection and characterization of beta-adrenergic receptors and adenylate cyclase in coated vesicles isolated from bovine brain. J Neurosci 1986;6(9):2578–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mullershausen F, Zecri F, Cetin C, Billich A, Guerini D, Seuwen K. Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat Chem Biol 2009;5(6):428–434. [DOI] [PubMed] [Google Scholar]
- 39.Calebiro D, Nikolaev VO, Gagliani MC, et al. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol 2009;7(8):e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferrandon S, Feinstein TN, Castro M, et al. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol 2009;5(10):734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feinstein TN, Wehbi VL, Ardura JA, et al. Retromer terminates the generation of cAMP by internalized PTH receptors. Nat Chem Biol 2011;7(5):278–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feinstein TN, Yui N, Webber MJ, et al. Noncanonical control of vasopressin receptor type 2 signaling by retromer and arrestin. J Biol Chem 2013;288(39):27849–27860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilliland CT, Taylor Gilliland C, Salanga CL, Kawamura T, Trejo J, Handel TM. The chemokine receptor CCR1 is constitutively active, which leads to G protein-independent, β-Arrestin-mediated internalization. J Biol Chem 2013;288(45):32194–32210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gidon A, Al-Bataineh MM, Jean-Alphonse FG, et al. Endosomal GPCR signaling turned off by negative feedback actions of PKA and v-ATPase. Nat Chem Biol 2014;10(9):707–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lyga S, Volpe S, Werthmann RC, et al. Persistent cAMP signaling by internalized LH receptors in ovarian follicles. Endocrinology 2016; 157(4):1613–1621. [DOI] [PubMed] [Google Scholar]
- 46.Irannejad R, Tomshine JC, Tomshine JR, et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature 2013; 495(7442):534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kotowski SJ, Hopf FW, Seif T, Bonci A, von Zastrow M. Endocytosis promotes rapid dopaminergic signaling. Neuron 2011;71(2):278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomsen ARB, Plouffe B, Cahill TJ 3rd, et al. GPCR-G protein-β-Arrestin super-complex mediates sustained G protein signaling. Cell 2016;166(4):907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jensen DD, Lieu T, Halls ML, et al. Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief. Sci Transl Med 2017;9(392): eaal3447. 10.1126/scitranslmed.aal3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yarwood RE, Imlach WL, Lieu T, et al. Endosomal signaling of the receptor for calcitonin gene-related peptide mediates pain transmission. Proc Natl Acad Sci U S A 2017;114(46):12309–12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jimenez-Vargas NN, Pattison LA, Zhao P, et al. Protease-activated receptor-2 in endosomes signals persistent pain of irritable bowel syndrome. Proc Natl Acad Sci U S A 2018;115(31):E7438–E7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.English EJ, Mahn SA, Marchese A. Endocytosis is required for CC chemokine receptor type 4 (CXCR4)-mediated Akt activation and antiapoptotic signaling. J Biol Chem 2018;293(29):11470–11480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sposini S, Jean-Alphonse FG, Ayoub MA, et al. Integration of GPCR signaling and sorting from very early endosomes via opposing APPL1 mechanisms. Cell Rep 2017;21(10):2855–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stoeber M, Jullié D, Lobingier BT, et al. A genetically encoded biosensor reveals location Bias of opioid drug action. Neuron 2018; 98(5):963–976.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parsons RL, May V. PACAP-induced PAC1 receptor internalization and recruitment of Endosomal signaling regulate cardiac neuron excitability. J Mol Neurosci 2018. 10.1007/s12031-018-1127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Godbole A, Lyga S, Lohse MJ, Calebiro D. Internalized TSH receptors en route to the TGN induce local Gs-protein signaling and gene transcription. Nat Commun 2017;8(1):443 10.1038/s41467-017-00357-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bowman SL, Shiwarski DJ, Puthenveedu MA. Distinct G protein– coupled receptor recycling pathways allow spatial control of down-stream G protein signaling. J Cell Biol 2016;214(7):797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neumann S, Geras-Raaka E, Marcus-Samuels B, Gershengorn MC. Persistent cAMP signaling by thyrotropin (TSH) receptors is not dependent on internalization. FASEB J 2010;24(10):3992–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hothersall JD, Bussey CE, Brown AJ, Scott JS, Dale I, Rawlins P. Sustained wash-resistant receptor activation responses of GPR119 agonists. Eur J Pharmacol 2015;762:430–442. [DOI] [PubMed] [Google Scholar]
- 60.Hothersall JD, Guo D, Sarda S, et al. Structure-activity relationships of the sustained effects of adenosine A2A receptor agonists driven by slow dissociation kinetics. Mol Pharmacol 2017;91(1):25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Griffiths G, Back R, Marsh M. A quantitative analysis of the endocytic pathway in baby hamster kidney cells. J Cell Biol 1989;109(6 Pt 1): 2703–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mellman I Endocytosis and molecular sorting. Annu Rev Cell Dev Biol 1996;12:575–625. [DOI] [PubMed] [Google Scholar]
- 63.Kaplan J, Moskowitz M. Studies on the turnover of plasma membranes in cultured mammalian cells II Demonstration of heterogeneous rates of turnover for plasma membrane proteins and glycoproteins. Biochim Biophys Acta 1975;389(2):306–313. [DOI] [PubMed] [Google Scholar]
- 64.Chu FF, Doyle D. Turnover of plasma membrane proteins in rat hepatoma cells and primary cultures of rat hepatocytes. J Biol Chem 1985;260(5):3097–3107. [PubMed] [Google Scholar]
- 65.Warren R, Doyle D. Turnover of the surface proteins and the receptor for serum asialoglycoproteins in primary cultures of rat hepatocytes. J Biol Chem 1981;256(3):1346–1355. [PubMed] [Google Scholar]
- 66.Preta G, Lotti V, Cronin JG, Sheldon IM. Protective role of the dynamin inhibitor Dynasore against the cholesterol-dependent cytolysin of Trueperella pyogenes. FASEB J 2015;29(4):1516–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park RJ, Shen H, Liu L, Liu X, Ferguson SM, De Camilli P. Dynamin triple knockout cells reveal off target effects of commonly used dynamin inhibitors. J Cell Sci 2013;126(Pt 22):5305–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Persaud A, Cormerais Y, Pouyssegur J, Rotin D. Dynamin inhibitors block activation of mTORC1 by amino acids independently of dynamin. J Cell Sci 2018;131(1):jcs211755. 10.1242/jcs.211755. [DOI] [PubMed] [Google Scholar]
- 69.Irannejad R, Pessino V, Mika D, et al. Functional selectivity of GPCR-directed drug action through location bias. Nat Chem Biol 2017; 13(7):799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hothersall JD, Brown AJ, Dale I, Rawlins P. Can residence time offer a useful strategy to target agonist drugs for sustained GPCR responses? Drug Discov Today 2016;21(1):90–96. [DOI] [PubMed] [Google Scholar]
- 71.Tsvetanova NG, von Zastrow M. Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol 2014;10(12): 1061–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jean-Alphonse F, Bowersox S, Chen S, Beard G, Puthenveedu MA, Hanyaloglu AC. Spatially restricted G protein-coupled receptor activity via divergent endocytic compartments. J Biol Chem 2014;289(7): 3960–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsvetanova NG, Trester-Zedlitz M, Newton BW, et al. G protein-coupled receptor endocytosis confers uniformity in responses to chemically distinct ligands. Mol Pharmacol 2017;91(2):145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giri L, Patel AK, Karunarathne WKA, Kalyanaraman V, Venkatesh KV, Gautam N. A G-protein subunit translocation embedded network motif underlies GPCR regulation of calcium oscillations. Biophys J 2014;107(1):242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wedegaertner PB. G protein trafficking. Subcell Biochem 2012;63: 193–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lobingier BT, Hüttenhain R, Eichel K, et al. An approach to spatiotemporally resolve protein interaction networks in living cells. Cell 2017;169(2):350–360.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Paek J, Kalocsay M, Staus DP, et al. Multidimensional tracking of GPCR signaling via peroxidase-catalyzed proximity labeling. Cell 2017;169(2):338–349.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Agarwal SR, Clancy CE, Harvey RD. Mechanisms restricting diffusion of intracellular cAMP. Sci Rep 2016;6:19577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lohse C, Bock A, Maiellaro I, et al. Experimental and mathematical analysis of cAMP nanodomains. PLoS One 2017;12(4):e0174856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Esseltine JL, Scott JD. AKAP signaling complexes: pointing towards the next generation of therapeutic targets? Trends Pharmacol Sci 2013;34(12):648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Conti M, Mika D, Richter W. Cyclic AMP compartments and signaling specificity: role of cyclic nucleotide phosphodiesterases. J Gen Physiol 2014;143(1):29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sibley DR, Strasser RH, Benovic JL, Daniel K, Lefkowitz RJ. Phosphorylation/dephosphorylation of the beta-adrenergic receptor regulates its functional coupling to adenylate cyclase and subcellular distribution. Proc Natl Acad Sci U S A 1986;83(24):9408–9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tran TM, Friedman J, Baameur F, Knoll BJ, Moore RH, Clark RB. Characterization of beta2-adrenergic receptor dephosphorylation: comparison with the rate of resensitization. Mol Pharmacol 2007; 71(1):47–60. [DOI] [PubMed] [Google Scholar]
- 84.Hislop JN, Henry AG, Marchese A, von Zastrow M. Ubiquitination regulates proteolytic processing of G protein-coupled receptors after their sorting to lysosomes. J Biol Chem 2009;284(29):19361–19370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Whistler JL, Enquist J, Marley A, et al. Modulation of postendocytic sorting of G protein-coupled receptors. Science 2002;297(5581): 615–620. [DOI] [PubMed] [Google Scholar]
- 86.Tanowitz M, Von Zastrow M. Ubiquitination-independent trafficking of G protein-coupled receptors to lysosomes. J Biol Chem 2002; 277(52):50219–50222. [DOI] [PubMed] [Google Scholar]
- 87.Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated beta-2 adrenergic receptor and beta-arrestin. Science 2001;294(5545):1307–1313. [DOI] [PubMed] [Google Scholar]
- 88.Dores MR, Chen B, Lin H, et al. ALIX binds a YPX(3)L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J Cell Biol 2012;197(3):407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Marchese A, Raiborg C, Santini F, Keen JH, Stenmark H, Benovic JL. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev Cell 2003;5(5):709–722. [DOI] [PubMed] [Google Scholar]
- 90.Mzhavia N, Pan H, Che F-Y, Fricker LD, Devi LA. Characterization of endothelin-converting enzyme-2. Implication for a role in the non-classical processing of regulatory peptides. J Biol Chem 2003; 278(17):14704–14711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pan H, Mzhavia N, Devi LA. Endothelin converting enzyme-2: a processing enzyme involved in the generation of novel neuropeptides. Protein Pept Lett 2004;11(5):461–469. [DOI] [PubMed] [Google Scholar]
- 92.Gupta A, Gomes I, Wardman J, Devi LA. Opioid receptor function is regulated by post-endocytic peptide processing. J Biol Chem 2014; 289(28):19613–19626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tian X, Irannejad R, Bowman SL, et al. The α-arrestin ARRDC3 regulates the endosomal residence time and intracellular signaling of the β2-adrenergic receptor. J Biol Chem 2016;291(28):14510–14525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Varandas KC, Irannejad R, von Zastrow M. Retromer endosome exit domains serve multiple trafficking destinations and regulate local G protein activation by GPCRs. Curr Biol 2016;26(23):3129–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yudowski GA, Puthenveedu MA, Henry AG, von Zastrow M. Cargomediated regulation of a rapid Rab4-dependent recycling pathway. Mol Biol Cell 2009;20(11):2774–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Puthenveedu MA, Lauffer B, Temkin P, et al. Sequence-dependent sorting of recycling proteins by actin-stabilized endosomal microdomains. Cell 2010;143(5):761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vistein R, Puthenveedu MA. Reprogramming of G protein-coupled receptor recycling and signaling by a kinase switch. Proc Natl Acad Sci U S A 2013;110(38):15289–15294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jong Y-JI, Kumar V, Kingston AE, Romano C, O’Malley KL. Functional metabotropic glutamate receptors on nuclei from brain and primary cultured striatal neurons. Role of transporters in delivering ligand. J Biol Chem 2005;280(34):30469–30480. [DOI] [PubMed] [Google Scholar]
- 99.Jong YJ, Kumar V, O’Malley KL. Intracellular metabotropic glutamate receptor 5 (mGluR5) activates signaling cascades distinct from cell surface counterparts. J Biol Chem 2009;284(51):35827–35838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jong YJ, Sergin I, Purgert CA, O’Malley KL. Location-dependent signaling of the group 1 metabotropic glutamate receptor mGlu5. Mol Pharmacol 2014;86(6):774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Luttrell LM, Wang J, Plouffe B, et al. Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci Signal 2018;11(549):eaat7650. 10.1126/scisignal.aat7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Patriarchi T, Cho JR, Merten K, et al. Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science 2018;360(6396):eaat4422. 10.1126/science.aat4422. [DOI] [PMC free article] [PubMed] [Google Scholar]