

Abstract
The function and fate of cellular RNAs are often governed by the phosphorylation state at the 5′ end or the identity of whatever cap may be present there. Here we describe methods for examining these important 5′-terminal features on any cellular or synthetic RNA of interest that can be detected by Northern blotting. One such method, PABLO, is a splinted ligation assay that makes it possible to accurately quantify the percentage of 5′ ends that are monophosphorylated. Another, PACO, is a capping assay that reveals the percentage of 5′ ends that are diphosphorylated. A third, boronate gel electrophoresis in conjunction with deoxyribozyme-mediated cleavage, enables different types of caps (e.g., m7Gppp caps versus NAD caps) to be distinguished from one another and the percentage of each to be determined. After completing all three tests, the percentage of 5′ ends that are triphosphorylated can be deduced by process of elimination. Together, this battery of assays allows the 5′ terminus of an RNA to be profiled in unprecedented detail.
Keywords: monophosphate, diphosphate, triphosphate, DNAzyme, guanylyltransferase, Pce1
1. Introduction
5′-terminal modifications have long been known to play pivotal roles in the maturation, function, and turnover of cellular RNAs. For example, bacterial transcripts are protected from degradation by the triphosphate initially present at the 5′ end. Its sequential conversion to a diphosphate and then a monophosphate, a process dependent on the RNA pyrophosphohydrolase RppH, renders those transcripts vulnerable to rapid attack by either the endonuclease RNase E or the 5′ exonuclease RNase J, both of which prefer RNA substrates whose 5′ terminus is monophosphorylated [1–4]. The influence of the m7Gppp cap at the 5′ end of eukaryotic mRNAs is even more wide-ranging, as this modification is important for a variety of nuclear and cytoplasmic events such as splicing, polyadenylation, nuclear export, and translation as well as for protecting messages from degradation [5]. It is added to pre-mRNAs in a multistep process that involves conversion of the initial 5′ triphosphate to a diphosphate that is then capped by an RNA guanylyltransferase and a methyltransferase, and eventually it is removed by cytoplasmic decapping enzymes to generate a monophosphorylated 5′ terminus susceptible to exonucleolytic attack [6–8]. Finally, the redox cofactor nicotinamide adenine dinucleotide (NAD) has been found to occasionally become incorporated into bacterial and eukaryotic RNAs as a noncanonical 5′ cap whose removal requires a distinct decapping enzyme [9–12].
Elucidating the interconversion and function of these diverse 5′ termini requires methods for distinguishing them from one another and for accurately determining their relative abundance. In this report, we describe assays that allow monophosphorylated, diphosphorylated, and various types of capped 5′ ends on individual cellular RNAs to be differentiated and quantified. These assays are applicable to any RNA that can be detected by Northern blotting.
2. Detection of monophosphorylated 5′ ends by PABLO
PABLO (Phosphorylation Assay By Ligation of Oligonucleotides) is a method for quantifying the percentage of 5′ ends that are monophosphorylated on any individual RNA of interest [13, 14]. This splinted ligation assay is based on the ability of T4 DNA ligase to covalently join the 3′ end of a DNA oligonucleotide (oligo X) to the 5′ terminus of RNA when the RNA is monophosphorylated and the two ends are juxtaposed by simultaneous base pairing to a bridging DNA oligonucleotide (oligo Y). The lower electrophoretic mobility of the denatured product of ligation enables it to be distinguished from its unligated counterpart by Northern blotting (Figure 1). By using a 10–23 deoxyribozyme (DNAzyme) [15] to cleave the RNA site-specifically 50–250 nt from the 5′ end, the ligated and unligated reaction products can readily be resolved on a polyacrylamide gel even if the original RNA is very long. Because the cleavage-site specificity of such DNAzymes is determined by sequence complementarity, they can be designed to cut an RNA at almost any location where a purine (A or G) is followed by a pyrimidine (preferably U) [16]. The sequences of the complementary arms that define the specificity of a 10–23 DNAzyme are target-dependent and typically each 10–11 nucleotides long, whereas the catalytic region at its center (GGCTAGCTACAACGA) is invariant (Figure 2).
Figure 1. Detection of monophosphorylated 5′ ends by PABLO.
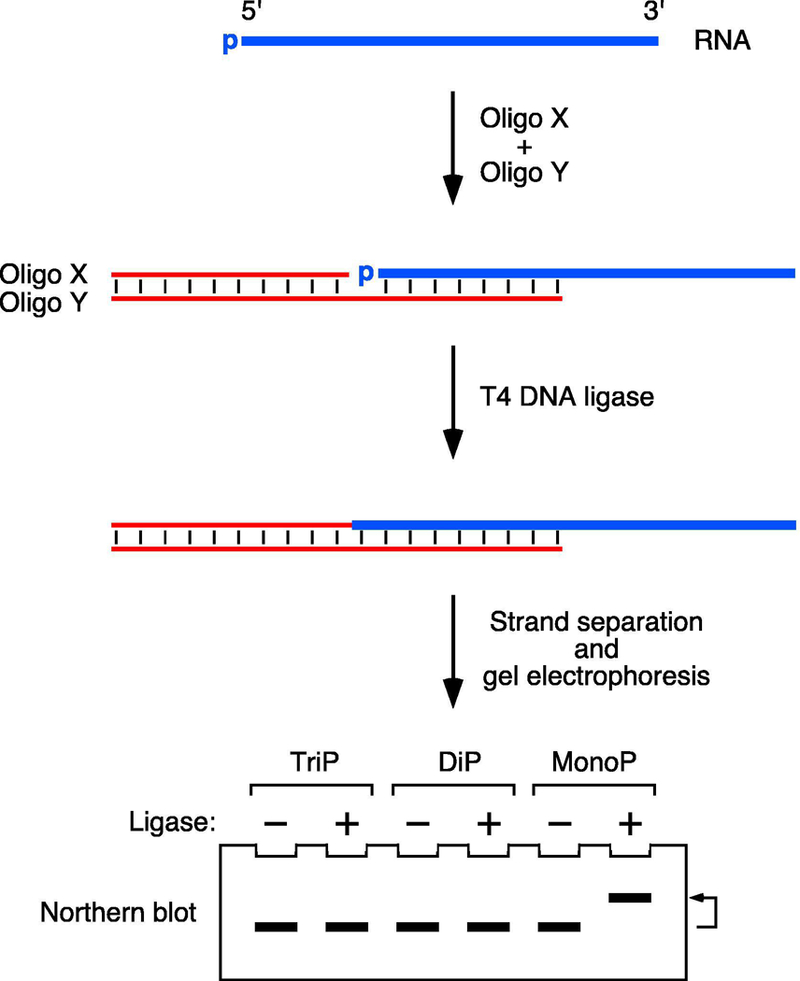
PABLO is a splinted ligation assay in which monophosphorylated RNA 5′ ends are selectively ligated to the 3′ end of a DNA oligonucleotide (oligo X) when the two are juxtaposed by simultaneous base pairing to a bridging DNA oligonucleotide (oligo Y) and treated with T4 DNA ligase. The ligation product and its unligated counterpart are then separated by electrophoresis on a denaturing gel and detected by Northern blotting. The differential mobility of these two bands can be enhanced by site-specific RNA cleavage with a DNAzyme (not shown). TriP, triphosphorylated RNA; DiP, diphosphorylated RNA; MonoP, monophosphorylated RNA.
Figure 2. RNA cleavage by 10–23 deoxyribozymes.
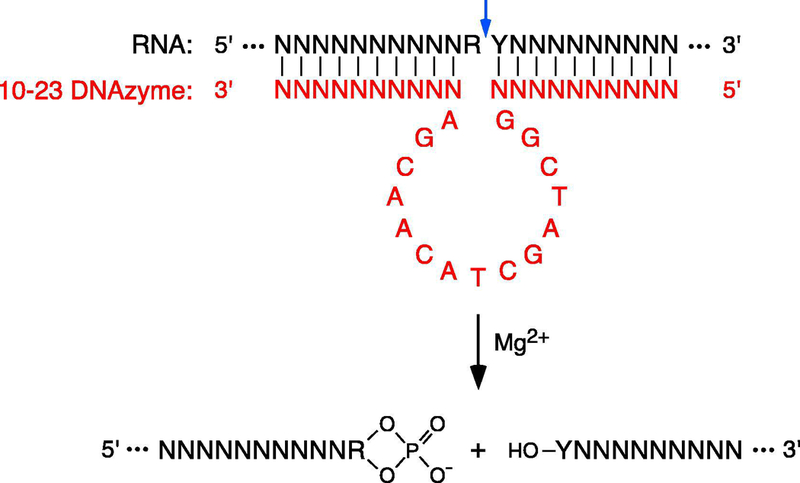
In the presence of Mg2+, the duplex formed by an RNA of interest (black) and a complementary 10–23 DNAzyme (red) [15] undergoes RNA cleavage at a unique site (blue arrow) between a purine (R) and a pyrimidine (Y) to generate a 5′ fragment bearing a 2′,3′ cyclic phosphate (PO4) at its 3′ end and a 3′ fragment bearing a hydroxyl (HO) at its 5′ end. N, any complementary nucleotide.
For transcripts or DNAzyme-cleavage products up to 250 nt in length, an oligo X that is 20–40 nt long is generally adequate, irrespective of its sequence (e.g., X22: 5′-GAACAATATGAATGATAACTTG-3′ orX32:5′-AAAAAAAAAAGAACAATATGAATGATAACTTG-3′). This oligonucleotide should be purified before use to eliminate any heterogeneity in length that may arise during its synthesis. The bridging oligonucleotide (oligo Y) must precisely juxtapose the 3′ end of oligo X and the RNA 5′ end of interest upon base pairing with both. Therefore, before designing oligo Y, the 5′ end of the RNA must be mapped at single-nucleotide resolution by a reliable method such as RLM-RACE (RNA ligase-mediated rapid amplification of cDNA ends) [17]. Typically, oligo Y is complementary to the 5′-terminal 18–22 nt of the RNA and the 3′-terminal 22 nt of oligo X. Strong secondary structure near the RNA 5′ end is best avoided, as it can interfere with PABLO by making it difficult for the bridging oligonucleotide to anneal.
Due to the finite efficiency of splinted ligation, PABLO reactions generally do not proceed all the way to completion. Therefore, the PABLO ligation yield, defined as the fraction of a particular 5′ end that becomes covalently joined to oligo X, reflects not only the percentage of that end that is monophosphorylated but also its intrinsic ligation efficiency [18]. The fraction of a given RNA 5′ end that is monophosphorylated can nevertheless be calculated by normalizing its ligation yield to that of a fully monophosphorylated but otherwise identical RNA standard analyzed in parallel (Figure 3):
| (1) |
where M is the fraction of monophosphorylated 5′ ends, YL is the measured PABLO ligation yield of the RNA under investigation, and ELM is the measured ligation efficiency of the fully monophosphorylated standard, on a scale from 0 (no ligation) to 1 (complete ligation). This standard can be conveniently generated by treating an equal amount of the RNA sample of interest with an excess of E. coli RppH, a pyrophosphatase able to convert a 5′ triphosphate, diphosphate, Gppp cap, or m7Gppp cap to a monophosphate [2, 4, 19].
Figure 3. Representative analysis by PABLO.
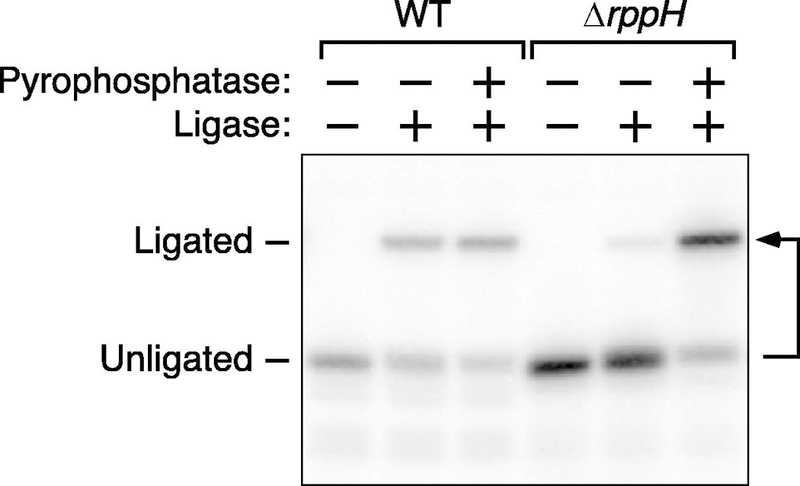
Total E. coli RNA was analyzed by PABLO to determine the percentage of yeiP mRNA 5′ ends that are monophosphorylated in wild-type cells (WT) and in an isogenic mutant lacking the rppH gene, which encodes an RNA pyrophosphohydrolase important for generating monophosphorylated 5′ ends in E. coli. For comparison, the ligation efficiency of fully monophosphorylated yeiP mRNA was measured in parallel by PABLO analysis of total RNA that had been treated with an excess of purified RppH (pyrophosphatase). The image of this blot is reproduced from Figure 3 of reference [4].
2.1. Protocol: PABLO
Three identical RNA samples should be analyzed in parallel: one to which DNA ligase is added, one to which DNA ligase is not added (mock-ligated negative control), and one to which DNA ligase is added after treatment with excess RppH (200 nM for 2 hr at 37°C in HEPES, pH 7.6 (20 mM), MgCl2 (10 mM), dithiothreitol (1 mM), glycerol (1%), and rRNasin (2 units/µl; Promega) to generate a fully monophosphorylated 5′ end for measuring ELM. For statistical purposes, each should be examined in triplicate.
Splinted ligation of RNAs 20–500 nt long can be examined without the need for DNAzyme cleavage by using an oligo X of proportionate length and by adjusting the percentage of polyacrylamide in the gel.
Recombinant T4 DNA ligase and E. coli RppH can be purchased commercially or purified from E. coli as previously described [4].
Combine total cellular RNA (10 µg) with oligonucleotide X (0.44 µM), oligonucleotide Y (0.09 µM), and a transcript-specific 10–23 DNAzyme (8.9 µM) in a total volume of 45 µl.
Heat the mixture to 85°C for 5 min, slowly cool it to 30°C over a period of 60–90 min, and then place it on ice.
Add a solution (35 µl) containing T4 DNA ligase (2,500 cohesive end units if purchased from New England Biolabs or 1.3 µM if purified in-house) in Tris-HCl, pH 7.5 (114 mM), MgCl2 (23 mM), dithiothreitol (23 mM), ATP (2.7 mM), and rRNasin (1 unit/µl; Promega).
Allow ligation and DNAzyme cleavage to proceed for 2 hr at 37°C.
Stop the reactions by adding EDTA (150 µl of an 8.3 mM solution). Phenol extract and ethanol precipitate the reaction products.
Dissolve the reaction products in 10 µl of water and an equal volume of loading solution (95% formamide, 20 mM EDTA, 0.0125% (w/v) bromophenol blue, 0.0125% (w/v) xylene cyanol), heat them to 95°C for 5 min, and subject them to electrophoresis on a denaturing 5–6% polyacrylamide, 7 M urea gel.
Electroblot the gel onto a membrane (Hybond-XL, GE Healthcare or Immobilon-NY+, Millipore), UV-crosslink the blot, and probe the blot with a 32P-labeled oligonucleotide complementary to the RNA of interest.
Visualize the radioactive bands on the blot with a phosphorimager, and quantify their relative intensities.
3. Detection of diphosphorylated 5′ ends by PACO
Another assay, PACO (Phosphorylation Assay by Capping Outcome), makes it possible to determine the percentage of a particular RNA 5′ end that is diphosphorylated. It relies on the specificity of the RNA guanylyltransferase Pce1, which preferentially caps diphosphorylated 5′ termini by transferring guanylate from GTP [20]. As originally conceived [4], this assay took advantage of the ability of the resulting unmethylated Gppp caps to protect the adjacent phosphates from removal by alkaline phosphatase, which will selectively convert the 5′ ends of the RNAs that remain uncapped (triphosphorylated and monophosphorylated RNAs) to 5′ hydroxyls. Therefore, diphosphorylated 5′ ends can be detected by treating RNA sequentially with Pce1, alkaline phosphatase, and a pyrophosphatase such as RppH to generate a monophosphorylated product that can be detected and quantified by PABLO.
An improved version of PACO avoids the need for phosphatase treatment, decapping, and ligation, thereby simplifying the assay. Instead, the diphosphorylated RNA capped by Pce1 is detected directly by boronate gel electrophoresis and Northern blotting (Figure 4). The 3-acrylamidophenylboronic acid integrated into the polyacrylamide matrix of a boronate gel transiently forms a covalent adduct with vicinal diols that pass by, selectively reducing the electrophoretic mobility of nucleic acids that contain such a chemical moiety, which is a distinctive characteristic of 5′ caps but not of uncapped 5′ ends [21, 22] (Figure 5). Two complications of using boronate gel electrophoresis to distinguish capped from uncapped mRNAs are the ubiquitous presence of a vicinal diol at mRNA 3′ ends, irrespective of 5′ capping, and the size of most mRNAs, which are typically too long to enter a polyacrylamide gel. Both can be overcome by site-specific cleavage with a 10–23 DNAzyme to generate a short (50–120 nt) 5′ fragment bearing a 2′,3′-cyclic phosphate instead of a vicinal diol at its 3′ terminus [15] (Figure 2). The migration of this RNA fragment on a boronate gel will be retarded only if its 5′ end is capped, which can be confirmed by its faster migration prior to capping or upon decapping.
Figure 4. Detection of diphosphorylated 5′ ends by PACO.
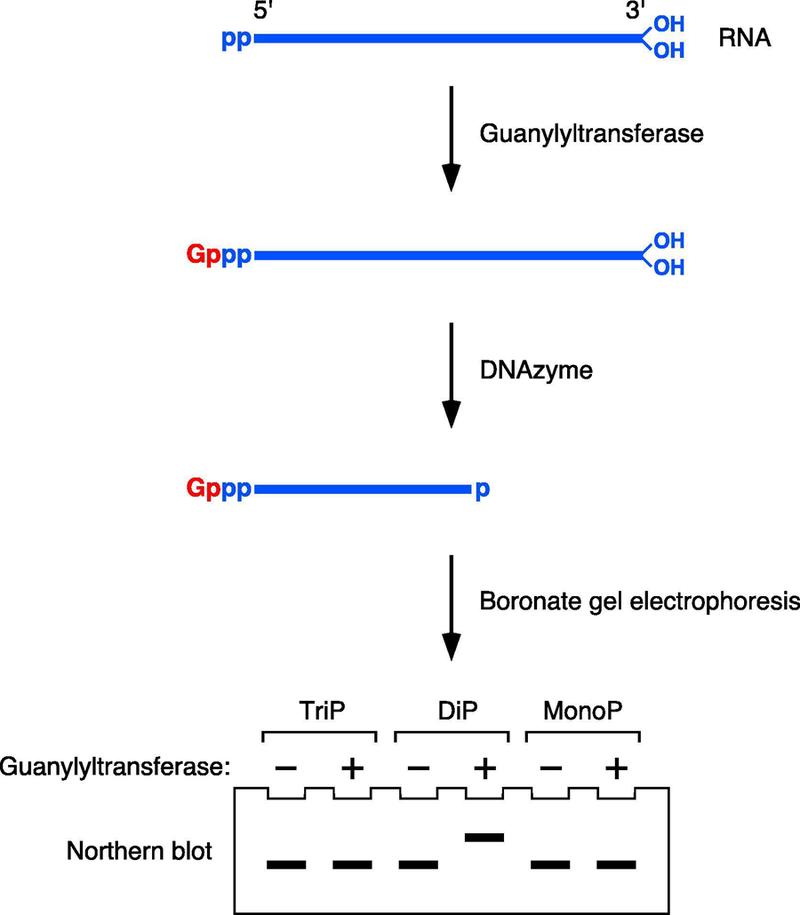
PACO detects diphosphorylated RNA 5′ ends on the basis of their reactivity with Schizosaccharomyces pombe Pce1, a guanylyltransferase that preferentially caps RNA 5′ ends that bear two phosphates rather than three or one. The cap added by Pce1 is detected by its ability to retard the electrophoretic migration of RNA on a boronate gel after site-specific cleavage with a DNAzyme to generate a 5′-terminal fragment bearing a 2′,3′ cyclic phosphate at its 3′ end. TriP, triphosphorylated RNA; DiP, diphosphorylated RNA; MonoP, monophosphorylated RNA. p, phosphate; Gppp, unmethylated cap; OH, hydroxyl.
Figure 5. Covalent adduct transiently formed between a cap diol and the matrix of a boronate gel.
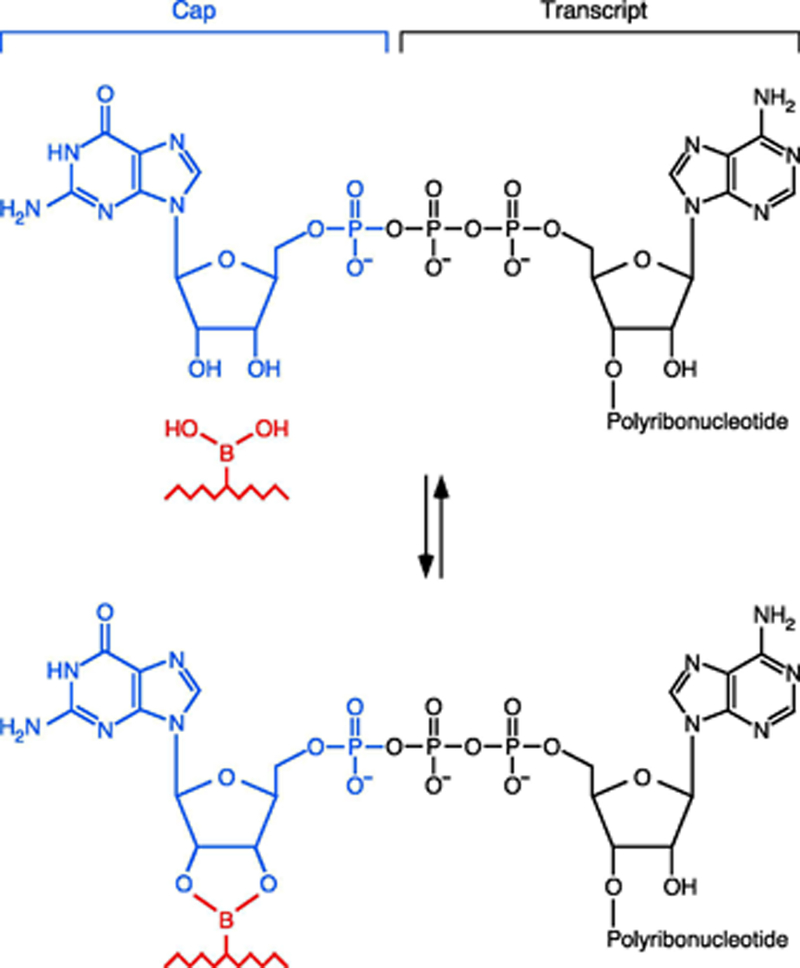
The boronic acid side chains in the polyacrylamide matrix of a boronate gel react reversibly with vicinal diols to form a cyclic diester, thereby reducing the electrophoretic mobility of nucleic acids able to form such a transient adduct. The only vicinal diols in RNA are in the 3′-terminal ribose and any 5′-terminal cap that may be present. Removal of the 3′-terminal vicinal diol by cleavage with a 10–23 DNAzyme generates a 5′ RNA fragment whose electrophoretic migration on a boronate gel will be retarded only if it is capped. Illustrated here is the reversible reaction of a boronic acid side chain of the gel matrix (red) with an unmethylated Gppp cap added by the RNA guanylyltransferase Pce1.
Unfortunately, Pce1 can also react, albeit more slowly, with triphosphorylated 5′ ends to add a Gpppp cap, a property shared by all other RNA guanylyltransferases that have been tested [4, 23, 24]. Therefore, to measure the percentage of a particular cellular RNA that is diphosphorylated, the PACO capping yield must be compared with that for each member of a set of triphosphorylated, diphosphorylated, and monophosphorylated RNA standards identical to the cellular transcript under investigation (Figure 6) so that the small contribution of the other 5′ termini to the overall capping yield can be subtracted mathematically. These standards are synthesized by in vitro transcription with T7 RNA polymerase and then combined with total RNA from a strain that lacks the transcript of interest to simulate the analysis of the endogenous cellular transcript by PACO. The following equation is then used to calculate the fraction of the cellular RNA that is diphosphorylated:
| (2) |
where D is the fraction of diphosphorylated 5′ ends, M is the fraction of monophosphorylated 5′ ends (as determined by PABLO), YC is the measured PACO capping yield, and ECT, ECD, and ECM are the measured capping efficiencies of the triphosphorylated, diphosphorylated, and monophosphorylated standards, respectively, on a scale from 0 (no capping) to 1 (complete capping), as determined from the PACO capping yield for each [4]. A detailed description of how ECT, ECD, and ECM can be calculated and then used to determine D is provided below in section 4.
Figure 6. Representative analysis by PACO based on boronate gel electrophoresis.
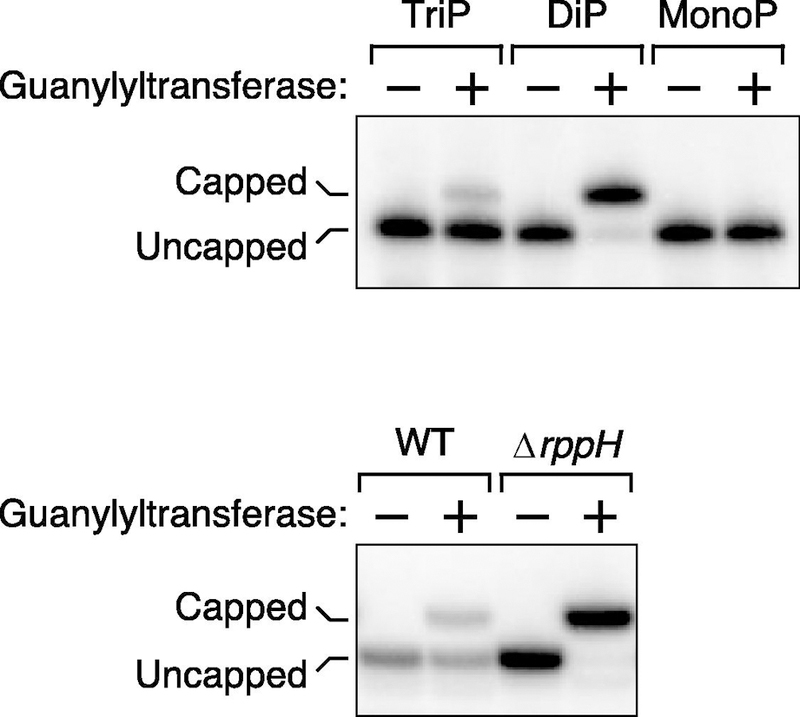
To determine the percentage of E. coli yeiP mRNA that is diphosphorylated, the ability of its 5′ end to be capped by the purified guanylyltransferase Pce1 was deduced from the capacity of such a cap to retard the electrophoretic migration of a 5′ fragment of that transcript on a boronate gel after site-specific cleavage with a DNAzyme.
(Top) Analysis of triphosphorylated (TriP), diphosphorylated (DiP), and monophosphorylated (MonoP) yeiP standards. Each was synthesized by in vitro transcription and analyzed in the presence of total RNA from E. coli cells lacking the yeiP gene to simulate an assay of the endogenous E. coli transcript.
(Bottom) Analysis of cellular RNA. The presence of diphosphorylated yeiP mRNA was examined in wild-type E. coli (WT) and an isogenic mutant lacking the rppH gene, which encodes a protein needed to convert diphosphorylated 5′ ends to monophosphorylated 5′ ends in E. coli.
For reasons that are not understood, the substrate specificity of Pce1 appears to be greater when examined by boronate gel electrophoresis rather than phosphatase protection, as judged from the relative capping efficiency of the RNA standards (ECD >> ECT and ECM = 0 by this improved method). As a result, it is possible to obtain a crude estimate of the fraction of diphosphorylated 5′ ends by using a simplified version of equation 2 that disregards the potential for anything other than diphosphorylated RNA to become capped:
| (3) |
where the fractional capping yield of the diphosphorylated standard can be used as an inexact approximation of ECD. This equation overestimates the fraction of an RNA of interest that is diphosphorylated by (ECT / ECD)(T), where T is the triphosphorylated fraction, a discrepancy that is insignificant when ECT << ECD or T << D.
Equation 2 assumes that all of the RNA of interest is either monophosphorylated, diphosphorylated, or triphosphorylated. If any is capped in vivo, the equation must be modified accordingly:
| (4) |
where C is the fraction of 5′ ends capped in vivo and YC is the fraction of 5′ ends capped by Pce1 in vitro. A negative PACO control in which the cellular RNA sample is subjected to boronate gel electrophoresis without prior treatment with Pce1 will reveal whether any is capped in vivo while also quantifying that fraction.
3.1. Protocol: PACO
Two identical cellular RNA samples should be analyzed in parallel: one to which Pce1 is added and one to which it is not added (negative control). A cognate set of triphosphorylated, diphosphorylated, and monophosphorylated RNA standards synthesized by in vitro transcription must also be analyzed. For statistical purposes, each should be examined in triplicate.
Capping of RNAs just 30–60 nt in length can be examined on a boronate gel containing a high percentage of polyacrylamide without the need for DNAzyme cleavage.
Recombinant Schizosaccharomyces pombe Pce1 can be purified from E. coli as previously described [4, 25].
3-acrylamidophenylboronic acid can be purchased commercially or chemically synthesized from 3-aminophenylboronic acid and acryloyl chloride [26].
Combine total cellular RNA (5 μg) with Pce1 (100 nM) in a solution (20 μl) containing Tris-HCl, pH 7.5 (50 mM), dithiothreitol (5 mM), GTP (1 mM), MgCl2 (1 mM), and rRNasin (2 units/μl; Promega). In parallel, substitute a triphosphorylated, diphosphorylated, or monophosphorylated RNA standard (5 ng) mixed with total cellular RNA (5 μg) from a mutant strain that either lacks the RNA of interest or, if the gene is essential, contains a translationally synonymous mutation that prevents DNAzyme cleavage of the endogenous transcript.
Warm the mixture to 37°C, and allow capping to proceed for 4 min.
Stop the reaction by adding EDTA (195 μl of a 4 mM solution). Phenol extract and ethanol precipitate the reaction products. cleavage products.
Combine the reaction products with a transcript-specific 10–23 DNAzyme (11 μM) in a total volume of 36 μl.
Heat the mixture to 85°C for 5 min, slowly cool it to 30°C over a period of 60–90 min, and then place it on ice.
Add a solution (4 μl) containing Tris-HCl, pH 7.5 (500 mM) and MgCl2 (100 mM).
Allow DNAzyme cleavage to proceed for 2 hr at 37°C.
Stop the reaction by adding EDTA (195 μl of a 4 mM solution), and ethanol precipitate the cleavage products.
Dissolve the DNAzyme cleavage products in 10 μl of water and an equal volume of gel loading solution (95% formamide, 20 mM EDTA, 0.0125% (w/v) bromophenol blue, and 0.0125% (w/v) xylene cyanol), heat them to 95°C for 5 min, and subject them to electrophoresis on a boronate gel [22] polymerized from 5.6% acrylamide:bisacrylamide (19:1) and 0.25% 3- acrylamidophenylboronic acid in 0.1 M Tris-acetate (pH 9.0) and 7 M urea. Use 0.1 M Trisacetate (pH 9.0) as the running buffer, and apply a low voltage (~8 V/cm) to prevent the gel from overheating.
Electroblot the gel onto a membrane (Hybond-XL, GE Healthcare or Immobilon-NY+, Millipore), UV-crosslink the blot, and probe the blot with a 32 P-labeled oligonucleotide complementary to the RNA of interest.
Visualize the radioactive bands on the blot with a phosphorimager, and quantify their relative intensities.
3.2. Protocol: synthesis of RNA standards
These in vitro transcripts should be identical in length and sequence to the cellular transcript for which they are serving as standards.
The template for in vitro transcription with T7 RNA polymerase must encode an RNA in which G is the first or second nucleotide. If neither is G in the cellular transcript of interest, substitute G at the second position of the standards.
For G-initiated RNAs, a T7 ϕ6.5 promoter should be used. For RNAs that begin with a nucleotide other than G, a T7 ϕ2.5 promoter should be used [27].
-
1a.
Triphosphorylated RNA is synthesized by in vitro transcription of the DNA template (10–20 nM) with T7 RNA polymerase (150 units; New England Biolabs) in a solution (40 µl) containing Tris-HCl, pH 7.9 (40 mM), MgCl2 (6 mM), spermidine (2 mM), and dithiothreitol (1 mM), as well as ATP, GTP, CTP, and UTP (1 mM each). To facilitate visualization during subsequent gel purification, a small amount of fluorescein-12-UTP can also be added.
-
1b.
Diphosphorylated or monophosphorylated RNA is synthesized by in vitro transcription with T7 RNA polymerase in the presence of an 18–30 fold molar excess of XDP (4.4–7.5 mM) or XMP (7.5 mM), respectively, over XTP (0.25 mM), where X is the first nucleotide of the transcript. The concentration of the other three NTPs should remain 1 mM each.
-
2.
Allow transcription to proceed for 8 hr at 37°C before quenching the reaction with an equal amount (40 µl) of gel loading solution (95% formamide, 20 mM EDTA, 0.0125% (w/v) bromophenol blue, and 0.0125% (w/v) xylene cyanol). Heat the samples to 95°C for 5 min, and subject them to electrophoresis on a 4.5–6.0% polyacrylamide, 7 M urea gel.
-
3.
Visualize the RNA band (e.g., by UV illumination to detect its fluorescein label), and excise a gel fragment containing the RNA.
-
4.
Crush the gel fragment, and extract it overnight at room temperature in 1 mM EDTA (500 µl).
-
5.
Separate the aqueous phase, and ethanol precipitate the RNA.
4. Quantitative analysis of PACO data
To analyze data obtained by PACO, one must first calculate the capping efficiencies of the triphosphorylated, diphosphorylated, and monophosphorylated standards (ECT, ECD, and ECM, respectively). Those values can then be used with equation 2 or 4 to determine the percentage of the corresponding cellular RNA that is disphosphorylated.
Capping efficiencies.
When determined by boronate gel electrophoresis, the capping efficiencies of the standards are calculated as follows:
| (5) |
| (6) |
| (7) |
where YCT, YCD, and YCM are the PACO capping yields measured for the triphosphorylated, diphosphorylated, and monophosphorylated standards, respectively, and DS and MS are the fraction of those standards that are diphosphorylated or monophosphorylated, as estimated from the molar ratio of NDP or NMP to the corresponding NTP during their synthesis by in vitro transcription [4].
By using this set of equations, the capping efficiencies of the triphosphorylated, diphosphorylated, and monophosphorylated standards can be determined by direct calculation. However, the complexity of equations 6 and 7 makes it difficult to calculate the standard deviations of ECD and ECM directly from the mean values and standard deviations of the original measurements. These errors can nevertheless be determined by Monte Carlo simulation, as follows. The mean values and standard deviations of the measurements are first used to generate random Gaussian sample sets of 100,000 values for each of the variables, which are then used to calculate ECT, ECD, and ECM. For each simulation, a histogram is constructed and fit to a spline function, and the mode (peak value) is determined. Because of the asymmetry of the distribution, an asymmetrical error equivalent to one standard deviation can then be calculated by integrating outward from the mode until a confidence level of 68.3% is attained.
Fraction of an RNA that is diphosphorylated.
Once the capping efficiencies of the standards have been calculated, they and the data from PACO and PABLO measurements on cellular RNA can be used with equation 2 or 4 (see above) to determine the fraction of the corresponding cellular transcript that is diphosphorylated. The fraction of diphosphorylated 5′ ends can be calculated even more precisely by taking into account the small percentage of those diphosphorylated termini that undergo PABLO ligation (typically <5%) and are therefore mistakenly tallied as monophosphorylated ends:
| (8) |
where M is the fraction of monophosphorylated 5′ ends, D is the fraction of diphosphorylated 5′ ends, YL is the measured PABLO ligation yield, ELM is the ligation efficiency of the monophosphorylated standard, and ELD is the much smaller ligation efficiency of the diphosphorylated standard [4]. As a result of this refinement, the D and M calculations become interdependent, making it necessary to calculate these values iteratively until each achieves convergence (YL / ELM can be used as an initial estimate of M).
In this manner, the fraction of 5′ ends that are diphosphorylated or monophosphorylated can be determined by direct calculation, but once again Monte Carlo simulation is needed to obtain errors for these values. This can be achieved as described for the capping efficiencies by using the distribution of values calculated for ECT, ECD, and ECM and random Gaussian sample sets generated from the means and standard deviations of the measured parameters to determine the mode and asymmetrical standard deviation of D and M.
Fraction of an RNA that is triphosphorylated.
By process of elimination, any 5′ ends found not to be monophosphorylated, diphosphorylated, or capped are likely to bear a triphosphate or, less commonly, a hydroxyl. Triphosphorylated 5′ termini can be distinguished from their hydroxylated counterparts by their ability to undergo splinted ligation after pyrophosphatase treatment.
5. Comparison of phosphate enumeration methods
The various methods for obtaining and analyzing PACO and PABLO data are compared in Figure 7 for a representative RNA, the E. coli yeiP transcript. The percentage of this mRNA that is monophosphorylated or diphosphorylated in wild-type E. coli and in an isogenic mutant lacking the rppH gene was calculated from the experimental results shown in Figures 3 and 6 either directly or by Monte Carlo simulation, using equations 2 and 8. These percentages were also calculated by making the simplifying assumption that the splinted ligation and capping reactions are entirely specific for monophosphorylated or diphosphorylated RNA, respectively, which allowed equations 1 and 3 to be used without iteration or Monte Carlo simulation. Also graphed are the percentages of monophosphorylated and diphosphorylated yeiP previously determined from PACO analyses based on phosphatase protection [4].
Figure 7. Comparison of methods for acquiring and analyzing PABLO and PACO data.
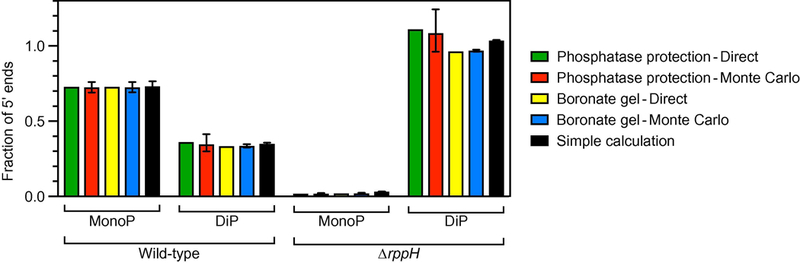
The phosphorylation state of the 5′ end of yeiP mRNA with a U-to-G substitution at the second nucleotide was examined in wild-type E. coli (WT) and an isogenic ∆rppH mutant by PABLO (Figure 3) and by two PACO procedures based on phosphatase protection [4] or boronate gel electrophoresis (Figure 6). In each case, the percentage of cellular transcripts that were monophosphorylated (MonoP) or diphosphorylated (DiP) was determined by iterative direct calculation (green and yellow bars) or Monte Carlo simulation (red and blue bars), only the latter of which allowed errors to be calculated (error bar = one standard deviation). In addition, the percentage of yeiP mRNA that was monophosphorylated or diphosphorylated was calculated from the data acquired by PABLO and PACO (boronate gel electrophoresis only) by using equations 1 and 3 (black bars), simplified formulas that disregard small deficiencies in the specificity of splinted ligation and capping.
Similar results were obtained in every case, demonstrating both the interchangeability of the two PACO procedures and the feasibility of the streamlined approach to data analysis. The high specificity of splinted ligation (i.e., the negligible ligation of diphosphorylated RNA) makes it possible to confidently calculate the percentage of monophosphorylated 5′ ends by the simple approach (M = YL / ELM) without the need for further refinement. The improved method for PACO based on boronate gel electrophoresis is more accurate than the original method based on phosphatase protection, and the specificity of this new procedure for diphosphorylated RNA is sufficiently high that the simple approach to data analysis (D ≈ YC / ECD, where the fractional capping yield of the diphosphorylated standard serves as an approximation of ECD) provides a satisfactory if imperfect estimate of the percentage of 5′ ends that are diphosphorylated, especially if the percentage of triphosphorylated 5′ ends is small. Moreover, the simplicity of the ratios (Y/E) in equations 1 and 3 allows standard deviations (σ) for M and D to be calculated from the raw data by straightforward error propagation, obviating the need for Monte Carlo simulation:
| (9) |
6. Distinguishing NAD caps from m7Gppp caps by boronate gel electrophoresis
The recent discovery of NAD caps (more precisely, nicotinamide mononucleotide caps) on eukaryotic transcripts [10, 11] makes it important to be able to distinguish them easily from conventional m7Gppp caps in the same cells. For individual RNAs, this can conveniently be accomplished by boronate gel electrophoresis after site-specific DNAzyme cleavage to generate a short (50–120 nt) 5′ fragment with a 2′,3′-cyclic phosphate at its 3′ end (see section 3). Although both types of cap contain a vicinal diol that can transiently form a covalent adduct with the boronic acid side chains of the gel matrix, thereby reducing electrophoretic mobility, NAD caps are more effective than m7Gppp caps at retarding migration on such gels (Figure 8). Consequently, the percentage of a given cellular transcript that is NAD-capped, m7Gppp-capped, or uncapped can be determined very accurately from the ratio of band intensities in a single lane of a boronate gel examined by Northern blotting beside a set of standards synthesized by in vitro transcription.
Figure 8. Differential electrophoretic mobility of RNA bearing an m7Gppp cap or NAD cap on a boronate gel.
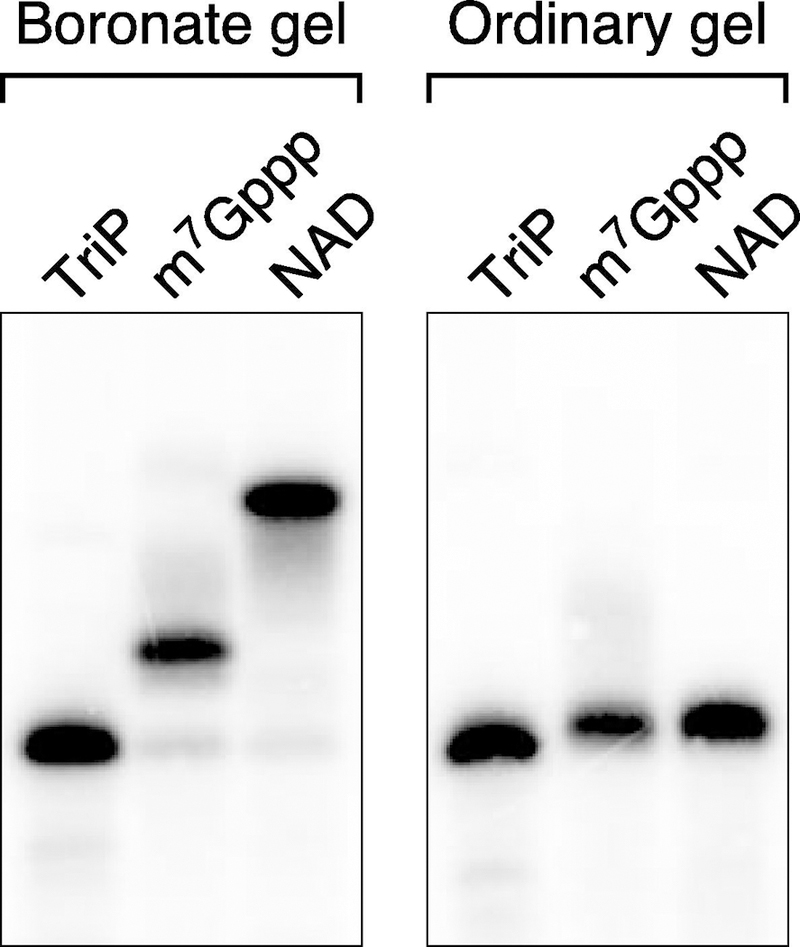
Otherwise identical yeiP RNAs bearing a 5′ triphosphate (TriP), m7 Gppp cap, or NAD cap were synthesized by in vitro transcription, cleaved 69 nt from the 5′ end with the same DNAzyme, and analyzed by Northern blotting after electrophoresis on a boronate gel (left) or an ordinary polyacrylamide gel (right).
6.1. Protocol: distinguishing caps by boronate gel electrophoresis
To ensure accurate band designation, the cellular RNA of interest should be analyzed beside a set of cognate RNA standards bearing an m7 Gppp cap, NAD cap, or 5′ triphosphate. The capped standards can be synthesized by in vitro transcription with T7 RNA polymerase in the presence of m7 GpppA, m7 GpppG, or NAD (see protocol 3.2). For statistical purposes, the cellular RNA should be examined in triplicate.
To distinguish the electrophoretic effects of capping from those caused by heterogeneity in the site of transcription initiation, the DNAzyme-cleaved RNA should also be examined on an ordinary polyacrylamide gel.
Capped RNAs just 30–60 nt in length can be examined on a boronate gel containing a high percentage of polyacrylamide without the need for DNAzyme cleavage.
3-acrylamidophenylboronic acid can be purchased commercially or chemically synthesized from 3-aminophenylboronic acid and acryloyl chloride [26].
Combine total cellular RNA (5–10 µg) with a transcript-specific 10–23 DNAzyme (11 µM) and water in a volume of 36 µl.
Heat the mixture to 85°C for 5 min, slowly cool it to 30°C over a period of 60–90 min, and then place it on ice.
Add a solution (4 µl) containing Tris-HCl, pH 7.5 (500 mM), MgCl2 (100 mM), and dithiothreitol (100 mM).
Allow DNAzyme cleavage to proceed for 2 hr at 37°C.
Stop the reaction by adding EDTA (195 µl of a 4 mM solution), and ethanol precipitate the cleavage products.
Dissolve the DNAzyme cleavage products in 10 µl of water and an equal volume of gel loading solution (95% formamide, 20 mM EDTA, 0.0125% (w/v) bromophenol blue, and 0.0125% (w/v) xylene cyanol), heat them to 95°C for 5 min, and subject them to electrophoresis on a boronate gel [22] polymerized from 5.6% acrylamide:bisacrylamide (19:1) and 0.25% 3-acrylamidophenylboronic acid in 0.1 M Tris-acetate (pH 9.0) and 7 M urea. Use 0.1 M Tris-acetate (pH 9.0) as the running buffer, and apply a low voltage (~8 V/cm) to prevent the gel from overheating.
Electroblot the gel onto a membrane (Hybond-XL, GE Healthcare or Immobilon-NY+, Millipore), UV-crosslink the blot, and probe the blot with a 32P-labeled oligonucleotide complementary to the RNA of interest.
Visualize the radioactive bands on the blot with a phosphorimager, and quantify their relative intensities.
7. Concluding remarks
The methods described above make it possible to thoroughly characterize the 5′ end of individual RNAs of interest, revealing the number of phosphates or type of cap there. The precision of these methods allows the detailed profiling even of 5′ termini whose location or state of phosphorylation/capping is heterogeneous. By disregarding minor deficiencies in the specificity of PABLO and PACO, data analysis can be simplified considerably without greatly impairing the accuracy of the calculations. Requiring little expertise beyond Northern blotting, in vitro transcription, and recombinant protein purification by affinity chromatography, these procedures should be experimentally accessible to a variety of researchers with diverse backgrounds.
Highlights.
5′-terminal modifications are critical for the function and decay of cellular RNAs.
The percentage of monophosphorylated 5′ ends can be quantified by splinted ligation.
The percentage of diphosphorylated 5′ ends can be quantified by in vitro capping.
Different types of 5′ caps can be distinguished by boronate gel electrophoresis.
Together, these assays allow the detailed characterization of any RNA 5′ end.
Acknowledgments
We are grateful to Helena Celesnik for her pioneering development of PABLO methodology and to Kevin Belasco for devising methods for Monte Carlo simulation. This research was supported by a fellowship to D.J.L. (T32AI007180) and grants to J.G.B. (R01GM035769 and R01GM123124) from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Mackie GA, Ribonuclease E is a 5’-end-dependent endonuclease, Nature 395 (1998) 720–723. [DOI] [PubMed] [Google Scholar]
- [2].Deana A, Celesnik H, Belasco JG, The bacterial enzyme RppH triggers messenger RNA degradation by 5’ pyrophosphate removal, Nature 451 (2008) 355–358. [DOI] [PubMed] [Google Scholar]
- [3].Richards J, Liu Q, Pellegrini O, Celesnik H, Yao S, Bechhofer DH, Condon C, Belasco JG, An RNA pyrophosphohydrolase triggers 5’-exonucleolytic degradation of mRNA in Bacillus subtilis, Mol. Cell 43 (2011) 940–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Luciano DJ, Vasilyev N, Richards J, Serganov A, Belasco JG, A novel RNA phosphorylation state enables 5’ end-dependent degradation in Escherichia coli, Mol. Cell 67 (2017) 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ramanathan A, Robb GB, Chan SH, mRNA capping: biological functions and applications, Nucleic Acids Res 44 (2016) 7511–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shuman S, Structure, mechanism, and evolution of the mRNA capping apparatus, Prog. Nucleic Acid Res. Mol. Biol 66 (2001) 1–40. [DOI] [PubMed] [Google Scholar]
- [7].Nagarajan VK, Jones CI, Newbury SF, Green PJ, XRN 5’→3’ exoribonucleases: structure, mechanisms and functions, Biochim. Biophys. Acta 1829 (2013) 590–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Grudzien-Nogalska E, Kiledjian M, New insights into decapping enzymes and selective mRNA decay, WIREs RNA 8 (2017) e1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cahová H, Winz ML, Höfer K, Nübel G, Jäschke A, NAD captureSeq indicates NAD as a bacterial cap for a subset of regulatory RNAs, Nature 519 (2015) 374–377. [DOI] [PubMed] [Google Scholar]
- [10].Walters RW, Matheny T, Mizoue LS, Rao BS, Muhlrad D, Parker R, Identification of NAD+ capped mRNAs in Saccharomyces cerevisiae, Proc. Natl. Acad. Sci. USA 114 (2017) 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jiao X, Doamekpor SK, Bird JG, Nickels BE, Tong L, Hart RP, Kiledjian M, 5’ end nicotinamide adenine dinucleotide cap in human cells promotes RNA decay through DXO-mediated deNADding, Cell 168 (2017) 1015–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Frindert J, Zhang Y, Nübel G, Kahloon M, Kolmar L, Hotz-Wagenblatt A, Burhenne J, Haefeli WE, Jäschke A, Identification, biosynthesis, and decapping of NAD-capped RNAs in B. subtilis, Cell Rep 24 (2018) 1890–1901. [DOI] [PubMed] [Google Scholar]
- [13].Celesnik H, Deana A, Belasco JG, Initiation of RNA decay in Escherichia coli by 5’ pyrophosphate removal, Mol. Cell 27 (2007) 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Celesnik H, Deana A, Belasco JG, PABLO analysis of RNA: 5’-phosphorylation state and 5’-end mapping, Methods Enzymol 447 (2008) 83–98. [DOI] [PubMed] [Google Scholar]
- [15].Santoro SW, Joyce GF, A general purpose RNA-cleaving DNA enzyme, Proc. Natl. Acad. Sci. USA 94 (1997) 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cairns MJ, King A, Sun LQ, Optimisation of the 10–23 DNAzyme-substrate pairing interactions enhanced RNA cleavage activity at purine-cytosine target sites, Nucleic Acids Res 31 (2003) 2883–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu X, Gorovsky MA, Mapping the 5’ and 3’ ends of Tetrahymena thermophila mRNAs using RNA ligase mediated amplification of cDNA ends (RLM-RACE), Nucleic Acids Res 21 (1993) 4954–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Richards J, Luciano DJ, Belasco JG, Influence of translation on RppH-dependent mRNA degradation in Escherichia coli, Mol. Microbiol 86 (2012) 1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Song MG, Bail S, Kiledjian M, Multiple Nudix family proteins possess mRNA decapping activity, RNA 19 (2013) 390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pei Y, Shuman S, Interactions between fission yeast mRNA capping enzymes and elongation factor Spt5, J. Biol. Chem 277 (2002) 19639–19648. [DOI] [PubMed] [Google Scholar]
- [21].Igloi GL, Kössel H, Affinity electrophoresis for monitoring terminal phosphorylation and the presence of queuosine in RNA. Application of polyacrylamide containing a covalently bound boronic acid, Nucleic Acids Res 13 (1985) 6881–6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Igloi GL, Kössel H, Use of boronate-containing gels for electrophoretic analysis of both ends of RNA molecules, Methods Enzymol 155 (1987) 433–448. [DOI] [PubMed] [Google Scholar]
- [23].Yu L, Shuman S, Mutational analysis of the RNA triphosphatase component of vaccinia virus mRNA capping enzyme, J. Virol 70 (1996) 6162–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen D, Luongo CL, Nibert ML, Patton JT, Rotavirus open cores catalyze 5’-capping and methylation of exogenous RNA: evidence that VP3 is a methyltransferase, Virology 265 (1999) 120–130. [DOI] [PubMed] [Google Scholar]
- [25].Doamekpor SK, Sanchez AM, Schwer B, Shuman S, Lima CD, How an mRNA capping enzyme reads distinct RNA polymerase II and Spt5 CTD phosphorylation codes, Genes Dev 28 (2014) 1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ivanov AE, Larsson H, Galaev IY, Mattiasson B, Synthesis of boronate-containing copolymers of N,N-dimethylacrylamide, their interaction with poly(vinyl alcohol) and rheological behaviour of the gels, Polymer 45 (2004) 2495–2505. [Google Scholar]
- [27].Coleman TM, Wang G, Huang F, Superior 5’ homogeneity of RNA from ATP-initiated transcription under the T7 φ2.5 promoter, Nucleic Acids Res 32 (2004) e14. [DOI] [PMC free article] [PubMed] [Google Scholar]