

Abstract
Noninvasive biomarkers are needed to monitor stable patients after kidney transplant (KT), because subclinical acute rejection (subAR), currently detectable only with surveillance biopsies, can lead to chronic rejection and graft loss. We conducted a multicenter study to develop a blood‐based molecular biomarker for subAR using peripheral blood paired with surveillance biopsies and strict clinical phenotyping algorithms for discovery and validation. At a predefined threshold, 72% to 75% of KT recipients achieved a negative biomarker test correlating with the absence of subAR (negative predictive value: 78%‐88%), while a positive test was obtained in 25% to 28% correlating with the presence of subAR (positive predictive value: 47%‐61%). The clinical phenotype and biomarker independently and statistically correlated with a composite clinical endpoint (renal function, biopsy‐proved acute rejection, ≥grade 2 interstitial fibrosis, and tubular atrophy), as well as with de novo donor-specific antibodies. We also found that <50% showed histologic improvement of subAR on follow-up biopsies despite treatment and that the biomarker could predict this outcome. Our data suggest that a blood-based biomarker that reduces the need for the indiscriminate use of invasive surveillance biopsies and that correlates with transplant outcomes could be used to monitor KT recipients with stable renal function, including after treatment for subAR, potentially improving KT outcomes.
Keywords: alloantibody, biomarker, clinical research/practice, clinical trial, genomics, kidney transplantation/nephrology, rejection: subclinical, translational research/science
1. INTRODUCTION
Kidney transplant (KT) remains the treatment of choice for most patients with end-stage kidney disease (ESRD),1,2 but long-term outcomes remain suboptimal.3,4 After KT, clinically unsuspected subclinical acute rejection (subAR) occurs in 20% to 25% of patients in the first 12 to 24 months and is associated with de novo donor-specific antibody (dnDSA) formation, interstitial fibrosis/tubular atrophy (IFTA), chronic rejection, and graft loss.5–13 Serum creatinine and immunosuppression levels, used almost exclusively to monitor KT recipients, are both insensitive and nonspecific.14 Surveillance biopsies can be used to monitor patients with stable renal function, but biopsies are invasive and associated with sampling error and there is a lack of consensus around both histologic interpretation (especially for “borderline changes”) and the effectiveness of treatment.5,15–18 Moreover, the vast majority (75%−80%) of surveillance biopsy specimens show normal histology (ie, the absence of subAR) and, therefore, expose patients to unnecessary biopsy risks. As a result, the current standard of care in monitoring patients after KT ranges from not using surveillance biopsies at all, to using them selectively in “high-immunologic risk” patients, to routine use in all patients.19 There is, therefore, a clear need to better detect the presence or absence of subAR, and genomic biomarkers in the blood or urine may provide useful noninvasive monitoring of KT recipients.17,20,21
The Clinical Trials in Organ Transplantation 08 (CT0T-08; NCT01289717) study was designed to develop molecular biomarkers for a number of clinical phenotypes in KT recipients. The focus of the current study was to develop and evaluate the performance and clinical validity of a novel gene expression profile biomarker that correlates with the presence or absence of subAR in the peripheral blood in patients with stable renal function after KT.
2. MATERIALS AND METHODS
We enrolled 307 subjects after KT into a multicenter 24-month observational study (CTOT-08) between March 2011 and May 2014. KT recipients underwent surveillance biopsies at 2 to 6, 12, and 24 months after KT as well as for-cause biopsies for acute renal dysfunction (Figure S1). Participating sites that routinely perform surveillance biopsies were geographically selected to provide racial and ethnic diversity. Study inclusion criteria were: male or female KT recipients (negative pregnancy test within 6 weeks of enrollment), age ≥18 years, ability to provide informed consent, and recipient of a first or subsequent KT from either a deceased or a living donor. Subject with combined or “en bloc” kidney grafts and subjects with HIV or hepatitis C virus infection were excluded.
We contemporaneously enrolled KT recipients into the Northwestern University (NU) transplant program’s biorepository study (NCT01531257), with eligibility criteria identical to those of CT0T-08. Patients undergo surveillance biopsies at NU with a frequency similar to that of CT0T-08. Patients who did not participate in CT0T-08 were enrolled into the NU biorepository study.
Clinical care followed a standard practice at each participating center. However, CT0T-08 subjects diagnosed with subAR on a surveillance biopsy, who were managed based on each site’s interpretation of the histopathology results, also underwent an intensive monitoring protocol consisting of blood sample collection every 2 weeks for laboratory and biomarker determination and a repeat biopsy at week 8. The intense monitoring (IM) was limited to 1 subAR episode per subject due to the need for a repeat biopsy.
All biopsy specimens were processed locally for routine histology, Simian virus 40, and C4d staining and were centrally read and interpreted by a pathologist using Banff 2007 criteria who was blinded to the clinical course.22 Clinical phenotypes (CPs) were assigned by the Data Coordinating Center (DCC at Rho Federal Systems) for both the discovery and validation cohorts for each paired sample by using the following predefined algorithm:
SubAR: histology on a surveillance biopsy consistent with acute rejection (≥Banff borderline cellular rejection and/or antibody-mediated rejection) AND stable renal function, defined as serum creatinine <2.3 mg/dL and <20% increase in creatinine compared with a minimum of 2 or 3 prior values over a mean period and range of 132 and 75–187 days, respectively.
Transplant excellent (TX [ie, no subAR]): normal histology on surveillance biopsy (no evidence of rejection: Banff i = 0 and t = 0, g = 0, ptc = 0; ci = 0 or 1 and ct = 0 or 1) AND stable renal function as defined in item 1.
While a previous study has shown that KT recipients with a serum creatinine level >1.5 mg/dL in the first 12 months have worse outcomes compared with those with levels of <1.5 mg/dL,23 we chose an upper limit of 2.3 mg/dL for 3 reasons: (1) we included a second criterion to ensure stability (<20% change in serum creatinine compared with the minimum of the previous 2 or 3 samples), (2) the follow-up in our study was 24 vs 12 months, and (3) there were very few samples in the group with higher creatinine levels, but all met our histology criteria (Banff ci and ct score = 0 or 1, respectively, and no other findings), eliminating other ongoing causes of renal injury. For these reasons, we did not perform sensitivity analyses for this small group but thought that these patients should be included because they reflect “real-life” patients and because eliminating them from our analyses might have introduced selection bias.
Thus, peripheral blood samples were collected at the time of each surveillance biopsy from CTOT-08 subjects. At the time of CTOT-08 analysis, we searched the biorepository for, and identified all peripheral blood samples paired with, surveillance biopsies available for validation studies in this independent cohort. All blood samples were drawn directly into PAXgene (BD BioSciences, San Jose, CA) tubes and processed as previously described24 in batches by using Affymetrix HT HG-U133+PM Array Plates on the Gene Titan MC instrument (Thermo Fisher Scientific, Waltham, MA) (GEO Accession No. GSE107509). Background correction based on the discovery data set were saved and applied to all discovery and validation samples by using frozen robust multiarray analysis.25 Figure S2 illustrates the workflow used for the discovery of the gene expression profile (GEP) and for assessment of biologic relevance.26–33 Threshold selection was based on “out-of-bag” performance metrics of the discovery cohort. Based on dichotomous outcomes (either positive or negative predicted probabilities above or below the threshold), profiles were compared with the clinical phenotypes to determine the performance of the classifiers. The independent NU biorepository cohort was used to externally validate the locked model/threshold discovered on the CTOT-08 cohort.
While sample-level predefined algorithms were used to define the CPs of either subAR or TX for each paired sample outlined here, CTOT-08 subjects underwent multiple surveillance biopsies during the 24-month study. Subjects demonstrated either subAR or TX phenotypes for any given sample but may have demonstrated instances of each phenotype in different samples over time. Thus, for the purpose of correlating biomarker results to CP, we classified samples used for biomarker development as either subAR or TX (sample level), whereas for the purpose of correlating both the CP and the biomarker classification to clinical outcomes, we stratified subjects into 3 phenotypic groups (subject level): subjects with surveillance biopsy specimens demonstrating subAR only (no TX), subjects with surveillance biopsy specimens demonstrating TX only (no subAR), and subjects with individual biopsy specimens demonstrating either subAR or TX. This third group, therefore, consisted of subjects who had experienced ≥1 instance of subAR and ≥1 instance of TX during the study period. Nonsurveillance biopsies (for cause) and surveillance biopsies with other findings (eg, recurrent disease, infections) were not included in this analysis.
To assess whether subjects who experienced subAR or who had a positive biomarker test had worse transplant outcomes, we used a primary clinical composite endpoint (CCE) consisting of 3 separate validated endpoints, all previously used in other studies to measure transplant outcomes:
24-Month biopsy (central read) showing evidence of chronic injury—IFTA (Banff grade ≥II IFTA [ci ≥2 or ct ≥2], OR
BPAR on any “for-cause biopsy” (central read), OR
Decrease in estimated glomerular filtration rate by >10 mL/ min/1.73 m2 (Chronic Kidney Disease Epidemiology Collaboration) between 4 and 24 months posttransplant
We also measured dnDSAs for both class I and II, known to associate with transplant outcome, as determined by each participating site per their practice; these were recorded as either positive or negative according to each site’s cutoff values. The study protocol required determinations at the time of the 12-and 24-month biopsies, but other values obtained at any time during the study were also used for our analyses.
To assess the impact of both CP and GEP on transplant outcome in the first 12 months (clinical composite or individual endpoints), at 24 months, we used odds ratios (ORs) and the Fisher exact test. The 2-sample t test was used to assess the ability of GEP-predicted probabilities during IM to detect resolution of subAR based on the repeat biopsy. Analysis of covariance was used to adjust for differences in predicted probabilities at baseline.
The CTOT-08 and NU biorepository studies were both subject to IRB approval, and informed consent was obtained from all patients. Oversight by the DCC included development of the study protocol, classification of CPs at the sample and patient levels, review of clinical site visits, monitoring of clinical data for integrity, review of clinical site visits, independent validation of all analyses related to clinical profile and GEP, associations between the CPs and endpoints, and manuscript preparation.
3. RESULTS
Table 1 shows the donor and recipient subject-level demographics by CP for both the CTOT-08 and NU biorepository patient cohorts. There were no discernable differences in demographics including type of immunosuppression between the 2 cohorts. Of the CTOT-08 subjects, 13.0% demonstrated only subAR (no TX), 57.7% demonstrated only TX (no subAR), and 29.2% demonstrated individual phenotypic instances of either subAR or TX (ie, ≥1 instance of subAR during the 24-month study). Thus, at the subject level, the CTOT-08, Clinical Trials in Organ Transplantation 08; NU, Northwestern University; subAR, subclinical acute rejection; TX, transplant excellent; ESRD, end-stage renal disease; PKD, polycystic kidney disease; CMV, cytomegalovirus; MMF, mycophenolate mofetil; mTOR, mammalian target of rapamycin.
TABLE 1.
Donor and recipient patient-level demographics and prevalence of clinical phenotypes for discovery and validation cohorts
| Demographics | CTOT-08 cohort (N = 253) |
NU cohort (N= 129) |
|||
|---|---|---|---|---|---|
| subAR, no TX (n = 33) | TX, no subAR (n = 146) | subAR and TX (n = 74) | subAR, no TX (n = 36) | TX, no subAR (n = 93) | |
| Donors | |||||
| Age, y | |||||
| Mean ± SD | 39.0 ± 15.57 | 38.1 ± 13.49 | 43.1 ± 13.30 | 40.7 ± 13.63 | 38.6 ± 13.28 |
| Range | 10–66 | 8–71 | 6–71 | 13–73 | 13–73 |
| Male sex | 17 (51.5) | 75 (51.4) | 39 (52.7) | 23 (63.9) | 46 (49.5) |
| Race | |||||
| White | 26 (78.8) | 98 (67.1) | 59 (79.7) | 23 (63.9) | 52 (55.9) |
| Black or African American | 2 (6.1) | 23 (15.8) | 2 (2.7) | 5 (13.9) | 15 (16.1) |
| Other | 1 (3.0) | 6 (4.1) | 4 (5.4) | 8 (22.2) | 25 (26.9) |
| Unknown or not reported | 4 (12.1) | 19 (13.0) | 9 (12.2) | 0 | 1 (1.1) |
| Ethnicity | |||||
| Hispanic or Latino | 5 (15.2) | 19 (13.0) | 11 (14.9) | 6 (16.7) | 21 (22.6) |
| Not Hispanic or Latino | 25 (75.8) | 110 (75.3) | 56 (75.7) | 30 (83.3) | 71 (76.3) |
| Unknown or not reported | 3 (9.1) | 17 (11.6) | 7 (9.5) | 0 | 1 (1.1) |
| Recipients | |||||
| Age, y | |||||
| Mean ± SD | 50.1 ± 14.76 | 50.2 ± 13.69 | 53.4 ± 13.53 | 52.1 ± 13.15 | 53.0 ± 12.67 |
| Range | 19–75 | 21–78 | 21–78 | 22–72 | 25–75 |
| Male sex | 22 (66.7) | 94 (64.4) | 51 (68.9) | 22 (61.1) | 52 (55.9) |
| Race | |||||
| White | 23 (69.7) | 87 (59.6) | 51 (68.9) | 21 (58.3) | 49 (52.7) |
| Black or African American | 6 (18.2) | 34 (23.3) | 8 (10.8) | 6 (16.7) | 18 (19.4) |
| Other | 4 (12.1) | 11 (7.5) | 5 (6.8) | 9 (25.0) | 26 (28.0) |
| Unknown or not reported | 0 | 14 (9.6) | 10 (13.5) | 0 | 0 |
| Ethnicity | |||||
| Hispanic or Latino | 2 (6.1) | 27 (18.5) | 12 (16.2) | 7 (19.4) | 15 (16.1) |
| Not Hispanic or Latino | 30 (90.9) | 112 (76.7) | 57 (77.0) | 28 (77.8) | 74 (79.6) |
| Unknown or not reported | 1 (3.0) | 7 (4.8) | 5 (6.8) | 1 (2.8) | 4 (4.3) |
| Deceased donor | 22 (66.7) | 60 (41.1) | 26 (35.1) | 19 (52.8) | 30 (32.3) |
| Primary reason for ESRD | |||||
| Cystic (includes PKD) | 2 (6.1) | 13 (8.9) | 14 (18.9) | 4 (11.1) | 10 (10.8) |
| Diabetes mellitus | 8 (24.2) | 30 (20.5) | 15 (20.3) | 10 (27.8) | 23 (24.7) |
| Glomerulonephritis | 9 (27.3) | 47 (32.2) | 13 (17.6) | 8 (22.2) | 28 (30.1) |
| Hypertension | 4 (12.1) | 29 (19.9) | 12 (16.2) | 7 (19.4) | 18 (19.4) |
| Other | 10 (30.3) | 27 (18.5) | 20 (27.0) | 7 (19.4) | 14 (15.1) |
| Secondary reason for ESRD | |||||
| Cystic (includes PKD) | 0 | 1 (0.7) | 0 | 0 | 1 (1.1) |
| Diabetes mellitus | 0 | 7 (4.8) | 1 (1.4) | 2(5.6) | 2(2.2) |
| Glomerulonephritis | 0 | 7 (4.8) | 2 (2.7) | 3(8.3) | 5 (5.4) |
| Hypertension | 6(18.2) | 14 (9.6) | 2 (2.7) | 4 (11.1) | 15 (16.1) |
| Other | 0 | 9 (6.2) | 2 (2.7) | 0 | 1 (1.1) |
| None reported | 27 (81.8) | 108 (74.0) | 67 (90.5) | 27 (75.0) | 69 (74.2) |
| Recipient PRA at transplant | |||||
| PRA class I % | |||||
| n | 29 | 1O7 | 62 | 36 | 93 |
| Mean ± SD | 7.4 ± 20.59 | 7.9 ± 20.85 | 6.9 ± 20.48 | 20.3 ± 29.41 | 19.5 ± 31.13 |
| Range | 0–100 | 0–100 | 0–96 | 0–89 | 0–99 |
| PRA class II % | |||||
| n | 29 | 1O7 | 61 | 36 | 93 |
| Mean ± SD | 11.3 ± 29.03 | 7.6 ± 21.29 | 6.1 ± 18.52 | 17.4 ± 31.36 | 12.9 ± 25.54 |
| Range | 0–100 | 0–100 | 0–80 | 0–100 | 0–100 |
| PRA single antigen cPRA % | |||||
| n | 26 | 86 | 46 | 36 | 93 |
| Mean ± SD | 32.8 ± 42.06 | 29.4 ± 35.82 | 25.9 ± 35.46 | 18.1 ± 28.51 | 11.9 ± 28.19 |
| Range | 0–100 | 0–99 | 0–100 | 0–91 | 0–98 |
| Donor and recipient CMV status | |||||
| D−, R+ | 3 (9.1) | 25 (17.1) | 16 (21.6) | 11 (30.6) | 18 (19.4) |
| D+, R− | 10 (30.3) | 23 (15.8) | 13 (17.6) | 7 (19.4) | 22 (23.7) |
| D−, R− | 7 (21.2) | 33 (22.6) | 21 (28.4) | 5 (13.9) | 16 (17.2) |
| D+, R+ | 11 (33.3) | 60 (41.1) | 20 (27.0) | 13 (36.1) | 36 (38.7) |
| Donor, recipient, or both not tested | 2 (6.1) | 5 (3.4) | 4 (5.4) | 0 | 1 (1.1) |
| Use of induction therapy | |||||
| Alemtuzumab | 19 (57.6) | 74 (50.7) | 42 (56.8) | 29 (80.6) | 80 (86.0) |
| Antithymocyte globulin | 12 (36.4) | 40 (27.4) | 14 (18.9) | 0 | 0 |
| Basiliximab | 3 (9.1) | 25 (17.1) | 18 (24.3) | 7 (19.4) | 11 (11.8) |
| Use of desensitization therapy | |||||
| Received any desensitization therapy | 0 | 9 (6.2) | 7 (9.5) | 4 (11.1) | 6 (6.5) |
| Use of maintenance therapy | |||||
| Steroid | 24 (72.7) | 71 (48.6) | 50 (67.6) | 13 (36.1) | 27 (29.0) |
| Tacrolimus | 33 (100) | 145 (99.3) | 74 (100) | 30 (83.3) | 89 (95.7) |
| Cyclosporine | 3 (9.1) | 7 (4.8) | 4 (5.4) | 3(8.3) | 2(2.2) |
| Azathioprine | 1 (3.0) | 0 | 0 | 1 (2.8) | 0 |
| MMF | 33 (100) | 143 (97.9) | 74 (100) | 35 (97.2) | 92 (98.9) |
| mTOR inhibitor | 1 (3.0) | 11 (7.5) | 5 (6.8) | 3(8.3) | 2(2.2) |
| Leflunomide | 0 | 2 (1.4) | 2 (2.7) | 0 | 0 |
| Belatacept | 0 | 1 (0.7) | 0 | 0 | 0 |
Values are given as number (%) unless otherwise specified. Of the 253 precisely phenotyped CTOT-08 subjects with stable renal function who underwent ≥1 surveillance biopsy, 33 (13.0%) demonstrated only subAR (no TX), 146 subjects (57.7%) demonstrated only TX (no subAR), and 74 (29.2%) demonstrated individual instances of either subAR or TX (ie, ≥1 instance of subAR during the 24-month study). The subAR only (no instances of TX per surveillance biopsies during the study period) and the subAR or TX groups collectively represent subjects with ≥1 episode of subAR (≥1 subAR). At the patient level, the prevalent incidence of ≥1 biopsyproved instance(s) of subAR was 42.3% (107/253) versus 57.7% for TX only. Subjects in the NU biorepository did not undergo serial sampling, and therefore there were only 2 groups: the sample-level prevalent incidence of subAR was 27.9% (36/129) compared with 72.1% for TX (93/129). prevalent incidence of ≥1 biopsy-proved instance(s) of subAR was 42.3% (107/253) versus 57.7% for TX only. Subjects in the NU biorepository did not undergo serial sampling, and thus there were only 2 groups: the sample-level prevalent incidence of subAR was 27.9% (36/129) compared with 72.1% for TX (93/129).
Figure S3A illustrates the sample-level disposition of paired samples for both discovery and validation cohorts. Of 283 CTOT-08 subjects, 253 had sufficient data to define the CP of either subAR or TX (ie, no subAR). In addition, 138 NU biorepository subjects had undergone surveillance biopsies and met the clinical definitions of either subAR or TX; 129 of 138 met the strict phenotypic algorithm used for CTOT-08 subjects. Figure S3B illustrates the subject-level disposition for both the CP and the GEP used to assess the impact of each on the clinical endpoints.
Figures S4A and S4B illustrate the ingenuity pathway analysis (IPA) results for the CTOT-08 (530) discovery and NU biorepository (129/138) validation cohorts, respectively, demonstrating biologic relevance of the differentially expressed genes used to populate the biomarker model (see later) and shared pathways between the discovery and validation cohorts. Also, in the CTOT-08 data set, Database for Annotation, Visualization and Integrated Discovery (DAVID) was used to identify the T cell receptor pathway as significant (P < .0001) by the Gene Ontology (GO) biological process as well as the canonical T cell receptor Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (P < .001). In both the CT0T-08 and the NU data sets, DAVID was also used to identify the B cell receptor, T cell receptor, and intereukin-2 receptor β-chain pathways as significant by the canonical KEGG pathways (P = .0002, .01, and .03, respectively). In addition, Tables S1A and S1B and Figures S5A and S5B illustrate the preranked gene set enrichment analysis for the CT0T-08 and NU biorepository data sets, further demonstrating biologic relevance of the genes populating the model.
Figure 1 illustrates the characteristics and performance of the random forests discovery model used to develop the biomarker and to select the threshold. We selected a random forests model optimizing for area under the curve (AUC; 0.85); the AUC after bootstrap was 0.84. We then selected a predicted probability threshold of 0.375 based on best overall performance, favoring specificity and negative predictive value (NPV; 87% and 88%) over sensitivity and positive predictive value (PPV; 64% and 61%, respectively). The classifier consisted of 61 probe sets that mapped to 57 genes (Figure S6). We then locked the model at the defined threshold in a blinded fashion in order to externally validate their ability to predict the phenotype of the NU biorepository samples. The results were interpreted dichotomously as either “positive” (ie, correlating with a clinical phenotype of subAR) if the probability exceeded the 0.375 threshold or “negative” (ie, correlating with TX) if ≤0.375. We first validated the classifier on 138 subjects from the NU biorepository (validation set 1) who had undergone surveillance biopsies (NPV 78%, PPV 51%), and we also validated the same locked classifier/threshold on a subset of 129 of 138 subjects who met the strict phenotypic algorithm used for the CT0T-08 study (NPV 80%, PPV 47%) (Table 2). To translate the performance of the biomarker into a narrative more relevant to clinical application, we made a negative call in 72% to 75% of the patients (NPV 78%−88%) versus a positive call (subAR) 25% to 28% of the time (PPV 47%−61%). 0f note, the total number of positive or negative tests closely mirrored the prevalent incidence of the subAR and TX CPs in the validation cohorts.
FIGURE 1.
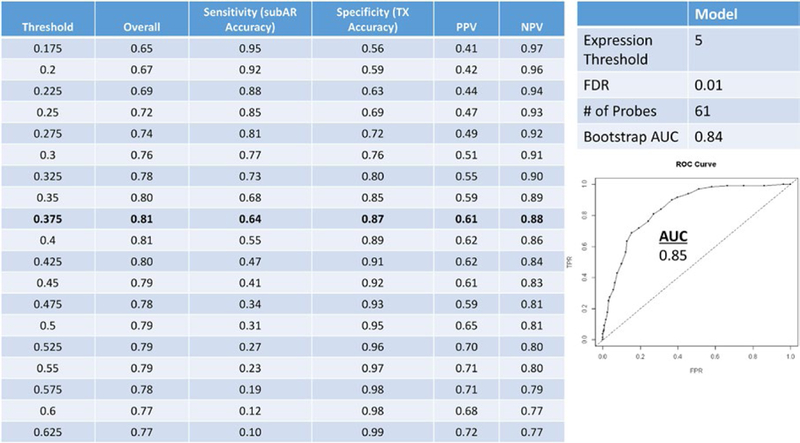
Discovery on 530 Clinical Trials in Organ Transplantation 08 (CTOT‐08) paired peripheral blood and surveillance biopsy samples cohort. We ran the top random forests model from the differentially expressed gene data with threshold selection and predictive metrics on 530 paired samples from the CTOT‐08 (400 [75.5%] transplant excellent [TX]; 130 [24.5%] subclinical acute rejection [subAR]) discovery training set cohort (100000 trees, expression threshold of 5, and false discovery rate [FDR] 0.01), optimizing for area under the curve (AUC; 0.85 [0.84 after internal validation by resampling bootstrap]). A predicted probability threshold of 0.375 was selected, yielding an overall accuracy of 0.81, specificity and negative predictive value (NPV) (87% and 88%, respectively) over sensitivity, and positive predictive value (PPV) (64% and 61%, respectively). The classifiers consisted of 61 probe sets mapping to 57 genes
TABLE 2.
Classifier Performance on Discovery and Validation Sets by Locked Classifier/Threshold Following Random Forests Model Selection
| Data Set | Paied Samples, n |
TX: subAR (% subAR prevalence) |
Probability Threshold |
% Negative (spared biopsy) |
NPV | True Negative | False Negative | % Positive (pickup subAR) |
PPV | True Positive | False Positive |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Discovery Set | 530 | 400:130 (24.5) |
0.375 | 74.7 | 88 | 349 | 42 | 25.3 | 61 | 83 | 51 |
| Validation set 1 | 138 | 96:42(30.4) | 0.375 | 71.7 | 78 | 77 | 22 | 28.3 | 51 | 20 | 19 |
| Validation set 2 | 129/138 | 93:36(27.9) | 0.375 | 72.1 | 80 | 74 | 19 | 27.9 | 47 | 17 | 19 |
We tested the locked model classifiers at the defined threshold (0.375) first on 138 subjects from the Northwestern University (NU) biorepository (validation set 1) who had undergone surveillance biopsies (subclinical acute rejection [subAR] 42 [30.4%]: transplant excellent [TX] 96). Performance metrics consisted of a negative predictive value (NPV) of 78% and a positive predictive value (PPV) of 51%. We then tested the same locked model/ threshold on a subset of 129/138 (subAR 36 [27.9%]: TX 93) participants who met the strict study CTOT–08 criteria for the clinical phenotype definitions of subAR and TX (validation set 2); performance metrics consisted of NPV of 80% and PPV of 47%. The biomarker test results were interpreted dichotomously as “positive” (ie, correlating with a clinical phenotype of subAR) if the probability exceeded the 0.375 threshold and “negative” (ie, correlating with TX) if <0.375. To translate the performance of the biomarker into a narrative more relevant to clinical application, we sought to calculate our ability to diagnose the presence or absence of subAR in any given sample, taking into consideration the prevalent incidence of both subAR and TX compared with the frequency of a correct positive versus negative biomarker test result. Accordingly, we made a negative call (no subAR) in 72% to 75% of the patients (NPV 78%–88%) versus a positive call (subAR) in 25% to 28% of the patients (PPV 47%–61%).
Table S2A-D show the clinical impact of both the CP and the GEP of subAR within the first 12 months on the CCE, as well as the association between the CP and GEP both within 12 and 24 months after transplant and the development of dnDSAs by the end of the study period (24 months). The data are presented according to 3 distinct groups of subjects who met the following criteria either within the first year or within the study period (2 years) after KT: (1) subAR or positive GEP only, (2) no subAR (TX) or negative GEP only, and (3) ≥1 instance of subAR or a positive GEP with ≥1 TX or negative GEP. These data are also graphically represented in Figure 2A-D. Figure 2A (CP and CCE) shows values of 73.9% (group 1) versus 35.5% (group 2) (0R 5.1, 95% CI 1.7–16.9, P < .001) and of 53.2% (group 3) versus 35.5% (group 1) (0R 2.1, 95% CI 1.1–4.0, P = .027). When individual components of the CCE (IFTA, BPAR, or Δ estimated glomerular filtration rate) were examined, only BPAR demonstrated significance in a comparison of groups 1 and 2 (P < .001). Similarly, within the first year after KT, 239 subjects met criteria defining the GEP as either a positive or negative biomarker test determination at both 12 and 24 months (182/239 [76.2%]), distributed equally among the 3 groups, and had sufficient clinical data to also define the CCE. Figure 2C (GEP and CCE) shows values of 66.7% (group 1) versus 37.3% (group 2) (0R 3.4, 95% CI 1.3–9.3, P = .009) and 48.6% (group 3) versus 37.3% (group 2) (0R 1.6, 95% CI 0.8–3.0, P = .17). An analysis of the individual components of the CCE revealed that only BPAR showed a significant difference when comparing subjects in groups 2 and 1 (P = .003).
FIGURE 2.
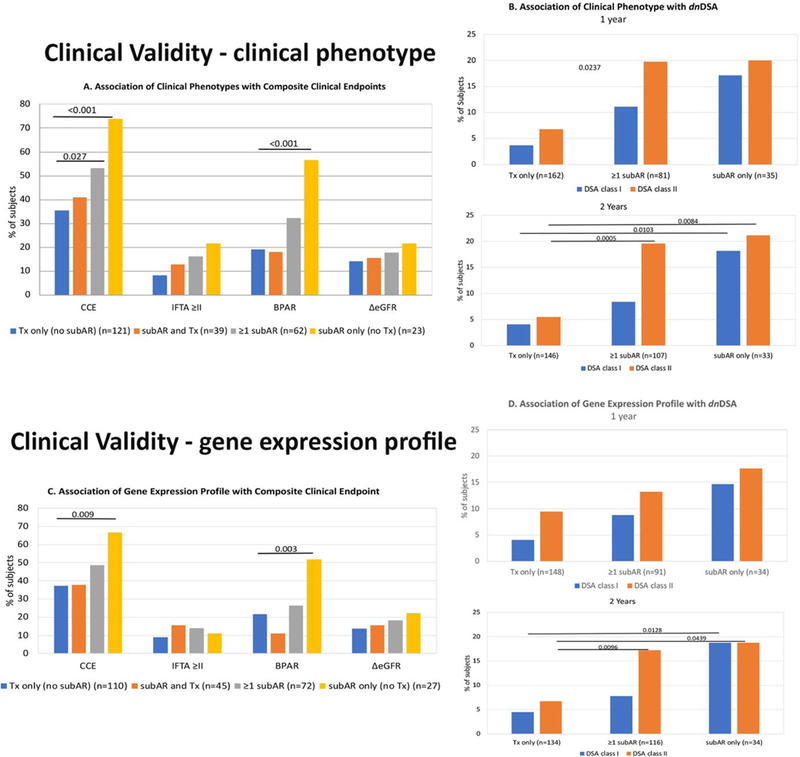
Clinical validity is demonstrated by the clinical significance of both the clinical phenotype (CP) and the gene expression profile (GEP) of subclinical acute rejection (subAR) within the first 12 months on the composite clinical endpoint (CCE), as well as the association between the CP and GEP both within 12 and 24 months after kidney transplant (KT) and the development of de novo DSAs (dnDSAs) by the end of the study period (24 months). The data are presented according to 3 distinct groups of subjects who met the following criteria within either the first year or the study period (2 years) after KT: (1) subAR or positive biomarker only, (2) no subAR (transplant excellent [TX]) or negative biomarker only, and (3) ≥1 instance of subAR or a positive biomarker with ≥1 TX or negative biomarker. A. Association of CP with CCE. Shown are the percentage of subjects who reached an endpoint (either the CCE) or each individual component of the CCE (grade ≥2 interstitial fibrosis/tubular atrophy [IFTA] on 24-month biopsy, any episode of biopsy-proved acute rejection [BPAR], or drop in glomerular filtration rate >10 mL/min/1.73 m2 between months 4 and 24). Subjects are divided based on CP (those with only TX on biopsies [blue bars], those with either subAR or TX [orange bars], those with ≥1 episode of subAR [gray bars], and those with only subAR [yellow bars] on surveillance biopsies). B. Association of CP with dnDSAs. 1. Percentage of subjects who developed dnDSAs at any time during the study, either class I (blue bars) or class II (orange bars), based on their CP group in the 24-month trial (subjects who had TX only on biopsies, ≥1 episode of subAR on biopsy, or only subAR on surveillance biopsy). 2. Similar depiction as panel 1 for the association between dnDSAs and CPs but limited to biopsy results obtained in the first year posttransplant. C. Association of GEP with CCE. Similar to A, shown are the percentage of subjects who reached an endpoint (either CCE) or each individual component of the CCE (grade ≥2 IFTA on 24-month biopsy, any episode of BPAR, or drop in glomerular filtration rate >10 mL/min/1.73 m2 between months 4 and 24). Subjects are divided by their GEP tests results: those subjects who had only TX on GEP (blue bars), those with either subAR or TX (orange bars), those with ≥1 test with subAR (gray bars), and those who had only subAR tests (yellow bars). D. Association of GEP with dnDSAs. 1. Association between the gene GEP test and the development of dnDSAs at any time posttransplant. This includes GEP tests done within the 24-month study period. Shown are the percentage of subjects who developed dnDSAs, both class I (blue bars) and class II (orange bars) grouped based on GEP tests. The subject groups are those with only TX blood tests, ≥1 subAR blood test, or only subAR blood tests. All blood tests were paired with surveillance biopsies. 2. Similar depiction as panel 1 with the association between dnDSAs and the GEP but limited to GEP blood test results obtained in the first year posttransplant.
We also conducted an analysis of the association between the CP and GEP at 12 and 24 months and the development of dnDSAs by the end of the study period (24 months after KT). Figure 2B (CP and dnDSAs) shows that at 12 months, there was a significant difference between groups 1 and 2 (class I P < .01, class II P = .02) and between groups 3 and 2 (class I P = .02, class II P < .01). At 24 months, differences were noted between groups 1 and 2 (class I P = .01, class II P = .01) and between groups 3 and 2 (class II P < .01). Figure 2D (GEP and dnDSAs) shows that at 12 months, dnDSA class I was significantly higher in group 1 than in group 2 (P = .03). At 24 months, differences were noted between groups 1 and 2 (class I P = .01, class II P = .04) and between groups 3 and 2 (class II P = .01).
Figure 3 shows the results of the IM protocol just described. While the local histology report for the surveillance biopsy was used to determine management, central reports were used to compare the baseline with the subsequent 8-week biopsies. Twenty-three subjects had the requisite serial data to be included in this analysis; 11 (47.8%) (3 untreated; 9 borderline, 2 grade 2A) showed histologic resolution and 12 (52.2%) (1 untreated; 6 borderline, 3 grade 1A, 2 antibody-mediated rejection [AMR], 1 borderline plus AMR) showed persistent or worsening rejection, including 11 (58%) of 19 who underwent treatment. Significant differences between the 2 groups in the predicted probability were observed at 4 (P = .014) and 8 (P = .015) weeks. When values were adjusted for differences in baseline probabilities, these comparisons remained significant. Changes in the change in probability scores (slope) between baseline and 4 (P = .045) and 8 weeks (P = .023) also differed between the 2 groups. We were interested to note that the differences in predicted probability scores between the 2 groups did not reach statistical significance (P = .073), consistent with the finding that 7 of 11 biopsies with histologic subAR at baseline were below the 0.375 threshold (biomarker negative) in the “resolved” group that included 3 untreated subjects, whereas 8 of 12 were above the threshold (biomarker positive) in the “unresolved” group.
FIGURE 3.
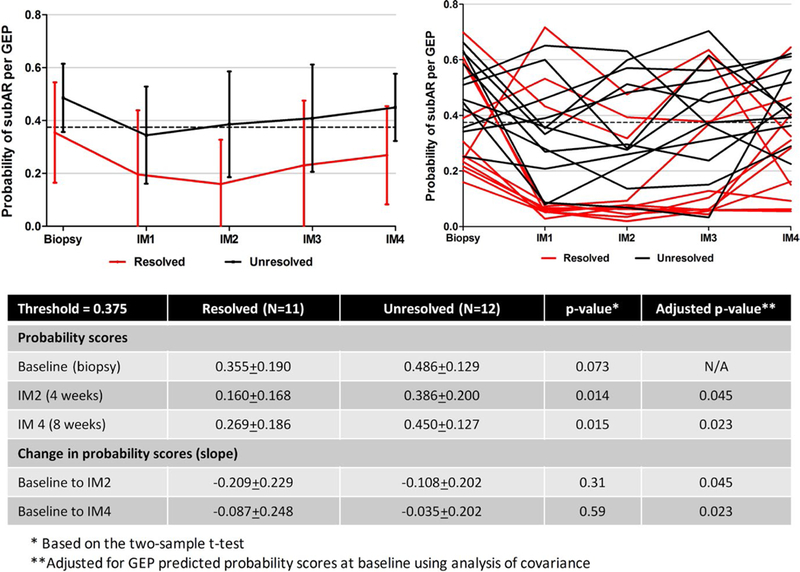
Predictive probabilities after diagnosis and treatment of subclinical acute rejection (subAR). Gene expression profile (GEP)–based probability scores for intense monitoring (IM) subjects (n = 23) were determined every 2 weeks (IM visits 1‐4) after the diagnosis of subAR. While treatment was triggered by the local biopsy report in 19 of 23 subjects, resolution of subAR was assessed by comparing the centrally read histologic findings of the baseline biopsy to the repeat biopsy done at 8 weeks. Of 23 subjects (8 treated), 11 showed improvement of the biopsy (resolved [red lines]), whereas 12 (11 treated) showed either no improvement or worsening (unresolved [black lines]) rejection. Trends in probability scores are shown for the 2 groups on the left and for individual subjects on the right. There were differences in baseline predictive probabilities that did not reach significance (P = .073). Significant differences were seen between the group with resolution of subAR compared with the unresolved group in the predicted probabilities of the subAR gene expression profile at 4 (P = .014) and 8 (P = .015) weeks. When baseline values were adjusted, these differences remained significant between the 2 groups, as did the slope between baseline and 4 (P = .045) and 8 weeks (P = .023)
4. DISCUSSION
We developed a blood-based molecular biomarker that correlates with the presence or absence of subAR on surveillance biopsies. Our data also show that this novel biomarker, derived from biologically relevant differentially expressed genes between the CPs of subAR and TX, also correlates with transplant outcomes.
Several studies have previously shown that GEPs in both urine and peripheral blood can detect KT rejection in the setting of graft dysfunction. The CT0T-04 study34 reported a 3-gene signature in urine samples capable of detecting acute KT rejection. While the authors included analyses of samples obtained before the clinical event, suggesting the potential to detect subAR, urine samples paired with surveillance biopsies were not specifically studied. Similarly, the CT0T-01 study35 reported that urine CXCL9 protein, in the absence of coincident infection, was able to detect clinically evident acute rejection (Banff grade ≥I). These authors also noted that the presence of this protein frequently preceded renal dysfunction, but subAR was not the focus of the study. Others have also reported GEPs in the peripheral blood diagnostic of acute KT rejection24,36 only in the context of renal dysfunction. A more recent study reported that donor-derived cell-free DNA37 in the peripheral blood of KT recipients was able to detect severe graft injury secondary but was not specific to rejection in patients who underwent a biopsy to investigate renal dysfunction.38
Molecular detection of subAR in peripheral blood has been more specifically addressed in recent studies. The kidney Solid Organ Response Test (kSORT), used in both the AART39 and ESCAPE40 studies, primarily predicted acute KT rejection; in the multicenter AART study, only a proportion of blood samples were paired with surveillance biopsies. When paired biopsies were used, central histology reports were not used to assign CPs when training the models. Moreover, the authors used best-fitting models, indeterminate values, and nonprevalent cohorts, potentially skewing performance metrics.41,42 The single-center ESCAPE study used blood samples paired with surveillance biopsies at 6 months posttransplant to show that kSORT, when used in combination with pretransplant enzyme-linked immunospot (ELISpot) assays, a measure of donor alloreac-tivity, predicted subAR. However, in a subsequent report, the same authors combined surveillance biopsies with serial ELISpot assays in a separate patient cohort, but not kSORT, to predict subAR as well as worse renal function and the emergence of dnDSAs.43 Another study, which paired blood samples with biopsies, was able to discover GEPs for subAR in both the blood and graft compartments, but these were not subjected to external validation using independent cohorts. Instead, the authors showed in a proof-of-concept study that orthogonal validation could be achieved across different genomic technologies and platforms.44
Our current study used a multicenter prospective design with standardized peripheral blood sample collection from prevalent patient populations undergoing surveillance biopsies, with the exclusive use of paired samples with central histology reports for both discovery and validation of the biomarker. We used both open source and custom-developed diagnostics pipelines and well-established bioinformatics methodologies to select the classifier for the model. We used a genome-wide discovery approach rather than a more targeted approach using only genes known to be up-regulated in correlation with a phenotype. It is interesting to note that while the differentially expressed genes from both data sets mapped to molecular pathways biologically relevant to rejection, only 7 of the 57 classifier genes showed biologic relevance to known rejection pathways and only 2 of 7 were up-regulated, suggesting that down-regulated genes may also be important in rejection. From a clinical application perspective, our data show that a negative test result, obtained in 72% to 75% of KT recipients, correlated with the clinical absence of subAR, while a positive test result, noted in 25% to 28%, correlated, albeit less strongly, with the clinical presence of subAR. Of note, the proportion of positive and negative test results closely approximated the prevalent incidence of the CPs in both discovery and validation cohorts.
The clinical significance of Banff borderline changes has been debated in the past45 and remains a topic of active deliberation.18 Using both a CCE and dnDSAs, our study clearly shows a significant association between the CP of subAR with worse transplant outcomes at 24 months, despite the fact that the vast majority of subAR found on our surveillance biopsies consisted of Banff borderline changes. We also noted that the majority of subAR was associated with T cell-mediated inflammation. This is mechanistically important given that some continue to argue that T cell-mediated acute rejection is not a major factor in the development of IFTA and antibody-mediated chronic rejection,46 while others continue to defend the causative role of these effector cells in the development of IFTA and chronic rejection,47–49 including the most recent Banff consensus report.50 An important determinant of the clinical validity of a biomarker is to demonstrate a biologically plausible association with a clinically significant outcome.51,52 Therefore, our study sought to determine whether a blood-based molecular biomarker that correlates with subAR could also associate independently with worse 24-month transplant outcomes. To our knowledge, no previous peripheral blood or urinary biomarker studies have demonstrated an impact on transplant outcomes; these associations have been made instead by inference. Our study clearly shows that a GEP that correlates with the absence or presence of histologic subAR independently associates with worse 24-month transplant outcomes and the development of dnDSAs, demonstrating clinical validity. Moreover, while the sample size was relatively small, data from our IM protocol suggest that biomarker probability correlated statistically with histologic resolution of subAR and that, in the majority of patients, the biomarker at baseline and at 4 and 8 weeks after the initial surveillance biopsy predicted resolution, suggesting potential clinical utility to monitor treatment of subAR in the context of a stable creatinine level. These novel data, including the lack of histologic response in the majority of patients treated for subAR, may help explain why a previous single-center randomized controlled study failed to show that treatment of subAR was effective.53
The findings that our biomarker associates with transplant outcomes independent of the CP despite the occasional discordance with the histologic phenotype, including the IM, have caused us to question the role of a paired biopsy as the “gold standard” for measuring the performance of a blood-based molecular biomarkers. This is especially relevant for borderline changes, the predominant histologic finding in patients with subAR. Biopsies are known to be associated with sampling error and variability in interobserver interpretation, especially for borderline changes, despite the use of standardized classifications. We interpret a positive blood-based biomarker test as a potentially actionable signal of alloimmune activation and impending graft injury that correlates with outcomes but that may have some degree of variation in correlation with histology based on chronology and on differences in the immunologic compartment.
Our study has several potential limitations. While the CTOT-08 study aims to develop biomarkers for a number of CPs, the stated focus of the current study was subAR in patients with stable renal function. We anticipate that the current biomarker will be used to monitor stable patients while using all other standard clinical and laboratory information, including viral assays for BK virus and graft biopsies when indicated. We arbitrarily selected a probability threshold with a balanced emphasis on NPV over PPV in order to optimally reduce the indiscriminate use of invasive surveillance biopsies in patients stratified as having a lower risk of harboring subAR. Despite important differences, an analogous stratification approach is currently used in clinical practice to monitor heart transplant patients.54 Also, validation in a separate cohort using a locked model/threshold resulted in a slight expected reduction in the predictive performance of the classifier. A comparable effect has been previously observed in a similar study,34 but not in others, when less stringent validation methods are used.41,55 We tested the performance of the biomarker in the validation cohort at a single defined probability threshold defined as a single point of the receiver operating characteristic and AUC rather than allowing for the use of a distribution of several “fit-for-purpose” thresholds along the curve. There are other potential limitations inherent to observational studies. To mitigate these, we selected centers with diverse populations, remained agnostic to immunologic risk or immunosuppression regimen, used clinical algorithms blinded to biomarker development, included many confounders known to corrupt primary analyses, used real-time monitoring by an independent DCC of sample collection to identify and resolve sources of missing data, applied central biopsy reports, and used ComBat adjustment26,27 for batch variation.
Taken together, our data suggest that the serial use of a novel biomarker that correlates with the absence of subAR and that demonstrates clinical validity may reduce the need for the routine and indiscriminate use of invasive surveillance biopsies in the majority of stable patients after KT, allowing for a biomarker stratified approach for those at a higher risk of harboring subAR. By providing molecular information that can inform personalized clinical management, such a monitoring strategy could potentially lead to improved outcomes after KT.
Supplementary Material
ACKNOWLEDGMENTS
We would like to acknowledge our patients, transplant coordinators, research services cores and the many care providers at each transplant center who helped achieve the successful completion of the CTOT-08 study. Specifically, Michael Leonard (Mayo Scottsdale), Jennifer Czerr (Cleveland Clinic), Gail Johnson and Christina Hurman (MUSC), Jaquelene Peysakhovich (Northwestern University), and Shannon Mount and Michele Meyer (Scripps Health) were instrumental in helping us achieve enrollment and capture of data and samples. We thank Drs Richard H. Scheuermann and Brian D. Aevermann (Craig Venter Institute, La Jolla, CA) for their guidance on biomarker development and validation. We also thank the CTOT Steering Committee for their guidance in protocol development, monitoring of study outcomes, and review of the data. We also thank the CTOT Steering Committee, particularly Dr Nancy D. Bridges (National Institute of Allergy and Infectious Diseases), for their guidance in protocol development, monitoring of study outcomes, and review of the data.
Funding information
We also acknowledge financial support in the form of grants from the National Institutes of Health (U01 AI084146, 3 U01 AI063594–07S1, 1U01AI088635, 2U19 AI063603, R34 AI118493) and from Transplant Genomics Inc, as well as from NU to the Comprehensive Transplant Center.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Abbreviations:
- AMR
antibody-mediated rejection
- AUC
area under the curve
- BPAR
biopsy-proved acute rejection
- CCE
clinical composite endpoint
- CMV
cytomegalovirus
- CP
clinical phenotype
- CTOT-O8
Clinical Trials in Organ Transplantation 08
- DAVID
Database for Annotation Visualization and Integrated Discovery
- DCC
Data Coordinating Center
- DEG
differentially expressed genes
- dnDSA
de novo donor-specific antibody
- ELISpot
enzyme-linked immunospot
- ESRD
end-stage renal disease
- GEP
gene expression profile
- GO
Gene Ontology
- IFTA
interstitial fibrosis and tubular atrophy
- IM
intense monitoring
- IPA
ingenuity pathway analysis
- IPA
ingenuity pathway analysis
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- kSORT
kidney Solid Organ Response Test
- KT
kidney transplant
- MMF
mycophenolate mofetil
- mTOR
mammalian target of rapamycin
- NPV
negative predictive value
- NU
Northwestern University
- PCR
polymerase chain reaction
- PKD
polycystic kidney disease
- PPV
positive predictive value
- ROC
receiver operating characteristic
- subAR
subclinical acute rejection
- TX
transplant excellent
Footnotes
DISCLOSURE
The authors of this manuscript have conflicts of interest to disclose as described by the American Journal of Transplantation. Drs Friedewald, Kurian, Whisenant, and Abecassis are paid consultants of and Drs Friedewald, Kurian, and Abecassis have equity interests in Transplant Genomics, Inc.
AWARDED OR FILED PATENTS PERTAINING TO RESULTS PRESENTED IN THE PAPER
“Gene expression profiles associated with subclinical kidney transplant rejections.” United States Application Number US15/358390 filed on November 22, 2016. Pending.
DATA AND MATERIALS AVAILABILITY
GEO Accession No. GSE107509.
REFERENCES
- 1.Tonelli M, Wiebe N, Knoll G, et al. Systematic review: kidney trans¬plantation compared with dialysis in clinically relevant outcomes. Am J Transplant. 2011;11(10):2093–2109. [DOI] [PubMed] [Google Scholar]
- 2.US Renal Data System. 2016 USRDS annual data report: Epidemiology of kidney disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2016. [Google Scholar]
- 3.Hart A, Smith JM, Skeans MA, et al. Kidney. Am J Transplant. 2016;16(Suppl 2):11–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meier-Kriesche HU, Schold JD, Srinivas TR, Kaplan B. Lack of improvement in renal allograft survival despite a marked decrease in acute rejection rates over the most recent era. Am J Transplant. 2004;4(3):378–383. [DOI] [PubMed] [Google Scholar]
- 5.Nankivell BJ, Chapman JR. The significance of subclinical rejection and the value of protocol biopsies. Am J Transplant. 2006;6(9):2006–2012. [DOI] [PubMed] [Google Scholar]
- 6.Kee TY, Chapman JR, O’Connell PJ, et al. Treatment of subclini-cal rejection diagnosed by protocol biopsy of kidney transplants. Transplantation. 2006;82(1):36–42. [DOI] [PubMed] [Google Scholar]
- 7.Heilman RL, Devarapalli Y, Chakkera HA, et al. Impact of subclinical inflammation on the development of interstitial fibrosis and tubular atrophy in kidney transplant recipients. Am J Transplant. 2010;10(3):563–570. [DOI] [PubMed] [Google Scholar]
- 8.Loupy A, Vernerey D, Tinel C, et al. Subclinical rejection phenotypes at 1 year post-transplant and outcome of kidney allografts. J Am Soc Nephrol. 2015;26(7):1721–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehta R, Bhusal S, Randhawa P, et al. Short-term adverse effects of early subclinical allograft inflammation in kidney transplant recipients with a rapid steroid withdrawal protocol. Am J Transplant. 2018;18(7):1710–1717. [DOI] [PubMed] [Google Scholar]
- 10.Parajuli S, Reville PK, Ellis TM, Djamali A, Mandelbrot DA. Utility of protocol kidney biopsies for de novo donor-specific antibodies. Am J Transplant. 2017;17(12):3210–3218. [DOI] [PubMed] [Google Scholar]
- 11.El-Zoghby ZM, Stegall MD, Lager DJ, et al. Identifying specific causes of kidney allograft loss. Am J Transplant. 2009;9(3):527–535. [DOI] [PubMed] [Google Scholar]
- 12.Gourishankar S, Leduc R, Connett J, et al. Pathological and clinical characterization of the ‘troubled transplant’: data from the DeKAF study. Am J Transplant. 2010;10(2):324–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El Ters M, Grande JP, Keddis MT, et al. Kidney allograft survival after acute rejection: the value of follow-up biopsies. Am J Transplant. 2013;13(9):2334–2341. [DOI] [PubMed] [Google Scholar]
- 14.Bouamar R, Shuker N, Hesselink DA, et al. Tacrolimus predose concentrations do not predict the risk of acute rejection after renal transplantation: a pooled analysis from three randomized-con-trolled clinical trials (dagger). Am J Transplant. 2013;13:1253–1261. [DOI] [PubMed] [Google Scholar]
- 15.Seron D, Moreso F. Protocol biopsies in renal transplantation: prognostic value of structural monitoring. Kidney Int. 2007;72(6): 690–697. [DOI] [PubMed] [Google Scholar]
- 16.Morgan TA, Chandran S, Burger IM, Zhang CA, Goldstein RB. Complications of ultrasound-guided renal transplant biopsies. Am J Transplant. 2016;16(4):1298–1305. [DOI] [PubMed] [Google Scholar]
- 17.Mehta R, Sood P, Hariharan S. Subclinical rejection in renal transplantation: reappraised. Transplantation. 2016;100(8):1610–1618. [DOI] [PubMed] [Google Scholar]
- 18.Becker JU, Chang A, Nickeleit V, Randhawa P, Roufosse C. Banff borderline changes suspicious for acute T cell-mediated rejection: where do we stand? Am J Transplant. 2016;16(9):2654–2660. [DOI] [PubMed] [Google Scholar]
- 19.Mehta R, Cherikh W, Sood P, Hariharan S. Kidney allograft surveillance biopsy practices across US transplant centers: a UNOS survey. Clin Transplant. 2017;31(5):e12945. [DOI] [PubMed] [Google Scholar]
- 20.Lo DJ, Kaplan B, Kirk AD. Biomarkers for kidney transplant rejection. Nat Rev Nephrol. 2014;10(4):215–225. [DOI] [PubMed] [Google Scholar]
- 21.Menon MC, Murphy B, Heeger PS. Moving biomarkers toward clinical implementation in kidney transplantation. J Am Soc Nephrol. 2017;28(3):735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solez K, Colvin RB, Racusen LC, Haas M, Sis B, Mengel M, et al. Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant. 2008;8(4):753–760. [DOI] [PubMed] [Google Scholar]
- 23.Hariharan S, McBride MA, Cherikh WS, Tolleris CB, Bresnahan BA, Johnson CP. Post-transplant renal function in the first year predicts long-term kidney transplant survival. Kidney Int. 2002;62(1):311–318. [DOI] [PubMed] [Google Scholar]
- 24.Kurian SM, Williams AN, Gelbart T, Campbell D, Mondala TS, Head SR, et al. Molecular classifiers for acute kidney transplant rejection in peripheral blood by whole genome gene expression profiling. Am J Transplant. 2014;14(5):1164–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA). Biostatistics. 2010;11(2):242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–127. [DOI] [PubMed] [Google Scholar]
- 27.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The SVA package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smyth GK. limma: linear models for microarray data In: Gentleman R, Carey VJ, Huber W, Irizzary RA, Dudoit S, eds. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. New York, NY: Statistics for Biology and Health; 2005:397–420. [Google Scholar]
- 30.Analysis IP. https://www.qiagenbioinformatics.com/products/in-genuity-pathway-analysis. Published 2017. Accessed December 12, 2017.
- 31.Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, et al. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. GenomeBiol. 2007;8(9):R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrell FE Jr, Lee KL, Mark DB. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med. 1996;15(4):361–387. [DOI] [PubMed] [Google Scholar]
- 34.Suthanthiran M, Schwartz JE, Ding R, Abecassis M, Dadhania D, Samstein B, et al. Urinary-cell mRNA profile and acute cellular rejection in kidney allografts. N Engl J Med. 2013;369(1):20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hricik DE, Nickerson P, Formica RN, et al. Multicenter validation of urinary CXCL9 as a risk-stratifying biomarker for kidney transplant injury. Am J Transplant. 2013;13(10):2634–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li L, Khatri P, Sigdel TK, et al. A peripheral blood diagnostic test for acute rejection in renal transplantation. Am J Transplant. 2012;12:2710–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snyder TM, Khush KK, Valantine HA, Quake SR. Universal noninvasive detection of solid organ transplant rejection. Proc Natl Acad Sci. 2011;108(15):6229–6234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bloom RD, Bromberg JS, Poggio ED, et al. Cell-free DNA and active rejection in kidney allografts. J Am Soc Nephrol. 2017;28(7):2221–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roedder S, Sigdel T, Salomonis N, et al. The kSORT assay to detect renal transplant patients at high risk for acute rejection: results of the multicenter AART study. PLoS Medicine. 2014;11(11):e1001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crespo E, Roedder S, Sigdel T, et al. Molecular and functional noninvasive immune monitoring in the ESCAPE Study for prediction of subclinical renal allograft rejection. Transplantation. 2017;101(6):1400–1409. [DOI] [PubMed] [Google Scholar]
- 41.Abecassis M, Kaplan B. Transplantation: Biomarkers in transplantation: the devil is in the detail. Nat Rev Nephrol. 2015;11(4):204–205. [DOI] [PubMed] [Google Scholar]
- 42.Kurian SM, Whisenant T, Mas V, et al. Biomarker guidelines for high¬dimensional genomic studies in transplantation: adding method to the madness. Transplantation. 2017;101(3):457–463. [DOI] [PubMed] [Google Scholar]
- 43.Crespo E, Cravedi P, Martorell J, et al. Posttransplant peripheral blood donor-specific interferon-gamma enzyme-linked immune spot assay differentiates risk of subclinical rejection and de novo donor-specific alloantibodies in kidney transplant recipients. Kidney Int. 2017;92(1):201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurian SM, Velazquez E, Thompson R, et al. Orthogonal comparison of molecular signatures of kidney transplants with sub-clinical and clinical acute rejection: equivalent performance is agnostic to both technology and platform. Am J Transplant. 2017;17(8):2103–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Veronese FV, Manfro RC, Roman FR, et al. Reproducibility of the Banff classification in subclinical kidney transplant rejection. Clin Transplant. 2005;19(4):518–521. [DOI] [PubMed] [Google Scholar]
- 46.Famulski KS, Halloran PF. Letter to AJT editor re: Nankivell et al. Am J Transplant. 2018;18(3):765–766. [DOI] [PubMed] [Google Scholar]
- 47.Modena BD, Kurian SM, Gaber LW, et al. Gene expression in biopsies of acute rejection and interstitial fibrosis/tubular atrophy reveals highly shared mechanisms that correlate with worse longterm outcomes. Am J Transplant. 2016;16(7):1982–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nankivell BJ. Reply: i-IFTA is better appreciated by its pathology rather than molecules. Am J Transplant. 2018;18(3):769–770. [DOI] [PubMed] [Google Scholar]
- 49.Nankivell BJ, Shingde M, Keung KL, et al. The causes, significance and consequences of inflammatory fibrosis in kidney transplantation: the Banff i-IFTA lesion. Am J Transplant. 2018;18(2):364–376. [DOI] [PubMed] [Google Scholar]
- 50.Haas M, Loupy A, Lefaucheur C, et al. The Banff 2017 kidney meeting report: revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am J Transplant. 2018;18(2):293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deng MC, Eisen HJ, Mehra MR. Methodological challenges of genomic research: the CARGO study. Am J Transplant. 2006;6(5 Pt 1):1086–1087. [DOI] [PubMed] [Google Scholar]
- 52.Halloran PF, Reeve J, Kaplan B. Lies, damn lies, and statistics: the perils of the P value. Am J Transplant. 2006;6(1):10–11. [DOI] [PubMed] [Google Scholar]
- 53.Rush D, Arlen D, Boucher A, et al. Lack of benefit of early protocol biopsies in renal transplant patients receiving TAC and MMF: a randomized study. Am J Transplant. 2007;7(11):2538–2545. [DOI] [PubMed] [Google Scholar]
- 54.Pham MX, Teuteberg JJ, Kfoury AG, et al. Gene-expression profiling for rejection surveillance after cardiac transplantation. N Engl J Med. 2010;362(20):1890–1900. [DOI] [PubMed] [Google Scholar]
- 55.Reeve J, Halloran PF. Biopsy transcriptome expression profiling: proper validation is key. Lancet. 2017;389(10069):600–601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.