

Abstract
Pulmonary neuroendocrine cells (PNECs) are rare airway epithelial cells whose function is poorly understood. Here we show that Ascl1-mutant mice which have no PNECs exhibit severely blunted mucosal type 2 response in models of allergic asthma. PNECs reside in close proximity to group 2 innate lymphoid cells (ILC2s) near airway branch points. PNECs act through calcitonin gene-related peptide (CGRP) to stimulate ILC2s and elicit downstream immune responses. In addition, PNECs act through neurotransmitter gamma-aminobutyric acid (GABA) to induce goblet-cell hyperplasia. The instillation of a mixture of CGRP and GABA in Ascl1-mutant airways restored both immune and goblet-cell responses. In accordance, lungs from human asthmatics show increased PNECs. These findings demonstrate that the PNEC–ILC2 neuroimmunological modules function at airway branch points to amplify allergic asthma responses.
One Sentence Summary:
PNEC–ILC2 axis elaborates asthma responses.
A current CDC report estimates that 24.6 million people in the US have asthma (1). Although much attention has been placed on immune cells as the source of symptoms, in recent years, the concept that non-hematopoietic tissues are a critical source of cytokines has gained support (2, 3). For example, lung epithelial cells such as alveolar type 2 cells produce IL-33, which in turn activates immune cells, including residential group 2 innate lymphoid cells (ILC2s) (4, 5). ILC2s secrete type 2 cytokines, including IL-5 and IL-13, which promote smooth muscle contraction, eosinophil infiltration, and goblet cell hyperplasia, key features of an asthmatic response (6, 7).
PNECs are a unique and poorly understood cell population in lung. They are rare, endoderm-derived epithelial cells, which constitute ~1% of the airway cell population (8, 9). They are the earliest specified lung epithelial cells, expressing Ascl1, its defining marker, starting at embryonic day (E) 12.5. In rodents, a majority of the PNECs form clusters, which are called neuroepithelial bodies (NEBs). NEBs are highly innervated by both afferent and efferent neurons (10). PNECs contain dense core vesicles filled with neuropeptides, amines, and neurotransmitters. In vitro, PNECs can be stimulated by oxygen, mechanical stretch, and chemical stimuli and release their vesicular content (11). Excess PNECs have been reported in lungs with various diseases, including chronic obstructive pulmonary disease (COPD), sudden infant death syndrome (SIDS), bronchopulmonary dysplasia (BPD), and small-cell lung cancer (12–16). Recently, we have shown that proper clustering of PNECs is essential for restricting immune cell number in the naïve neonatal lung (17). However, the precise in vivo role of PNECs in lung pathogenesis remains unknown.
Results
PNECs are required for goblet-cell hyperplasia in asthma models
To generate a mouse mutant lacking pulmonary neuroendocrine cells, we inactivated Ascl1, which encodes a transcription factor that is essential for their formation as shown in the Ascl1 global null mutants (18, 19). To bypass perinatal lethality of the global nulls, we inactivated Ascl1 in precursors of PNECs in the airway epithelium using Shhcre (a cre insertion into the sonic hedgehog gene) at the onset of lung development (hereafter Ascl1CKO for conditional knockout) (Fig. S1) (20). This early inactivation led to a complete absence of PNECs, as indicated by the lack of CGRP or Synaptophysin-positive cells in the airway epithelium (Fig. 1A-D, A’-D’). Synaptophysin also labels the nerves that normally innervate PNECs. In the mutant, these nerves remained present subjacent to the airway even though they no longer intercalate into the epithelium (Fig. 1C, D, C’, D’). In contrast to the absent PNECs, the intrinsic neurons, which are also dependent on Ascl1, remained as indicated by TUJ1 (Tubulin β3) expression, presumably because Shhcre is not active in these cells to delete Ascl1 (Fig. S2).
Figure 1.
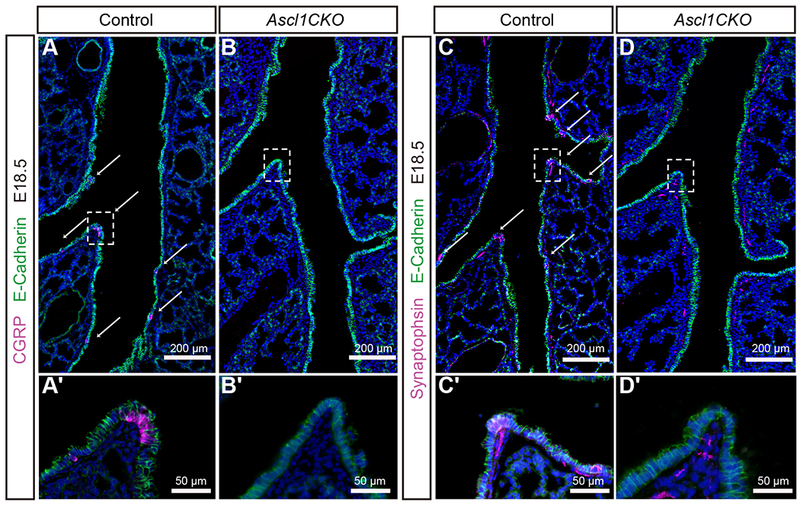
Inactivation of Ascl1 by Shhcre in lung epithelium prevents PNEC formation whereas nerves remain subjacent to airways.
(A-D, A’-D’) At E18.5, CGRP staining outlines PNECs in E-Cadherin-positive epithelium. Synaptophysin staining outlines PNECs and the nerves, including those that innervate PNECs. Arrows indicate PNECs in the control airway. No CGRP or Synaptophysin staining is detected in the epithelium in Ascl1CKO mice, indicating a complete absence of PNECs. Boxed areas are magnified in corresponding panels below. In the mutant, although the nerves no longer innervate epithelial cells (compare C’ to D’), the nerve tracks are still detected nearby. Data are representative of sections from n = 3 mice of each genotype.
Ascl1CKO mutants were viable at birth, which is counter to the postulation that PNECs are required for transition from intra-uterine environment to air breathing. The mutant lungs showed normal branching morphogenesis and alveolar morphology (Fig. S3). Based on immunofluorescent staining as well as qRT-PCR of key cell-type markers, no apparent defects were found in airway club and ciliated cells, or in type I and type II alveolar cells. No goblet cells were detected in the naïve mutant airways, similar to the control. These data indicate that despite being the first differentiated cell type in the airway, the absence of PNECs has no gross effects on the development of other cell types in the lung at baseline.
To test whether disruption of PNEC may impact lung responses to environmental cues, we used an established model to induce allergic asthma-like responses by sensitizing early postnatal mice to ovalbumin (OVA) injected in combination with alum. Mice were then challenged with OVA inhalation (Fig. 2A) (21). As predicted, control mice showed robust goblet-cell hyperplasia along the airways (Fig. 2B). All MUC5AC-positive goblet cells were also positive for SCGB1A1, a club-cell marker, suggesting that there was a partial conversion of club cells into mucus-producing cells (Fig. S4). In contrast, Ascl1CKO mice showed substantially reduced goblet-cell hyperplasia, which registered at approximately 10% of the control response based on a histological mucin index (Fig. 2C-F). This observation was corroborated by drastically reduced expression of Muc5ac and goblet-cell-promoting factors including Spdef and Foxa3 (Fig. 2G). Markers for other airway epithelial cell types, club cells and ciliated cells, remain normal in Ascl1CKO mice following OVA (Fig. S4).
Figure 2.
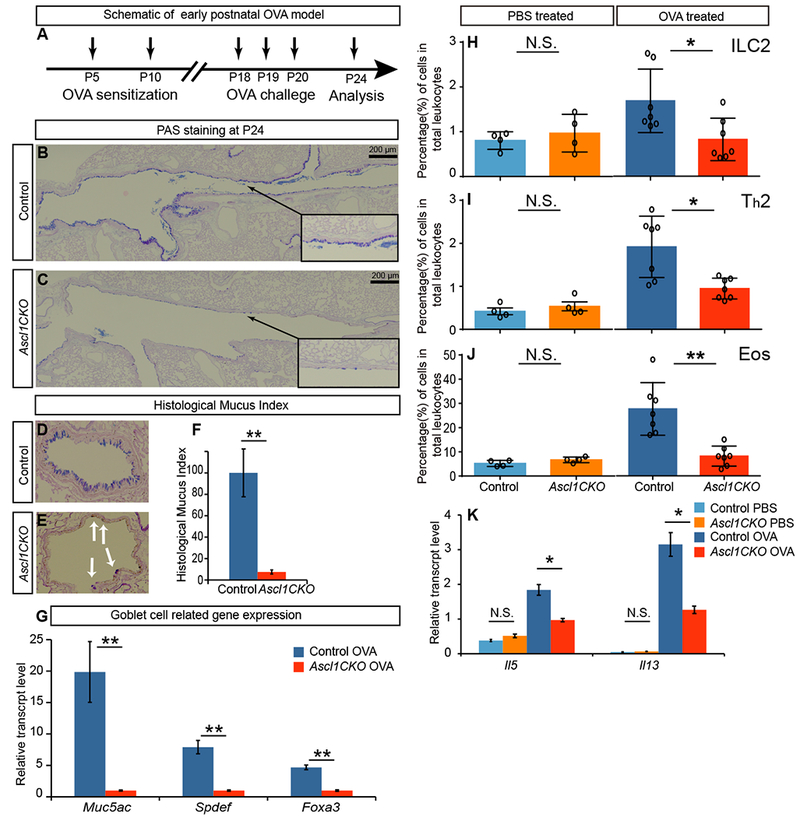
PNECs are required for goblet-cell hyperplasia and immune-cell infiltration in a model of asthma.
(A) Regime of early postnatal OVA model indicating postnatal time points for sensitization, challenge and analysis. (B and C) Representative PAS staining of P24 longitudinal airway sections of OVA challenged control and Ascl1CKO mice. Arrows from insets point to areas that are magnified. (D-F) Representative PAS staining of transverse sections, from which histological mucus index was calculated by linear percent of epithelium positive for PAS stained mucus (n = 3 for each). Student’s t-test was used. Arrows in E indicate residual PAS staining. (G) Goblet-cell-related gene expression as assayed by qRT-PCR of P24 whole lungs after OVA challenge (n = 3 for each). Student’s t-test was used. (H-J) Flow cytometry analysis of immune cells from whole lungs, after either control PBS or OVA challenge as labeled. Mann–Whitney U test was used (n = 4 for PBS groups and n = 7 for OVA groups). (K) Cytokine gene expression as assayed by qRT-PCR of P24 whole lungs (n = 3 each). Student’s t-test was used. N.S. for not significant p ≥ 0.05, * for p < 0.05, ** for p < 0.01. Data are representative of three experiments. Error bars represent mean ± SEM.
To determine if PNECs are also required for allergen-induced goblet cell hyperplasia in the adult lung, we exposed mutant and control mice to house dust mites (HDM), a common allergen that also causes asthma in humans (22). By both PAS staining and quantification of Muc5ac transcripts, we found that the goblet-cell hyperplasia response was reduced in the Ascl1CKO mutants compared to control in the HDM model (Fig. S5). Thus, PNECs amplify airway goblet-cell hyperplasia in the asthma models tested.
PNECs are required for type 2 immune responses in asthma models
A cardinal feature of asthma is eosinophillic infiltration and type 2 T helper (Th2) cell priming in the lung (7). To further explore the role of PNECs in this aspect of the asthmatic response, we compared immune cell frequencies in neonatal OVA/Alum -treated Ascl1CKO mice and control mice. The Ascl1CKO mice exhibited lower numbers of ILC2s, eosinophils, and Th2 cells than control mice after OVA challenge (Fig. 2H-J, S6). These changes were further supported by reduced Il5 and Il13 expression (Fig. 2K). These differences were not due to baseline immune cell differences, as PBS-treated Ascl1CKO mice and control mice had similar levels of immune cells (Fig. 2H-J, S6).
To determine if PNECs are required for allergen-induced immune response in the adult lung, we returned to the adult HDM model. In this adult model, Ascl1CKO mice showed decreased immune-cell infiltration compared to controls (Fig. S7). We also depleted PNECs in the postnatal stage using a different genetic model. We used Shhcre to activate diphtheria toxin receptor expression under the transcriptional control of a PNEC product calcitonin gene-related peptide (CGRP, encoded by Calca gene) in a CalcaloxP-GFP-loxP-DTR strain (23). The introduction of diphtheria toxin into the lung resulted in ~70% reduction in PNECs. When challenged with HDM, these mice also showed a dampened immune response, although to a lesser extent compared to the Ascl1CKO mutant, likely due to residual PNECs in the DTR model (Fig. S8). Thus, PNECs amplify allergen-induced type 2 immune responses in the asthma models tested.
Absence of PNECs leads to the reduction of key neuropeptides and the neurotransmitter GABA
In a normal lung, type 2 immune responses are led by changes in cytokines such as IL-25, IL-33, and TSLP (24–26). In Ascl1CKO lungs, these cytokines are detected at similar levels as compared to control after each of the OVA challenges (Fig. S9). These results suggested that these epithelium-derived cytokines may not explain why type 2 immune responses are different when PNECs are absent.
A key feature of PNECs is their production and secretion of neuropeptides and neurotransmitters (11). To test the hypothesis that PNECs control type 2 responses through some of these molecules, we assayed the expression of several neuropeptides that have been implicated in inflammatory diseases. These include vasoactive intestinal peptide (Vip), Calca (encoding CGRP), chromogranin A (Chga), and neuropeptide Y (Npy) (11, 27–29). In control lungs, Calca, Chga, Npy, and Vip levels were increased after OVA challenge compared to PBS, with Calca showing the largest fold increase. In Ascl1CKO lungs, Calca, Chga, Npy, but not Vip were significantly lower than control mice following OVA challenge (Fig. 3A). The relative fold changes in Calca and Chga were larger compared to that of Npy, possibly due to the concentrated expression of Calca and Chga, but not Npy, in PNECs (11, 27).
Figure 3.
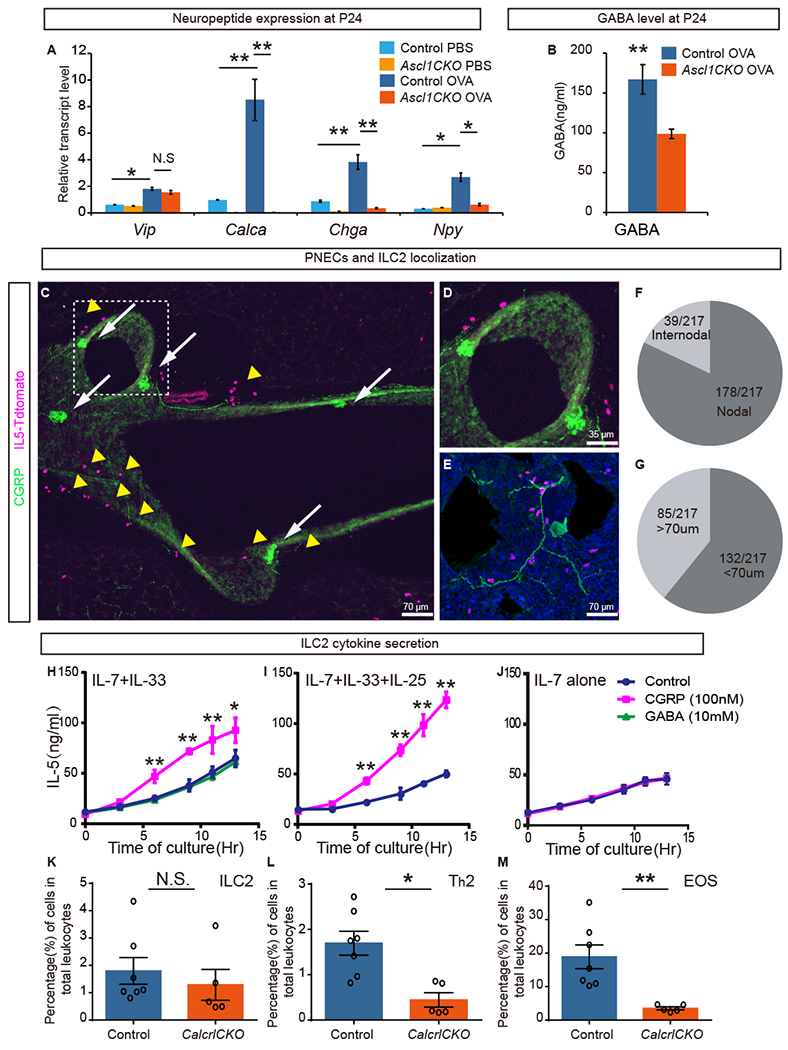
PNECs reside in proximity to ILC2s and stimulate their cytokine production through CGRP.
(A) Neuropeptide gene expression as assayed by qRT-PCR in P24 whole lungs after PBS or OVA challenge (n = 3 each). Student’s t-test was used. (B) GABA level as assayed by ELISA in P24 whole lungs after OVA challenge (n = 3 each). Student’s t-test was used. (C-E) From naïve mice, representative images of PNEC (green, anti-CGRP antibody, arrows) and ILC2 (magenta, Il5-tdTomato reporter, arrowheads) localization near branch points in longitudinal (C, D) or transverse (E) vibratome sections of airways. Boxed area in C is magnified in D. Anti-CGRP antibody also labels sensory nerve (E). (F) Of the total 217 ILC2s analyzed, 178 (82%) localized within 70 μm of airway branch (nodal) points. (G) Of the total of 217 ILC2s analyzed, 132 (61%) of them are localized within 70 μm of PNECs. (H-J) Effects of CGRP or GABA on IL-5 production by sorted primary ILC2s in culture, in the presence of cytokines indicated on the top left. CGRP, but not GABA, can increase IL-5 secretion as assayed by ELISA (n = 3 each). Student’s t-test was used. (K-M) Data from flow cytometry analysis of immune cells from whole lung of HDM treated adult control and mutant mice as labeled. Mann–Whitney U test was used. n = 7 for each group. N.S. for not significant p ≥ 0.05, * for p < 0.05, ** for p < 0.01. Error bars represent mean ± SEM.
To further investigate the source of neuropeptide change, we focused on CGRP as an example. CGRP is expressed in the airway epithelium as well as in lung sensory nerves with cell bodies located in the vagal ganglia. By qRT-PCR, we found that Calca expression in the vagal ganglia was not statistically different in wild-type mice treated with OVA compared to PBS controls under the regime that we used (Fig. S10A). The expression is also similar in the vagal ganglia of OVA-treated Ascl1CKO mice compared to genotype control (Fig. S10B). Within the lung epithelium, by immunofluorescent staining, we found that CGRP expression remained restricted to PNECs following challenge (Fig. S11). There was no increase in PNEC proliferation (Fig. S11). Rather, CGRP signal was detected at a notably higher intensity, suggesting that this PNEC-specific increase contributes to the overall increase of CGRP levels in the lung (Fig. S11).
In addition to neuropeptides, we also assayed for the levels of GABA, a PNEC-produced neurotransmitter that can stimulate goblet cell hyperplasia (30, 31). GABA levels were reduced in Ascl1CKO mutants, possibly due to loss of GAD1, a rate-limiting enzyme in GABA biosynthesis, and VGAT, the vesicular GABA transporter, which are both specifically expressed in PNECs in the lung (Fig. 3B) (31). Together, these differences in neuropeptides and neurotransmitter suggest that they may be responsible for the reduced immune-cell infiltration and goblet-cell hyperplasia in the Ascl1CKO mutant.
PNECs reside in proximity to ILC2s and can stimulate ILC2 cytokine production
To identify possible direct targets of PNEC signaling, we turned to a distinct feature of these cells: that PNECs are preferentially localized to airway branch junctions (8, 9). We searched for immune cells residing in close proximity to these junctions. ILC2s are a population of tissue-resident immune cells that have been shown to play a central role in pulmonary type 2 responses (32, 33). We investigated the localization of ILC2s using a mouse reporter line with tdTomato knocked into the Il5 locus (28). In naïve mice, in addition to confirming the previous finding that ILC2s are localized near the basement membrane subjacent to the airway epithelium (11, 27, 28, 34), we also found that a majority of ILC2s (178/217 total cells counted, or 82%) reside within 70 μm of airway branch junctions, also called nodal points (Fig. 3C-F) (9). By labeling PNEC with anti-CGRP antibody in the Il5-tdTomato background, we found that ILC2s were preferentially localized within 70 μm of PNECs (132/217 total cells counted, or 61%) (Fig. 3G). Thus, ILC2s may be a possible direct target of PNEC signaling.
To test if ILC2s can perceive PNEC signals, we first addressed whether they expressed the appropriate receptors. Existing RNA-seq data indicate that ILC2s express the CGRP co-receptors Calcrl and Ramp1, and the GABA receptor Gabrr1, but not the CHGA receptor gene (35). We confirmed CGRP and GABA receptor gene expression by qPCR assay in primary ILC2s sorted from lungs of naïve wild-type mice (Fig. S12).
To test if ILC2s could be stimulated by PNEC signals, we cultured primary lung ILC2s from naïve lungs and addressed their response to CGRP or GABA. ILC2s respond to cytokines such as IL-33 (36). Consistent with this, following in vivo OVA challenge, there was an increase of IL-33 receptor genes St2, but no change in a more general IL-1 family signal mediator Il-1/Rap gene, in sorted primary ILC2s, (Fig. S13). CGRP further increased IL-5 production from ILC2s when cultured in the presence of IL-33. However, GABA had a negligible effect (Fig. 3H). Furthermore, CGRP further enhanced IL-5 production in the presence of both IL-25 and IL-33 (Fig. 3I) (36). CGRP stimulation did not occur in the absence of IL-25 or IL-33 (Fig. 3J). To complement data from IL-5 ELISAs, we also assayed an array of candidate cytokines using the Biolegend LEGENDplex Th2 system. We confirmed that IL-5 as well as IL-6 were increased when ILC2s were cultured in the presence of CGRP (Fig. S14).
Based on CFSE staining, there was little change in ILC2 division, indicating that CGRP promotes ILC2 cytokine production, but not proliferation (Fig. S15). IL-33 did not appear to alter CGRP receptor gene expression level in ILC2s, suggesting that CGRP and IL-33 may work in parallel to stimulate ILC2s (Table. S1). We also tested CGRP and GABA function on sorted Th2 cells, but observed no change in IL-5 production (Fig. S16). Furthermore, the addition of CGRP to sorted eosinophils led to only a slight increase of leukotriene C4 production, a minor change compared to the effect of CGRP on ILC2s at the same concentration (Fig. S17). Thus, CGRP can directly act on ILC2s to promote their maturation and production of cytokines such as IL-5. Activated ILC2s, in turn, have been shown to recruit eosinophils and trigger a cascade of Th2 responses (32, 33).
To further validate the PNEC/CGRP/ILC2 axis in vivo, we deleted calcitonin receptor-like (Calcrl), in ILC2 cells using Il5cre. When treated with HDM, mutants showed reduced immune cell infiltration compared to control, consistent with the notion that CGRP signaling through ILC2s is required for a full Th2 immune response (Fig. 3K-M, S18).
Inactivation of GABA synthesis or transport disrupts goblet cell hyperplasia without affecting immune responses
Increased expression of GABA receptors in asthmatic patient lung epithelial cells has been reported, and intranasal administration of GABA inhibitor can suppress OVA-induced goblet cell hyperplasia in mice (30). Thus, even though GABA had no effect on ILC2 stimulation, we tested the hypothesis that GABA signaling from PNECs is required for goblet-cell hyperplasia in vivo. We disrupted GABA signaling by inactivating Gad1, which encodes a GABA synthesis protein, or Vgat, which encodes GABA transporter. As PNECs are the only cells in lung that express these genes, inactivation by Shhcre (hereafter Gad1CKO or VgatCKO mice) led to the specific disruption of GABA production from PNECs (31). Airway epithelial cell development remained normal in these mutants (Fig. S19). However, when treated with OVA as neonates, both mutants showed a striking deficit in goblet-cell hyperplasia (Fig. 4A-D), confirming recent findings (37). To test if GABA is also required in adults for the goblet-cell hyperplasia response, we subjected adult Gad1CKO mice to the adult OVA model and found dampened goblet-cell production of mucus (Fig. S20). To further validate that disrupting GABA production in the lung was responsible for reduced goblet cell hyperplasia, we inactivated Gad1 in the lung epithelium using Nkx2-1creERT2 (generating Nkx2-1creERT2 Gad1fl/fl, or Nkx2-1creER;Gad1 mutant). Nkx2-1creER;Gad1 mutants also showed a deficit in goblet-cell hyperplasia (Fig. S21). These results suggest that GABA production from the PNECs are essential for the goblet-cell hyperplasia response.
Figure 4.
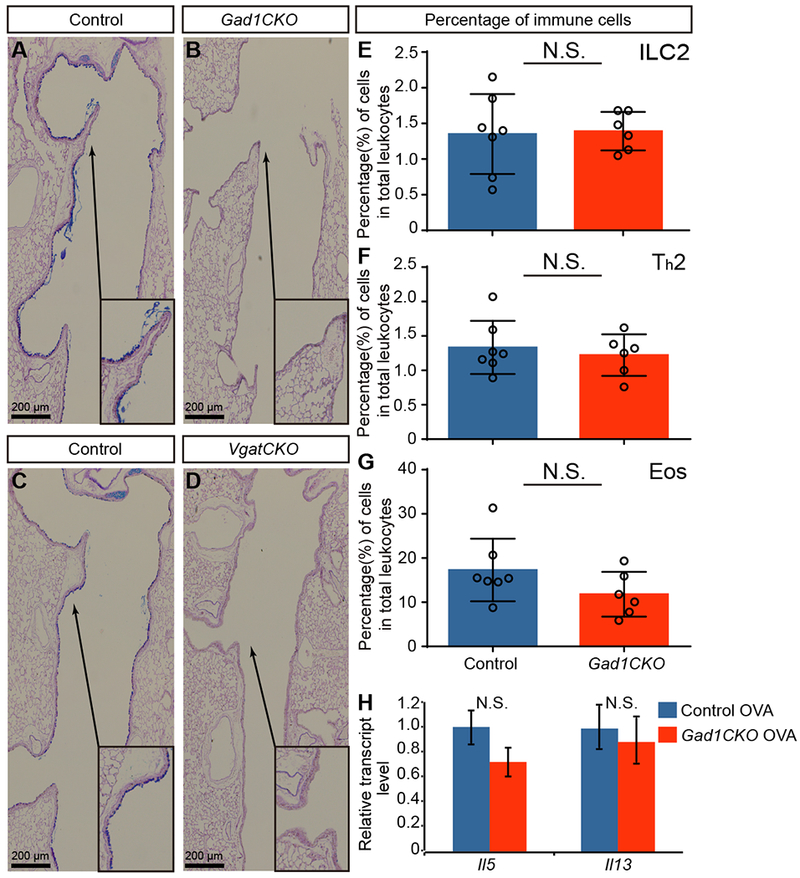
GABA from PNECs is essential for goblet-cell hyperplasia. (A-D) Representative PAS staining of OVA-challenged airways at P24. Arrows from insets point to areas that are magnified. (E-G) Percentage of immune cells in whole lungs after OVA challenge (n = 7 for control and n = 6 for mutant groups). Mann–Whitney U test was used. (H) Cytokine gene expression as assayed by qRT-PCR in whole lung after OVA challenge. Student’s t-test was used (n = 3 for each group). N.S. for not significant p ≥ 0.05, * for p < 0.05, ** for p < 0.01. Error bars represent mean ± SEM.
Next, we addressed if the goblet-cell phenotype in the GABA-signaling mutants is associated with a disruption of immune-cell infiltration and cytokine production in vivo. We found that the Gad1CKO mice had similar levels of ILC2s, Th2 cells, and eosinophils compared to controls upon OVA challenge, unlike in the Ascl1CKO mutant (Fig. 4E-G, S22). Il5 and Il13 levels were not significantly different. Thus, consistent with in vitro data (Fig. 3H), GABA signaling from PNECs is essential for goblet-cell hyperplasia without affecting type 2 immune cell infiltration in the OVA asthma model.
Instillation of CGRP and GABA in the Ascl1CKO mutant reverses defective goblet-cell hyperplasia and immune-cell infiltration
To address if CGRP and GABA are sufficient to reverse the defective asthma-like response in the absence of PNECs, we introduced a mixture of CGRP and GABA via intratracheal administration immediately after each OVA challenge to mimic the in vivo role of PNECs. Based on previously published doses, we used a 40-μl mixture of 100 nM CGRP and 10 mM GABA (38, 39). We found that the exogenous administration of CGRP and GABA was sufficient to restore asthma-like phenotypes in Ascl1CKO mice, including goblet-cell hyperplasia, immune-cell infiltration, and type 2 cytokine expression (Fig. 5, S23). Other airway cell types were not perturbed (Fig. S24). As a control, we also administered the same mixture to wild-type mice that were sensitized but not challenged with OVA. GABA and CGRP did not induce goblet-cell hyperplasia in these mice, indicating that these PNEC products collaborate with immune cells to mount goblet-cell hyperplasia triggered by allergen challenge (Fig. S25). Together, these findings offer an in vivo demonstration that CGRP and GABA are key bioactive products from PNECs that can drive lung allergic responses in the absence of these cells.
Figure 5.
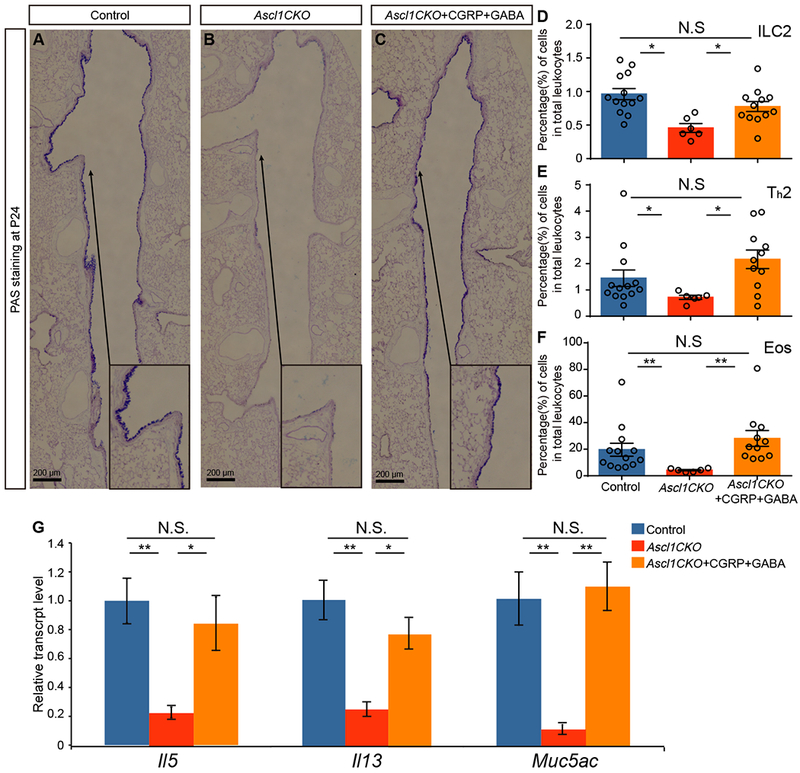
In vivo restoration of OVA responses in Ascl1CKO mutants with intra-tracheal instillation of CGRP and GABA.
(A-C) PAS staining after OVA challenge. Arrows point to areas that are magnified in the insets. (D-F) Immune-cell numbers from whole lungs after OVA challenge (n = 13 for control, n = 6 for Ascl1CKO, and n = 11 for Ascl1CKO+CGRP+GABA groups). Mann–Whitney U test was used. (G) Gene expression as assayed by qRT-PCR in whole lungs after OVA challenge. Student’s t-test was used (n = 3 for each group). N.S. for not significant p ≥ 0.05, * for p < 0.05, ** for p < 0.01. Error bars represent mean ± SEM.
PNECs are increased in the airways of human asthma patients
Although PNEC pathology has been documented in a number of lung diseases, whether they change in human asthma has not been directly addressed. To investigate this, we stained lung sections from asthmatics and age-matched controls using anti-Bombesin antibody, which is commonly used to outline all human PNECs. We also used anti-CGRP antibody, which like in mice, outlines the subset of CGRP-producing PNECs (Fig. 6A-D, Table S1 for patient characteristics, treatment regime, and primary autopsy findings). We found that by quantifying either total PNECs, PNECs in the proximal bronchiole or distal respiratory bronchiole, or PNEC cluster size, there was an increase in asthmatic samples compared to controls following each of these comparisons (Fig. 6E-L). The increase in the CGRP-positive PNEC subset was more significant than the Bombesin-positive total PNECs (p = 0.0485 for total Bombesin, p = 0.0052 for total CGRP). These findings in humans are consistent with our findings in mice, and are suggestive of a trend that an increase in PNECs, in particular CGRP-expressing PNECs, may contribute to allergic asthma.
Figure 6.
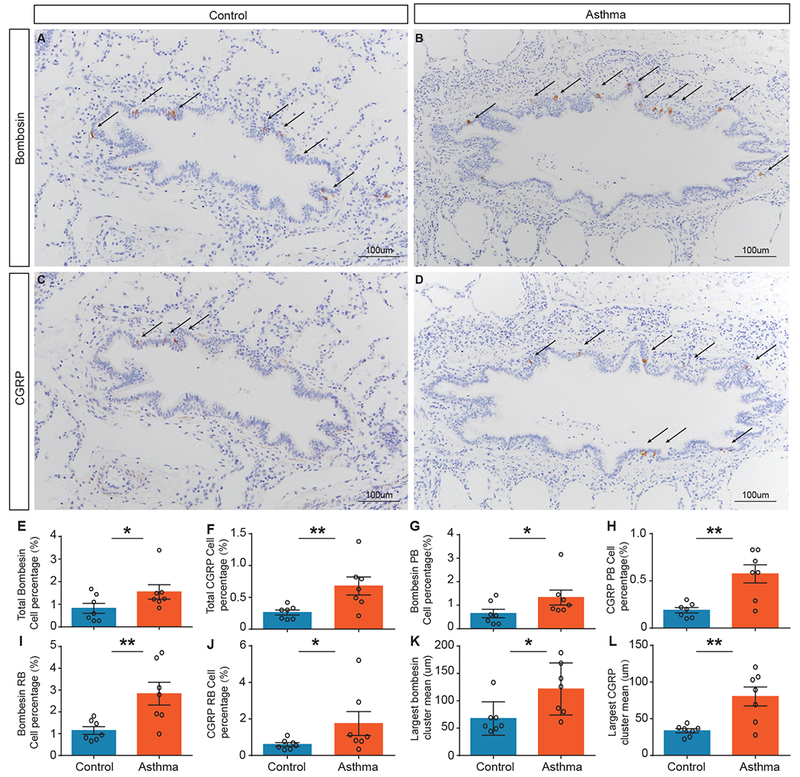
PNECs are increased in number in asthmatic lungs.
(A-D) Representative images of control and asthma autopsy lung sections stained with anti-Bombesin antibody or anti-CGRP antibody to delineate PNECs expressing each marker. (E-J) Quantification of percentage of Bombesin- or CGRP-positive epithelia area for total, or proximal bronchiole (PB) versus distal respiratory bronchiole (RB). There is an increase in asthma samples with both Bombesin and CGRP. Bombesin: total p = 0.0485, PB p = 0.0274, and RB p = 0.0037. CGRP: total p = 0.0052, PB p = 0.0076, and RB p = 0.0274. (K and L) Quantification of the mean large Bombesin- or CGRP-positive cluster size. There is an increase in asthma samples with both Bombesin and CGRP. Bombesin: p = 0.010, and CGRP p = 0.0075. Mann–Whitney U test was used (n = 7 controls and n = 7 asthmatics). Ages: 16 months to 13 years (see Table S2 for age match and other clinical data). Error bars represent mean ± SEM.
Discussion
The precise in vivo function of PNECs has been a long-standing question in lung biology. Here we show that PNECs are critical for mounting type 2 immune responses in mouse models of asthma. They act through secreted neuropeptides and neurotransmitters, including CGRP and GABA (Fig. 7A). PNECs signal directly to ILC2s, and together they form a neuroimmunological module to sense and respond to environmental stimuli that enter the airway (Fig. 7B).
Figure 7. PNEC–ILC2 axis functions at airway branch-point junctions to amplify the allergic asthma response.
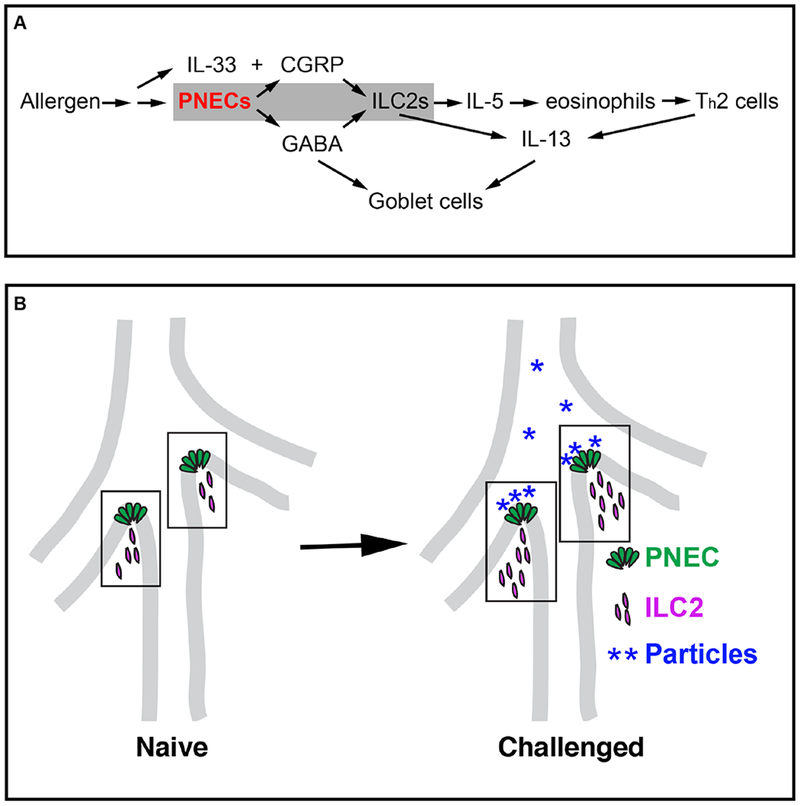
(A) A model for PNEC function in asthmatic responses. PNECs act through CGRP to stimulate ILC2s, and GABA to stimulate goblet cell hyperplasia. (B) A diagram illustrating PNEC–ILC2 interactions in neuroimmunological modules (boxed areas) at airway branch-point junctions, where particles carrying allergen or other antigens congregate.
The finding that the collaboration between PNECs and ILCs occurs preferentially at airway branch points is intriguing because computational modeling of particle dynamics shows that over time, particles that enter the airway concentrate at branch points (40). This correlation suggests that cells that reside at these sites are at a prime location to sample diverse inputs, including chemosignals (Fig. 7B). This preferential positioning of PNECs is established in utero, poising these cells to respond to the environment at first breath. In line with growing evidence supporting the impact of early-life exposures on the immune system (41), we propose that in the early postnatal environment, with every stimulation, the PNEC/ILC2 neuroimmunological modules mature into signaling centers that are critical in airway defense against pathogens and tissue damage.
Our results show that PNECs act through distinct secreted products to control different aspects of the responses to allergen (Fig. 7A). Although GABA synthesis and secretion are essential for goblet-cell hyperplasia (37), GABA regulates neither cytokine levels nor immune-cell infiltration. In contrast, CGRP can stimulate residential ILC2s at close range, inducing them to release IL-5, which can in turn recruit eosinophils and elicit a cascade of immune responses (32, 33, 42). In the absence of PNECs and consequently CGRP from these cells, the ILC2 response is severely dampened even though IL-33 is still present. Consistent with this, global inactivation of either Calcrl or Ramp1, genes encoding co-receptors for CGRP, results in diminished lung inflammation upon OVA challenge (29). Furthermore, our data showed that Il5cre-mediated inactivation of Calcrl also resulted in a deficit in the immune response. These results demonstrate that signals from PNECs are important triggers of ILC2 activation in the two allergic asthma models that we have used.
It remains unclear how PNECs are activated following allergen challenge. Our data suggest that the increase of CGRP production in the lung is not due to an increase in PNEC cells, but rather an increase in CGRP level per PNEC. Recent data indicate that innervation of PNEC is required for the increased production of GABA, suggesting that stimulating efferent neurons that innervate PNECs may be a mechanism that can activate PNECs (37). It is also possible that allergen-induced upstream immune cells and signals may activate PNECs.
In addition to PNECs, neurons are also a source of neuropeptides in the lung. While genetic ablation of TRPV1-expressing sensory neurons did not disrupt an OVA-induced immune response, ablation of a larger pool of NAV1.8 nociceptor-expressing sensory neurons did blunt eosinophil and T-cell responses (43, 44). Several recent studies also showed that neuron-derived neuromedin can directly interact with ILC2s and regulate immune responses in both the intestine and lung (45–47). Another current study showed that TRPV1-neurons, acting through CGRP, regulates protective immunity against lethal Staphylococcus aureus pneumonia (48). With the increasing awareness that neuropeptides are important regulators of lung immune responses, our findings here establish PNECs as a crucial source of signals.
PNECs represent the first specialized cell type that forms within the lung epithelium, suggesting that early establishment of this sensory population is of prime importance to building a functional organ that is responsive to environmental inputs. Our finding that PNECs are increased in asthmatic patient lungs raise the possibility that this increase may contribute to disease. Given the evolutionary conservation of these cells, it is unlikely that PNECs act as the lung “appendix”, a site of pathogenesis with no apparent role in homeostasis. Rather, PNECs are likely essential for lung defense, in addition to conferring responses to allergens as demonstrated here. Elucidating the full capacity of PNECs is important to understanding how blocking their function, or the function of their secreted products, can be used safely and effectively to treat allergic lung diseases such as asthma. Similar cells such as PNECs are found in other tissues, such as the enteric endocrine cells in the intestine. These neuroendocrine cells may prove to play critical roles in translating organ-specific environmental inputs into appropriate tissue responses.
Materials and Methods
Animals
All mice were housed and all experimental procedures were carried out in American Association for Accreditation of Laboratory Animal Care accredited facilities and labs at either the University of Wisconsin-Madison or University of California, San Diego. This study followed the Guide for the Care and Use of Laboratory Animals. Shhcre, Ascl1fl, Vgatfl, Gad1fl, Calcrlfl, Il5tdTomato-cre Nkx2-1creERT2, and Calcalox-Gfp-lox-Dta mice have all been described previously (20, 23, 28, 49–51). Il5tdTomato-cre, Gad1fl, Calcrlfl and Ascl1fl mice were on the B6 background. Shhcre , Nkx2-1creERT2, Calcafl-Gfp-fl-Dta, and Vgatfl mice were back-crossed to the B6 background for at least three generations starting from a mixed background. Littermates were used as controls to minimize potential genetic background effects.
Allergen-induced asthma model
For the early postnatal OVA asthma model, pups were sensitized by intraperitoneal injections of 10 μg of ovalbumin in 10 μl of PBS (OVA, A5503; Sigma) with 10 μl Imject alum (ThermoScientific) on postnatal day (P) 5 and P10, followed by three 15-minutes challenges with 3% aerosolized OVA solution on P18, P19, and P20. For controls in this protocol, all pups were injected on P5 and P10 with OVA and alum, and challenged on P18, P19 and P20 with PBS. Mice were sacrificed on P24 for analysis.
For the adult OVA asthma model, both mutant and control mice were sensitized by intraperitoneal injections of 50 μg of ovalbumin (OVA, A5503; Sigma) with 10 μl Imject alum (ThermoScientific) at 6 weeks of age on day 0 and day 7, followed by five 15-minutes challenges with 3% aerosolized OVA solution on days 15, 16, 17, 18, and 19. Mice were sacrificed on day 23 for analysis.
For the adult HDM model, 50 μg of HDM extract (Dermatophagoides pteronyssinus, Greer Labs) was introduced intranasally at 4 weeks of age on days 0, 7, 17, and 21. For controls in this protocol, PBS was administered intranasally on the same schedule instead of HDM. Mice were sacrificed on day 24 for analysis.
In Shhcre;Calcalox-Gfp-lox-DTR mutant and corresponding controls, Diphtheria toxin (List labs) at 10 ng/g body weight was administered intranasally at P18 and P22. Treated mice were rested for at least four days before HDM treatment.
In pregnant females used to generate Nkx2-1creERT2;Gad1fl/fl mutant and corresponding controls, tamoxifen was injected at embryonic days (E) 11 and 12 to induce cre for the inactivation of Gad1.
Histology, mean linear intercept (MLI), and histological mucus index (HMI) analysis
Mice were euthanized using CO2. Lungs were inflated with 4% PFA (paraformaldehyde) at 35 cmH2O airway pressure, and then fixed overnight. Lungs were then prepared for paraffin (8 µm), cryo (10 µm), or vibratome (100 µm) sectioning. Goblet cells were stained using a periodic acid–Schiff (PAS) staining kit (Sigma). The histological mucus index (the percentage of PAS-positive cells in the bronchial epithelium) was determined by using Image-J software (NIH). To quantify mean linear intercept (MLI), 20× H & E images were used. For each genotype, three mice, three sections per mouse and three independent fields per section were analyzed. Samples were compared using Student’s t-test, by MLI ± SEM with statistical significance set at p < 0.05.
Immunostaining
The following primary antibodies were used at the indicated final concentrations for immunofluorescence or immunohistological staining: rabbit anti-Synaptophysin plolyclonal antibody [5 μg/ml] (Fisher), rabbit anti-Bombesin polyclonal antibody [5 μg/ml] (ImmunoStar, Inc.), rabbit anti-Calcitonin gene-related peptide/CGRP polyclonal antibody [5 μg/ml] (LifeSpan BioSciences), rabbit anti-CGRP polyclonal antibody [2 μg/ml] (Sigma), rat anti-E-Cadherin polyclonal antibody [5 μg/ml] (Abcam), rabbit anti-SCGB1A1 polyclonal antibody [5 μg/ml] (Seven Hills Bioreagents), rabbit anti-SPC polyclonal antibody [5 μg/ml] (Seven Hills Bioreagents), mouse anti-MUC5AC monoclonal antibody [5 μg/ml] (MRQ-9, Sigma), and syrian hamster anti-T1alpha (PDPN) polyclonal antibody [5 μg/ml] (Developmental Studies Hybridoma Bank). The following secondary antibodies were used: Cy3-conjugated goat anti-rabbit IgG [2 μg/ml], FITC-conjugated goat anti-rabbit IgG [2 μg/ml], FITC-conjugated goat anti-rabbit IgG [2 μg/ml], goat anti-rat FITC [2 μg/ml] (all from Jackson Immunoresearch), HRP-conjugated goat anti-rabbit IgG [2 μg/ml] (Vector Laboratories), and ImmPACT DAB peroxidase (HRP) substrate following manufacturer’s protocol (Vector Laboratories). Images were acquired either by ZEISS LSM 880 with Airyscan (Figure 3C,D, or by ZEISS Axio Imager 2 (the rest of mouse images), or Nikon DSRi1 digital camera mounted on a Nikon Eclipse 80i microscope (human sample images).
EdU analysis of cell proliferation
For EdU analysis of cell proliferation, ~300 μl of 400 μM EdU solution (ThermoScientific) was intraperitoneally injected. Mice were sacrificed one hour after EdU injection. Lungs were fixed in 4% PFA overnight and prepared for cryo sectioning (20 µm). EdU was detected using the Click-iT EdU Kit with Alexa Fluor 488 (Invitrogen).
Tissue processing for cell sorting and flow cytometry
After euthanization and before tissue harvest, mice were transcardially perfused with 5 ml of cold PBS. Whole lungs were digested by gentle shaking in 5 ml of HBSS with 0.1 Wünsch units (WU) ml of Liberase (Roche) and 25 mg DNase I (Roche) for 60 min at 37 °C, and then mechanically dissociated using GentleMACS C tubes (Miltenyi Biotec) followed by straining through a 70-μm filter. The cells were then re-suspended in 40% Percoll, underlined with 66% Percoll and centrifuged. Hematopoietic cells were isolated from the interphase for sorting and analysis.
Cell sorting and flow cytometry
The single-cell suspensions from above were pelleted, aliquoted at ~1 × 106 cells per tube, and stained with Fc blocking antibody (5 μg/ml, BD) and Live/Dead Fixable Dead Cell Stain Kit (Invitrogen) at room temperature for 10 min. The cells were washed and then incubated with surface marker antibody cocktail for 45 min at 4 °C. For sorting ILC2s and Th2 cells, the following antibodies were used (all antibodies are from Biolegend, used at 2 μg/ml, unless otherwise specified): BV785-conjugated anti-CD90.2 (30/H12), AF700-conjugated anti-B220 (RA36B2), AF700-conjugated anti-CD11b (M1/70), AF700-conjugated anti-CD11c (N418), AF700-conjugated anti-Nk1.1 (PK136), BV510-conjugated anti-CD4 (GK1.5), PE-Cy7-conjugated anti-TCR beta (H57-597), FITC-conjugated anti-TCR delta (GL3), and PE-conjugated anti-ST2 (DIH9). For sorting eosinophils, the following antibodies were used: PE-conjugated anti-CD64 (X54-5/7.1), PE-Cy7-conjugated anti-CD11c (N418), and BB515 anti-Siglec F (2 μg/ml, E50-2440, BD). Samples were analyzed on an LSR II (BD Biosciences) with four lasers (405 nm, 488 nm, 561 nm, and 635 nm). Data were analyzed with FlowJo software (Treestar).
Cell culture
Freshly sorted ILC2s were cultured in RPMI culture media (Sigma) with 10% (vol/vol) heat-inactivated FBS (from Japan BioSerum), 100 U/ml penicillin/streptomycin, 10 mM HEPES buffer solution, 1× MEM nonessential amino acids, 1 mM sodium pyruvate, 50 μM 2-mercaptoethanol and 50 μg/ml gentamycin sulfate. Cells were cultured with IL-7 (10 ng/ml) and additional indicated cytokines (each at 10 ng/ml) for 12 hr. Then either CGRP (Sigma, 100 nM) or GABA (Sigma, 10 mM) were added. Freshly sorted Th2 cells were cultured with immobilized anti-CD3 antibody (Biolegend, 145-2C11, 10 μg/mL) with IL-33 (R&D Systems, 10 ng/ml) and IL-7 (R&D Systems, 10 ng/ml), and either CGRP (100 nM) or GABA (100mM). Freshly sorted eosinophils were cultured with IL-5 (R&D Systems, 10 ng/ml), and with or without CGRP (100 nM). Cytokine production were determined by using IL-5 ELISA kit from eBioscience or the Biolegend LEGENDplex Th2 system. Leukotriene C4 production were measure by using the LTC4 ELISA kit (Cayman Chemical).
Human asthma study population
After Institutional Review Board approval, the pathology database at Seattle Children’s Hospital was queried from 1960-2016 to identify autopsies in which asthma was documented in the final report. An age-matched control was obtained from the same year as each asthma case.
Human sample morphometric analysis
All slides were stained, imaged, and quantified together. For each subject, immunostaining for CGRP and Bombesin was quantified in up to 12 random peripheral (non-cartilage containing) airways. Airways were classified as either proximal (comprising membranous and terminal bronchioles with complete muscular walls) or respiratory bronchioles (air passages composed of part muscular bronchial wall and part alveoli). Airways were visualized and captured with a Nikon DSRi1 digital camera mounted on a Nikon Eclipse 80i microscope and analyzed using NIS-Elements Advanced Research Software v4.13 (Nikon Instruments Inc., Melville, NY). All cases were photographed and analyzed using the same magnification, exposure time, lamp intensity and camera gain. No image processing was carried out prior to intensity analysis. In each airway, the immunostained area was expressed as a percentage of the total airway epithelial area (cell %). The largest immune-positive cluster size was documented for each airway and divided by total airways counted to obtain mean large cluster size.
Statistical Methods
As indicated in the figure legends, Student’s t-test was used for parametric data and the Mann–Whitney U test was used for non-parametric data. All statistical analyses were performed using Prism 6 (GraphPad).
Primers
All primers are listed in Table S3.
Supplementary Material
Acknowledgments
We would like to thank Dr. Q. Wu, Dr. K. Caron, and Dr. S.Han for sharing mouse strains, Dr. J. Whitsett for providing antibody, Dr. V. Nizet for the use of a flow cytometer, Dr. D. Broide for assistance with the HDM model, Dr. L. Nantie for help on imaging, Dr. S. Gong and Y. Wang for consultation on the delivery of peptides, and Dr. H. Hoffman, Dr. G. Haddad, and Dr. M. Kronenberg for advice. We would also like to thank the University of Wisconsin Carbone Cancer Center Flow Cytometry Laboratory for assistance with the use of flow cytometers.
Funding: This work was supported by NIH OT2OD023857, R01HL113870, R01HL119946, R01HL122406, and Wisconsin Partnership Program grant 2897 (to X.S.).
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: All data are presented either in the body of the paper or in the Supplementary Materials.
Supplementary Materials
Fig. S1-S25
Table S1 to S3
References
- 1.Akinbami LJ, Moorman JE, Liu X, Asthma prevalence, health care use, and mortality: United States, 2005-2009. Natl Health Stat Report, 1–14 (2011). [PubMed] [Google Scholar]
- 2.Hardman CS, Panova V, McKenzie AN, IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. Eur J Immunol 43, 488–498 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ziegler SF, Artis D, Sensing the outside world: TSLP regulates barrier immunity. Nat Immunol 11, 289–293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molofsky AB, Savage AK, Locksley RM, Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 42, 1005–1019 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byers DE et al. , Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest 123, 3967–3982 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein Wolterink RG et al. , Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol 42, 1106–1116 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Lambrecht BN, Hammad H, The immunology of asthma. Nat Immunol 16, 45–56 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Kuo CS, Krasnow MA, Formation of a Neurosensory Organ by Epithelial Cell Slithering. Cell 163, 394–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noguchi M, Sumiyama K, Morimoto M, Directed Migration of Pulmonary Neuroendocrine Cells toward Airway Branches Organizes the Stereotypic Location of Neuroepithelial Bodies. Cell Rep 13, 2679–2686 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Brouns I et al. , Neurochemical pattern of the complex innervation of neuroepithelial bodies in mouse lungs. Histochem Cell Biol 131, 55–74 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Cutz E, Pan J, Yeger H, Domnik NJ, Fisher JT, Recent advances and contraversies on the role of pulmonary neuroepithelial bodies as airway sensors. Seminars in Cell & Developmental Biology 24, 40–50 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Gu X et al. , Chemosensory functions for pulmonary neuroendocrine cells. Am J Respir Cell Mol Biol 50, 637–646 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cutz E, Perrin DG, Pan J, Haas EA, Krous HF, Pulmonary neuroendocrine cells and neuroepithelial bodies in sudden infant death syndrome: potential markers of airway chemoreceptor dysfunction. Pediatr Dev Pathol 10, 106–116 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Gillan JE, Cutz E, Abnormal pulmonary bombesin immunoreactive cells in Wilson-Mikity syndrome (pulmonary dysmaturity) and bronchopulmonary dysplasia. Pediatr Pathol 13, 165–180 (1993). [DOI] [PubMed] [Google Scholar]
- 15.Young LR et al. , Neuroendocrine cell distribution and frequency distinguish neuroendocrine cell hyperplasia of infancy from other pulmonary disorders. Chest 139, 1060–1071 (2011). [DOI] [PubMed] [Google Scholar]
- 16.van Meerbeeck JP, Fennell DA, De Ruysscher DKM, Small-cell lung cancer. Lancet 378, 1741–1755 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Branchfield K et al. , Pulmonary neuroendocrine cells function as airway sensors to control lung immune response. Science 351, 707–710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito T et al. , Basic helix-loop-helix transcription factors regulate the neuroendocrine differentiation of fetal mouse pulmonary epithelium. Development 127, 3913–3921 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Borges M et al. , An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature 386, 852–855 (1997). [DOI] [PubMed] [Google Scholar]
- 20.Harris-Johnson KS, Domyan ET, Vezina CM, Sun X, beta-Catenin promotes respiratory progenitor identity in mouse foregut. Proc Natl Acad Sci U S A 106, 16287–16292 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aven L et al. , An NT4/TrkB-dependent increase in innervation links early-life allergen exposure to persistent airway hyperreactivity. Faseb J 28, 897–907 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calderon MA et al. , Respiratory allergy caused by house dust mites: What do we really know? J Allergy Clin Immunol 136, 38–48 (2015). [DOI] [PubMed] [Google Scholar]
- 23.McCoy ES et al. , Peptidergic CGRPalpha primary sensory neurons encode heat and itch and tonically suppress sensitivity to cold. Neuron 78, 138–151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurowska-Stolarska M et al. , IL-33 induces antigen-specific IL-5(+) T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol 181, 4780–4790 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Zhou BH et al. , Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol 6, 1047–1053 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Ballantyne SJ et al. , Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J Allergy Clin Immunol 120, 1324–1331 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Li S et al. , Epithelium-generated neuropeptide Y induces smooth muscle contraction to promote airway hyperresponsiveness. J Clin Invest 126, 1978–1982 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nussbaum JC et al. , Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502, 245–248 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li M et al. , Deficiency of RAMP1 attenuates antigen-induced airway hyperresponsiveness in mice. PLoS One 9, e102356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiang YY et al. , A GABAergic system in airway epithelium is essential for mucus overproduction in asthma. Nat Med 13, 862–867 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Schnorbusch K et al. , GABAergic signaling in the pulmonary neuroepithelial body microenvironment: functional imaging in GAD67-GFP mice. Histochem Cell Biol 140, 549–566 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Halim TYF et al. , Group 2 Innate Lymphoid Cells Are Critical for the Initiation of Adaptive T Helper 2 Cell-Mediated Allergic Lung Inflammation. Immunity 40, 425–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bartemes KR et al. , IL-33-Responsive Lineage(−)CD25(+)CD44(hi) Lymphoid Cells Mediate Innate Type 2 Immunity and Allergic Inflammation in the Lungs. J Immunol 188, 1503–1513 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li MY et al. , Deficiency of RAMP1 Attenuates Antigen-Induced Airway Hyperresponsiveness in Mice. PLoS One 9, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moro K et al. , Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol 17, 76–86 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Moro K et al. , Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Barrios J et al. , Early life allergen-induced mucus overproduction requires augmented neural stimulation of pulmonary neuroendocrine cell secretion. Faseb J 31, 4117–4128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young AB, Chu D, Distribution of Gaba-a and Gaba-B Receptors in Mammalian Brain - Potential Targets for Drug Development. Drug Develop Res 21, 161–167 (1990). [Google Scholar]
- 39.Mason BN et al. , Induction of Migraine-Like Photophobic Behavior in Mice by Both Peripheral and Central CGRP Mechanisms. Journal of Neuroscience 37, 204–216 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoegger MJ et al. , Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 345, 818–822 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gollwitzer ES, Marsland BJ, Impact of Early-Life Exposures on Immune Maturation and Susceptibility to Disease. Trends Immunol 36, 684–696 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Halim TY et al. , Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trankner D, Hahne N, Sugino K, Hoon MA, Zuker C, Population of sensory neurons essential for asthmatic hyperreactivity of inflamed airways. Proc Natl Acad Sci U S A 111, 11515–11520 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Talbot S et al. , Silencing Nociceptor Neurons Reduces Allergic Airway Inflammation. Neuron 87, 341–354 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wallrapp A et al. , The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549, 351–356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klose CSN et al. , The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 549, 282–286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cardoso V et al. , Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 549, 277–281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baral P et al. , Nociceptor sensory neurons suppress neutrophil and gammadelta T cell responses in bacterial lung infections and lethal pneumonia. Nat Med, doi: 10.1038/nm.4501 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pacary E et al. , Proneural transcription factors regulate different steps of cortical neuron migration through Rnd-mediated inhibition of RhoA signaling. Neuron 69, 1069–1084 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB, Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci 11, 998–1000 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chattopadhyaya B et al. , GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron 54, 889–903 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.