

Abstract
Fusarium Head Blight (FHB) is one of the most devastating diseases of cereals worldwide, threatening both crop production by affecting cereal grain development, and human and animal health by contaminating grains with mycotoxins. Despite that maize residues constitute the primary source of inoculum for Fusarium pathogenic species, the structure and diversity of Fusarium spp. and microbial communities in maize residues have received much less attention than in grains. In this study, a metabarcoding approach was used to study the bacterial, fungal and Fusarium communities encountered in maize stalks collected from 8 fields in Brittany, France, after maize harvest during fall 2015. Some predominant genera found in maize residues were cereal or maize pathogens, such as the fungal Fusarium, Acremonium, and Phoma genera, and the bacterial Pseudomonas and Erwinia genera. Furthermore, a high predominance of genera with previously reported biocontrol activity was found, including the bacterial Sphingomonas, Pedobacter, Flavobacterium, Pseudomonas, and Janthinobacterium genera; and the fungal Epicoccum, Articulospora, Exophiala, and Sarocladium genera. Among Fusarium spp., F. graminearum and F. avenaceum were dominant. We also found that the maize cultivar and previous crop could influence the structure of microbial communities. Using SparCC co-occurrence network analysis, significant negative correlations were obtained between Fusarium spp. responsible for FHB (including F. graminearum and F. avenaceum) and bacterial OTUs classified as Sphingomonas and fungal OTUs classified as Sarocladium and Epicoccum. Considering that isolates belonging to these taxa have already been associated with antagonist effect against different Fusarium spp. and/or other pathogenic microorganisms and due to their predominance and negative associations with Fusarium spp., they may be good candidates as biocontrol agents. Combining the use of Fusarium-specific primers with universal primers for bacteria and fungi allowed us to study the microbial communities, but also to track correlations between Fusarium spp. and other bacterial and fungal genera, using co-occurrence network analysis. Such approach could be a useful tool as part of a screening strategy for novel antagonist candidates against toxigenic Fusarium spp., allowing the selection of taxa of interest.
Keywords: maize residues, bacterial communities, fungal communities, Fusarium communities, biocontrol agents, co-occurrence network
Introduction
Fusarium Head Blight (FHB) of cereals (Nazari et al., 2014) is caused by several Fusarium species among which F. graminearum, F. culmorum, F. avenaceum, and F. poae are the main causal agents in Europe (Xu et al., 2005; Hellin et al., 2016). FHB is one of the most important diseases affecting cereals worldwide (Ramirez et al., 2006; Bateman et al., 2007; Gong et al., 2015) and represents a threat to human and animal health due to the possible production of mycotoxins by Fusarium species (Desjardins and Proctor, 2007). Crop rotation, and in particular maize as previous crop, can increase the risk of FHB incidence as previous crop infected residues are the primary source of pathogenic species (Shaner, 2003; Bateman et al., 2007; Fernandez et al., 2008). A high incidence of Fusarium species was found in the first internode-stalk of maize plants (Scauflaire et al., 2011), which is usually left in the field, turning into a main inoculum source for the following crop (Maiorano et al., 2008). Current crop, cropping history and tillage system have a significant influence on Fusarium and fungal communities of crop residues (Fernandez et al., 2008), on maize rhizospheric microbiome (Benitez et al., 2017) and on bulk soil microbial communities (Legrand et al., 2018). Although the plant genotype affects the rhizosphere microbial communities in maize (Aira et al., 2010), no studies have focused on how maize genotype affect the phyllosphere or the crop residue microbiome.
The low efficacy of current control strategies, mainly based on agricultural practices including tillage and the use of less sensitive cultivars, is prompting the scientific community to seek alternatives. Among them, the application of biocontrol agent against Fusarium species has been one of the major focuses of current research due to their compliance with environmental standards. Several candidate antagonists have been developed after isolation of microbial strains from different parts of cereals, such as root rhizosphere from maize (Abiala et al., 2015) and barley (Abd El Daim et al., 2015), wheat anthers (Palazzini et al., 2007), seed endophytes from wheat (Díaz Herrera et al., 2016), endophytes from maize (Mousa et al., 2015), or even from maize residues (Luongo et al., 2005; Singh et al., 2009), agricultural soils (He et al., 2009), silages and forest soils (Baffoni et al., 2015). Generally, the isolation of antagonistic candidates is empirical and needs confrontation tests under laboratory conditions which are used to screen a high number of candidates before field evaluations (He et al., 2009; Schöneberg et al., 2015). The efficacy of antagonists is usually reduced under field conditions compared to laboratory conditions (Luongo et al., 2005; Crane et al., 2013; Schisler et al., 2015; Legrand et al., 2017), mainly because of the complex interactions of antagonists with their biotic and abiotic environment in the field. Alternatively, this stepwise approach may also result in the possible loss of isolates that does not pass laboratory selection step but have good efficacy under field conditions (Schöneberg et al., 2015). In the latter study, they found that Clonostachys rosea, a weak competitor in in vitro co-culture with two F. graminearum and one F. crookwellense strains, showed the best antagonist potential of the total 12 strains screened in the field and was the only one able to reduce FHB incidence when inoculated after the pathogen. To increase the efficacy, recent studies demonstrated a synergistic/antagonistic activity of cocktail strains, such as the use of seven species isolated from maize roots to increase the efficacy of protection against F. verticillioides in maize kernels (Niu et al., 2017); similar results have been reached using a consortium of individually non-antagonistic bacteria of F. oxysporum in Arabidopsis thaliana (Fujiwara et al., 2016). Such isolation approach, however, is time-consuming, and still lack of efficacy. Despite some promising results, only a limited number of FHB biocontrol agents are commercially available (Legrand et al., 2017).
These approaches could undoubtedly benefit from the use of -omics technologies to better describe the microbial community functioning and improve the screening of antagonist organisms. Indeed, we must first gain a deeper understanding of the microbiota to which pathogens are confronted, and study the diversity and structure of pathogens themselves, especially for complex pathosystems such as FHB. Such knowledge may help select more appropriate biocontrol strategies, adapted to the Fusarium and microbial communities, which may vary according to the pedo-climatic environment of the agroecosystem. Specific primers designed to track Fusarium communities in soils and in wheat kernels using Next Generation Sequencing (NGS) approaches have already been developed (Edel-Hermann et al., 2015; Karlsson et al., 2016) but no studies combined this approach with the use of universal primers for fungal and/or bacterial species. A few metabarcoding studies aimed at describing the microbial communities in maize crop bulk soils and/or rhizospheric soils (Peiffer et al., 2013; Li et al., 2014; Zhao et al., 2016), while other focused on the influence of the presence of maize residues on soil microbiota (Chen et al., 2015; De la Cruz-Barrón et al., 2017) but none were dedicated to the microbial communities found on maize crop residues. Yet, since the primary source of Fusarium spp. inoculum originates from infected maize crop residues, it is important to deepen the knowledge of microbial communities associated with maize stalks. Those communities may contribute to soil suppressiveness against Fusarium pathogenic species and may be a good source of potential antagonists. Such approaches have already been undertaken in suppressive soils to vanilla and banana Fusarium wilt disease (Fu et al., 2017; Xiong et al., 2017) and in suppressive maize stalks to Fusarium ear rot (Köhl et al., 2015).
In this context, the aims of the present study were to (i) describe the bacterial, fungal and Fusarium communities found in maize stalks collected from fields after harvest in Brittany, France, using a metabarcoding approach; (ii) determine whether agronomic factors including the cultivar and/or the previous crop influence the microbial community structure and diversity; (iii) correlate the abundance of Fusarium pathogenic species with other bacterial and fungal taxa, using SparCC co-occurrence network analysis, as a preliminary step to identify potential antagonist microorganisms.
Materials and Methods
Maize Stalk Sampling
Maize stalks were collected from eight agricultural fields across Brittany, France in November 2015. In total, seven fields were surveyed in Finistère (Gouesnou, Saint-Renan, Locmaria-Plouzané and Plouzané) and one in Ille-Et-Vilaine (Gennes sur Seiche) located approximately 300 km away from the other fields (Figure 1). Field characteristics including the maize types and varieties, the previous crop and tillage practices were recorded (Figure 1). In each field, the above-ground parts of 15 maize stalks with nodal region were randomly sampled. Stalks were sampled within 3 days after maize harvest and were stored at -80°C until DNA extraction, except the one in Ille-Et-Vilaine, which was sampled within a month after maize harvest. P1 was chosen as an outgroup to help interpret the degree of variability observed within the maize kernel silage fields from Finistère.
FIGURE 1.

Field characteristics. (A) Location of the sampled fields; (B) agronomic characteristics of the fields.
DNA Extraction
For each field, 15 stalks were randomly chosen and 3 groups of 3 stalks were randomly selected at laboratory with each group corresponding to a biological replicate. For each replicate, four different portions of approximately 1 cm (nodal, internodal without leave, internodal with leave, and the external part) were cut with a sterilized scalpel from each stalk, mixed altogether and ground with liquid nitrogen in an autoclaved mortar and pestle. The pulverized tissues were stored in 1.5 mL Eppendorf tubes at 4°C until DNA extraction, performed within 4 h. DNA was extracted from 200 mg of pulverized maize stalks using FastDNA®SPIN kit (MP Biomedicals, Santa Ana, CA, United States) following the manufacturer’s instructions. Quality and concentration of purified DNA were determined using a UV spectrophotometer (NanoDrop 1000, Thermo Scientific, United States), and dilutions of at least 10 ng/μl were prepared for each DNA sample.
PCR Amplification and MiSeq Sequencing
A total of 24 samples (8 fields × 3 replicates) were selected for amplicon PCRs and high-throughput sequencing. Preparation of 16S rRNA, ITS and TEF1 libraries, and Illumina MiSeq 300 PE sequencing were performed at the McGill University and Génome Québec Innovation Centre, Montreal, Canada. Primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGGTATCTAATCC-3′) (Herlemann et al., 2011) were used to amplify the variable regions V3 and V4 of the 16S rRNA gene; primers ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) (White et al., 1990; Gardes and Bruns, 1993) to amplify the internal transcribed spacer; and primers TEF_FUS_F6 (5′-CCGGTCACTTGATCTACCAG-3′) and TEF_FUS_R7 (5′-ATGACGGTGACATAGTAGCG-3′) (Cobo-Diaz, Baroncelli, Le Floch, Picot, unpublished) to amplify a 430 bp region of the translation elongation factor (TEF1) of Fusarium species.
16S rRNA Read Filtering
The raw sequences were processed and analyzed with QIIME v1.9.1 (Quantitative Insights Into Microbial Ecology) (Caporaso et al., 2010). After joining the paired-end reads using the multiple_join_paired_ends.py and multiple_split_libraries_fastq.py scripts with default parameters, the chimeric sequences were then removed using UCHIME algorithm (Edgar et al., 2011) implemented in VSEARCH v1.1.31 against the ChimeraSlayer reference database (Haas et al., 2011). UCLUST algorithm (Edgar, 2010) was used for OTU picking and taxonomic assignment, which was made against GreenGenes v13.5 database (McDonald et al., 2012). To minimize the inflation of rare OTUs in the community analysis, we include only OTUs with sequence count greater than 10 (Brown et al., 2015; Oliver et al., 2015). Also, chloroplast, mitochondria and “No assigned” OTUs were discarded.
ITS Read Filtering
Although expected, a low level of joined paired-end reads was obtained for the ITS dataset, leading us to choose a different approach using QIIME v1.9.1 (Caporaso et al., 2010). The forward and reverse files were merged independently, using multiple_split_libraries_fastq.py. ITS1 and ITS2 regions were first extracted separately from forward and reverse non-chimera-fasta files, respectively, using ITSx v1.0.11 (Bengtsson-Palme et al., 2013) before being concatenated in a new file. A chimera filtering was made on concatenated file using the UCHIME algorithm (Edgar et al., 2011) with VSEARCH v1.1.3 see text footnote1 and a modified version of the UNITE/INSDC representative/reference sequences version 7.2 (UNITE Community, 2017) as reference database. The modification consisted in extracting ITS1 and ITS2 regions by ITSx software and concatenated them in the modified version of the database.
The ITS1-ITS2 concatenated file of non-chimeric sequences was used for OTU picking running the QIIME script pick_open_reference_otus.py, with BLAST (Altschul et al., 1990) as taxonomic assignment method and a modified version of UNITE plus INSD non-redundant ITS database version 7.1 (Kõljalg et al., 2013). Again, the modified version consisted in concatenating ITS1 and ITS2 regions after extracting them using ITSx software.
To minimize the overestimation of rare OTUs in the community analysis, we include only OTUs with sequence count greater than 10 (Brown et al., 2015; Oliver et al., 2015). Only OTUs assigned to kingdom Fungi were used for further analysis. The taxonomy for fungi known to have both sexual and asexual stages was replaced by accepted names according to Chen et al. (2018).
TEF1 Read Filtering
Paired-end reads were processed with QIIME (Caporaso et al., 2010), using the multiple_join_paired_ends.py and multiple_split_libraries_fastq.py scripts with default parameters. Pick de novo strategy was then employed to cluster the sequences into OTUs using pick_de_novo_otus.py, at 97% similarity cutoff. A first taxonomic assignment was performed using BLAST (Altschul et al., 1990) against NCBI non-redundant nucleotide database (nt)2. Only sequences assigned to Fusarium or the teleomorph name (Gibberella and Nectria), longer than 360 bp and with a percentage of identity higher than 97% were selected for further analysis, and only OTUs with sequence count greater than 10 were selected to minimize the inflation of rare OTUs in the community analysis (Brown et al., 2015; Oliver et al., 2015).
A second step of taxonomic assignment was done using the Fusarium MLST database web3, with the “pairse DNA alignments” tool, and compared with that provided by nt database. TEF1 sequences obtained along with references were aligned using MAFFT v7.304 (Katoh et al., 2017). Multiple sequence alignments were exported to MEGA7 (Kumar et al., 2016) and the best-fit substitution model was calculated for each separate sequence dataset. Using MrBayes 3.2.6 (Ronquist et al., 2012), the Markov chain Monte Carlo (MCMC) algorithm was performed to generate phylogenetic trees with Bayesian posterior probabilities for combined sequence datasets using the nucleotide substitution models determined by MEGA7 (Kimura 2-parameter with gamma distributed rate variation among sites [K2-G]). Four MCMC chains were run simultaneously for random trees for 2,000,000 generations (standard deviation of split frequencies between runs reached <0.01). Samples were taken every 500 generations. The first 25% of trees were discarded as burn-in phase of each analysis and posterior probabilities were determined from the remaining trees.
Alpha and Beta-Diversity Analysis
Metabarcoding datasets obtained after filtering (V3-V4 region of 16S rRNA, ITS1-ITS2 concatenated regions and Fusarium TEF1 sequences) were processed equally. A single rarefaction, based on the sample with the lowest number of reads, was used for alpha-diversity analysis using single_rarefaction.py QIIME script. OTUs richness (observed_otus) and evenness (equitability or Pielou’s index) were calculated with alpha_diversity.py QIIME script. The statistical software R v2.9.10 was used to perform one-way ANOVA with Tukey HSD post hoc test, for statistical analysis. Differences with p < 0.05 were regarded as statistically significant.
Taxa relative abundances across samples were compared with STAMP (Statistical Analysis of Metagenomic Profiles) bioinformatics software v 2.1.3 (Parks et al., 2014), using the OTU table from QIIME pipeline without any rarefaction. Statistical significance of the differences between multiple group-samples were calculated using ANOVA test, Tukey-Kramer post hoc test at 0.95 confidence interval, and corresponding p-values were corrected by Benjamini-Hochberg FDR (Benjamini and Hochberg, 2000).
Canonical Correspondence Analysis (CCA) were made at OTU level using four different datasets: (i) grouping the replicates by field samples, (ii) removing sample P1, (iii) grouping only maize varieties Telexx and P8400, and (iv) grouping previous crop forage rapeseed, wheat and mustard; using R package Vegan. Analyses of variance (ANOVA) were made between fields, variety or previous crop.
Significant correlations between the relative abundance of bacterial, fungal and Fusarium OTUs were made using Sparse Correlations for Compositional data algorithm implemented in SparCC python module (Friedman and Alm, 2012) and corresponding networks were plotted using the R package qgraph (Epskamp et al., 2012). Only correlations with a R-corr absolute value greater than 0.3 and p-value less than 0.05 were plotted. For correlation analysis, the relative abundance per bacterial, fungal or Fusarium OTUs was calculated by dividing the number of sequences per OTU by the total number of amplicon sequences for each sample. Additionally, relative abundance of Fusarium OTUs obtained with TEF1 primers, was also divided by the percentage of ITS sequences assigned to Fusarium genus, in order to have an estimated relative abundance (percentage) of each Fusarium species (determined by TEF1) in the total fungal community (determined by ITS).
Accession Numbers
All the raw reads have been deposited at the NCBI and are available under the Bioproject ID PRJNA3940634, with BioSample accession numbers from SAMN07348271 to SAMN07348278.
Results
Microbial Community Structure
A total of 1,041,456 sequences of 16S rRNA gene were clustered into 2,334 OTUs after filtering raw reads from 24 maize residue samples (8 fields × 3 biological replicates) and 12,936 sequences per sample were randomly extracted for alpha-diversity analysis. Only 1.98 and 4.54 % of the sequences were assigned to mitochondria and chloroplast, respectively, and removed along filtering step. Richness and evenness indices were significantly found the lowest in P1, with 488 observed OTUs, vs. 771 to 1,025 in the other samples, and with evenness value of 0.65 vs. 0.74 to 0.83. P2 also had significantly lower values compared to P3 and P4 for richness (771 OTUs vs. 964 OTUs in P4 and 1,025 in P3) and compared to P7 for evenness (0.74 vs. 0.83 in P7) (Figure 2A,B).
FIGURE 2.

Alpha-diversity indices. Richness index (Observed OTUs) and evenness index (equitability or Pielou’s index) per sample for (A,B) 16S rRNA data, (C,D) ITS data, and (E,F) TEF1 data. Letters indicate statistical differences between samples (p < 0.05). Telexx variety samples are plotted in green and P8600 variety samples are plotted in blue.
For ITS, 1,129,203 sequences were clustered into 455 OTUs and 29,319 sequences per sample were randomly extracted for alpha-diversity analysis. No sequences belonging to plants were detected. There were no significant differences for alpha-diversity indices between fields, with average values of richness between 88 and 159 OTUs, and 0.49 to 0.70 for equitability, except for the significantly higher values of observed richness found in P3 compared to P1 (159 vs. 88 OTUs, respectively) (Figure 2C,D).
In the case of TEF1 sequences assigned to Fusarium, a total of 1,023,229 sequences were clustered into 48 OTUs after raw read filtering. The percentage of sequences not assigned to the Fusarium genus was very low and only represented 0.039 % of the total sequences after removing those corresponding to phage phiX174, used for quality controls in Illumina sequencing. Between 33,688 and 56,829 sequences per sample (except replicate 1 from P1, which was removed for further analysis because it only had 15 sequences) were obtained, and 33,693 sequences per sample were randomly extracted for alpha-diversity analysis. Although P1 showed a higher value of evenness (0.55) and richness (28 OTUs) than other fields (evenness from 0.34 to 0.43 and richness from 11 to 19 OTUs), these differences were not statistically significant (Figure 2E,F).
Bacterial Community Composition
A total of 17 phyla and 143 genera were detected based on 16S rRNA sequences. The most abundant phyla were Proteobacteria (50.5–80.6%), Bacteroidetes (13.6–31.9%), Verrucomicrobia (0.8–8.0%), Actinobacteria (2.7–5.1%), and TM7 (0.2–4.4%), with Alphaproteobacteria (23.4–42.8%), Gammaproteobacteria (7.7–32.2%), and Betaproteobacteria (6.6–14.8%) as the main proteobacteria classes (Figure 3A). P1 had a significantly higher abundance of Proteobacteria (80.6% vs. 50.5 to 67.7%) and a lower abundance of Betaproteobacteria (6.6% vs. 11.4 to 31.9%), Bacteroidetes (13.6% vs. 21.9 to 31.9%), and Verrucomicrobia (1.3 and 0.8, vs. 4.1 to 5.1%) compared to the other fields. Alpha and Gamma-proteobacteria were also found to be higher in P1 although differences were not significant. Other significant differences were found in P3, where a higher abundance of Proteobacteria (67.7% vs. 50.5 to 62.8%) and Gammaproteobacteria (28.8% vs. 7.7 to 18.3%) and a lower abundance of Bacteroidetes (21.9% vs. 24.1 to 31.9%) were observed compared to the other samples, except P1 (Figure 3A).
FIGURE 3.
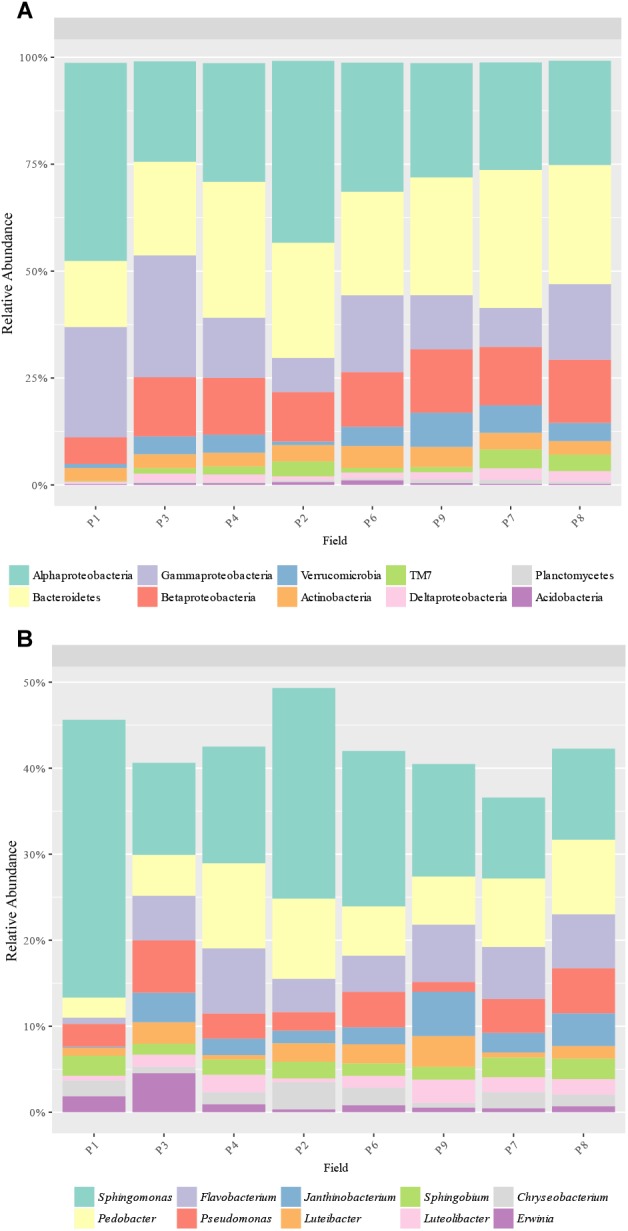
Bacterial taxa distribution. Relative abundance of the predominant (A) bacterial phyla (and Proteobacteria classes) and (B) genera obtained by 16S rRNA amplicons.
The most abundant genera were 4 proteobacteria: Sphingomonas (9.6–27.9%), Pseudomonas (1.1–6.2%), Janthinobacterium (0.1–5.2%) and Sphingobium (1.3–2.4%); and 2 bacteroidetes: Pedobacter (2.1–9.8%) and Flavobacterium (0.6–7.7%) (Figure 3B). In total, the abundance of 18 genera was found to be significantly different between samples, with some genera more abundant in P1, such as Adhaeribacter, Phormidium, Stenotrophomonas, and Skermanella (Figure 4). Some genera were also found to be significantly more abundant in field P3 (Rodhoferax, Sporocytophaga, Buchnera, and Sediminibacterium), P4 (Segetibacter and Paracoccus), P6 (Paenibacillus and Mycobacterium), P7 (Polaromonas, Bdellovibrio, and Gemmatimonas), and P9 (Kaistia) (Figure 4).
FIGURE 4.

Heatmap of bacterial genera with significant differences (p < 0.05) between samples. Relative abundance data were z-scored normalized by row. Plot was made using pheatmap R package with the default parameters. Sample names have the format Pij, where i refers to the field site (i = 1–8) and j refers to the replicate within the field (j = 1–3).
Fungal Community Composition
A total of 16 classes and 124 genera were detected from ITS sequences. The most abundant classes were Sordariomycetes (20.3–63.1%), Dothideomycetes (12.5–39.6%), Leotiomycetes (3.6–18.0%), Eurotiomycetes (1.8–13.8%), Tremellomycetes (2.4–9.9%), and Saccharomycetes (0.0–33.0%), with Sordariomycetes being the most abundant class in all samples except in P1, which had Saccharomycetes as the main class (Figure 5A). In addition, Saccharomycetes was the unique class with different relative abundances between samples, which was more abundant in P1 than the others (33.0% vs. up to 2.3%). The most abundant genera obtained were Fusarium (6.1–44.8%), Epicoccum (1.4–27.1%), Articulospora (3.5–16.6%), Microdochium (0.3–24.6%), Exophiala (1.0–12.9%), Sarocladium (0.4–14.5%), Cryptococcus (2.1–7.7%), Candida (0.0–30.0%), Acremonium (0.1–11.5%), and Phoma (0.0–9.2%) (Figure 5B). Only 2 genera had significant different abundances between fields: Candida (class Saccharomycetes) and Phoma (class Dothideomycetes) were more abundant in P1 than other fields (30.0% vs. up to 2.4% and 9.2 vs. up to 5.4%, respectively) (Figure 5B).
FIGURE 5.
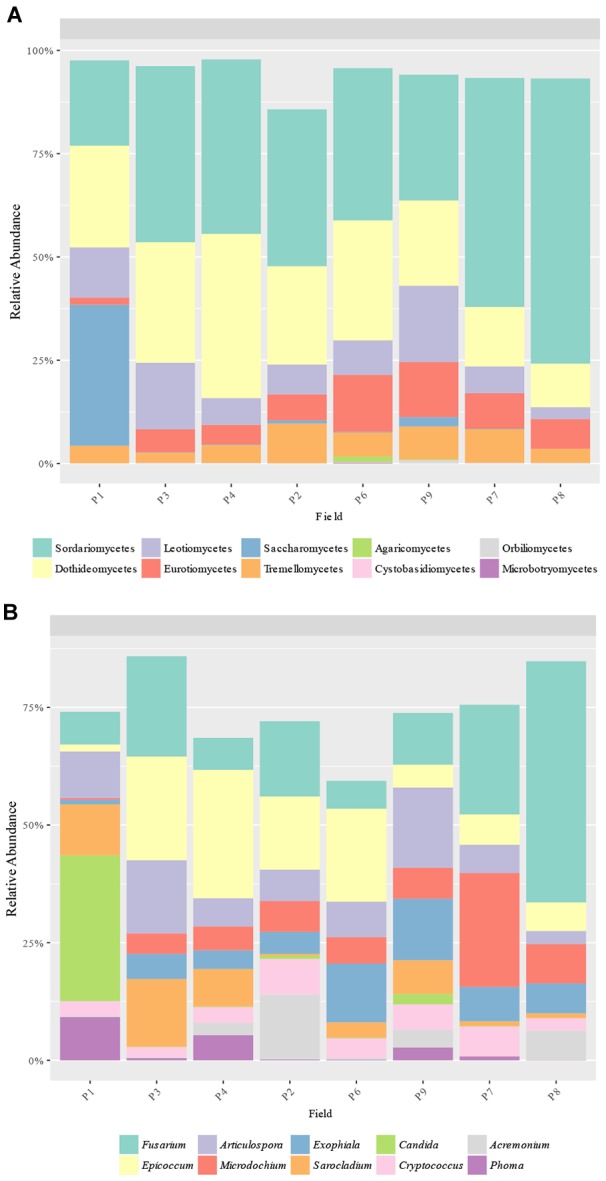
Fungal taxa distribution. Relative abundance of the predominant fungal (A) classes and (B) genera obtained by ITS amplicons.
Fusarium Community Composition
A total of 15 Fusarium species were detected according to taxonomic assignment performed by phylogenetic tree, although 5 OTUs were not clearly assigned to species level (Figure 6 and Supplementary Table S1). Three of them (denovo419, denovo339, and denovo314) were named as Fusarium sp. for further analysis, while denovo380 and denovo779 were grouped with the 2 OTUs assigned to F. equiseti (denovo921 and denovo438) inside Fusarium sp. FIESC (Fusarium incarnatum-equiseti species complex) for further analysis.
FIGURE 6.

Phylogenetic tree of TEF1 sequences from Fusarium OTUs and reference isolates, which accession number was indicated. Bayesian posterior probability (BPP) values (above 0.50) are shown at the nodes. The thickened nodes represent BPP values higher than 0.9.
Major species were F. graminearum (18.9–71.9%), F. avenaceum (15.7–53.8%), F. poae (0.0–18.6%), F. oxysporum (0.002–12.5%), F. verticillioides (0.0–13.4%), F. temperatum (0–10.1%), and F. sporotrichioides (0.0–11.1%), which covered between 80.2 to 100% of the total Fusarium sequences per field (Figure 7). The abundance of these species was unevenly distributed across fields. However, whatever the field, the most abundant species were F. graminearum and F. avenaceum, which sum accounted for 68.9 to 90.1% of the total Fusarium sequences per field, except in P1 (46.6%), and they were also the 2 Fusarium species present in all the fields. It is noteworthy that F. avenaceum outnumbered F. graminearum only in the three fields that had Telexx as maize variety (P2, P6, and P9). Moreover, other species were also subdominant depending on the field: F. poae in P4, P6, P7, and P8 (from 9.2 to 18.6%); F. oxysporum in P1 and P8 (15.4 and 11.8%, respectively); F. sporotrichioides and F. temperatum in P1 (16.6 and 15.2%, respectively); and F. verticillioides in P9 (13.4%); although there were not significant differences in relative abundance of Fusarium species between samples (Figure 7).
FIGURE 7.
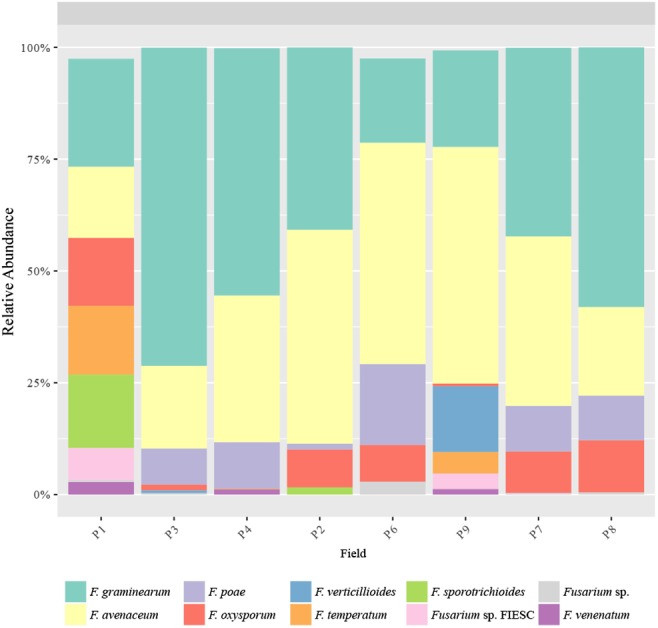
Fusarium species distribution. Relative abundance of the predominant Fusarium species obtained by TEF1 amplicons.
Canonical Correspondence Analysis
Unsurprisingly, Canonical Correspondence Analysis (CCA) showed that the composition and distribution of microbial communities in the outgroup P1 was different from the others samples. Bacterial, fungal and Fusarium communities presented significant differences between fields (ANOVA: F = 1.6308, Pr < 0.001 for bacteria; F = 2.0422, Pr < 0.001 for fungi; F = 1.7723, Pr < 0.001 for Fusarium), with P1 as the most different field in all 3 cases; and also P2, P3, and P9 for bacteria, and P9 for Fusarium communities (Figure 8A–C). The CCA excluding P1 showed similar degree of significance between samples (F = 1.4219 and 1.6738, for bacterial and fungal, respectively, Pr < 0.001), except for Fusarium (F = 1.3428, Pr = 0.038). A clear separation of almost all fields was observed for bacterial communities; P4 and P6 for fungal communities; and P6 and P9 for Fusarium communities (Figure 8D–F). The same analysis was done selecting the fields which had Telexx and P8400 as maize varieties, obtaining significant differences between varieties in bacterial (F = 1.5128, Pr = 0.002) and fungal (F = 1.7022, Pr = 0.015) communities but not in Fusarium communities (F = 1.2672, Pr = 0.214) (Figure 8G–I). Regarding the previous crop, only forage rapeseed, mustard and wheat were analyzed, resulting in significant differences between groups in bacterial (F = 1.4201, Pr = 0.001), fungal (F = 1.7673, Pr = 0.001) and Fusarium communities (F = 1.716, Pr = 0.01) (Figure 8J–L).
FIGURE 8.

Canonical correspondence analysis (CCA) calculated using OTUs relative abundance. Each dot represents a sample replicate for (A,D,G,J) bacterial, (B,E,H,K) fungal, and (C,F,I,L) Fusarium communities, using (A,B,C) all sites, (D,E,F) all sites except P1, (G,H,I) samples grouped by maize variety and (J,K,L) by previous crop.
SparCC Correlation Network
In total, 26 positive and 13 negative significant correlations (|coefficient correlation (=corr)|>0.3 and p-value < 0.05) were found between 27 OTUs: 6 from 16SrRNA, 19 from ITS and 4 from TEF1 (Figure 9A). The highest positive correlations were obviously found between F01 and Fs01, both assigned to F. graminearum by ITS and TEF1 sequencing, respectively (corr = 0.82); and between F02 and Fs02, both assigned to F. avenaceum by ITS and TEF1 sequencing (corr = 0.51). There were also significant positive correlations between Fs02 and the 2 F. graminearum OTUs (F01 and Fs01, corr = 0.40 for both) (Figure 9A). One fungal and one bacteria OTUs presented negative correlations with some Fusarium OTU(s): F04, assigned to Sarocladium strictum, against Fs03, assigned to F. oxysporum (corr = -0.30); and B01, assigned to Sphingomonas, against F01, Fs01, and Fs04, assigned to F. graminearum by ITS and TEF1, and to F. poae by TEF1 (corr = -0.36, -032 and -0.32, respectively). Moreover, B01 also presented negative correlations with B02, B03, B04, F06, and F07, assigned to Pseudomonas, Luteolibacter, family Xanthomonadaceae, Monographella cucumerina, and F. poae, respectively (Figure 9A).
FIGURE 9.

SparCC correlation networks observed between OTUs, obtained with 16S rRNA gene (Bacteria), ITS (Fungi), and TEF1 (Fusarium) sequences. Nodes correspond to OTUs, and connecting edges indicate correlations between them. Only nodes with negative or positive correlations, with values less than –0.30 (red) or larger than 0.30 (green), were represented. (A) SparCC correlation network using all the samples; (B) SparCC correlation network without the outgroup P1. OTUs marked with asterisk had been reassigned taxonomically using blastn against nr/nt-NCBI database and Fusarium MLST web database (Supplementary Table S2).
The same SparCC correlation analysis was done excluding the results from the outgroup field P1, which fungal and bacterial communities and distribution greatly varied from the other fields. In total 28 OTUs (7 from 16SrRNA, 18 from ITS and 3 from TEF1) presented 20 positive and 16 negative significant correlations between them (Figure 9B). As happened in the previous network analysis, the highest positive correlations were between F01 and Fs01 (corr = 0.83), assigned to F. graminearum by ITS and TEF1, respectively; between F02 and Fs02 (corr = 0.53), assigned to F. avenaceum by ITS and TEF, respectively; and between F02 and the two OTUs assigned to F. graminearum (corr = 0.41 for both TEF1 and ITS). Two fungal OTUs had negative correlations vs. some Fusarium OTU: the same Sarocladium OTU identified before (F04) vs. Fs03 and F08, assigned to F. oxysporum by TEF1 and ITS sequences (corr = -0.31 and -0.35, respectively); and F05, assigned to Epicoccum nigrum, vs. Fs04 and F10, assigned to F. temperatum and Candida sake (corr = -0.34 and -0.35, respectively) (Figure 9B).
Discussion
By sequencing three different amplicons from 24 maize samples, a total of 2,334 bacterial OTUs, 1,428 fungal OTUs and 48 Fusarium OTUs were obtained. This study is one of the first metabarcoding studies on maize residues, resulting in a higher diversity than previously found in other NGS studies of the maize rhizosphere (Peiffer et al., 2013; Li et al., 2014).
Proteobacteria (mainly alpha and gamma-proteobacteria), Bacteroidetes and Actinobacteria were found as the most abundant phyla in maize stalk surface, the first two of which have already been reported as the most abundant in the maize rhizosphere (Peiffer et al., 2013; Li et al., 2014), in soil samples after maize harvesting (Chen et al., 2015) and in the first stages of maize straw decomposition (Sun et al., 2013). Although maize residues can be considered as a separate compartment and a particular ecological niche compared to soil samples, some of the more abundant bacterial genera in the present study had also been found as predominant in maize rhizospheric soils, including Sphingobium (Peiffer et al., 2013; Li et al., 2014), Flavobacterium (Li et al., 2014; Yang et al., 2017; Correa-Galeote et al., 2016) and Sphingomonas (Correa-Galeote et al., 2018). Apart from these genera, the diversity of bacterial communities found in our maize residues generally differed from that of maize rhizospheric soils found in other studies (Peiffer et al., 2013; Li et al., 2014; Correa-Galeote et al., 2016; Yang et al., 2017). It should also be underlined that 2 of the over-represented genera in P1, Adhaeribacter and Stenotrophomonas, were also significantly more abundant in Fusarium wilt suppressive soils (Siegel-Hertz et al., 2018).
Some of the most abundant fungal genera found in the present study, including Fusarium, Epicoccum, Acremonium, Sarocladium, and Phoma had been reported as endophytes isolated from maize (Pan and May, 2009; Amin, 2013; Xing et al., 2018). In particular, Fusarium, Epicoccum, and Phoma were reported as the most abundant endophytes isolated from maize leaves, using two different lineages (Szilagyi-Zecchin et al., 2016), while Acremonium was detected exclusively in Fusarium wilt suppressive soils, but not in conducive soils (Siegel-Hertz et al., 2018). It is important to highlight that, in our maize samples, genera with reported biocontrol activity (Table 1) were predominant and included several bacterial genera such as Sphingomonas, Pedobacter, Flavobacterium, Pseudomonas, Janthinobacterium, Sphingobium, Chryseobacterium, Luteibacter, Dyadobacter, and Rhizobium; and fungal genera such as Epicoccum, Articulospora, Exophiala, Sarocladium, Cryptococcus, Candida, Acremonium, and Metschnikowia. On the other hand, only two bacterial genera, Pseudomonas and Erwinia, and three fungal genera, Fusarium, Acremonium, and Phoma, within the most abundant ones are known to include maize or cereal pathogens. A priori, although maize residues have been reported as the primary source of pathogenic species (Shaner, 2003; Bateman et al., 2007; Fernandez et al., 2008), microbial communities obtained from maize crop residues presented an important amount of organisms that could increase the protection of future rhizospheric complexes against pathogens. This hypothesis was corroborated with the presence of one bacterial and two fungal OTUs, assigned to Sphingomonas, Epicoccum nigrum, and Sarocladium strictum, negatively correlated to some Fusarium OTUs; these three genera were within the most abundant ones in the microbial communities studied. Likewise, a lower increase of Fusarium spp. colonization of maize stalks has also been reported when Sphingomonas species were more abundant (Köhl et al., 2015) and a strain of Sphingomonas was found to be antagonistic against F. avenaceum, F. culmorum, F. tricinctum, and F. graminearum (Wachowska et al., 2013). Acremonium spp. (basionym of Sarocladium) (Summerbell et al., 2011) were also found to be more abundant in maize stalks characterized by a lower increase in Fusarium colonization, rendering it as a potential antagonist of Fusarium spp. (Köhl et al., 2015). A better taxonomic classification of the Acremonium species present in our samples might be necessary as some species belonging to this genera have been reported to be pathogenic on maize (Tagne et al., 2002). Several Acremonium strains isolated from maize were able to inhibit the growth of some pathogens, such as Pythium ultimum, Sclerotium oryzae, Rhizoctonia solani, and Pyricularia oryzae (Potshangbam et al., 2017) or produce pyrrocidines A and B, which induce host defense mechanisms against microbial pathogens (Wicklow and Poling, 2009). Some Sarocladium endophytes isolated from wheat were also reported to inhibit F. graminearum and F. culmorum growth (Comby et al., 2017). It was also found that some strains of Epicoccum nigrum were able to reduce the mycelial growth of F. graminearum, F. avenaceum, and F. oxysporum on PDA (Ogórek and Plaskowska, 2011) and also in sterile wheat grain assays with F. graminearum (Jensen et al., 2016); or to reduce the sporulation of F. culmorum and F. graminearum on wheat straw (Luongo et al., 2005). Furthermore, E. nigrum has been used as a biological control on peaches and nectarines orchards against Monilinia spp. (De Cal et al., 2009) and against Pythium debaryanum and P. ultimum on cotton seedlings (Hashem and Ali, 2004). Overall, the high presence of these genera, previously reported as antagonists and negatively correlated to toxigenic Fusarium species or other pathogenic organisms, suggests that such taxa may be of interest as part of biocontrol strategies against toxigenic Fusarium spp.
Table 1.
Pathogen and antagonistic characteristics of species or strains belonging to the most abundant genus obtained in maize residues.
| % of total | Plant | Wheat | Maize | Biocontrol | ||
|---|---|---|---|---|---|---|
| sequences | pathogen | pathogen | pathogen | activity | Reference for biocontrol activity | |
| Bacterial genus | ||||||
| Sphingomonas | 16.0 | Yes | No | No | Yes | Wachowska et al., 2013 |
| Pedobacter | 6.7 | No | No | No | Yes | De Boer et al., 2007; Song et al., 2017 |
| Flavobacterium | 5.1 | No | No | No | Yes | Gunasinghe et al., 2004 |
| Pseudomonas | 3.6 | Yes | Yes | Yes | Yes | Hennessy et al., 2017 |
| Janthinobacterium | 2.5 | No | No | No | Yes | Berg et al., 2001 |
| Sphingobium | 1.9 | Yes | No | No | Yes | van Bruggen et al., 2014; Fu et al., 2017 |
| Chryseobacterium | 1.7 | Yes | No | No | Yes | Yin et al., 2013; Sang et al., 2018 |
| Luteibacter | 1.7 | No | No | No | Yes | De Boer et al., 2007 |
| Luteolibacter | 1.5 | No | No | No | No | |
| Erwinia | 1.3 | Yes | Yes | Yes | No | |
| Agrobacterium | 1.3 | Yes | No | No | (1) | |
| Hymenobacter | 1.2 | No | No | No | No | |
| Dyadobacter | 1.0 | No | No | No | Yes (2) | Fu et al., 2017 |
| Rhizobium | 0.8 | No | No | No | Yes (3) | Al-Ani et al., 2012 |
| Fungal genus | ||||||
| Fusarium | 17.1 | Yes | Yes | Yes | Yes (3,4) | Ghini et al., 2000; Boari and Vurro, 2004 |
| Epicoccum | 13.1 | Yes | No | No | Yes | Luongo et al., 2005 |
| Articulospora | 9.1 | No | No | No | Yes | Sugahara et al., 2008 |
| Microdochium | 7.6 | Yes | Yes | No | No | |
| Exophiala | 7.0 | No | No | No | Yes | Duvick et al., 1998 |
| Sarocladium | 5.7 | Yes | No | No | Yes | Comby et al., 2017 |
| Cryptococcus | 4.4 | Yes | No | No | Yes | Schisler et al., 2015 |
| Candida | 4.2 | No | No | No | Yes | Calvo-Garrido et al., 2013 |
| Acremonium | 3.1 | Yes | Yes | Yes | Yes (3) | Rajakumar et al., 2005 |
| Phoma | 2.4 | Yes | Yes | Yes | No | |
| Xenobotryosphaeria | 2.2 | No | No | No | No | |
| Pyrenochaetopsis | 2.2 | No | No | No | No | |
| Ramularia | 0.6 | Yes | No | No | No | |
| Hannaella | 0.5 | Yes | No | No | No | |
| Metschnikowia | 0.4 | No | No | No | Yes | Manso and Nunes, 2011 |
(1) Used for Agrobacterium-mediated plant transformation (Genetically Modified Organisms).
(2) Associated to disease suppressiveness.
(3) Over-represented in Fusarium wilt suppressive soils (Siegel-Hertz et al., 2018).
(4) Non-pathogenic strains.
The most abundant Fusarium species found in our maize residues were F. graminearum, F. avenaceum, and F. poae, with 41.3, 35.4, and 7.1% of the total sequences, respectively, which are one of the main causal agents responsible for FHB (Parry et al., 1995; Bottalico and Perrone, 2002). Likewise, F. graminearum and F. avenaceum were also described as the predominant species on maize stalks after a 6-month exposure period in the field, as evaluated by qPCR (Köhl et al., 2015) and are commonly found as the main Fusarium species in wheat, using TEF1 as well as Fusarium spp. specific primers (Karlsson et al., 2016, 2017) or using culture-dependent approaches (Xu et al., 2005; Nicolaisen et al., 2014; Basler, 2016). F. avenaceum was also found as the dominant Fusarium species in some soil samples associated to perennial plants (LeBlanc et al., 2017). In opposite to our findings, F. culmorum is commonly described as a dominant Fusarium species in maize or cereals (Scauflaire et al., 2011; Basler, 2016; Hellin et al., 2016). Mutual exclusion between strains of F. graminearum and F. culmorum had been previously demonstrated (Siou et al., 2015). This competition could account for the low abundance of F. culmorum (0.05%) in maize stalks in our present study. In addition, shifts from F. culmorum to F. graminearum on wheat have been described in different European countries, such as England and Wales (Jennings et al., 2004), Netherlands (Waalwijk et al., 2003), Denmark (Nielsen et al., 2011), and Belgium for maize ears and stalks (Scauflaire et al., 2011). Climatic changes were the main hypothesis put forward, although the increase in maize-wheat rotation crops may also contribute to this increase in F. graminearum and decrease in F. culmorum (Dill-Macky and Jones, 2000; Cromey et al., 2002). The distribution of the Fusarium communities also strongly depends on the environmental conditions which would favor some species over the others. For instance, Dorn et al. (2011) found that F. graminearum, F. verticillioides, and F. proliferatum were the most dominant Fusarium species isolated from maize kernels in Switzerland (22.3–81.6, 2.5–41.8, and 0.6–22.0% of occurrence in kernels, respectively), while F. equiseti, F. proliferatum, and F. verticillioides were the main Fusarium species in stalks (43.6, 16.3, and 11.5% of occurrence in nodes, and 27.6, 41.5, and 15.5% in internodes, respectively). In our study, F. graminearum and F. avenaceum seem to be more adapted than species from the Fusarium fujikuroi species complex (FFSC), including F. verticillioides and F. temperatum, which were dominant in maize kernels in Poland (Czembor et al., 2015) and in maize kernels from Southern to Central Europe (Logrieco et al., 2002). The mild and humid climatic conditions found in Brittany, which are known to be favorable to F. graminearum and F. avenaceum (Xu et al., 2008) may account for such observations. In contrast, higher prevalence of Fusarium ear rot caused by FFSC including F. verticillioides occurs under hot and dry conditions. For instance, F. verticillioides incidence was found negatively correlated to rainfall values in maize fields in Argentina (Pereira et al., 2011) and to kernel moisture in maize fields in United States (Bush et al., 2004). Moreover, we found that Telexx maize variety had highest abundance of F. avenaceum than F. graminearum, while the opposite was found for the other varieties used in the study. These differences in Fusarium composition could also induce differences in mycotoxin concentrations, as deoxynivalenol and zearalenone are produced by F. graminearum while moniliformin, enniatins and beauvericin can be produced by F. avenaceum (Ferrigo et al., 2016).
Microbial communities found in the maize residues on field P1 was the most different compared to the other fields. In general, this field was characterized by both significant lower bacterial and a higher Fusarium alpha-diversity indices, with a higher abundance of the fungal Candida. Several factors differed from the outgroup P1 compared to the other fields (Table 1) including the maize type and cultivar, the agricultural practices, the previous crop and the location. In addition, these samples were collected 1 month after harvest, suggesting that maize residues were already in the process of degradation. Maize genotype has already been reported to influence the microbial communities in rhizospheric samples (Li et al., 2014), it could also have a strong influence in the microbial communities on others parts of the plant including the residues. But due to bias in the sampling design (because this study was rather designed to estimate the diversity found in maize residues from various fields in Brittany), we cannot conclude which factor(s) contributed mainly to these differences between P1 and the other fields. Additional sampling will be undertaken to further clarify which factor(s) has(have) the higher influence on microbial communities, with an emphasis to maize microbial dynamics over the course of maize residue degradation.
The significance of this study first lies in its design of a new specific pair of primers to identify Fusarium species with metabarcoding approach. This new culture-independent approach for Fusarium species identification could be adapted to other genera, by the design of specific primers for Illumina metabarcoding. In addition, the combined used of these primers with universal primers for fungi and bacteria allowed, not only to provide an accurate description of the microbiota as well as the pathogenic Fusarium spp. under various agronomic practices (maize cultivar, previous crop), but also to assess the potential relationships between microorganisms using co-occurrence network analysis. More particularly, we could identify predominant taxa negatively correlated to toxigenic Fusarium spp. Therefore, such approach could be used as a pre-filtering for the selection of potential antagonists as part of biocontrol strategies. Following this investigation, culture-dependent approaches must be done to determine the antagonistic potential of species identified by the co-occurrence network analysis, both in laboratory and field experiments. Illumina technology allows putting more than one amplicon type and dozens of samples in only one run (Herbold et al., 2015). This approach is time-saving compared to the empirical BCA isolation strategies, and could have more importance in the screening of antagonists.
Based on the results of this preliminary study, we also suggest focusing on the microbial dynamics throughout the plant cultivation cycle in maize-wheat rotations, taking also into account the influence of plant cultivar on microbial communities.
Author Contributions
AP and GLF contributed to the conception and design of the study. AP performed sampling. JC-D performed DNA extraction and shipping, read filtering and OTU table filtering, and wrote the first draft of the manuscript. JC-D and GLF performed statistical analysis. RB performed phylogenetic tree. All authors contributed to manuscript revision, read and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully thank the farmers for kindly giving us access to their field and providing information about their field management.
Funding. This work was supported by the Brittany Region (Grant #9097 MycoRes) and the Institut Brestois Santé-Agro-Matière (IBSAM).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00261/full#supplementary-material
Taxonomic assignment of representative TEF1 sequences for each Fusarium OTU obtained, using phylogenetic tree, blastn vs. nt-NCBI database, and pairse alignment vs. FUSARIUM-MLST database.
Taxonomic assignment of representative TEF1 and ITS sequences for each Fusarium and unclassified genus OTUs plotted in network analysis, using blastn vs. nt-NCBI database, and pairse alignment vs. FUSARIUM-MLST database.
References
- Abd El Daim I. A., Häggblom P., Karlsson M., Stenström E., Timmusk S. (2015). Paenibacillus polymyxa A26 Sfp-type PPTase inactivation limits bacterial antagonism against Fusarium graminearum but not of F. culmorum in kernel assay. Front. Plant Sci. 6:368. 10.3389/fpls.2015.00368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abiala M. A., Odebode A. C., Hsu S. F., Blackwood C. B. (2015). Phytobeneficial properties of bacteria isolated from the rhizosphere of maize in Southwestern Nigerian soils. Appl. Environ. Microbiol. 81 4736–4743. 10.1128/AEM.00570-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aira M., Gómez-Brandón M., Lazcano C., Bååth E., Domínguez J. (2010). Plant genotype strongly modifies the structure and growth of maize rhizosphere microbial communities. Soil Biol. Biochem. 42 2276–2281. 10.1016/j.soilbio.2010.08.029 [DOI] [Google Scholar]
- Al-Ani R. A., Adhab M. A., Mahdi M. H., Abood H. M. (2012). Rhizobium japonicum as a biocontrol agent of soybean root rot disease caused by Fusarium solani and Macrophomina phaseolina. Plant Protect. Sci. 4 149–1155. 10.17221/16/2012-PPS [DOI] [Google Scholar]
- Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Amin N. (2013). Diversity of endophytic fungi from root of maize var. pulut (waxy corn local variety of South Sulawesi, Indonesia). Int. J. Curr. Microbiol. Appl. Sci. 2 148–154. [Google Scholar]
- Baffoni L., Gaggia F., Dalanaj N., Prodi A., Nipoti P., Pisi A., et al. (2015). Microbial inoculants for the biocontrol of Fusarium spp. in durum wheat. BMC Microbiol. 15:242. 10.1186/s12866-015-0573-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler R. (2016). Diversity of Fusarium species isolated from UK forage maize and the population structure of F. graminearum from maize and wheat. PeerJ 4:e2143. 10.7717/peerj.2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman G. L., Gutteridge R. J., Gherbawy Y., Thomsett M. A., Nicholson P. (2007). Infection of stem bases and grains of winter wheat by Fusarium culmorum and F. graminearum and effects of tillage method and maize-stalk residues. Plant Pathol. 56 604–615. 10.1111/j.1365-3059.2007.01577.x [DOI] [Google Scholar]
- Bengtsson-Palme J., Ryberg M., Hartmann M., Branco S., Wang Z., Godhe A., et al. (2013). Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 4 914–919. 10.1111/2041-210X.12073 [DOI] [Google Scholar]
- Benitez M.-S., Osborne S. L., Lehman R. M. (2017). Previous crop and rotation history effects on maize seedling health and associated rhizosphere microbiome. Sci. Rep. 7:15709. 10.1038/s41598-017-15955-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (2000). On the adaptive control of the false discovery rate in multiple testing with independent statistics. J. Educ. Behav. Statist. 25 60–83. 10.3102/10769986025001060 14584715 [DOI] [Google Scholar]
- Berg G., Fritze A., Roskot N., Smalla K. (2001). Evaluation of potential biocontrol rhizobacteria from different host plants of Verticillium dahliae Kleb. J. Appl. Microbiol. 91 963–971. 10.1046/j.1365-2672.2001.01462.x [DOI] [PubMed] [Google Scholar]
- Boari A., Vurro M. (2004). Evaluation of Fusarium spp. and other fungi as biological control agents of broomrape (Orobanche ramosa). Biol. Control 30 212–219. 10.1016/j.biocontrol.2003.12.003 [DOI] [Google Scholar]
- Bottalico A., Perrone G. (2002). Toxigenic Fusarium species and mycotoxins associated with head blight in small-grain cereals in Europe. Eur. J. Plant Pathol. 108 611–624. 10.1023/A:1020635214971 15645174 [DOI] [Google Scholar]
- Brown S. P., Veach A. M., Rigdon-Huss A. R., Grond K., Lickteig S. K., Lothamer K., et al. (2015). Scraping the bottom of the barrel: are rare high throughput sequences artifacts? Fungal Ecol. 13 221–225. 10.1016/j.funeco.2014.08.006 [DOI] [Google Scholar]
- Bush B. J., Carson M. L., Cubeta M. A., Hagler W. M., Payne G. A. (2004). Infection and fumonisin production by Fusarium verticillioides in developing maize kernels. Phytopathology 94 88–93. 10.1094/PHYTO.2004.94.1.88 [DOI] [PubMed] [Google Scholar]
- Calvo-Garrido C., Elmer P. A. G., Viñas I., Usall J., Bartra E., Teixidó N. (2013). Biological control of Botrytis bunch rot in organic wine grapes with the yeast antagonist Candida sake CPA-1. Plant Pathol. 62 510–519. 10.1111/j.1365-3059.2012.02684.x [DOI] [Google Scholar]
- Caporaso J. G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Zhang J., Zhao B., Zhou G., Ruan L. (2015). Bacterial community structure in maize stubble-amended soils with different moisture levels estimated by bar-coded pyrosequencing. Appl. Soil Ecol. 86 62–70. 10.1016/j.apsoil.2014.09.011 [DOI] [Google Scholar]
- Chen W., Hambleton S., Seifert K. A., Carisse O., Diarra M. S., Peters R. D., et al. (2018). Assessing performance of spore samplers in monitoring aeromycobiota and fungal plant pathogen diversity in Canada. Appl. Environ. Microbiol. 84 e2601–e2617. 10.1128/AEM.02601-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comby M., Gacoin M., Robineau M., Rabenoelina F., Ptas S., Dupont J., et al. (2017). Screening of wheat endophytes as biological control agents against Fusarium head blight using two different in vitro tests. Microbiol. Res. 202 11–20. 10.1016/j.micres.2017.04.014 [DOI] [PubMed] [Google Scholar]
- Correa-Galeote D., Bedmar E. J., Arone G. J. (2018). Maize endophytic bacterial diversity as affected by soil cultivation history. Front. Microbiol. 9:484. 10.3389/fmicb.2018.00484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa-Galeote D., Bedmar E. J., Fernández-González A. J., Fernández-López M., Arone G. J. (2016). Bacterial communities in the rhizosphere of amilaceous maize (Zea mays L.) as assessed by pyrosequencing. Front. Plant Sci. 7:1016. 10.3389/fpls.2016.01016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane J. M., Gibson D. M., Vaughan R. H., Bergstrom G. C. (2013). Iturin levels on wheat spikes linked to biological control of Fusarium head blight by Bacillus amyloliquefaciens. Phytopathology 103 146–155. 10.1094/PHYTO-07-12-0154-R [DOI] [PubMed] [Google Scholar]
- Cromey M. G., Shorter S. C., Lauren D. R., Sinclair K. I. (2002). Cultivar and crop management influences on Fusarium head blight and mycotoxins in spring wheat (Triticum aestivum) in New Zealand. N. Z. J. Crop Hortic. Sci. 30 235–247. 10.1080/01140671.2002.9514220 [DOI] [Google Scholar]
- Czembor E., Stȩpień Ł., Waśkiewicz A. (2015). Effect of environmental factors on Fusarium species and associated mycotoxins in maize grain grown in Poland. PLoS One 10:e0133644. 10.1371/journal.pone.0133644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boer W., Wagenaar A.-M., Klein Gunnewiek P. J. A., Van Veen J. A. (2007). In vitro suppression of fungi caused by combinations of apparently non-antagonistic soil bacteria: suppression of fungi by non-antagonistic soil bacteria. FEMS Microbiol. Ecol. 59 177–185. 10.1111/j.1574-6941.2006.00197.x [DOI] [PubMed] [Google Scholar]
- De Cal A., Larena I., Liñán M., Torres R., Lamarca N., Usall J., et al. (2009). Population dynamics of Epicoccum nigrum, a biocontrol agent against brown rot in stone fruit. J. Appl. Microbiol. 106 592–605. 10.1111/j.1365-2672.2008.04030.x [DOI] [PubMed] [Google Scholar]
- De la Cruz-Barrón M., Cruz-Mendoza A., Navarro–Noya Y. E., Ruiz-Valdiviezo V. M., Ortíz-Gutiérrez D., Ramírez-Villanueva D. A., et al. (2017). The bacterial community structure and dynamics of carbon and nitrogen when maize (Zea mays L.) and its neutral detergent fibre were added to soil from Zimbabwe with contrasting management practices. Microb. Ecol. 73 135–152. 10.1007/s00248-016-0807-8 [DOI] [PubMed] [Google Scholar]
- Desjardins A. E., Proctor R. H. (2007). Molecular biology of Fusarium mycotoxins. Int. J. Food Microbiol. 119 47–50. 10.1016/j.ijfoodmicro.2007.07.024 [DOI] [PubMed] [Google Scholar]
- Díaz Herrera S., Grossi C., Zawoznik M., Groppa M. D. (2016). Wheat seeds promoters harbour bacterial endophytes with potential as plant growth of biocontrol agents Fusarium graminearum. Microbiol. Res. 18 37–43. 10.1016/j.micres.2016.03.002 [DOI] [PubMed] [Google Scholar]
- Dill-Macky R., Jones R. K. (2000). The effect of previous crop residues and tillage on Fusarium head blight of wheat. Plant Dis. 84 71–76. 10.1094/PDIS.2000.84.1.71 [DOI] [PubMed] [Google Scholar]
- Dorn B., Forrer H. R., Jenny E., Wettstein F. E., Bucheli T. D., Vogelgsang S. (2011). Fusarium species complex and mycotoxins in grain maize from maize hybrid trials and from grower’s fields. J. Appl. Microbiol. 111 693–706. 10.1111/j.1365-2672.2011.05091.x [DOI] [PubMed] [Google Scholar]
- Duvick J., Rood T., Maddox J., Gilliam J. (1998). “Detoxification of mycotoxins in planta as a strategy for improving grain quality and disease resistance: identification of fumonisin-degrading microbes from maize,” in Molecular Genetics of Host-Specific Toxins in Plant Disease, eds Kohmoto K., Yoder O. C. (Dordrecht: Springer; ), 369–381. 10.1007/978-94-011-5218-1_41 [DOI] [Google Scholar]
- Edel-Hermann V., Gautheron N., Mounier A., Steinberg C. (2015). Fusarium diversity in soil using a specific molecular approach and a cultural approach. J. Microbiol. Methods 111 64–71. 10.1016/j.mimet.2015.01.026 [DOI] [PubMed] [Google Scholar]
- Edgar R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26 2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- Edgar R. C., Haas B. J., Clemente J. C., Quince C., Knight R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27 2194–2200. 10.1093/bioinformatics/btr381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epskamp S., Cramer A. O. J., Waldorp L. J., Schmittmann V. D., Borsboom D. (2012). qgraph: network visualizations of relationships in psychometric data. J. Statist. Softw. 48 10.18637/jss.v048.i04 [DOI] [Google Scholar]
- Fernandez M., Huber D., Basnyat P., Zentner R. (2008). Impact of agronomic practices on populations of Fusarium and other fungi in cereal and noncereal crop residues on the Canadian Prairies. Soil Tillage Res. 100 60–71. 10.1016/j.still.2008.04.008 [DOI] [Google Scholar]
- Ferrigo D., Raiola A., Causin R. (2016). Fusarium toxins in cereals: occurrence, legislation, factors promoting the appearance and their management. Molecules 21:627. 10.3390/molecules21050627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J., Alm E. J. (2012). Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8:e1002687. 10.1371/journal.pcbi.1002687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L., Penton C. R., Ruan Y., Shen Z., Xue C., Li R., et al. (2017). Inducing the rhizosphere microbiome by biofertilizer application to suppress banana Fusarium wilt disease. Soil Biol. Biochem. 104 39–48. 10.1016/j.soilbio.2016.10.008 [DOI] [Google Scholar]
- Fujiwara K., Iida Y., Someya N., Takano M., Ohnishi J., Terami F., et al. (2016). Emergence of antagonism against the pathogenic fungus Fusarium oxysporum by interplay among non-antagonistic bacteria in a hydroponics using multiple parallel mineralization. J. Phytopathol. 164 853–862. 10.1111/jph.12504 [DOI] [Google Scholar]
- Gardes M., Bruns T. D. (1993). ITS primers with enhanced specificity for basidiomycetes – Application to the identification of mycorrhizae and rusts. Mol. Ecol. 2 113–118. 10.1111/j.1365-294X.1993.tb00005.x [DOI] [PubMed] [Google Scholar]
- Ghini R., Mezzalama M., Ambrosoli R., Barberis E., Garibaldi A., Piedade S. M. S. (2000). Fusarium oxysporum strains as biocontrol agents against Fusarium wilt: effects on soil microbial biomass and activity. Pesquisa Agropecuária Brasileira 35 93–101. 10.1590/S0100-204X2000000100012 [DOI] [Google Scholar]
- Gong A.-D., Li H.-P., Yuan Q.-S., Song X.-S., Yao W., He W.-J., et al. (2015). Antagonistic mechanism of iturin A and plipastatin A from Bacillus amyloliquefaciens S76-3 from wheat spikes against Fusarium graminearum. PLoS One 10:e0116871. 10.1371/journal.pone.0116871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasinghe R. N., Ikiriwatte C. J., Karunaratne A. M. (2004). The use of Pantoea agglomerans and Flavobacterium sp. to control banana pathogens. J. Hortic. Sci. Biotechnol. 79 1002–1006. 10.1080/14620316.2004.11511852 [DOI] [Google Scholar]
- Haas B. J., Gevers D., Earl A. M., Feldgarden M., Ward D. V., Giannoukos G., et al. (2011). Chimeric 16S rRNA sequence formation and detection in sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21 494–504. 10.1101/gr.112730.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashem M., Ali E. (2004). Epicoccum Nigrum as biocontrol agent of Pythium damping-off and root-rot of cotton seedlings. Arch. Phytopathol. Plant Protect. 37 283–297. 10.1080/03235400310001612955 [DOI] [Google Scholar]
- He J., Boland G. J., Zhou T. (2009). Concurrent selection for microbial suppression of Fusarium graminearum, Fusarium head blight and deoxynivalenol in wheat. J. Appl. Microbiol. 106 1805–1817. 10.1111/j.1365-2672.2009.04147.x [DOI] [PubMed] [Google Scholar]
- Hellin P., Dedeurwaerder G., Duvivier M., Scauflaire J., Huybrechts B., Callebaut A., et al. (2016). Relationship between Fusarium spp. diversity and mycotoxin contents of mature grains in southern Belgium. Food Addit. Contam. Part A 33 1228–1240. 10.1080/19440049.2016.1185900 [DOI] [PubMed] [Google Scholar]
- Hennessy R. C., Glaring M. A., Olsson S., Stougaard P. (2017). Transcriptomic profiling of microbe–microbe interactions reveals the specific response of the biocontrol strain P. fluorescens In5 to the phytopathogen Rhizoctonia solani. BMC Res. Notes 10:376. 10.1186/s13104-017-2704-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbold C. W., Pelikan C., Kuzyk O., Hausmann B., Angel R., Berry D., et al. (2015). A flexible and economical barcoding approach for highly multiplexed amplicon sequencing of diverse target genes. Front. Microbiol. 6:731 10.3389/fmicb.2015.00731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlemann D. P., Labrenz M., Jürgens K., Bertilsson S., Waniek J. J., Andersson A. F. (2011). Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5 1571–1579. 10.1038/ismej.2011.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings P., Coates M. E., Walsh K., Turner J. A., Nicholson P. (2004). Determination of deoxynivalenol- and nivalenol-producing chemotypes of Fusarium graminearum isolated from wheat crops in England and Wales. Plant Pathol. 53 643–652. 10.1111/j.0032-0862.2004.01061.x [DOI] [Google Scholar]
- Jensen B. D., Knorr K., Nicolaisen M. (2016). In vitro competition between Fusarium graminearum and Epicoccum nigrum on media and wheat grains. Eur. J. Plant Pathol. 146 657–670. 10.1007/s10658-016-0950-6 [DOI] [Google Scholar]
- Karlsson I., Edel-Hermann V., Gautheron N., Durling M. B., Kolseth A.-K., Steinberg C., et al. (2016). Genus-specific primers for study of Fusarium communities in field samples. Appl. Environ. Microbiol. 82 491–501. 10.1128/AEM.02748-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson I., Friberg H., Kolseth A.-K., Steinberg C., Persson P. (2017). Agricultural factors affecting Fusarium communities in wheat kernels. Int. J. Food Microbiol. 252 53–60. 10.1016/j.ijfoodmicro.2017.04.011 [DOI] [PubMed] [Google Scholar]
- Katoh K., Rozewicki J., Yamada K. D. (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Briefings Bioinform. bbx 108. 10.1093/bib/bbx108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhl J., Lombaers C., Moretti A., Bandyopadhyay R., Somma S., Kastelein P. (2015). Analysis of microbial taxonomical groups present in maize stalks suppressive to colonization by toxigenic Fusarium spp.: a strategy for the identification of potential antagonists. Biol. Control 83 20–28. 10.1016/j.biocontrol.2014.12.007 [DOI] [Google Scholar]
- Kõljalg U., Nilsson R. H., Abarenkov K., Tedersoo L., Taylor A. F. S., Bahram M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22 5271–5277. 10.1111/mec.12481 [DOI] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Tamura K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc N., Kinkel L., Kistler H. C. (2017). Plant diversity and plant identity influence Fusarium communities in soil. Mycologia 109 128–139. 10.1080/00275514.2017.1281697 [DOI] [PubMed] [Google Scholar]
- Legrand F., Picot A., Cobo-Díaz J. F., Chen W., Le Floch G. (2017). Challenges facing the biological control strategies for the management of Fusarium head blight of cereals caused by F. graminearum. Biol. Control 113 26–38. 10.1016/j.biocontrol.2017.06.011 [DOI] [Google Scholar]
- Legrand F., Picot A., Cobo-Díaz J. F., Carof M., Chen W., Le Floch G. (2018). Effect of tillage and static abiotic soil properties on microbial diversity. Appl. Soil Ecol. 132 135–145. 10.1016/j.apsoil.2018.08.016 [DOI] [Google Scholar]
- Li X., Rui J., Mao Y., Yannarell A., Mackie R. (2014). Dynamics of the bacterial community structure in the rhizosphere of a maize cultivar. Soil Biol. Biochem. 68 392–401. 10.1016/j.soilbio.2013.10.017 [DOI] [Google Scholar]
- Logrieco A., Mulè G., Moretti A., Bottalico A. (2002). Toxigenic Fusarium species and mycotoxins associated with maize ear rot in Europe. Eur. J. Plant Pathol. 108:597. 10.1023/A:1020679029993 2615796 [DOI] [Google Scholar]
- Luongo L., Galli M., Corazza L., Meekes E., Haas L. D., Van Der Plas C. L., et al. (2005). Potential of fungal antagonists for biocontrol of Fusarium spp. in wheat and maize through competition in crop debris. Biocontrol Sci. Technol. 15 229–242. 10.1080/09583150400016852 [DOI] [Google Scholar]
- Maiorano A., Blandino M., Reyneri A., Vanara F. (2008). Effects of maize residues on the Fusarium spp. infection and deoxynivalenol (DON) contamination of wheat grain. Crop Protect. 27 182–188. 10.1016/j.cropro.2007.05.004 [DOI] [Google Scholar]
- Manso T., Nunes C. (2011). Metschnikowia andauensis as a new biocontrol agent of fruit postharvest diseases. Postharv. Biol. Technol. 61 64–71. 10.1016/j.postharvbio.2011.02.004 [DOI] [Google Scholar]
- McDonald D., Price M. N., Goodrich J., Nawrocki E. P., DeSantis T. Z., Probst A., et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6 610–618. 10.1038/ismej.2011.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousa W. K., Shearer C. R., Limay-Rios V., Zhou T., Raizada M. N. (2015). Bacterial endophytes from wild maize suppress Fusarium graminearum in modern maize and inhibit mycotoxin accumulation. Front. Plant Sci. 6:805. 10.3389/fpls.2015.00805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazari L., Pattori E., Terzi V., Morcia C., Rossi V. (2014). Influence of temperature on infection, growth, and mycotoxin production by Fusarium langsethiae and F. sporotrichioides in durum wheat. Food Microbiol. 39 19–26. 10.1016/j.fm.2013.10.009 [DOI] [PubMed] [Google Scholar]
- Nicolaisen M., Justesen A. F., Knorr K., Wang J., Pinnschmidt H. O. (2014). Fungal communities in wheat grain show significant co-existence patterns among species. Fungal Ecol. 11 145–153. 10.1016/j.funeco.2014.06.002 [DOI] [Google Scholar]
- Nielsen L. K., Jensen J. D., Nielsen G. C., Jensen J. E., Spliid N. H., Thomsen I. K., et al. (2011). Fusarium head blight of cereals in Denmark: species complex and related mycotoxins. Phytopathology 101 960–969. 10.1094/PHYTO-07-10-0188 [DOI] [PubMed] [Google Scholar]
- Niu B., Paulson J. N., Zheng X., Kolter R. (2017). Simplified and representative bacterial community of maize roots. Proc. Natl. Acad. Sci. U.S.A. 114 E2450–E2459. 10.1073/pnas.1616148114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogórek R., Plaskowska E. (2011). Epicoccum nigrum for biocontrol agents in vitro of plant fungal pathogens. Commun. Agric. Appl. Biol. Sci. 76 691–697. [PubMed] [Google Scholar]
- Oliver A. K., Brown S. P., Callaham M. A., Jumpponen A. (2015). Polymerase matters: non-proofreading enzymes inflate fungal community richness estimates by up to 15 %. Fungal Ecol. 15 86–89. 10.1016/j.funeco.2015.03.003 [DOI] [Google Scholar]
- Palazzini J. M., Ramirez M. L., Torres A. M., Chulze S. N. (2007). Potential biocontrol agents for Fusarium head blight and deoxynivalenol production in wheat. Crop Protect. 26 1702–1710. 10.1016/j.cropro.2007.03.004 [DOI] [Google Scholar]
- Pan J. J., May G. (2009). Fungal-fungal associations affect the assembly of endophyte communities in maize (Zea mays). Microb. Ecol. 58 668–678. 10.1007/s00248-009-9543-7 [DOI] [PubMed] [Google Scholar]
- Parks D. H., Tyson G. W., Hugenholtz P., Beiko R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30 3123–3124. 10.1093/bioinformatics/btu494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry D. W., Jenkinson P., McLEOD L. (1995). Fusarium ear blight (scab) in small grain cereals? – A review. Plant Pathol. 44 207–238. 10.1111/j.1365-3059.1995.tb02773.x [DOI] [Google Scholar]
- Peiffer J. A., Spor A., Koren O., Jin Z., Tringe S. G., Dangl J. L., et al. (2013). Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. U.S.A. 110 6548–6553. 10.1073/pnas.1302837110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira P., Nesci A., Castillo C., Etcheverry M. (2011). Field studies on the relationship between Fusarium verticillioides and maize (Zea mays L.): effect of biocontrol agents on fungal infection and toxin content of grains at harvest. Int. J. Agron. 2011:486914 10.1155/2011/486914 [DOI] [Google Scholar]
- Potshangbam M., Devi S. I., Sahoo D., Strobel G. A. (2017). Functional characterization of endophytic fungal community associated with Oryza sativa L. and Zea mays L. Front. Microbiol. 8:325. 10.3389/fmicb.2017.00325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakumar E., Aggarwal R., Singh B. (2005). Fungal antagonists for the biological control of Ascochyta blight of chickpea. Acta Phytopathol. Entomol. Hungarica 40 35–42. 10.1556/APhyt.40.2005.1-2.5 [DOI] [Google Scholar]
- Ramirez M. L., Chulze S., Magan N. (2006). Temperature and water activity effects on growth and temporal deoxynivalenol production by two Argentinean strains of Fusarium graminearum on irradiated wheat grain. Int. J. Food Microbiol. 106 291–296. 10.1016/j.ijfoodmicro.2005.09.004 [DOI] [PubMed] [Google Scholar]
- Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Höhna S., et al. (2012). MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61 539–542. 10.1093/sysbio/sys029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang M. K., Jeong J.-J., Kim J., Kim K. D. (2018). Growth promotion and root colonisation in pepper plants by phosphate-solubilising Chryseobacterium sp. strain ISE14 that suppresses Phytophthora blight: biocontrol and plant growth by phosphate-solubilising Chryseobacterium. Ann. Appl. Biol. 172 208–223. 10.1111/aab.12413 [DOI] [Google Scholar]
- Scauflaire J., Mahieu O., Louvieaux J., Foucart G., Renard F., Munaut F. (2011). Biodiversity of Fusarium species in ears and stalks of maize plants in Belgium. Eur. J. Plant Pathol. 131 59–66. 10.1007/s10658-011-9787-1 [DOI] [Google Scholar]
- Schisler D. A., Boehm M. J., Paul P. A., Rooney A. P., Dunlap C. A. (2015). Reduction of Fusarium head blight using prothioconazole and prothioconazole-tolerant variants of the Fusarium head blight antagonist Cryptococcus flavescens OH 182.9. Biol. Control 86 36–45. 10.1016/j.biocontrol.2015.04.002 [DOI] [Google Scholar]
- Schöneberg A., Musa T., Voegele R. T., Vogelgsang S. (2015). The potential of antagonistic fungi for control of Fusarium graminearum and Fusarium crookwellense varies depending on the experimental approach. J. Appl. Microbiol. 118 1165–1179. 10.1111/jam.12775 [DOI] [PubMed] [Google Scholar]
- Shaner G. (2003). “Epidemiology of Fusarium head blight of small grain cereals in North America”, in Fusarium Head Blight of Wheat and Barley, eds Leonard K. J., Bushnell W. R. (St. Paul, MN: The American Phytopathological Society; ), 84–119. [Google Scholar]
- Siegel-Hertz K., Edel-Hermann V., Chapelle E., Terrat S., Raaijmakers J. M., Steinberg C. (2018). Comparative microbiome analysis of a Fusarium wilt suppressive soil and a Fusarium wilt conducive soil from the Châteaurenard Region. Front. Microbiol. 9:568 10.3389/fmicb.2018.00568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D. P., Backhouse D., Kristiansen P. (2009). Interactions of temperature and water potential in displacement of Fusarium pseudograminearum from cereal residues by fungal antagonists. Biol. Control 48 188–195. 10.1016/j.biocontrol.2008.10.015 [DOI] [Google Scholar]
- Siou D., Gélisse S., Laval V., Suffert F., Lannou C. (2015). Mutual exclusion between fungal species of the Fusarium head blight complex in a wheat spike. Appl. Environ. Microbiol. 81 4682–4689. 10.1128/AEM.00525-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.-S., Seo D.-J., Jung W.-J. (2017). Identification, purification, and expression patterns of chitinase from psychrotolerant Pedobacter sp. PR-M6 and antifungal activity in vitro. Microb. Pathogen. 107 62–68. 10.1016/j.micpath.2017.03.018 [DOI] [PubMed] [Google Scholar]
- Sugahara H., Kondo T., Okada M., Ikeda Y., Kaida K., Fudou R., et al. (2008). Articulospora sp. produces Art1, an inhibitor of bacterial histidine kinase. Biosci. Biotechnol. Biochem. 72 2521–2525. 10.1271/bbb.80021 [DOI] [PubMed] [Google Scholar]
- Summerbell R. C., Gueidan C., Schroers H.-J., de Hoog G. S., Starink M., Rosete Y. A., et al. (2011). Acremonium phylogenetic overview and revision of Gliomastix, Sarocladium, and Trichothecium. Stud. Mycol. 68 139–162. 10.3114/sim.2011.68.06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B., Wang X., Wang F., Jiang Y., Zhang X.-X. (2013). Assessing the relative effects of geographic location and soil type on microbial communities associated with straw decomposition. Appl. Environ. Microbiol. 79 3327–3335. 10.1128/AEM.00083-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilagyi-Zecchin V. J., Adamoski D., Gomes R. R., Hungria M., Ikeda A. C., Kava-Cordeiro V., et al. (2016). Composition of endophytic fungal community associated with leaves of maize cultivated in south Brazilian field. Acta Microbiol. Immunol. Hungar. 63 449–466. 10.1556/030.63.2016.020 [DOI] [PubMed] [Google Scholar]
- Tagne A., Neergaard E., Hansen H. J., The C. (2002). Studies of host–pathogen interaction between maize and Acremonium strictum from Cameroon. Eur. J. Plant Pathol. 108 93–102. 10.1023/A:1015092030874 [DOI] [Google Scholar]
- UNITE Community (2017). UNITE USEARCH/UTAX Release. Available at: https://search.datacite.org/works/10.15156/bio/587476 [Google Scholar]
- van Bruggen A. H. C., Francis I. M., Jochimsen K. N. (2014). Non-pathogenic rhizosphere bacteria belonging to the genera Rhizorhapis and Sphingobium provide specific control of lettuce corky root disease caused by species of the same bacterial genera. Plant Pathol. 63 1384–1394. 10.1111/ppa.12212 [DOI] [Google Scholar]
- Waalwijk C., Kastekein P., de Vries I., Kerényi Z., Van Der Lee T., Hesselink T., et al. (2003). Major changes in Fusarium spp. in wheat in the Netherlands. Eur. J. Plant Pathol. 109 743–754. 10.1023/A:1026086510156 [DOI] [Google Scholar]
- Wachowska U., Kucharska K., Jȩdryczka M., Łobik N. (2013). Microorganisms as biological control agents against Fusarium pathogens in winter wheat. Polish J. Environ. Stud. 22 591–597. [Google Scholar]
- White T. J., Bruns T. D., Lee S. B., Taylor J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols – A Guide to Methods and Applications, eds Innis M. A., Gelfand D. H., Sninsky J. J., White T. J. (San Diego, CA: Academic Press; ), 315–322. [Google Scholar]
- Wicklow D. T., Poling S. M. (2009). Antimicrobial activity of pyrrocidines from Acremonium zeae against endophytes and pathogens of maize. Phytopathology 99 109–115. 10.1094/PHYTO-99-1-0109 [DOI] [PubMed] [Google Scholar]
- Xing H.-Q., Ma J.-C., Xu B.-L., Zhang S.-W., Wang J., Cao L., et al. (2018). Mycobiota of maize seeds revealed by rDNA-ITS sequence analysis of samples with varying storage times. Microbiology Open 7:e00609. 10.1002/mbo3.609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W., Li R., Ren Y., Liu C., Zhao Q., Wu H., et al. (2017). Distinct roles for soil fungal and bacterial communities associated with the suppression of vanilla Fusarium wilt disease. Soil Biol. Biochem. 107 198–207. 10.1016/j.soilbio.2017.01.010 [DOI] [Google Scholar]
- Xu X.-M., Nicholson P., Thomsett M. A., Simpson D., Cooke B. M., Doohan F. M., et al. (2008). Relationship between the fungal complex causing Fusarium head blight of wheat and environmental conditions. Phytopathology 98 69–78. 10.1094/PHYTO-98-1-0069 [DOI] [PubMed] [Google Scholar]
- Xu X.-M., Parry D. W., Nicholson P., Thomsett M. A., Simpson D., Edwards S. G., et al. (2005). Predominance and association of pathogenic fungi causing Fusarium ear blightin wheat in four European countries. Eur. J. Plant Pathol. 112 143–154. 10.1007/s10658-005-2446-7 [DOI] [Google Scholar]
- Yang Y., Wang N., Guo X., Zhang Y., Ye B. (2017). Comparative analysis of bacterial community structure in the rhizosphere of maize by high-throughput pyrosequencing. PLoS One 12:e0178425. 10.1371/journal.pone.0178425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin C., Hulbert S. H., Schroeder K. L., Mavrodi O., Mavrodi D., Dhingra A., et al. (2013). Role of bacterial communities in the natural suppression of Rhizoctonia solani bare patch disease of wheat (Triticum aestivum L.). Appl. Environ. Microbiol. 79 7428–7438. 10.1128/AEM.01610-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M., Sun B., Wu L., Gao Q., Wang F., Wen C., et al. (2016). Zonal soil type determines soil microbial responses to maize cropping and fertilization. mSystems 1:e00075–16. 10.1128/mSystems.00075-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Taxonomic assignment of representative TEF1 sequences for each Fusarium OTU obtained, using phylogenetic tree, blastn vs. nt-NCBI database, and pairse alignment vs. FUSARIUM-MLST database.
Taxonomic assignment of representative TEF1 and ITS sequences for each Fusarium and unclassified genus OTUs plotted in network analysis, using blastn vs. nt-NCBI database, and pairse alignment vs. FUSARIUM-MLST database.