

Abstract
Background
The brain bioavailability of novel small molecules developed to address central nervous system disease is classically documented through ex vivo or in vivo analyses conducted in rodent models. Data acquired in rodent models are, however, not easily transferrable to human as the pharmacokinetic and pharmacodynamics profiles of the species are quite different.
Methods
Using drugs selected for their differential transport across the blood‐brain barrier, we here demonstrate the feasibility of brain microdialysis in normal vigil macaque monkey by measuring brain extracellular fluid bioavailability of carbamazepine, digoxin, oxycodone, and quinidine.
Results
All drugs, but digoxin, were found in dialysate samples. Drugs that are substrate of P‐glycoprotein show a difference of bioavailability or brain pharmacokinetic parameters between rodents and primates.
Conclusion
Data suggest that brain microdialysis in vigil macaque monkey, the species of choice for classic pharmacokinetic/pharmacodynamics studies could help predicting human brain bioavailability of a small molecule depending on the protein involved in the efflux transport from the brain.
Keywords: awake, bioavailability, blood brain barrier, brain, central nervous system, crossing, methods, microdialysis, non‐human primate, pharmacokinetic
1. INTRODUCTION
The definition of pharmacokinetics and pharmacodynamics is an essential step in the development of a new treatment. The vast majority of blood‐brain barrier (BBB) crossing investigations is performed in ex vivo rodent models using a brain uptake index,1 in situ brain perfusion,2 brain/plasma ratio,3 brain homogenate technique,4 or autoradiography.5 While several in vivo methods such as cerebrospinal fluid (CSF)6, 7 or extracellular fluid (ECF) sampling8 are available, different imaging techniques such as positron emission tomography9 or the single‐photon emission computed tomography10 can also be used, but these imaging techniques are limited by the need for a radioactive isoform of the drug of interest.
While the CSF sampling technique is routinely used, it is limited as it only addresses the drug bioavailability in one specific compartment, most often the spinal CSF. This technique allows for estimating the unbound brain concentration of compounds rapidly penetrating the brain that cross the BBB by passive diffusion.11 However, CSF sampling is not an accurate procedure when several processes are involved in BBB crossing.7 On the other hand, ECF sampling can be used to define the pharmacologically active concentration of a drug in a specific anatomical region.4 Microdialysis is the gold standard when it comes to sampling endogenous or exogenous compounds solubilized in ECF in a specific brain area, thanks to passive diffusion through a dialysis membrane.12, 13
Despite its advantages,14, 15, 16 brain microdialysis is not classically used for measuring drug bioavailability or for evaluating neurotoxic exposition. This technique is mostly performed in rodents while only few studies were conducted in non‐human primates.17, 18 The BBB structure is, however, knowingly different between rodents and primates.19 Such differences account for instance for differential efflux transporter expression20, 21, 22 or differential metabolism.23, 24 Thus, the use of microdialysis in the primate brain would provide the most useful information. Here, we demonstrate the usefulness and feasibility of the approach by measuring bioavailability of carbamazepine, digoxin, oxycodone, and quinidine in the striatum, a basal ganglia subcortical structure, of normal awake macaques. These drugs have been selected based upon their differential transport mechanisms across the BBB. For example, digoxin and quinidine are transported by P‐glycoprotein (Pgp).25, 26, 27 Carbamazepine28 is transported by the breast cancer resistance protein.29 All drugs cross BBB mainly by passive diffusion30, 31, 32 except oxycodone, which is taken up from blood by the organic cation transporter.33 However, carbamazepine, digoxin, and quinidine can be influxed into the brain by some influx transporters,29 organic anion transporters,34 and organic cation transporters,35, 36 respectively.
2. METHODS
2.1. Animals
No animal was terminated in the course of this study. Two adult (4 years old, 6.3 and 6.8 kg) male rhesus monkeys (Macaca mulatta, Xierxin, Beijing, P. R. China) were used in an AAALAC‐accredited facility. These animals were housed in individual primate cages under controlled conditions for humidity, temperature, and light (12‐hour light/12‐hour dark cycle, lights on at 8.00 am); food and water were available ad libitum. Animal care was supervised by veterinarians experienced in non‐human primates’ husbandry. Experiments were carried out in accordance with European Communities Council Directive (2010/63/EU) for the care of laboratory animals following acceptance of the study design by the Institute of Lab Animal Science (Chinese Academy of Science, Beijing, China) IACUC for non‐human primate experiments.
2.2. Surgery
All surgery was carried out under isoflurane anesthesia. Customized guide cannulas (CMA 11, Sweden) were placed bilaterally above the precentral cortex (from anterior commissure: anteroposterior +7 mm, lateral 13 mm, depth +12 mm) under stereotactic guidance,18, 37, 38, 39, 40 using ventriculography and postoperative X‐ray evaluation. Guide cannulas were permanently fixed to the skull with surgical screws and dental acrylic resin. After 1 week of recovery, microdialysis recordings commenced.
2.3. Microdialysis
Animals were trained for 8 weeks before surgery to sit in a primate restraint chair (Crist, USA).41, 42, 43 One week after surgery, concentric dialysis probes (CMA 11, cut‐off 6 kDa, membrane of 2 mm, 240 μm outer diameter, and shaft 16 mm for session 1 and 18 mm for session 2, Sweden) were lowered through the cannula guide to reach the target. Two different lengths were used to perform two dialyses with a 48‐hour interval in the same structure but in a tissue not affected by gliosis, for avoiding such confounding factor.14 Probes were perfused with artificial CSF (2 μL/min, Harvard Apparatus, France) for 60 minutes for stabilization. Thereafter, dialysates (40 μL) were collected on ice every 20 minutes for 320 minutes postdrug administration. Treatments were applied after stabilization and the four drugs (carbamazepine, digoxin, oxycodone, and quinidine) were concomitantly given by nasogastric gavage in order to control the administrated doses through the same route used in human patients. Treatment doses are shown in Table 1. The choice of doses was made in accordance to previous studies, Sessions 1 and 2 (dose X and 2X, respectively) took place with a washout of 48 hours. Blood samples were collected immediately before dosing (t = 0) and at 1, 3, 5, 9 and 24 hours after the drug was administered. Plasma was separated immediately by centrifugation at 1500 g for 10 minutes. Plasma and dialysis samples were immediately frozen after collection at 80°C until analysis. The in vivo recovery of the microdialysis probe for each compound was estimated using in vitro microdialysis. After in vivo microdialysis, CMA 11/2‐mm probes were immersed in an artificial CSF solution containing 80, 0.1, 60, and 60 μg/mL of carbamazepine, digoxin, oxycodone, and quinidine, respectively. A quantity of 1.5 mL of this model solution was placed at 37°C and perfused with artificial CSF solution at 2 μL/min for 2 hours. After 60 minutes of stabilization, the dialysate was collected every 20 minutes interval and stored at –80°C before analysis. The ratio of the concentration in the dialysate vs that in the solution was calculated as the in vitro recovery.
Table 1.
Drugs: Doses, suppliers, and supporting references
| Drug | Dose X (mg/kg) | Dose 2X (mg/kg) | Supplier | References |
|---|---|---|---|---|
| Carbamazepine | 7.5 | 15 | Tegretol, Novartis, France | (Lockard et al, 1974) |
| Digoxin | 0.05 | 0.1 | Digoxine nativelle, Teofarma, France | (Mayer et al, 1996; Nademanee et al, 1984; Ragueneau et al, 1999) |
| Oxycodone | 5.0 | 10 | Oxynorm, Mundipharma, France | (Hassan et al, 2007; Lalovic et al, 2006; Leow et al, 1992) |
| Quinidine | 12.5 | 25 | Quinidine anhydrous, Sigma Aldrich, France | (Phillips et al, 1985; Sindrup et al, 1996; Starling et al, 1997) |
2.4. Sample preparation
After thawing at room temperature for 1 hour, plasma samples or dialysate samples were mixed on a vortex mixer for 15 seconds. A quantity of 20 μL of plasma or CSF was added with 100 μL of the internal standard solution (glibenclamide at 0.02 μg/mL dissolved in methanol) then vortex‐mixed for 30 seconds. Samples were then centrifuged at 10 000 g for 10 minutes at room temperature and 3 μL of the supernatant aliquot was injected into the liquid chromatography coupled to mass spectrometry system.
A modified protocol was used to detect digoxin in plasma. After thawing at room temperature for 1 hour, plasma samples were mixed on the vortex mixer for 15 seconds. Aliquots of 1.0 mL of plasma samples were placed into 10‐mL conical plastic test tubes containing with 50 μL of the internal standard solution. Samples were vortex‐mixed for 15 seconds and then 5 mL of methyl tertiary butyl ether was added. Test tubes were then vortex‐mixed vigorously for 90 seconds and centrifuged at 4500 g for 10 minutes. The supernatant was dried under a stream of nitrogen. The residue was reconstituted in 50 μL of methanol, and then centrifuged at 10 000 g for 10 minutes. A quantity of 3 μL aliquots of the supernatant was then injected into the liquid chromatography coupled to mass spectrometry system.
2.5. Chemical information
Analytical grade carbamazepine, digoxin, oxycodone, quinidine, and ammonium acetate (Sigma‐Aldrich, Saint Louis, MO, USA), and glibenclamide (National Institute for the control of Pharmaceutical and Biological Products, Beijing, China) were used for calibration. High‐performance liquid chromatography grade methanol (Mallinckrodt Baker, Inc., Phillipsburg, NJ, USA) and acetonitrile (Mallinckrodt Baker, Inc., Phillipsburg, NJ, USA and Honeywell Burdick & Jackson, Muskegon, Ml, USA) was used for chromatography. Methyl tertiary butyl ether (Merck Inc., Darmstadt, Germany) was of analytical grade. Deionized water used throughout the study was purified by a Milli‐QR Academic water purification system (Merck Millipore, Billerica, MA, USA).
2.6. Sample analysis
The analysis was performed on an Agilent 6410B triple quadrupole liquid chromatography‐mass spectrometer system (Agilent Technologies, Santa Clara, CA, USA) consisting of an Agilent 1200 RRLC system connected to a triple quadrupole mass spectrometer with an electrospray ionization interface usable in either positive‐ionization or negative‐ionization mode. A MassHunter workstation was used for chromatography‐mass spectrometer control and data acquisition (Agilent Technologies).
The high‐performance liquid chromatography separations were achieved using a Capcell pak C18 MG II (2.0 mm × 50 mm, 3 μm, Shiseido, Tokyo, Japan) column. The mobile phase consisted of solvent A (water and acetonitrile (90:10), containing 10 mM ammonium acetate) and solvent B (water and acetonitrile (20:80), containing 10 mM ammonium acetate) delivered at a flow rate of 0.4 mL/min. The gradient elution started with 0% B and reached 20% B at 0.5 minutes, maintained 20% B for 0.8 minutes, then reached 50% B at 2 minutes, 100% B at 5 minutes, maintained 100% B for 1.0 minute, and then quickly returned to 0% B at 6.1 minutes, which was maintained for 1.9 minutes. The column temperature was maintained at 35°C, and the sample injection volume was 3 μL.
The optimum operating parameters of the electrospray ionization interface in the positive mode were as follows: nebulizer: 45 psi, dry gas: 10 L/min, dry temperature: 350°C, capillary: 4000 V, delta electron multiplier voltage: 600 V, 0‐1 minute: to waste, 1‐8 minutes: to mass spectrometer. Quantification was achieved using the multiple‐reaction‐monitoring mode. The following precursor‐to‐product ion transitions were subjected to multiple reaction monitoring: for carbamazepine, m/z 237.0 to m/z 194.0 (fragmentor, 120 V; collision energy, 18 V); for quinidine, m/z 325.0 to m/z 184.0 (160 V; 25 V); for lovastatin, m/z 405.0 to m/z 199.0 (90 V; 15 V); for simvastatin, m/z 419.0 to m/z 199.0 (90 V; 15 V); and for glibenclamide, m/z 494.0 to m/z 169.0 (90 V; 15 V).
The limit of quantification for carbamazepine, oxycodone, and quinidine was 2.5 ng/mL and was 0.25 ng/mL for digoxin. The limit of detection for all drugs was not measured.
2.7. Pharmacokinetics analysis
The area under the plasma or brain concentration‐time curve (AUC0‐24 h) was estimated by the trapezoidal rule integration using R software version 2.13.2.44 The half‐time (t 1/2) was calculated using the least‐squares linear regression analysis of the terminal part of the log concentration‐time curves, described by the two following equations: Equation 1: ; Equation 2: . With ke the elimination rate constant. The correlations between plasma and dialysate samples were drawn thanks to a linear regression passing through the origin. The coefficients of determination were calculated using R software version 2.13.2.44
3. RESULTS
3.1. Probes recovery
The recovery of the microdialysis probes was determined in vitro by measuring the gain in the dialysis solution for each compounds: 6.2 ± 0.1, 6.1 ± 0.1, and 3.4 ± 0.2 % (mean ± SEM) for carbamazepine, oxycodone, and quinidine, respectively (Table 2). For digoxine, the recovery was below 0.2 % (Table 2).
Table 2.
Recovery of microdialysis probes. Results are expressed as mean ± SEM
| Drug | Solution (μg/mL) | Dialysate (μg/mL) | Recovery (%) |
|---|---|---|---|
| Carbamazepin | 79.4 ± 1.4 | 4.9 ± 0.1 | 6.2 ± 0.1 |
| Digoxine | 101 ± 2 ng/mL | <0.20 ng/mL | <0.2 |
| Oxycodone | 59.1 ± 0.4 | 3.6 ± 0.1 | 6.1 ± 0.1 |
| Quinidin | 58.0 ± 4.7 | 1.91 ± 0.03 | 3.4 ± 0.2 |
3.2. Carbamazepine
Carbamazepine showed a large plasma and ECF bioavailability correlated with the dose (Figure 1) and a dialysate/plasma ratio of 1.139 ± 0.075 (Table 3). Plasma and dialysis t max were comparable (Table 3) while t 1/2 differed between both compartments, 3.6 ± 0.6 hours in plasma and shorter in dialysate with 2.3 ± 0.1 hours (Table 3).
Figure 1.
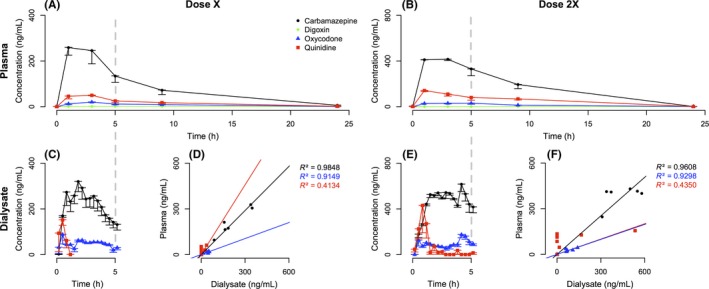
Plasma and brain pharmacokinetic of carbamazepine, digoxin, oxycodone, and quinidine. Analysis of plasma (A‐B) and microdialysis (C‐E) levels and of their relationship (D‐F) in awake normal macaques administered with carbamazepine (black dot), digoxin (green diamond), oxycodone (blue triangle), and quinidine (red square) at dose X (A‐C‐D) or dose 2X (B‐E‐F). Data are presented as mean ± with SEM
Table 3.
Plasma and brain pharmacokinetic parameters. Results expressed as mean ± SEM
| Plasma | ECF dialysate | Ratio | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drug | Dose (mg/kg) | Totale AUC0‐5 h (ng/mL.h) | Free AUC0‐5 h (ng/mL.h) | Totale Cmax (ng/mL) | Free Cmax (ng/mL) | t max (h) | t 1/2 (h) | AUC0‐5 h (ng/mL.h) | Cmax (ng/mL) | t max (h) | t 1/2 (h) | ECF/Free plasma AUC0‐5 h (%) |
| Carbamazepine | 7.5 | 3 217.0 ± 612.0 | 1 013.4 ± 192.8a | 856.5 ± 91.8 | 269.8 ± 28.9a | 2 ± 0.5 | 3.6 ± 0.6 | 1 070.5 ± 233.7 | 321.6 ± 45.3 | 2.7 ± 0.3 | 2.3 ± 0.1 | 113.9 ± 7.5 |
| 15 | 5 649.6 ± 259.4 | 1 779.9 ± 81.9a | 1 333.0 ± 16.5 | 419.9 ± 5.2a | 2 192.1 ± 43.3 | 693.4 ± 27.5 | ||||||
| Digoxin | 0.05 | 5.2 ± 0.5 | 2.5 ± 0.2b | 1.3 ± 0.1 | 0.6 ± 0.1b | 5 ± 1.2 | 30.9 ± 10.1 | / | / | / | / | / |
| 0.1 | 10.8 ± 2.2 | 5.1 ± 1.0b | 3.0 ± 0.3 | 1.4 ± 0.1b | / | / | ||||||
| Oxycodone | 5 | 127.7 ± 20.8 | 70.2 ± 11.4c | 36.4 ± 2.5 | 20.0 ± 1.4c | 3 ± 0.7 | 5.4 ± 0.3 | 244.2 ± 13.3 | 104.8 ± 34.4 | 1.8 ± 0.5 | 4.2 ± 1.1 | 368.1 ± 64.5 |
| 10 | 238.1 ± 35.2 | 131.0 ± 19.3c | 65.2 ± 7.2 | 35.9 ± 4.0c | 457.9 ± 12.5 | 232.6 ± 24.6 | ||||||
| Quinidine | 12.5 | 2 442.8 ± 238.4 | 193.0 ± 18.9d | 722.0 ± 28.0 | 57.0 ± 2.2b | 2 ± 0.5 | 3.7 ± 0.6 | 94.4 ± 23.6 | 190.9 ± 49.9 | 0.7 ± 0.1 | 0.6 ± 0.1 | 65.3 ± 24.3 |
| 25 | 6 496.8 ± 550.7 | 512.7 ± 43.9d | 1 836.0 ± 66.5 | 145.0 ± 5.3b | 377.7 ± 102.3 | 669.5 ± 135.1 | ||||||
All dialysate concentrations are corrected by the probe recovery factors showed in Table 2.
Data corrected by the free plasma factor showed in Ref. 49.
Data corrected by the free plasma factor showed in Ref. 47.
Data corrected by the free plasma factor showed in Ref. 48.
Data corrected by the free plasma factor showed in Ref. 63.
3.3. Digoxin
Digoxin showed a low plasma level and was not detected in dialysis samples (concentration was lower than 0.20 ng/mL) (Table 3). The bioavailability is correlated with the dose (Table 3).
3.4. Oxycodone
Oxycodone showed a low plasma level but a substantial level in dialysate (Figure 1) correlated with the dose and a dialysate/plasma ratio at 3.681 ± 0.645 (Table 3). Plasma and dialysis t 1/2 were comparable (Table 3) while t max differed between both compartments 3.0 ± 0.7 hours in plasma and shorter in dialysate with 1.8 ± 0.5 hours (Table 3).
3.5. Quinidine
Quinidine showed a low plasma level with a substantial level in dialysate (Figure 1). Interestingly, plasma and dialysate pharmacokinetics parameters were not comparable and not correlated with the dose (Table 3). Half‐time were 3.7 ± 0.6 and 0.6 ± 0.1 hours and t max were 2.0 ± 0.5 and 0.7 ± 0.1 hours in plasma and dialysis samples, respectively (Table 3).
4. DISCUSSION
We show that microdialysis in the awake normal macaque is a reliable method for the detection of small molecule bioavailability in the central nervous system. All drugs, except digoxin, were found in dialysate samples and showed a correlation to plasma levels (Figure 1). Because they were obtained in the awake monkey, and since no animals were terminated for the completion of this study, these data advance the definition of brain availability of drugs when compared to previous microdialysis studies performed in sedated rhesus monkeys.45 Sedation indeed modifies the hemodynamic parameters, inducing changes in brain chemistry and electrophysiology.15, 46
Although this brain pharmacokinetic model shows interesting perspectives, the study is limited for several reasons. First, we did not evaluate the plasmatic unbound fraction of drugs but used primate data from literature.47, 48, 49 Our main goal was not to analyze the plasma and brain pharmacokinetics parameters of the four selected drugs, but rather to demonstrate the feasibility and reliability of the approach in the awake macaque. A larger cohort would be required for measuring brain levels of new chemical entities or neurotoxicant in future studies. Given the interindividual variability observed in this study, a minimum of six animals would be required. However, the drugs being used in this study are well‐known drugs. The purpose of the study was to compare the human and macaque bioavailability, hence, the choice of well‐known drugs. The power was therefore sufficient for highlighting the similarities between human patients and non‐human primates.
Carbamazepine crosses the BBB by passive diffusion32 but some influx transporter may also be involved29 and is not efflued by Pgp28, 50, 51 (but see Ref. 52) but may be transported by adenosine triphosphate‐binding cassette transporters, such as the breast cancer resistance protein.29 Dialysate/plasma ratio of the carbamazepine was approximately 100% reported from human53 or from rat54 and is comparable to our data suggesting that there is no species‐related differences for this drug in BBB transport.
Digoxin was not found in dialysate samples as expected according to data suggesting digoxin enters CSF very slowly with a t max 10 hours later than the plasmatic peak and according to the low plasma level of free drug. Digoxin crosses the BBB by passive diffusion30 but is poorly lipid‐soluble and slowly enters the CSF55 and is a substrate of Pgp.25, 56 Moreover, it also seems digoxin crosses the BBB by active transport via organic anion‐transporting polypeptide 2.25, 34, 57 This transporter, also known as solute carrier 21A6,58 is expressed in rodent20, 57 but not in humans22 or non‐human primates.21 Moreover, the total clearance and bioavailability are comparable between human and monkey47 while total clearance was higher in rodent.34 These data support the fact that the non‐human primate model is closer to human physiology for estimating brain availability of drugs which, like digoxin, have clear species‐related differences in BBB transport.
Oxycodone crosses the BBB by active absorption through organic cation transporter33, 59 and is not a substrate of Pgp.26 Dialysate/plasma ratio is similar between macaque and rat, 368.1 ± 64.5 % (Table 3) and 300%,60 respectively, but to the best of our knowledge, no dialysis data is available for oxycodone in humans. Interestingly, brain bioavailability shows a large variability between rodent species.61, 62 Since the organic cation transporter involved in oxycodone transport across BBB is not clearly identified,59 it is therefore not possible to compare its expression between species and draw conclusions about pharmacokinetics differences based on such a differential expression as for digoxin.
Quinidine has a low molecular weight (324 g/mol), high lipophilicity, and besides passive diffusion,31 quinidine would cross the BBB by active transport through organic cation transporter, especially the organic cation/carnitine transporter.35, 36 It is also a substrate of Pgp27 accounting for a fast but short BBB crossing explaining why the brain/plasmatic ratio differs between techniques assessing brain quinidine bioavailability54 and why the coefficients of determination of brain/plasmatic ratio is low in transient condition (Figure 1). Dialysate/plasma ratio is lower in rat, 65.3 ± 24.3% (Table 3) and 3.83 ± 0.55%63 for macaque and rat, respectively, but, to the best of our knowledge, no dialysis data are available for humans. These data support the fact that there are species‐related differences in BBB transport of quinidine.
Drugs that are substrate of Pgp, like digoxine and quinidine, show a difference in bioavailability or brain pharmacokinetic parameters between rodents and primates. This can be explained by the level of Pbp expression in blood‐ECF barrier of macaques and rodents, 5.12 ± 0.91 fmol/μg prot21 and 14.1 ± 2.1 fmol/μg prot,22 respectively, while the expression level is 6.06 ± 1.69 fmol/μg prot for human.22 Pgp is the more abundant efflux transporter in rodent22 unlike primate for whom it is adenosine triphosphate‐binding cassette G2 (breast cancer resistance protein).21, 22
Our results suggest that depending on the protein involved in the efflux transport from the brain, the non‐human primate model can predict more reliably the human brain bioavailability. This in part because of the comparable efflux protein expression profiles in brain microvessels of the two species.21, 22 Other proteins are involved in the BBB function like enzymes64 and tight junction proteins.65, 66 These species‐specific differences in expression need to be more thoroughly investigated to make a rational choice of model of human brain pharmacokinetic.
5. CONCLUSION
Microdialysis in the awake non‐human primate allows to model human brain pharmacokinetics as accurately as possible with the advantage of dialyzing in the brain area of interest for the considered disease in highly relevant animal model. Such procedure is particularly relevant to the investigations of new chemical entities or putative neurotoxicants that cause neurological disorders.67 The brain pharmacokinetics and pharmacodynamics profiles of drugs are still inadequately addressed in most development programs and such a platform is needed for our knowledge of pharmacokinetic and pharmacodynamics properties in the brain.
CONFLICT OF INTEREST
None.
AUTHOR CONTRIBUTIONS
TT contributed to the experimental design, conducted the experiments, and had written the manuscript; CW performed liquid chromatography coupled to mass spectrometry; GP contributed to the experimental design, assisted TT for awake microdialysis; EB contributed to the experimental design, assisted TT for stereotaxic surgery, and supervised the study; QL contributed to the animals experiments; JZ supervised liquid chromatography coupled to mass spectrometry, HC contributed to the experimental design and supervised the study. All authors have read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We are grateful to Dr. Daniel Ko for proof‐reading the manuscript.
Thiollier T, Wu C, Porras G, et al. Microdialysis in awake macaque monkeys for central nervous system pharmacokinetics. Animal Model Exp Med. 2018;1:314–321. 10.1002/ame2.12046
REFERENCES
- 1. Oldendorf WH. Brain uptake of radiolabeled amino acids, amines, and hexoses after arterial injection. Am J Physiol. 1971;221:1629‐1639. [DOI] [PubMed] [Google Scholar]
- 2. Takasato Y, Rapoport SI, Smith QR. An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am J Physiol. 1984;247:H484‐H493. [DOI] [PubMed] [Google Scholar]
- 3. Doran A, Obach RS, Smith BJ, et al. The impact of P‐glycoprotein on the disposition of drugs targeted for indications of the central nervous system: evaluation using the MDR1A/1B knockout mouse model. Drug Metab Dispos. 2005;33:165‐174. [DOI] [PubMed] [Google Scholar]
- 4. Reichel A. Addressing central nervous system (CNS) penetration in drug discovery: basics and implications of the evolving new concept. Chem Biodivers. 2009;6:2030‐2049. [DOI] [PubMed] [Google Scholar]
- 5. Keller F, Waser PG. Brain pharmacokinetics of centrally acting drugs, a quantitative autoradiographic study. Arch Int Pharmacodyn Ther. 1984;267:200‐212. [PubMed] [Google Scholar]
- 6. Lin JH. CSF as a surrogate for assessing CNS exposure: an industrial perspective. Curr Drug Metab. 2008;9:46‐59. [DOI] [PubMed] [Google Scholar]
- 7. Shen DD, Artru AA, Adkison KK. Principles and applicability of CSF sampling for the assessment of CNS drug delivery and pharmacodynamics. Adv Drug Deliv Rev. 2004;56:1825‐1857. [DOI] [PubMed] [Google Scholar]
- 8. Alavijeh MS, Palmer AM. Measurement of the pharmacokinetics and pharmacodynamics of neuroactive compounds. Neurobiol Dis. 2010;37:38‐47. [DOI] [PubMed] [Google Scholar]
- 9. Syvänen S, Lindhe O, Palner M, et al. Species differences in blood‐brain barrier transport of three positron emission tomography radioligands with emphasis on P‐glycoprotein transport. Drug Metab Dispos. 2009;37:635‐643. [DOI] [PubMed] [Google Scholar]
- 10. Koizumi H, Fujisawa H, Kurokawa T, et al. Recovered neuronal viability revealed by Iodine‐123‐iomazenil SPECT following traumatic brain injury. J Cereb Blood Flow Metab. 2010;30:1673‐1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu X, Smith BJ, Chen C, et al. Evaluation of cerebrospinal fluid concentration and plasma free concentration as a surrogate measurement for brain free concentration. Drug Metab Dispos. 2006;34:1443‐1447. [DOI] [PubMed] [Google Scholar]
- 12. Deguchi Y, Morimoto K. Application of an in vivo brain microdialysis technique to studies of drug transport across the blood‐brain barrier. Curr Drug Metab. 2001;2:411‐423. [DOI] [PubMed] [Google Scholar]
- 13. Wang Y, Wong SL, Sawchuk RJ. Microdialysis calibration using retrodialysis and zero‐net flux: application to a study of the distribution of zidovudine to rabbit cerebrospinal fluid and thalamus. Pharm Res. 1993;10:1411‐1419. [DOI] [PubMed] [Google Scholar]
- 14. De Lange EC, de Boer AG, Breimer DD. Methodological issues in microdialysis sampling for pharmacokinetic studies. Adv Drug Deliv Rev. 2000;45:125‐148. [DOI] [PubMed] [Google Scholar]
- 15. Bourne JA. Intracerebral microdialysis: 30 years as a tool for the neuroscientist. Clin Exp Pharmacol Physiol. 2003;30:16‐24. [DOI] [PubMed] [Google Scholar]
- 16. Chaurasia CS, Müller M, Bashaw ED, et al. AAPS‐FDA workshop white paper: microdialysis principles, application and regulatory perspectives. Pharm Res. 2007;24:1014‐1025. [DOI] [PubMed] [Google Scholar]
- 17. Darvesh AS, Carroll RT, Geldenhuys WJ, et al. In vivo brain microdialysis: advances in neuropsychopharmacology and drug discovery. Expert Opin Drug Discov. 2011;6:109‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Porras G, De Deurwaerdere P, Li Q, et al. L‐dopa‐induced dyskinesia: beyond an excessive dopamine tone in the striatum. Sci Rep. 2014;4:3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Watson RE, DeSesso JM, Hurtt ME, Cappon GD. Postnatal growth and morphological development of the brain: a species comparison. Birth Defects Res B Dev Reprod Toxicol. 2006;77:471‐484. [DOI] [PubMed] [Google Scholar]
- 20. Kamiie J, Ohtsuki S, Iwase R, et al. Quantitative atlas of membrane transporter proteins: development and application of a highly sensitive simultaneous LC/MS/MS method combined with novel in‐silico peptide selection criteria. Pharm Res. 2008;25:1469‐1483. [DOI] [PubMed] [Google Scholar]
- 21. Ito K, Uchida Y, Ohtsuki S, et al. Quantitative membrane protein expression at the blood‐brain barrier of adult and younger cynomolgus monkeys. J Pharm Sci. 2011;100:3939‐3950. [DOI] [PubMed] [Google Scholar]
- 22. Uchida Y, Ohtsuki S, Katsukura Y, et al. Quantitative targeted absolute proteomics of human blood‐brain barrier transporters and receptors. J Neurochem. 2011;117:333‐345. [DOI] [PubMed] [Google Scholar]
- 23. Garrick NA, Murphy DL. Species differences in the deamination of dopamine and other substrates for monoamine oxidase in brain. Psychopharmacology. 1980;72:27‐33. [DOI] [PubMed] [Google Scholar]
- 24. Johannessen JN, Chiueh CC, Burns RS, Markey SP. Differences in the metabolism of MPTP in the rodent and primate parallel differences in sensitivity to its neurotoxic effects. Life Sci. 1985;36:219‐224. [DOI] [PubMed] [Google Scholar]
- 25. Mayer U, Wagenaar E, Dorobek B, Beijnen JH, Borst P, Schinkel AH. Full blockade of intestinal P‐glycoprotein and extensive inhibition of blood‐brain barrier P‐glycoprotein by oral treatment of mice with PSC833. J Clin Invest. 1997;100:2430‐2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boström E, Simonsson USH, Hammarlund‐Udenaes M. Oxycodone pharmacokinetics and pharmacodynamics in the rat in the presence of the P‐glycoprotein inhibitor PSC833. J Pharm Sci. 2005;94:1060‐1066. [DOI] [PubMed] [Google Scholar]
- 27. Syvänen S, Schenke M, van den Berg DJ, Voskuyl RA, de Lange EC. Alteration in P‐glycoprotein functionality affects intrabrain distribution of quinidine more than brain entry‐a study in rats subjected to status epilepticus by kainate. AAPS J. 2012;14:87‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Höcht C, Lazarowski A, Gonzalez NN, et al. Differential hippocampal pharmacokinetics of phenobarbital and carbamazepine in repetitive seizures induced by 3‐mercaptopropionic acid. Neurosci Lett. 2009;453:54‐57. [DOI] [PubMed] [Google Scholar]
- 29. Sun J, Xie L, Liu X. Transport of carbamazepine and drug interactions at blood‐brain barrier. Acta Pharmacol Sin. 2006;27:249‐253. [DOI] [PubMed] [Google Scholar]
- 30. Schinkel AH, Wagenaar E, van Deemter L, Mol CA, Borst P. Absence of the mdr1a P‐Glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J Clin Invest. 1995;96:1698‐1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kusuhara H, Suzuki H, Terasaki T, Kakee A, Lemaire M, Sugiyama Y. P‐Glycoprotein mediates the efflux of quinidine across the blood‐brain barrier. J Pharmacol Exp Ther. 1997;283:574‐580. [PubMed] [Google Scholar]
- 32. Barakat NS, Omar SA, Ahmed AA. Carbamazepine uptake into rat brain following intra‐olfactory transport. J Pharm Pharmacol. 2006;58:63‐72. [DOI] [PubMed] [Google Scholar]
- 33. André P, Debray M, Scherrmann JM, Cisternino S. Clonidine transport at the mouse blood‐brain barrier by a new H+ antiporter that interacts with addictive drugs. J Cereb Blood Flow Metab. 2009;29:1293‐1304. [DOI] [PubMed] [Google Scholar]
- 34. Kawahara M, Sakata A, Miyashita T, Tamai I, Tsuji A. Physiologically based pharmacokinetics of digoxin in mdr1a knockout mice. J Pharm Sci. 1999;88:1281‐1287. [DOI] [PubMed] [Google Scholar]
- 35. Ohashi R, Tamai I, Yabuuchi H, et al. Na(+)‐dependent carnitine transport by organic cation transporter (OCTN2): its pharmacological and toxicological relevance. J Pharmacol Exp Ther. 1999;291:778‐784. [PubMed] [Google Scholar]
- 36. Van Montfoort JE, Müller M, Groothuis GM, Meijer DK, Koepsell H, Meier PJ. Comparison of “type I” and “type II” organic cation transport by organic cation transporters and organic anion‐transporting polypeptides. J Pharmacol Exp Ther. 2001;298:110‐115. [PubMed] [Google Scholar]
- 37. Ahmed MR, Berthet A, Bychkov E, et al. Lentiviral overexpression of GRK6 alleviates L‐dopa‐induced dyskinesia in experimental Parkinson's disease. Sci Transl Med. 2010;2:28ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fasano S, Bezard E, D'Antoni A, et al. Inhibition of Ras‐guanine nucleotide‐releasing factor 1 (Ras‐GRF1) signaling in the striatum reverts motor symptoms associated with L‐dopa–induced dyskinesia. PNAS. 2010;107:21824‐21829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gold SJ, Hoang CV, Potts BW, et al. RGS9–2 negatively modulates l‐3,4‐dihydroxyphenylalanine‐induced dyskinesia in experimental Parkinson's disease. J Neurosci. 2007;27:14338‐14348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Porras G, Berthet A, Dehay B, et al. PSD‐95 expression controls L‐DOPA dyskinesia through dopamine D1 receptor trafficking. J Clin Invest. 2012;122:3977‐3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Boraud T, Bezard E, Bioulac B, Gross CE. Dopamine agonist‐induced dyskinesias are correlated to both firing pattern and frequency alterations of pallidal neurones in the MPTP‐treated monkey. Brain. 2001;124:546‐557. [DOI] [PubMed] [Google Scholar]
- 42. Leblois A, Meissner W, Bezard E, Bioulac B, Gross CE, Boraud T. Temporal and spatial alterations in GPi neuronal encoding might contribute to slow down movement in Parkinsonian monkeys. Eur J Neurosci. 2006;24:1201‐1208. [DOI] [PubMed] [Google Scholar]
- 43. Leblois A, Meissner W, Bioulac B, Gross CE, Hansel D, Boraud T. Late emergence of synchronized oscillatory activity in the pallidum during progressive Parkinsonism. Eur J Neurosci. 2007;26:1701‐1713. [DOI] [PubMed] [Google Scholar]
- 44. Ihaka R, Gentleman R. R: a language for data analysis and Graphics. J Comput Graph Stat. 1996;5:299‐314. [Google Scholar]
- 45. Li J, von Pföstl V, Zaldivar D, Zhang X, Logothetis N, Rauch A. Measuring multiple neurochemicals and related metabolites in blood and brain of the rhesus monkey by using dual microdialysis sampling and capillary hydrophilic interaction chromatography‐mass spectrometry. Anal Bioanal Chem. 2012;402:2545‐2554. [DOI] [PubMed] [Google Scholar]
- 46. Salord F, Keita H, Lecharny JB, Henzel D, Desmonts JM, Mantz J. Halothane and isoflurane differentially affect the regulation of dopamine and gamma‐aminobutyric acid release mediated by presynaptic acetylcholine receptors in the rat striatum. Anesthesiology. 1997;86:632‐641. [DOI] [PubMed] [Google Scholar]
- 47. Akabane T, Tabata K, Kadono K, Sakuda S, Terashita S, Teramura T. A comparison of pharmacokinetics between humans and monkeys. Drug Metab Dispos. 2010;38:308‐316. [DOI] [PubMed] [Google Scholar]
- 48. Leow KP, Wright AW, Cramond T, Smith MT. Determination of the serum protein binding of oxycodone and morphine using ultrafiltration. Ther Drug Monit. 1993;15:440‐447. [DOI] [PubMed] [Google Scholar]
- 49. Levy RH, Moreland TA, Morselli PL, Guyot M, Brachet‐Liermain A, Loiseau P. Carbamazepine/valproic acid interaction in man and rhesus monkey. Epilepsia. 1984;25:338‐345. [DOI] [PubMed] [Google Scholar]
- 50. Owen A, Pirmohamed M, Tettey JN, Morgan P, Chadwick D, Park BK. Carbamazepine is not a substrate for P‐glycoprotein. Br J Clin Pharmacol. 2001;51:345‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Luna‐Tortós C, Fedrowitz M, Löscher W. Several major antiepileptic drugs are substrates for human P‐glycoprotein. Neuropharmacology. 2008;55:1364‐1375. [DOI] [PubMed] [Google Scholar]
- 52. Potschka H, Fedrowitz M, Löscher W. P‐glycoprotein and multidrug resistance‐associated protein are involved in the regulation of extracellular levels of the major antiepileptic drug carbamazepine in the brain. NeuroReport. 2001;12:3557‐3560. [DOI] [PubMed] [Google Scholar]
- 53. Scheyer RD, During MJ, Hochholzer JM, Spencer DD, Cramer JA, Mattson RH. Phenytoin concentrations in the human brain: an in vivo microdialysis study. Epilepsy Res. 1994;18:227‐232. [DOI] [PubMed] [Google Scholar]
- 54. Liu X, Van Natta K, Yeo H, et al. Unbound drug concentration in brain homogenate and cerebral spinal fluid at steady state as a surrogate for unbound concentration in brain interstitial fluid. Drug Metab Dispos. 2009;37:787‐793. [DOI] [PubMed] [Google Scholar]
- 55. Allonen H, Anderson KE, Iisalo E, Kanto J, Strömblad LG, Wettrell G. Passage of digoxin into cerebrospinal fluid in man. Acta Pharmacol Toxicol (Copenh). 1977;41:193‐202. [DOI] [PubMed] [Google Scholar]
- 56. Mayer U, Wagenaar E, Beijnen JH, et al. Substantial excretion of digoxin via the intestinal mucosa and prevention of long‐term digoxin accumulation in the brain by the mdr 1a P‐glycoprotein. Br J Pharmacol. 1996;119:1038‐1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Noé B, Hagenbuch B, Stieger B, Meier PJ. Isolation of a multispecific organic anion and cardiac glycoside transporter from rat brain. Proc Natl Acad Sci U S A. 1997;94:10346‐10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cui Y, König J, Nies AT, et al. Detection of the human organic anion transporters SLC21A6 (OATP2) and SLC21A8 (OATP8) in liver and hepatocellular carcinoma. Lab Invest. 2003;83:527‐538. [DOI] [PubMed] [Google Scholar]
- 59. Okura T, Hattori A, Takano Y, et al. Involvement of the pyrilamine transporter, a putative organic cation transporter, in blood‐brain barrier transport of oxycodone. Drug Metab Dispos. 2008;36:2005‐2013. [DOI] [PubMed] [Google Scholar]
- 60. Boström E, Simonsson USH, Hammarlund‐Udenaes M. In vivo blood‐brain barrier transport of oxycodone in the rat: indications for active influx and implications for pharmacokinetics/pharmacodynamics. Drug Metab Dispos. 2006;34:1624‐1631. [DOI] [PubMed] [Google Scholar]
- 61. Lalovic B, Kharasch E, Hoffer C, Risler L, Liu‐Chen L‐Y, Shen DD. Pharmacokinetics and pharmacodynamics of oral oxycodone in healthy human subjects: role of circulating active metabolites. Clin Pharmacol Ther. 2006;79:461‐479. [DOI] [PubMed] [Google Scholar]
- 62. Hassan HE, Myers AL, Lee IJ, Coop A, Eddington ND. Oxycodone induces overexpression of P‐glycoprotein (ABCB1) and affects paclitaxel's tissue distribution in Sprague Dawley rats. J Pharm Sci. 2007;96:2494‐2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Starling JJ, Shepard RL, Cao J, et al. Pharmacological characterization of LY335979: a potent cyclopropyldibenzosuberane modulator of P‐glycoprotein. Adv Enzyme Regul. 1997;37:335‐347. [DOI] [PubMed] [Google Scholar]
- 64. Ghersi‐Egea JF, Leininger‐Muller B, Cecchelli R, Fenstermacher JD. Blood‐brain interfaces: relevance to cerebral drug metabolism. Toxicol Lett. 1995;82–83:645‐653. [DOI] [PubMed] [Google Scholar]
- 65. Kratzer I, Vasiljevic A, Rey C, et al. Complexity and developmental changes in the expression pattern of claudins at the blood‐CSF barrier. Histochem Cell Biol. 2012;138:861‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Virgintino D, Errede M, Robertson D, et al. Immunolocalization of tight junction proteins in the adult and developing human brain. Histochem Cell Biol. 2004;122:51‐59. [DOI] [PubMed] [Google Scholar]
- 67. Cannon JR, Greenamyre JT. The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol Sci. 2011;124:225‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]