

Abstract
The introduction of ectopic DNA, such as plasmids, into yeast cells has for decades been a critical protocol for the study of this eukaryotic model system. We describe here an efficient transformation procedure for use in the fission yeast Schizosaccharomyces pombe. This method relies on chemical agents (lithium acetate, and polyethylene glycol) and temperature stresses, which ultimately facilitate transfer of the genetic material through the cell wall and plasma membrane without significant impact to the transferred DNA or the recipient cell. Using this protocol, we consistently see transformation efficiencies between 1.0 × 103 and 1.0 × 104 transformants per microgram of the plasmid with 108 S. pombe cells. The principal benefits and advantages of this method are its simplicity, efficiency and relative speed of completion.
Keywords: Schizosaccharomyces pombe, fission yeast, lithium acetate, polyethylene glycol, transformation, pFL20
2. Introduction
Introduction of DNA into yeast is commonly referred to as transformation and is routinely used in vast and varied research applications, principally to modify cellular phenotypes by the controlled expression of exogenous gene products. Frederick Griffith (1928) first described “the transforming principle” by which non-virulent strains of Streptococcus pneumonia were transformed into virulent strains by the addition of what would only decades later be identified as genes or DNA [1,2]. In their modern form transformation methodologies have undergone regular optimization and customization.
At its simplest, transformation is the introduction of DNA into cells cultured in media that allows only cells with the new DNA to grow. Marker genes such as URA3 or LEU2 of S. cerevisiae are often inserted in DNA transformed into S. pombe with auxotrophic mutations in ura4 or leu1, respectively. Transformation can be induced by chemicals or electroporation [3–7]. Both methods are commonly employed in eukaryotic model organisms such as Sacchromyces cerevisiae, and Schizosaccharomyces pombe [8,9].
The fission yeast S. pombe has been a powerful model system for decades now, employed with great effectiveness and potency to address fundamental biological and biomedical questions. Here, we present a simple and efficient method of lithium acetate/polyethylene glycol (PEG) transformation that consistently produces transformation efficiencies between 1.0 × 103 and 1.0 × 104 colonies per microgram of plasmid with 108 Schizosaccharomyces pombe cells (Fig. 1).
Figure 1.
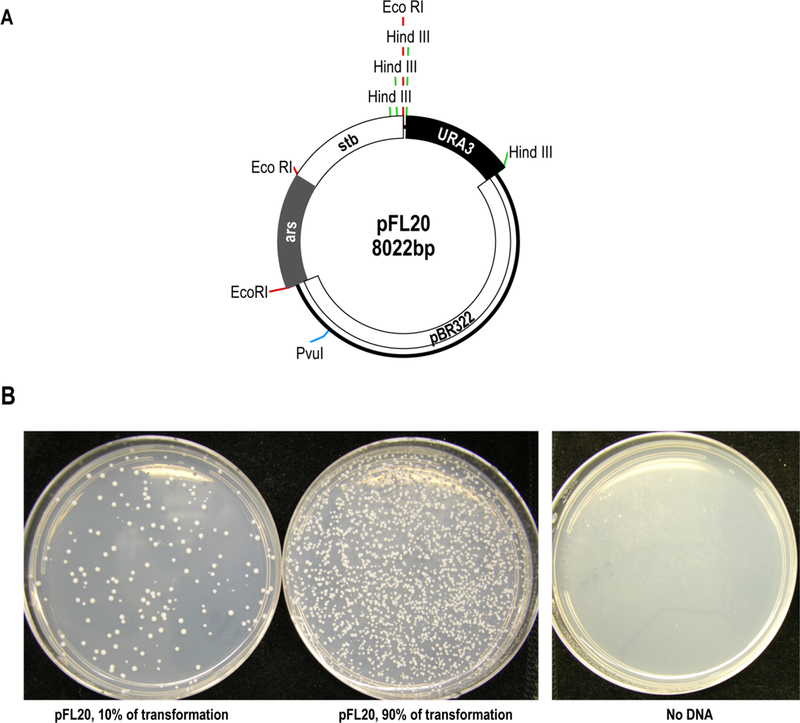
Lithium acetate transformation of S. pombe using pFL20 as a positive control. (A) A map of pFL20 showing the pBR322 backbone, the stabilization fragment (stb), autonomously replication sequence (ars), restriction sites, and selectable markers URA3 and Amp [12]. Eco RI sites are in red, Hind III in green, and the Pvu I site is in blue. (B) Results of transformation with pFL20 showing colonies on PMG –ura plates. Two amounts of the pFL20 transformation were plated, 10% and 90%. The no DNA control plate indicates there was no contaminating source of growth in the experiment.
3. Materials
3.1. Required Instruments
Variable speed centrifuges for both culture harvest (5 to 250 ml) and microfuge tube manipulations (0.5 to 2.0 mL); culture tube roller; spectrophotometer; variable temperature thermal incubator, as well as programmable water bath or heat block.
3.2. Preparation of Media
YES (yeast extract plus supplements) (per liter): 5 g yeast extract (Difco), 30 g glucose and 2 g complete dropout amino acid mix (see 3.3.1 below, and Note 1).
PMG (pombe glutamate medium) (per liter): 3 g potassium hydrogen phthalate [14.7 mM]; 2.2 g Na2PO4 [15.5 mM]; 3.75 g L-glutamic acid; monosodium salt (#G-5889, Sigma-Aldrich); 20 g glucose (2% w/v); 20 ml of 50x salt stock (see 3.3.2 below); 1 mL of 1000x vitamin stock (see 3.3.3 below); 0.1 mL of 10,000X mineral stock (see 3.3.4 below); 2 g of amino acid dropout mix lacking uracil and leucine (see Note 1).
PMG supplemented with leucine and vitamin B1 (PMG-U+L+B1): To PMG medium add (to a final concentration) 250 µg/mL L-Leucine, and 10 µM vitamin B1 (see Note 2).
3.3. Preparation of Stock Solutions (Table 1)
Table 1:
Transformation Solutions
| Solution | Stock Concentration | Working concentration |
|---|---|---|
| (A). Tris-EDTA with Lithium Acetate | ||
| 1. Tris-base ethylenediamine tetra acetic acid (dissodium salt dihydrate) |
100X (1M Tris-HCl, pH 8.0) (100 mM EDTA) |
1X (10 mM Tris-HCl, pH 8.0) (1 mM EDTA) |
| 2. Lithium Acetate | 1M | 100 mM |
| (B). Polyethylene Glycol with Lithium Acetate and Tris-EDTA | ||
| 1. Poly-ethylene Glycol (PEG) 3,350 | 44% | 40% |
| 2. Lithium Acetate | 1M | 100 mM |
| 3. Tris-EDTA | 100X | 1X |
Complete amino acid dropout powder: 5 g adenine sulfate and 2 g each of the following amino acids: alanine, arginine HCl, aspartic acid, asparagine H2O, cysteine HCl.H2O, glutamic acid, glutamine, glycine, histidine HCl.H2O, isoleucine, leucine, lysine HCl, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, uracil, and valine. Mix thoroughly (see Note 3). For addition to selective PMG media, exclude uracil and leucine components (or whatever selectable amino acid markers your experimental design dictates) as these will be produced by exogenous plasmid markers (see Note 1).
50x salt stock (per liter): 2 g Na2SO4 [14.1 mM], 50 g KCl [0.67 M], 0.735 g CaCl2.2H2O [4.99 mM], and 52.5 g MgCl2.6H2O [0.26 M]. Dissolve in deionized water and autoclave. May be stored at room temperature for years.
1000x vitamin stock (per liter): 1 g pantothenic acid [4.20 mM], 10 g nicotinic acid [81.2 mM], 10 g inositol [55.5 mM], and 10 mg biotin [40.8 µM]. Dissolve in deionized water and filter sterilize.
10,000x mineral stock (per liter): 5 g boric acid [80.9 mM], 4 g MnSO4 [23.7 mM], 4 g ZnSO4.7H2O [13.9 mM], 2 g FeCl2.6H2O [7.40 mM], 0.4 g molybdic acid [2.47 mM], 1 g KI [6.02 mM], 0.4 g CuSO4.5H2O [1.60 mM], and 10 g citric acid [47.6 mM]. Dissolve in deionized water and filter sterilize. May be aliquoted into 50 mL portions. Solution is light sensitive, so aliquots should be wrapped in foil.
3.4. Preparation of equilibrated Phenol:Chloroform:Isoamylalchol solution
UltraPure™ phenol:chloroform:isoamyl alcohol (25:24:1, v/v) is commercially available (ThermoFisher #15593049). If you make your own phenol:chloroform:isoamyl solution equilibrate it before use by adding an equal volume of 0.1 M Tris-HCl (pH 8.0); stir the mixture on a magnetic stirrer for 15 minutes, and then let sit to allow layers to separate. May be stored stably in a dark bottle for several months at 4°C. Take care to always use the phenolic (bottom) layer. (See Note 4).
3.5. Preparation of Carrier DNA
Commercial bulk DNA is available from various sources; we used Sigma Aldrich (D6898) herring sperm DNA. Dissolve 1 g DNA in 100 mL of 1x TE buffer overnight at 4˚C with slow constant stirring. (See Note 5).
Treat with Proteinase K (New England Biolabs #P8107S) [final concentration of 10 ug/mL] for 2 hours at 37˚C.
Precipitate DNA by adding 0.6 volumes isopropanol at room temperature and mix well.
Resuspend DNA in 50 mL TE buffer and extract with the 50 mL phenol:chloroform:isoamyl solution. Precipitate aqueous phase with 0.6 volumes isopropanol.
Wash pellet with 25 mL 70% cold ethanol to remove traces of phenol and isopropanol. Remove excess ethanol. Resuspend DNA in 50 mL TE buffer, determine concentration (1.0 OD at 260 nm is 50 mg/mL), and adjust to 10 mg/mL.
Place vessel containing herring sperm DNA in beaker with ice and sheer the DNA by sonication using a high power setting and cycles of 30 seconds on and 30 seconds off to prevent DNA from getting hot. After 6 minutes of sonication time determine the size of DNA using a 0.7% TBE agarose gel. The desired size range is approximately 2.0 kb to 6.0 kb.
When the desired size range is achieved aliquot DNA into 1 mL eppendorf tubes and store in freezer. Before using for transformation heat tubes at 95°C for 5 minutes. Once denatured there is no need to reboil for subsequent use.
3.6. Preparation of 100x Tris-EDTA (per liter)
Dissolve 121.1 g Tris base and 37.2 g Na2 EDTA (Ethylenediamine tetra acetic acid dissodium salt dihydrate) in 800ml distilled water; adjust pH to 7.6 with 2N Hydrochloric acid before bringing volume to final 1 liter. Aliquot into 100 mL bottles and autoclave to sterilize. (See Note 6).
3.7. Preparation of Lithium Acetate (per liter)
Dissolve 102 g of LiOAc (dihydrate, Sigma L-6883) in deionized water and autoclave sterilize. (See Note 6).
3.8. Preparation of 44 % Polyethylene Glycol-3350 (per liter)
Dissolve 440 g PEG 3350 (polyethylene glycol, Sigma P-4338) in deionized water and filter sterilize. Store in smaller volume aliquots. (See Notes 6 and 7).
3.9. Plasmids and strains
The lab strain of S. pombe used for transformation in the figures shown here is YHL912 (h-, ura4–294, leu1–32) [10]. The plasmid pFL20 was used for the positive control in this protocol because it is commonly used in S. pombe, transforms well, and because it contains a fragment of S. pombe sequence that causes stable transmission to daughter cells (Fig. 1A) [11,12].
4. Methods
Figure 2 provides a step-wise checklist for critical steps in the transformation protocol detailed below.
Figure 2.
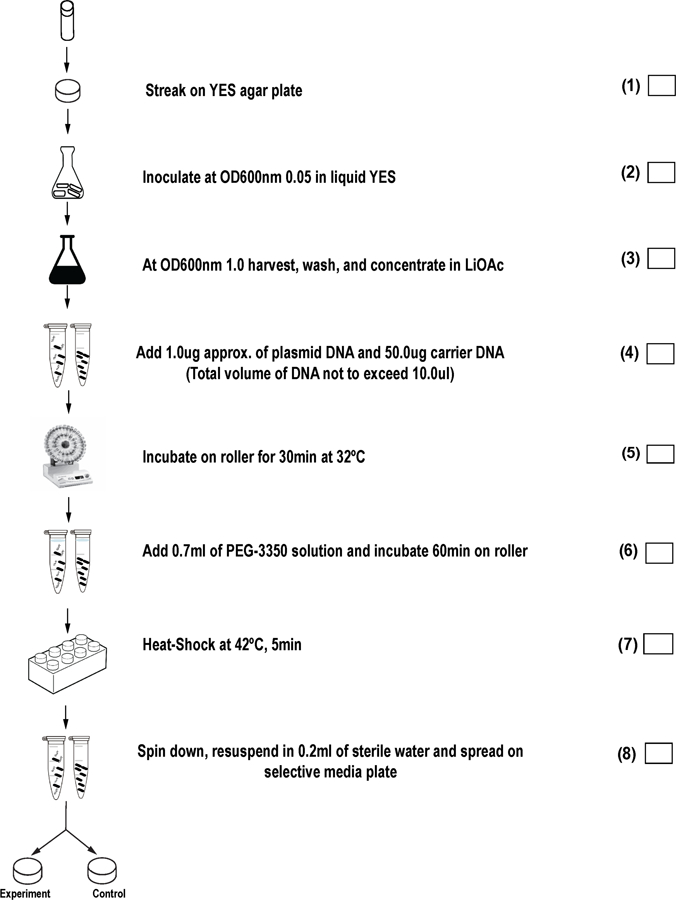
Transformation flowchart: Step by step checklist for lithium acetate transformation of S. pombe.
Part A: Preparation of Competent Cells
-
1
Revive the strains intended for transformation from colony purified, frozen glycerol stocks (“perms”) by streaking cells on rich media (non-selective, such as YES agar plates), and grow at 32°C for up to 3 days.
-
2
When cells have revived, use a sterile toothpick to harvest a match-head amount of cells from the thick part of the streak and suspend in 1.0 mL of sterile liquid media (rich); make sure cells are well suspended by vigorous vortexing. Determine the optical density of the 1.0 mL resuspension at 600nm using serial dilutions. Use this resuspension of known OD concentration to inoculate an over-night culture at a starting OD of 0.05 OD/mL units. Each 10 mL of over-night cell culture is sufficient for one transformation reaction.
-
3
Incubate inoculated cultures in orbital shaking incubator at 32˚C, 225 rpm over-night. Cultures should reach a final OD 600 of 0.5 to 1.0 so plan your inoculation and culture start time appropriately. (See Note 8).
-
4
Following overnight growth, measure the OD of experimental cultures. (See Note 9). When cultures are at the desired OD transfer cells to an appropriate centrifugation vessel (we use 15 mL to 250 mL screw-cap disposable tubes and bottles) and harvest cells with a 10-minute, 1,700 x g centrifugation (3,000 rpm Beckman, Model J6-MI, rotor JS-4.2 swinging-bucket rotor).
-
5
Following centrifugation, discard supernatant, and resuspend cell pellet in the original culture volume of sterile deionized water. Spin again at 1700 x g, this time for five minutes. Discard supernatant.
-
6
Resuspend cell pellet in half the original volume using 1x TE supplemented with 0.1M LiOAc. Centrifuge at 1700 x g for five minutes.
-
7
Resuspend cell pellet in 1/100th the original volume in 1x TE 0.1M LiOAc solution. Transfer to microfuge tube (1.5ml) and incubate using a roller drum at 32˚C for 1h.
-
8
Make 0.1ml aliquots in microfuge tubes each with 5 microliters of carrier DNA. (See Note 10).
Part B: Transformation of Competent Cells
-
9
Set up one 0.1 mL competent cell aliquot (pre-mixed with carrier DNA, as described in step 8) for each desired transformation reaction. Add approximately 1 microgram of the plasmid or fragment of DNA to be transformed (See Note 11). Be sure to include an aliquot with no transforming DNA as a negative control reaction, and make it the last sample manipulated, to test the sterility of technique. Also include a positive transformation reaction with a purified plasmid of known concentration (such as 1 microgram of pFL20, Figure 1A). The positive control measures the efficiency of the cells to be transformed (See Note 12). Two independent experimental plasmids or fragments may be transformed into the same strain, if both have distinguishable selective gene markers, and the strain is auxotrophic or sensitive for both selection genes. (See Note 13).
-
10
Incubate all samples (with carrier and experimental transformation DNA) at 32˚C for 30 minutes in a roller drum.
-
11
Add 0.7 ml of PEG solution (40% PEG 3,350 in 1x TE with 0.1M LiOAc) and thoroughly mix by gentle repeat pipetting.
-
12
Incubate all mixed samples for 60 minutes at 32˚C. (the roller drum is not necessary). (See Note 14).
-
13
Heat shock cells for 5 minutes at 42˚C. (See Note 15).
-
14
Pellet cells by spinning 2 minutes at 1,000 x g (3,000 rpm, Model Eppendorf 5430, Rotor: FA-45–30-11). Resuspend final cell pellet in 200µl of sterile deionized water or rich media with hand pipetting and/or vortexing.
-
15
Spread cells on agar plates containing selective medium (See Note 16). The medium can lack an amino acid if cells are auxotrophic and the appropriate selectable gene marker is present in the transformed DNA. Medium with an antibiotic can be used if a resistance marker is included in the DNA. (See Note 17). Sterile, disposable spreaders may be used but we prefer to use 3mm glass beads to spread cells (See Note 18). To each plate we add 10–30 sterile glass beads, replace the lid, and shake the plate from side-to-side by hand for about 30 seconds.
-
16
Transformation plates are wrapped with parafilm, inverted, and incubated up to 5 days at 32˚C. To calculate transformation frequency, count the number of colonies on the selective media and compare to the microgram amount of input DNA. The no DNA negative control plates should lack colonies (see Note 19, and Fig. 1B).
-
17
In addition to the notes provided below, Table 2 suggests trouble-shooting recommendations.
Table 2:
Transformation Trouble-Shooting Suggestions
| Problem | Possible Reason | Suggested Solution |
|---|---|---|
| No Transformant Colonies | Incorrect selection media | Confirm composition of selection medium and plasmid marker |
| Mistake in solution composition | Confirm transformation solution recipes and consider making all fresh | |
| Inaccurate incubation temperatures | Confirm that growth incubator temperatures are 30–32˚C and that heat-shock temperature is 42˚C | |
| Low Transformation Efficiency | Incorrect PEG concentration | Prepare fresh PEG |
| Concentration of carrier DNA | Confirm concentration | |
| Insufficient Heat-Shock |
|
|
| Over-growth or mold on selective transformation plates. Colonies with no DNA control. | Contamination |
|
Footnotes
Notes
Dropout mixture contains equal weights of all amino acids and uracil but for adenine use 2.5 times the amount of the other components [13]. Complete dropout mixture should be used in YES medium. For the selection of cells that take up the transforming DNA, the dropout mixture added to PMG (or EMM) excludes the component corresponding to the marker gene and auxotroph used. We find that using “complete” dropout mixture yields better transformation efficiency than the five amino acid supplements typically recommended for S. pombe.
Uracil is excluded from media to select for cells that have taken in the transformed expression plasmid marked with the URA3 gene marker.
The powders are mixed in a milling machine or with steel balls or ceramic pellets.
Wear lab coat, gloves and eye protection whenever handling phenol.
Following over-night stirring, inspect visually, to ensure that DNA is resuspended. The suspension will be viscous, but fibers should not be visible to the naked eye.
Works best to aliquot (we use 100 mL bottles for 100X TE and 50 mL screw cap disposable tubes for the PEG solution) for sterility, as unintended contamination of large volume solutions is a common problem.
By storing 45 mL aliquots in 50 mL disposable tubes the PEG transformation solution can easily be made by the addition of 5 mL 1M LiOAc and 0.5 mL 100 X TE.
Estimate that S. pombe cells double approximately every 3 hours, and often experience a short replication lag in the early doublings. Cultures harvested at OD600 higher than 1.5 have significantly reduced transformation efficiencies.
Make sure ODs are read within the linear range of your spectrophotometer, which is usually between 0.1 and 0.3. You will therefore need to dilute a small sample of your culture to get an accurate OD reading.
At this point, if desired, aliquots may be prepared for freezing and future use with the addition of glycerol to 15% and 5–10 ug of carrier DNA (we use 5 uL of a pre-made Herring Sperm DNA stock at 10 mg/mL. This produces competent S. pombe aliquots that can be stored at −80˚C for up to 6 months. However, freezing cells reduces transformation efficiency 50-fold.
10 microliters is the maximum volume that can be added without reducing transformation efficiency.
If the positive control produces high numbers of transformants and the experimental DNAs do not then one should evaluate the experimental DNAs for purity, concentration, and genetic selection. The no DNA control is necessary to test for contamination in the transformation solutions, the carrier DNA, and cultures.
Due to recombination between transformed DNAs in S. pombe, it is better to perform sequential transformation reactions, first transforming in one plasmid. When the single plasmid strain is grown for the second round of transformation, its best to grow the liquid culture in non-selective media to increase efficiency. The final selection plates lack the components necessary to select for both plasmids.
Rolling or otherwise agitating may help if transformation efficiency is unexpectedly low.
We use a heat-block because it results in less contamination that a water bath.
Because single colonies are desired we spread 180 microliters onto one plate and the remaining 20 microliters onto another plate.
We find that PMG media is superior, but this can also be accomplished with the more commonly used S. pombe EMM media.
Glass beads (3 mm, Sigma #Z143928–1EA) can be washed and re-used, so after use they should be stored in 70% ethanol until they can be cleaned. They should be acid washed before re-use, as well as before use from the manufacturer. We wash beads in a glass beaker by covering with sufficient volume of concentrated sulfuric or nitric acid, and soak 30 minutes to overnight. (Remember to wear gloves, lab coat, goggles and facemask when decanting acid). Following soaking, carefully decant the acid into an appropriate waste container; we do this by pouring the beads into a large Buchner funnel on a 3-legged stand, over the waste bin. Rinse the beads with dH2O, periodically measuring the pH of the run-off, until the run-off pH is close to that of water. Drain beads and autoclave portions in foil-covered 100 mL glass bottles. Keeping the plastic lids separate (as they will melt), heat the bottles in a hot drying oven for several days. Beads must be completely dry, but cooled to room temperature before use.
Wrap plates with parafilm and invert (agar-side down) plates during incubations. This helps to keep plates from drying out during the long growth incubation, and also helps to keep water that might condense onto the plate lid from dripping down onto the plate face, and smearing the transformation colonies.
6. References
- 1.Griffith F (1928) The Significance of Pneumococcal Types. J Hyg (Lond) 27 (2):113–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Downie AW (1972) Pneumococcal transformation-a backward view. Fourth Griffith Memorial Lecture. J Gen Microbiol 73 (1):1–11. doi: 10.1099/00221287-73-1-1 [DOI] [PubMed] [Google Scholar]
- 3.Cohen SN, Chang AC, Hsu L (1972) Nonchromosomal antibiotic resistance in bacteria: genetic transformation of Escherichia coli by R-factor DNA. Proc Natl Acad Sci U S A 69 (8):2110–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166 (4):557–580 [DOI] [PubMed] [Google Scholar]
- 5.Dower WJ, Miller JF, Ragsdale CW (1988) High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res 16 (13):6127–6145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wirth R, Friesenegger A, Fiedler S (1989) Transformation of various species of gram-negative bacteria belonging to 11 different genera by electroporation. Mol Gen Genet 216 (1):175–177 [DOI] [PubMed] [Google Scholar]
- 7.Miller JF, Dower WJ, Tompkins LS (1988) High–voltage electroporation of bacteria: genetic transformation of Campylobacter jejuni with plasmid DNA. Proc Natl Acad Sci U S A 85 (3):856–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinnen A, Hicks JB, Fink GR (1978) Transformation of yeast. Proc Natl Acad Sci U S A 75 (4):1929–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beach D, Nurse P (1981) High–frequency transformation of the fission yeast Schizosaccharomyces pombe. Nature 290 (5802):140–142 [DOI] [PubMed] [Google Scholar]
- 10.Levin HL (1995) A novel mechanism of self-primed reverse transcription defines a new family of retroelements. Mol Cell Biol 15 (6):3310–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heyer W, Sipiczki M, Kohli J (1986) Replicating plasmids in Schizosaccharomyces pombe: improvement of symmetric segregation by a new genetic element. Mol Cell Biol 6:80–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Losson R, Lacroute F (1983) Plasmids carrying the yeast OMP decarboxylase structural and regulatory genes: transcription regulation in a foreign environment. Cell 32:371–377 [DOI] [PubMed] [Google Scholar]
- 13.Sangesland M, Atwood-Moore A, Rai SK, Levin HL (2016) Qualitative and Quantitative Assays of Transposition and Homologous Recombination of the Retrotransposon Tf1 in Schizosaccharomyces pombe. Methods Mol Biol 1400:117–130 doi: 10.1007/978-1-4939-3372-3_8 [DOI] [PMC free article] [PubMed] [Google Scholar]