

Abstract
Covalent drug discovery has undergone a resurgence in recent years due to comprehensive optimization of the structure–activity relationship (SAR) and the structure–reactivity relationship (SRR) for covalent drug candidates. The natural product oridonin maintains an impressive pharmacological profile through its covalent enone warhead on the D-ring and has attracted substantial SAR studies to characterize its potential in the development of new molecular entities for the treatment of various human cancers and inflammation. Herein, for the first time, we report the excessive reactivity of this covalent warhead and mediation of the covalent binding capability through a Rh2(esp)2-catalyzed mild and concise regio- and stereospecific aziridination approach. Importantly, aziridonin 44 (YD0514), with a more-druglike irreversible covalent warhead, has been identified to significantly induce apoptosis and inhibit colony formation against triple-negative breast cancer with enhanced antitumor effects in vitro and in vivo while displaying lower toxicity to normal human mammary epithelial cells in comparison to oridonin.
Graphical Abstract
INTRODUCTION
Covalent drugs can possess exceptionally high potency, ligand efficiency, and long-lasting effects given that they directly react with targets to form covalent bonds and thereby generate corresponding bioactivities. Throughout the history of modern medicine, covalent drugs have been profoundly successful therapies for a wide array of human diseases. For example, from 1982–2009, 39 covalent drugs had been approved by the FDA, with the majority having been approved before the year 2000.1 However, due to potential toxicities and safety risks, electrophiles have been considered undruglike and were virtually nonexistent in modern target-specific drug discovery and development for many years.
Recently, the notion of exploring new generations of covalent drugs has resurged with the advent of targeted covalent inhibitors, promising a positive benefit-to-risk ratio.1–3 The electrophilic Michael acceptor enamides and ynamides (Figure 1, 1–5) are frequently utilized as covalent warheads in the design of novel synthetic kinase inhibitors to enhance the biological efficacy, extend the duration of action, and overcome drug resistance.4–8 The successful comeback of covalent drugs is partially ascribed to the optimal mix of structure–activity relationship (SAR) of entire molecules and structure–reactivity relationship (SRR) of attached electrophiles, aiming to acquire the most-suitable candidates.2 An additional support for covalent drug discovery is the broadly applied, simple, and efficient methodology for determining the SRR by the assessment of the reaction rate between covalent warheads and a biologically relevant substrate glutathione (GSH) in vitro.9–13 An optimal balance of warhead reactivity is necessary because the excess reactivity of covalent warheads may hinder the overall selectivity and safety of drugs, while reduced reactivity may lead to failed covalent binding to the target protein to produce the corresponding effect.2
Figure 1.

(A) Representative molecules bearing covalent warheads enamide and ynamide (highlighted in red). (B) Representative molecules with covalent warheads epoxide and aziridine (highlighted in red).
In addition to the above-mentioned covalent synthetic drugs, covalent natural products from aspirin found in 1899 to the sesquiterpenoid artemisinin have revolutionized medicine and have been an invaluable inspiration for the development of various therapeutic agents.1,14,15 In the kingdom of covalent natural products and derivatives, the enone, ethylene oxide, and aziridine groups are three representative classes of warheads (Figure 1, 6–12), all of which can react with nucleophilic groups of target proteins, such as the thiol of cysteine residues, and share a similar reaction mechanism to form covalent bonds.16–18
Besides these examples depicted in Figure 1, another covalent natural product, the kaurane-type diterpenoid oridonin (13 in Table 1), has attracted increased attention in recent years due to its high natural abundance, historic application in traditional herbal medicine (available over the counter as “Donglingcao Pian”), and impressive anti-inflammation and anticancer pharmacological activities.19,20 Thus, oridonin as a single drug ingredient is currently undergoing a clinical observational study in China (ChiCTR-OOB-16007883; http://www.chictr.org.cn/enIndex.aspx). Over the past decade, several independent research groups, including our team, have utilized oridonin as a chemical lead for the design and synthesis of novel oridonin derivatives, with these studies resulting in pivotal SAR information.21 Briefly, modifications on the A-ring, the C-14 position (Table 1, entry 1, highlighted in blue), appear to be tolerable for improving the biological efficacy and druglike profiles of oridonin. The enone system in D-ring (Table 1, entry 1, highlighted in red), as a classical covalent warhead, was identified to be a requisite Michael acceptor for biological activities,22 and removal of this enone system generally resulted in a dramatic loss of activity.21,23 Importantly, among the bioactive oridonin derivatives depicted in Table 1, 14 was advanced into a phase-I human clinical trial for the treatment of leukemia in 2015 (CTR20150246; www.chinadrugtrials.org.cn), while all of the others in Table 1 are currently in preclinical development.
Table 1.
Summary of the Preclinical and Clinical Development of Oridonin-like Compounds
| Entry | Compounds | Molecular targets regulated | Biological responds and clinical indications | Ref. |
|---|---|---|---|---|
| 1 | 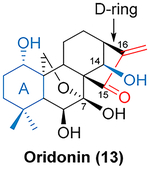 |
Nrf2, NF-kB, ROS, p53, p21 and mitochondrial apoptosis pathways | Antioxidant, cytoprotection and antiinflammation at low concentrations (e.g. 1.4 (μM); Anti-proliferation and pro-apoptosis at relative higher concentrations (e.g. 14 (μM) in vitro | 24–27 |
| Inhibition of tumor growth in t(8;21) leukemia murine model (7.5 and 15 mg/kg) | ||||
| Oridonin-containing herb “Donglingcao” is an over-the-counter (OTC) anti-inflammatory medicine in China | ||||
| 2 | 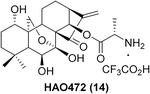 |
NF-kB | An alanine ester prodrug of oridonin as Phase I clinical candidate for the treatment of leukemia (80–320 mg/d, iv) | 28, 29 |
| 3 | 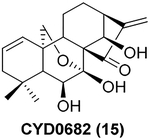 |
p53, p21 and caspase-3 | Antiproliferation against LX-2 cell line with IC50 value 0.49 μM, 10-fold more potent than oridonin in vitro | 30 |
| 4 | 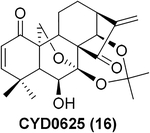 |
p21 and p16 | Significant antiproliferation against LX-2 cell line with 20-fold more potent activity than oridonin in vitro | 31, 32 |
| 5 | 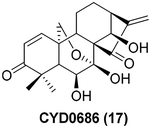 |
NF-kB, Bcl-2/Bax and PARP | Antiproliferation against both ER- positive breast cancer MCF-7 and triple-negative MDA-MB-231 cell lines with IC50 values 0.98 and 5.6 μM in vitro, respectively | 31 |
| Growth suppression of MDA-MB-231 xenograft tumors in vivo (5 mg/kg) | ||||
| 6 | 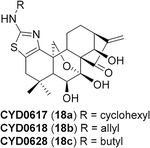 |
NF-kB, Bcl-2/Bax, PARP, DR5 and XBPl | Antiproliferation against various cancer cell lines with the IC50 values range from 0.2 ~ 2.0 μM in vitro | 33–35 |
| Growth suppression of MDA-MB-231 and HCC1806 xenograft tumors in vivo (5 mg/kg) |
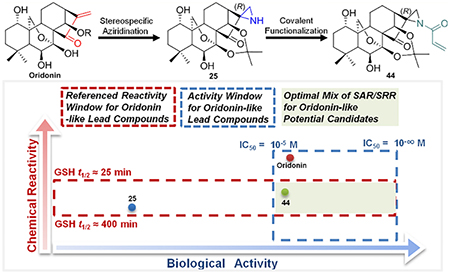
Inspired by recent advances of both synthetic and natural product covalent drugs, our approach reported here was guided by the idea that the exploring the SRR information on the covalent enone warhead in the D-ring is additionally beneficial to the further optimization for the biological activities, safety, and other druglike properties of oridonin. To our knowledge, this is the first attempt to measure the reactivity of the electrophile enone in D-ring with GSH. Based on our SRR, favorable and druglike covalent groups have been designed and incorporated to replace the overly reactive enone in the D-ring through a mild and concise homogeneous Rh2(esp)2-catalyzed direct aziridination in a regio- and stereospecific manner. As a continuation of our previous effort in the identification of novel oridonin analogues with the bioactive functionalities diversely installed in the A-ring, herein, we describe the discovery of a new analog (2R,3′S,3a′R,3a1′R,6a′R,7′S,11′S,11a′S)-1-acryloyl-7′,11′-dihydroxy-5′,5′,8′,8′-tetramethyldecahydro-2′ H-spiro[aziridine-2,15′-[6a,11a](epoxymethano)[3,3a1]ethanophenanthro[1,10-de][1,3]dioxin]-14′-one (aziridonin 44, YD0514) with a more-druglike irreversible covalent warhead capable of significantly inducing apoptosis and inhibiting colony formation against triple-negative breast cancer with comparable enhanced antitumor effects in vitro and in vivo while displaying lower toxicity to normal human mammary epithelial cells in comparison to oridonin.
RESULTS AND DISCUSSION
Activity and Reactivity of the Covalent Enone Warhead in the D-Ring.
GSH is viewed as a major reducing small molecule participating in cellular redox reactions with reactive oxygen and nitrogen species (ROS/RNS) and maintaining intracellular redox balance. Accumulating evidence indicates that treatment with oridonin could decrease the intracellular GSH concentration, resulting in the increase of ROS and ROS-induced apoptosis in cancer cell lines.25,36–39 The enone fragment in the D-ring of oridonin has been identified as a classical covalent warhead Michael acceptor, which reacts with various thiol groups and residues of small molecules (e.g., GSH) and target proteins (e.g., thioredoxin reductase) to afford irreversible inhibition.25 A similar mechanism was revealed in the studies of oridonin-like kaurane diterpenoids.22,40 Interestingly, the reduction of this enone by Pd/C-catalyzed hydrogenation significantly decreases antiproliferation effects.41 Thus, the efficacy of oridonin is believed to largely depend on this D-ring enone pharmacophore by Michael addition with GSH and other protein targets.
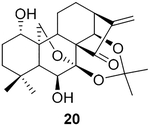
Given that systematic studies on the reactivities of various covalent groups with GSH and the corresponding SRR are useful for the development of novel covalent kinase inhibitors, we utilized nuclear magnetic resonance (NMR) spectroscopy to determine the experimental GSH reaction rates of oridonin and its C-14 position derivatives 14 (Table 1) and 20 (Table 3). The oridonin-like substrates were incubated at 37°C with 1.5–2 equiv of GSH in 0.5 mL of DMSO-d6 and 0.05 mL of sodium phosphate deuterium oxide buffer (pH 7.4). The samples were measured by 1H NMR spectrum at time points of 1st, 5th, and 10th min, respectively, for quantitatively describing the reactivities of the enones presented in these substrates with GSH. The reaction half-life (t1/2) of oridonin with GSH was found to be markedly less than 1 min (Figure 2). The resultant product was further validated with high-resolution mass spectrometry (HRMS), indicating that the transformation of oridonin to its adduct 19 was complete (see Figure S1). Additionally, the alanine ester at the C-14 position (14) may enhance the enone’s reactivity (see Figure S2). Introducing a sterically hindered acetonide group at the nearby C-7/C-14 positions (20) slightly weakens the reactivity of the enone; however, the t1/2 of the drug substrate is still less than 1 min (see Figure S3).
Table 3.
Practical Extension of Rh-Catalyzed Aziridination
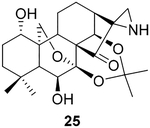
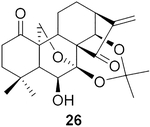
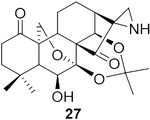
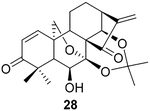
The data were based on isolated yields.
NR: no reaction.
Figure 2.

Reactivity of oridonin with GSH determined by 1H NMR analysis.
Previous studies on the development of covalent inhibitors for epidermal growth factor receptor (EGFR) suggest that the suitable reactivity window of a covalent drug, in terms of t1/2, is from 25 to 400 min.11 The half-life of the covalent drug dimethyl fumarate (Tecfidera), an antimultiple sclerosis (MS) blockbuster approved by the FDA in 2013, was identified to be approximately 50–60 min with 2 equiv of GSH at pH 7.4.42,43 Additionally, the binding of the natural covalent drug bardoxolone methyl (6) with GSH was confirmed to be mild and reversible.16,42,44 In comparison to the reactivity window of these reference compounds, oridonin displays a notably short half-life to form adducts and possesses highly irreversible binding with GSH, thereby leading to potential off-target effects and safety concerns for preclinical and clinical development. In particular, we found that the efficacious dose of oridonin-like compounds was between 5 and 15 mg/kg (Table 1) in vivo, while the median lethal dose (LD50) of oridonin was 37.5 mg/kg.45 A prodrug strategy, as utilized in human phase-I clinical trial candidate 14, could decrease the toxicity of oridonin and remedy the safety concerns.29 However, for continued development and safety we believe structural optimization of the enone remains imperative. From a chemistry standpoint, excessive reactivity, to a large degree, is ascribed to the exocyclic double bond of enone in a densely functionalized chemical microenvironment near the D-ring. Hence, we envisioned that the replacement of the enone with a more-druglike covalent warhead could maintain the original anticancer efficacy while decreasing the covalent binding reactivity to a suitable half-life value. As shown in Figure 1, epoxide and aziridine groups are two feasible covalent warheads for the replacement of the enone. Furthermore, aziridines are milder than epoxides in chemical reactions and have better aqueous solubility. Inspired by the approved, aziridinated drug 10 (Figure 1), the aziridine group was selected as a mild covalent warhead for our investigation on oridonin derivatives.
Chemistry for Aziridinated Oridonin Analogues.
Aziridines, the smallest nitrogenous heterocycles, are important synthetic target structures and building blocks.46,47 Although several methodologies are available for the synthesis of aziridines from olefins, the development of an efficient and stereospecific aziridination with conjugated olefins remains a challenge.48 Additionally, the oridonin skeleton is not stable for a relatively long time (hours) in strong acidic or basic media,49 and therefore, our ideal aziridination should be performed in a mild and near-neutral condition. Recently, Jat et al. published a moderate and scalable aziridination of olefins, utilizing O-(2,4-dinitrophenyl)hydroxylamine (DPH) as the aminating agent and Du Bois’s catalyst Rh(esp)2 at room temperature.50 However, the practical application of their new methodology was not extended to the aziridination of enone substrates. However, some researchers report that the aziridination of enones could be promoted by specific organic amine catalysts under basic conditions.51–53
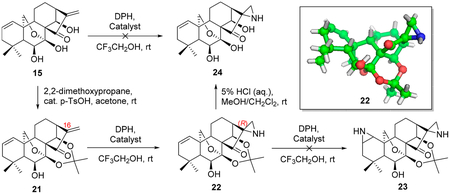
Given the chemical sensitivity of oridonin to strong acidic or basic conditions, our investigation began by subjecting mixtures of the representative oridonin-like substrates (15 and 21, Table 2) and DPH in the presence of 5% mol equivalent of various homogeneous rhodium catalysts at room temperature, referring to the method reported by Jat et al. The direct aziridination of 15 failed to proceed in our initial screening of rhodium catalysts. However, the transformation of its C-7 and C-14 acetonide- protected substrate 21 to the corresponding aziridine 22 as a sole regio-specific product could be efficiently promoted by Rh2(esp)2 or Rh2(OAc)4 under mild conditions (Table 2, entries 1–5). The equivalent amount of DPH was optimized from 2.2 to 1.5 for improving efficiency of the aminating agent and eliminating the serious dyeing effect (dark yellow) of DPH, further leading to a more scalable purification in a best yield of 66% (Table 2, entries 6–7). The structure of 22, obtained under the reaction condition illustrated in Table 2 (entry 7), was unambiguously determined by X-ray crystallographic analysis (CCDC no 1050339; see Table S3), indicating that the carbon atom of 21 at the C-16 position was stereospecifically converted to the R stereo configuration. Interestingly, the olefin at the C1–C2 position of 21 or 22 could not be further aziridinated to compound 23, even utilizing more aminating agent, higher temperature, or extended time (Table 2, entries 8–10). This selectivity of our aziridination may be owing to the steric hindrance of the oridonin skeleton, consequently inactivating the olefin at the A-ring. Hereafter, the deprotection of C1–C14 dihydroxyl of 22 with 5% HCl aqueous solution afforded compound 24 (Table 2).
Table 2.
Exploration and Optimization of Oridonin-Based Aziridinationa
| entries | DPH amount (equiv) | catalystb | temp/time | recovery of 21c | yield of productsc |
|---|---|---|---|---|---|
| 1 | 2.2 | Rh2(esp)2 | rt./12 h | trace | 22 (69%) |
| 2 | 2.2 | Rh2(OAc)4 | rt./12 h | trace | 22 (58%) |
| 3 | 2.2 | Rh2(TPA)4 | rt./12 h | >80% | 22 (trace) |
| 4 | 2.2 | Rh2(R-DOSP)4 | rt./12 h | >80% | 22 (NA)d |
| 5 | 2.2 | Rh2(S-DOSP)4 | rt./12 h | >80% | 22 (NA) |
| 6 | 1.2 | Rh2(esp)2 | rt./12 h | 18% | 22 (54%) |
| 7 | 1.5 | Rh2(esp)2 | rt./12 h | 5% | 22 (66%) |
| 8 | 4.4 | Rh2(esp)2 | rt./24 h | NA | 22 (71%) |
| 23 (NA) | |||||
| 9 | 4.4 | Rh2(esp)2 | 50°C/24 h | NA | 22 (66%) |
| 23 (NA) | |||||
| 10 | 4.4 | Rh2(esp)2 | reflux/48 h | NA | 22 (56%) |
| 23 (NA) |
All reaction conditions in this table were performed, utilizing 21 as the substrate.
The equivalent of catalyst is 5%.
The data were based on isolated yields.
NA: not available.
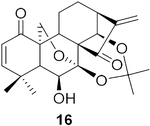
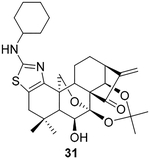
To further explore the practicality and scope of this aziridination reaction, several diverse and bioactive oridonin derivatives, identified from our previous efforts (e.g., 16, 20, 26, and 28), were selected as representative substrates and successfully converted to the corresponding oridonin-like aziridines (Table 3, entries 1–4). However, the thiazole analogue 31 failed to be transformed, likely due to the catalyst poisoning by sulfur in the thiazole moiety. Interestingly, the small regular cyclic or exocyclic enones (e.g., 32 and 33) also failed in the examination of the aziridination reaction (Table 3, entries 6–7).
In the structural analysis of the reactive fragment (Figure 3A, substructure of 21) and the similar inactive substrate (33), we turned our attention to the possible critical role of the neighboring substituted oxygen (Figure 3A, red) in the Rh2(esp)2-catalyzed regio- and stereospecific aziridination. The catalyst Rh2(esp)2, with large steric hindrance (Figure 3B), captures “:NH” from DPH to afford triplet-spin-state nitrene intermediate 34.50 We assumed that the electron density of 33’s olefin is decreased by the α-site ketone, thereby inducing chemical inactivation, while the intermediate 34 charges the corresponding olefin (Figure 3C). The neighboring substituted oxygen donates electron density to the olefin, leading to the reactivation of the olefin for aziridination. Meanwhile, this oxygen group anchors the “:NH” group of nitrene 34 at one side of the olefin via an intermolecular hydrogen bond, consequently leading to the regio- and stereospecific aziridination. Otherwise, the regio- and stereospecific properties of the aziridination may be alternatively ascribed to the interactions between the unique steric microenvironment of oridonin’s D-ring and nitrene intermediate 34.
Figure 3.

Plausible reaction mechanism for Rh2(esp)2 catalyzed regio- and stereospecific aziridination of oridonin-like substrates. (A) Analysis of the reactive substructure and substrate participating in the aziridination. (B) The structures of Rh2(esp)2. (C, D) The comparison of two different substrates (C) 33 vs (D) 21, assisted by the neighboring oxygen group participation in the Rh2(esp)2-promoted regio- and stereospecific aziridination.
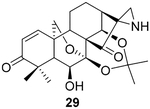
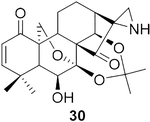
To pursue diverse aziridine derivatives and highlight the application potential of our chemistry, the follow-up synthesis commenced with the preparation of oridonin aziridinated analogs with selected substrates, as mentioned above. The deprotection of C1–C14 dihydroxyl of 25, 27, 29, or 30 with 5% HCl aqueous solution achieved the desired compounds 35–38, respectively (Scheme 1A). According to the previously established SAR, the fused thiazole moieties on A-ring could significantly enhance anticancer activities of oridonin (Table 1, entry 6). With the aim of further improving the anticancer profiles of oridonin aziridines, the representative target compound 43 was designed with an additional N-allyl substituted thiazole at the A-ring and synthesized from an aziridinated substrate 27 prior to the thiazole ring formation due to the aforementioned reason for aziridination failure of 31 directly (Table 3, entry 5). As depicted in Scheme 1B, protection of the secondary amine of aziridine 27 with Fmoc chloride in the presence of Na2CO3 provided 39 in 34% yield. Treatment of 39 with 5% aqueous HCl readily resulted in the deprotected derivative 40 in 75% yield. Subsequently, compound 40 was brominated by PyHBr3 in THF, followed by the Hantzsch reaction of 41, with N-allylthiourea yielding the thiazole 42. Finally, removal of the acetonide protection group with piperidine in DMF afforded the desired compound 43. Moreover, the secondary amine of aziridine provides an additional site for further structural modifications or the formation of salts to improve the bioactivities and druglike properties such as aqueous solubility. As presented in Scheme 1C, we could attach various functional fragments such as an additional enone to the aziridine ring leading to compound 44, and a noncovalent propionyl moiety leading to 45 for comparison through different conventional mild reactions. Such mediated covalent warheads at the secondary amine may directly exert bioactivities independent of the aziridine ring and, alternatively, regulate the covalent bond formation reactivities of the aziridine ring indirectly.
Scheme 1. Synthesis of the Aziridinated Oridonin Analoguesa.

a Reagents and conditions: (a) 5% HCl (aq), MeOH, CH2Cl2, 15 min, 28~62%; (b) Fmoc chloride, Na2CO3, dioxane, 0°C → rt, 12 h, 34%; (c) 5% HCl (aq), MeOH, CH2Cl2, 15 min, 75%; (d) PyHBr3, THF, 0°C, 2 h, 82%; (e) N-allylthiourea, EtOH, reflux, 5 h, 44%; (f) piperidine, DMF, rt, 1 h, 82%; (g) acrylic acid, HBTU, DIPEA, CH2Cl2, 0°C → rt, 12 h, 33%; (h) propionic acid, HBTU, DIPEA, CH2Cl2, 0°C → rt, 12 h, 74%.
Covalent Bond Formation Reactivity of the New Warheads.
From our initial investigation of reactions between aziridines and thiol groups from the SciFinder database, we found that the representative compound 2-aziridinecarboxylic acid, which is similar to the reactive fragments of our aziridinated oridonin analogs, could react with the thiol of cysteine in buffer at room temperature for 20 h in high yield (see Figure S4).54To determine the experimental GSH reaction rates of aziridinated oridonin analogs, compound 25 was incubated at 37°C with 1.5–2 equiv of GSH in 0.5 mL of d6-DMSO and 0.05 mL of sodium phosphate deuterium oxide buffer (pH 7.4). The sample was analyzed by 1H NMR at 11 time points (Figure 4) and indicated that the reactivity of the covalent warhead aziridine in the D-ring is much more moderate than the enone at the same position. Based on the integrated area of 1H NMR peak d generated from the reaction, t1/2 was identified as approximately 4–6 h. Additionally, the adduct product was also validated by HRMS analysis of the reaction at 5 h, indicating the dominant component of the sample as the adduct 46. Meanwhile, the signals of starting materials GSH and aziridine 25 were obviously observed in such analysis, suggesting that aziridinated oridonin analogs are much more stable compared to oridonin with exposure to GSH (see Figure S5). Moreover, compound 25 with the aziridination of the D-ring enone has higher microsomal and plasma stability compared to oridonin in vitro (see Table S2 and Figure S9), indicating that the D-ring aziridine is a druglike pharmacophore as well as an available site of modification for improving pharmacokinetics profiles.
Figure 4.

Reactivity of aziridinated oridonin analogue 25 with GSH determined by 1H NMR.
Under the same conditions, the acrylamide analogue 44 possesses a more reactive warhead in comparison to the aziridinated oridonin 25. Based on the integrated area of a 1H NMR peak disappearing during the reaction, the half-life is identified from 30 to 60 min (Figure 5). Similarly, the adduct product 47 was validated by HRMS analysis after the reaction at 1 h (see Figure S7). In conclusion, the aziridine or acrylamide aziridine attached to the D-ring of oridonin are milder covalent warheads with suitable reactivity in terms of the half-life values, which are in line with the reactivity window (25~400 min)11 suggested for covalent drugs.
Figure 5.

Reactivity of aziridinated oridonin analogue 44 with GSH determined by 1H NMR.
In Vitro Biological Activities against Breast Cancer Cell Lines.
The growth inhibitory effects of these newly synthesized oridonin aziridinated derivatives were initially evaluated in two breast cancer cell lines, MCF-7 (ER-positive) and MDA-MB-231 (ER-negative and triple-negative) using MTT assays as described in the in vitro screening protocol (see the Experimental section). The ability of these new analogs to inhibit the growth of cancer cells was summarized in Table S1 and compared to oridonin. Given that the original enone warhead in the D-ring is a critical pharmacophore, it is not surprising that the overall antiproliferative effects of these aziridinated oridonin molecules are overall lower than those lead compounds we previously reported.31,33,55,56 However, several aziridinated oridonin derivatives such as 42 and 43 displayed moderate activities. Compound 44 exhibited significantly improved antiproliferative effects against highly invasive triple-negative breast cancer MDA-MB-231 cells and MCF-7 cells with IC50 values of 8.32 and 9.39 μM, respectively, in comparison with oridonin (IC50 values of 29.4 μM and 6.72 μM against corresponding cell lines). Interestingly, replacement of the covalent warhead enamide of 44 with a noncovalent propionyl moiety (compound 45) resulted in a complete loss of activity, indicating the important presence of the covalent warhead for biological activity. Next, aziridinated oridonin analogue 44 was selected for the colony formation assay and was found to significantly inhibit the colony formation of highly invasive triple-negative breast cancer cells MDA-MB-231, SK-BR3, and MDA-MB-468 and also the ER-positive breast cancer cells MCF-7 at low micromolar concentrations of the compound (Figure 6A). As shown in Figure 6B, 44 also suppressed the proliferation of MDA-MB-231, SK-BR3, MDAMB-468, and MCF-7 breast cancer cells in a dose-dependent manner.
Figure 6.

Growth inhibitory effects of 44 against MDA-MB-231, SK-BR3, MDA-MB-468, and MCF-7 breast cancer cells. (A) 44 significantly inhibits colony formation of MDA-MB-231, SK-BR3, MDA-MB-468, and MCF-7 cells. (B) 44 suppresses the proliferation of MDA-MB-231, SK-BR3, MDA-MB-468, and MCF-7 cells. Data are presented as the mean ± SD of at least three independent experiments. Significance between different groups was determined by using one-way ANOVA; p = 0.0003 (MDA-MB-231), p = 0.0002 (MDA-MB-468), p < 0.0001 (SK-BR3), and p = 0.0012 (MCF-7).
Based on its antiproliferative effects shown in Figure 6, further preliminary mechanistic studies of 44 were explored to determine whether 44 can induce apoptosis of breast cancer cells. MDA-MB-231 cells were treated with 44 at varying concentrations (1, 5, 10, 15, or 20 μM) for 48 h and stained with FITC-Annexin V and propidium iodide (PI). The percentages of apoptotic MDA-MB-231 cells were determined by flow cytometry. As shown in Figure 7A,B, 44 can induce apoptosis in breast cancer cells at 10 μM (18.2%), 15 μM (26.8%), and 20 μM (47.6%) concentrations at 48 h (early and late apoptosis together) dose-dependently compared to vehicle control. Apparently, 44 induces apoptosis of MDA-MB-231 cells, which at least in part contributes to its antiproliferative effects. Previous studies have demonstrated that oridonin induces apoptosis of cancer cells by regulating a series of transcription factors and protein kinases as well as pro- and anti-apoptotic proteins such as NF-κB37,57,58 and Bcl-2.59,60 To elucidate the potential mechanisms contributing to apoptosis induction by 44, expression levels of several proteins related to apoptosis were determined by Western blotting. As shown in Figure 7C, treatment of MDA-MB-231 cells with 44 dose-dependently led to the down-regulation of NF-κB (p65) protein and up-regulation of cleaved PARP, with the latter as an apoptosis marker. In addition, 44 also reduced the ratio of Bcl-2 to Bax, which are important apoptotic marker proteins. These results suggest that 44 is capable of inducing apoptosis to decrease cellular proliferation. The parental chemotype, oridonin, has been reported to inhibit tumor cell proliferation and induce cancer cell death through cell cycle arrest,58,59,61 autophagy,37,57,62,63 and necrosis.64 Therefore, more-extensive mechanistic studies in terms of major drug targets and signaling pathways associated with 44 are under investigation, and the results will be reported in due course.
Figure 7.

Induction of apoptosis in MDA-MB-231 breast cancer cells by 44. (A) Flow-cytometry analysis of apoptotic MDA-MB-231 breast cancer cells induced by 44 at different concentrations. (B) Apoptotic ratio of different concentrations of 44 in MDA-MB-231 breast cancer cells. The values are means ± SD of at least three independent experiments. A single asterisk represents p < 0.05, two asterisks represent p < 0.01, and three asterisks represent p < 0.001 to 0 μM group (DMSO, vehicle control). (C) Western-blot analysis of biomarkers for apoptosis induced by 44 in MDA-MB-231 breast cancer cells at different concentrations (48 h).
In Vitro Growth Inhibition on Normal Mammary Epithelial Cells.
Selective toxicity for cancer, but not normal cells, is essential in the development of targeted cancer experimental therapeutics. To investigate whether the improved antiproliferative effects of 44 against breast cancer cells were attributed to undesired cell toxicities, we further examined its inhibitory effects on the growth of normal mammary epithelial cells MCF-10A. As shown in Figure 8, 44 exhibited comparable (0–10 μM) or significantly lower (15~100 μM) growth-inhibitory activity against MCF-10A cells at all tested concentrations while displaying significantly enhanced anti-cancer activities against triple-negative MDA-MB-231 cancer cells in comparison to oridonin. These results provide a proof of concept that designing novel covalent warheads in the D-ring of oridonin could be beneficial in lead optimization for improved biological activities, safety, and other druglike properties.
Figure 8.

Effect of 44 and oridonin on normal mammary epithelial cells (MCF-10A) proliferation. MCF-10A cells were treated with varying concentrations of 44 and oridonin for 48 h. Values are mean ± SD of three independent experiments. Statistical significance was determined using a Student t-test, in comparison to the corresponding value of oridonin treatment at the same concentration. ns indicates not significant, a single asterisk indicates p < 0.05, triple asterisks indicate p < 0.001, and quadruple asterisks indicate p < 0.0001.
Growth Suppression of 44 on Triple-Negative Breast Cancer Xenografts in Mice.
Compound 44 was further evaluated for its suppression of tumor growth in the triple-negative breast cancer MDA-MB-231 xenograft model. As shown in Figure 9, mice treated with 10 and 15 mg/kg of 44 (intraperitoneal) showed a significant inhibitory effect on tumor growth compared to the mice treated with vehicle. The dose of 15 mg/kg group with the treatment of 44 showed a better inhibitory effect than the oridonin group (one-way ANOVA, p = 0.0023), while both 44 and oridonin displayed a comparable efficacy at 10 mg/kg. These findings suggest that 44 is a potential anticancer drug candidate with effective antitumor activity and better druglike properties for further preclinical development.
Figure 9.

In vivo efficacy of 44 in suppressing the xenograft tumor growth of triple-negative breast cancer. The breast cancer cell line MDA-MB-231 was used to generate xenograft tumors in nude mice for 10 and 15 mg/kg doses via intraperitoneal injection, respectively. Data are presented as the mean ± SEM of tumor volume at each time point. Significance between different groups was determined by using one-way ANOVA; p = 0.0023.
CONCLUSIONS
From incidental discovery to rational design, the SRR of warheads has been recognized as a critical index in the development of covalent drugs. For the first time, SRR studies on the natural product oridonin have been implemented by 1H NMR and HRMS analyses monitoring its reaction with GSH and determining its D-ring electrophile enone reactivity. To explore more-druglike oridonin derivatives with optimized covalent warheads, we utilized a mild and efficient Rh2(esp)2-catalyzed reaction to regio- and stereospecifically construct new aziridinated oridonin analogues, replacing the overly reactive enone at the same site on D-ring. For the synthetic chemistry approach, this Rh2(esp)2 promoted reaction may undergo the neighboring oxygen group participation. Moreover, the reaction methodology was demonstrated to be feasible for the aziridination of various oridonin-like substrates. Furthermore, the reactivities of the representative aziridinated oridonin derivatives 25 and 44 have been successfully mediated to be much milder than oridonin. The initial GSH t1/2 values of oridonin, 25 and 44 are classified to be <1 min (excessively active), 240–360 min (relatively inactive) and 30–60 min (druglike), respectively, indicating that new aziridinated analogues have more-suitable druglike properties, in line with the referenced reactivity window of covalent drugs. Accordingly, aziridonin 44 has demonstrated significantly enhanced apoptosis induction and colony formation inhibition against triple-negative breast cancer in vitro and in vivo with lower toxicity to normal human mammary epithelial cells in comparison to oridonin. Taken together, our findings support that optimization of diverse bioactive functionalities at the A-ring, while moderation of the covalent warhead reactivities at the D-ring may be a viable approach to the discovery and development of effective and druglike oridonin derivatives in the future.
EXPERIMENTAL SECTION
General.
All commercially available starting materials and solvents were reagent grade and used without further purification. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60 and a particle size of 0.063–0.200 mm (70–230 mesh, flash). Analytical TLC was carried out employing silica gel 60 F254 plates (Merck, Darmstadt, Germany). Visualization of the developed chromatograms was performed with detection by UV (254 nm). NMR spectra were recorded on a Bruker-600 (1H, 300 MHz; 13C, 75 MHz) spectrometer. 1H and 13C NMR spectra were recorded with TMS as an internal reference. Chemical shifts down-field from TMS were expressed in parts per million, and J values were given in hertz. High-resolution mass spectra (HRMS) were obtained from Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: nano ESI spray voltage was 1.8 kV, capillary temperature was 275°C, and the resolution was 60 000; ionization was achieved by positive mode. Melting points were measured on a Thermo Scientific Electrothermal digital melting point apparatus and uncorrected. Purity of final compounds was determined by analytical HPLC, which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20AD SPD-20A UV–vis). HPLC analysis conditions: Waters μBondapak C18 (300 mm × 3.9 mm), flow rate 0.5 mL/min, and UV detection at 270 and 254 nm, linear gradient from 10% acetonitrile in water (0.1% TFA) to 100% acetonitrile (0.1% TFA) in 20 min, followed by 30 min of the last-named solvent. All biologically evaluated compounds are >95% pure.
(2R,3′S,3a′R,3a1′R,6a′R,7′S,11a′R)-7′-Hydroxy-5′,5′,8′,8′-tetramethyl-1′,3′,3a′,7′,7a′,8′,9′,11b′-octahydro-2′H-spiro[aziridine-2,15′-[6a,11a](epoxymethano)[3,3a1]ethanophenanthro[1,10-de]-[1,3]dioxin]-14′-one (22).
To a suspension of 2155 (28.2 mg, 0.07 mmol) in CF3CH2OH (0.7 mL) was added Rh2(esp)2 (1.1 mg) and DPH (17.5 mg, 0.09 mmol) at room temperature. The resulting mixture was stirred at room temperature for 24 h. Next, the solvent was removed under reduced pressure, and the residue was dissolved in CH2Cl2 (20 mL). The organic layer was washed with water (20 mL) and saturated brine (20 mL), dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using silica gel column; elution with 50% EtOAc in hexanes afforded the desired product 22 (21.6 mg, 74%) as a yellowish amorphous solid; (21.6 mg, 74%) as a yellowish amorphous solid; = −72.4 (c 0.20, MeOH); 1HNMR (300 MHz, CDCl3, δ ): 5.81(m, 1H), 5.24 (m, 1H), 5.11 (d, J = 11.7 Hz, 1H), 4.97 (d, J = 1.8 Hz, IH), 4.07−3.79 (m, 3h), 2.47−2.34 (m, 1H), 2.24 (d, J = 9.2 Hz, 2H), 2.04−1.72 (m, 6H), 1.69 (s, 3H), 1.64 (d, J = 6.2 Hz, 3H), 1.41(s, 3H), 1.18 (s, 3H), 1.07 (s, 3H); 13C NMR (75 MHz, CDCl3, δ ): 216.8, 130.4, 124.1, 101.4, 95.2, 72.0, 70.9, 65.0, 58.1, 56.6, 48.9, 48.7, 41.1, 38.5, 38.0, 32.3, 31.9, 31.1, 30.3, 26.5, 25.4, 22.1, 17.2; HRMS (ESI): [M + H]+ calcd for C23H32NO5, 402.2280; found, 402.2268. The stereochemistry of 22 was secured by X-ray crystallographic analysis. The data have been assigned at the Cambridge Crystallographic Data Centre to the deposition number CCDC 1050339.
(2R,5′S,6′S,6a′R,9′S,11 b′R,14′R)-5′,6′,14 ‘-Trihydroxy-4 ‘,4 ‘-dimethyl-4 ‘,4a’,5’,6’,9’,10’,11’,11’-octahydro-3 ‘H,7′H-spiro- [aziridine-2,8’-[6,11b](epoxymethano>[6a,9]methanocyclohepta- [a]nQphthalen¡−7’-one (24).
To a solution of 22 (101.0 mg, 0.24 mmol) in MeOH (2 mL) and CH2Cl2 (0.5 mL) was added hydrochloric acid aqueous solution (1 N, 0.5 mL) at room temperature. The resulting mixture was stirred at room temperature for 15 min. The solvent was removed under reduced pressure and the residue was dissolved in CH2Cl2 (20 mL) and saturated NaHCO3 aqueous solution (10 mL). The organic layer was washed with water (20 mL) and saturated brine (20 mL), dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using silica gel column; elution with 2.5% MeOH in CH2Cl2 afforded the desired product 24 (71.0 mg, 78%) as a white amorphous solid. = −102.5 (c 0.11, MeOH);1H NMR (300 MHz, CDCl3, δ ): 5.91−5. 77 (m, 1H), 5.25−5.14 (m, 1H), 4.95 (s, 1H), 4.78 (d, J = 11.3 Hz, 1H), 4.06−3.66 (m, 6H), 2.54−2.22 (m, 2H), 2.02−1.46 (m, 6H), 1.41 (m, 1H), 1.32−1.23 (m, 1H), 1.18 (s, 3H), 1.06 (s, 3H), 0.99−0.80 (m, 1H).13C NMR (75 MHz, CDCl3, δ ): 216.0, 130.9, 124.1, 97.2, 74.6, 72.8, 65.2, 63.7, 58.4, 52.3, 49.7, 43.7, 41.1, 38.4, 32.4, 30.9, 22.2, 22.1, 21.8, 16.2. HRMS [M + H]+: calcd for C20H28NO5, 362.1967; found, 362.1958.
(2R,3′S,3a′R,3a1′R,6a′R,7′S,11′S,11a′S)-7′,11′-Dihydroxy-5′,5′,8′,8′-tetramethyldecahydro-2′H-spiro[aziridine-2,15′-[6a,11a]-(epoxymethano)[3,3a1]ethanophenanthro[1,10-de][1,3]dioxin]-14’-one (25).
Compound 25 (211.6 mg) was prepared from 2055 in 76% yield by a procedure similar to that used to prepare compound 22. The title compound 25 was obtained as a yellowish amorphous solid. = − 102.8 (c 0.13, MeOH);1H NMR (300 MHz, CDCl3, δ ): 5.46 (d, J = 11.6 Hz, 1H), 4.94 (d, J = 1.7 Hz, 1H), 4.28 (dd, J = 9.9, 1.5 Hz, 1H), 4.06 (dd, J = 9.9, 1.2 Hz, 1H), 3.90 (dd, J = 11.6, 7.6 Hz, 1H), 3.50 (m, 1H), 2.39 (m, 1H), 2.23 (m, 2H), 1.92 (m, 3H), 1.82−1.21 (m, 15H), 1.20−1.11 (m, 6H). 13C NMR (75 MHz, CDCl3, δ ): 217.9, 101.1, 94.7, 74.5, 73.0, 70.9, 62.8, 59.0, 56.6, 50.5, 49.3, 40.7, 39.0, 38.2, 33.7, 33.3, 31.9, 30.4, 29.8, 26.8, 25.4, 22.6, 19.9. HRMS [M + H]+: calcd for C23H34NO6, 420.2386; found, 420.2371.
(2R,3′ S,3 a′ R,3a1 R,6a′ R,7′ S, 11a′ S)-7′-Hydroxy-5′,5′,8′,8′-tetra- methyloctahydro-2′ H-spiro [aziridine-2,15′-[6a,11a]- (epoxymethano)[3,3a1]ethanophenanthro[1,10-de][1,3]dioxine]- 11′,14′(7′H)-dione (27).
Compound 27 (20.1 mg) was prepared from 2656 in 82% yield by a procedure similar to that used to prepare compound 22. The title compound 27 was obtained as obtained as a yellowish amorphous solid. = −59.6 (c 0.18, MeOH); 1H NMR (300MHz, CDCl3, δ ): 4.97 (dd, J = 6.7, 5.0 Hz, 2H), 4.25 (dd, J = 10.2, 1.5 Hz, 1H), 4.06 (dd, J = 10.1,1.6 Hz, 1H), 3.94 (dd, J = 11.7, 8.7 Hz, 1H), 2.43 (m, 3H), 2.25 (m 2H), 2.11(m, 1H), 2.02–1.62 (m,10H), 1.54 (m, 1H), 1.40 (s, 3H), 1.22 (s, 3H), 1.07 (s, 3H); 13C NMR (75 MHz, CDCl3, δ ): 217.1, 211.5, 101.5, 95.6, 71.8, 70.8, 64.7, 59.9, 56.0, 48.9, 48.1, 46.5, 38.4, 38.3, 35.8, 33.0, 32.1, 31.0, 30.2, 26.4, 25.3, 23.1, 18.6; HRMS [M + H]+: calcd for C23H32NO6, 418.2230; found, 418.2215.
(2R,3′ S,3a′ R,3a1′ R,6a′ R,7′ S,11a′ R)-7′-Hydroxy-5′,5′,8′,8′-tetra- methyl-1′,3′,3a′,7a′,8′,11b′-hexahydro-2′ H-spiro[aziridine-2,15′- [6a, 11a](epoxymethano)[3,3a1]ethanophenanthro[1,10-de][1,3]- dioxine]-9′,14′ (7′H)-dione (29).
Compound 29 (29.1 mg) was prepared from 2831 in 61% yield by a procedure similar to that used to prepare compound 22. The title compound 29 was obtained as obtained as a yellowish amorphous solid. = −95.4(c 0.29,MeOH); 1H NMR (300 MHz, CDCl3, δ ): 6.33 (d, J = 10.2 Hz, 1H), 6.04 (d, J = 10.3 Hz, 1H), 5.23 (d, J = 11.8 Hz, 1H), 5.00 (d, J = 1.6 Hz, 1H), 4.11 (m, 3H), 2.44 (m, 1H), 2.27 (m, 2H), 2.06 (s, 1H), 2.02–1.53 (m, 8H), 1.50–1.07 (m, 10H); 13C NMR (75 MHz, CDCl3, δ ): 216.6, 203.2, 142.4, 129.9, 101.7, 95.2, 70.8, 70.7, 64.3, 56.2, 56.1, 48.8, 48.0, 44.6, 38.7, 32.1, 30.2, 26.3, 25.4, 23.9, 22.5, 17.1, 14.2; HRMS [M + H]+: calcd for C23H30NO6, 416.2073; found, 416.2058.
(2R,3′ S,3a′ R,3a1′ R,6a′ R,7′ S,11a′ S)-7′ -Hydroxy-5′,5′,8′,8′-tetra- methyl-1′,3 ′,3a ′,7a ′,8 ′,11b′-hexahydro-2′ H-spiro[aziridine-2,15′ - [6a, 11a](epoxymethano)[3,3a1]ethanophenanthro[1,10-de][1,3]- dioxine]-11′,14′(7′H)-dione (30).
Compound 30 (11.0 mg) was prepared from 1631 in 71% yield by a procedure similar to that used to prepare compound 22. The title compound 30 was obtained as obtained as a yellowish amorphous solid. = −111.6 (c 0.10, MeOH); 1H NMR (300 MHz, CDCl3, δ ): 6.81 (d, J = 10.1 Hz, 1H), 5.87 (d, J = 10.1 Hz, 1H), 5.17–4.96 (m, 2H), 4.27 (dd, J = 9.9, 1.6 Hz, 1H), 4.16–4.03 (m, 2H), 2.45–2.32 (m, 1H), 2.29–2.18 (m, 2H), 2.04 (d, J = 7.3 Hz, 2H), 1.97–1.85 (m, 2H), 1.77–1.65 (m, 6H), 1.41 (d, J = 2.2 Hz, 6H), 1.27 (d, J = 2.2 Hz, 3H); 13C NMR (75 MHz, CDCl3, δ ): 217.3, 196.3, 162.1, 126.7, 101.5, 95.5, 71.6, 70.8, 65.2, 56.6, 56.2,49.0, 47.1,45.7, 38.2,35.9,32.1, 30.4,30.2,26.5, 25.3,25.0, 19.1; HRMS [M + H]+: calcd for C23H30NO6, 416.2073; found, 416.2063.
(6S,6aR,6a1R,9aR, 10S,12bR)-2-(Cyclohexylamino)-6-hydroxy- 5,5,8,8-tetramethyl- 15-methylene-4,5,5a,6,9a,10,12,12a-octahy- dro-11H-6a,12b-(epoxymethano)-6a1,10-ethano[1,3]dioxino- [4′,5′,6′:8,9]phenanthro[4,3-d]thiazol-16-one (31).
To a solution of 18a33 (190.8 mg, 0.38 mmol) in acetone (10 mL) was added 2,2- dimethoxypropane (1.5 mL) and p-TsOH (10 mg) at room temperature. The resulting mixture was refluxed for 8 h. The solvent was removed under reduced pressure, and the residue was dissolved in CH2Cl2 (20 mL) and saturated NaHCO3 aqueous solution (10 mL). The organic layer was washed with water (20 mL) and saturated brine (20 mL), dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using silica gel column; elution with 25% EtOAc in hexane afforded the desired product 31 (168.1 mg, 82%) as a white amorphous solid. = −68.0 (c 0.10, MeOH); 1H NMR (300 MHz, CDCl3, δ ): 6.13 (d, J = 1.3 Hz, 1H), 5.54–5.47 (m, 2H), 4.92 (d, J = 1.7 Hz, 1H),4.81 (d, J = 7.8 Hz, 1H),4.33 (dd, J = 9.8, 1.5 Hz, 1H), 4.04–3.86 (m, 2H), 3.25–3.10 (m, 1H), 3.05 (dd, J = 8.9, 1.6 Hz, 1H), 2.57–2.44 (m, 2H), 2.29 (m, 1H), 2.10–1.94 (m, 4H), 1.92–1.83 (m, 1H), 1.78–1.69 (m, 3H), 1.64–1.55 (m, 2H), 1.36 (s, 4H), 1.27 (m, 6H), 1.19 (m, 3H), 1.01 (s, 3H), 0.88 (s, 1H); 13C NMR (75 MHz, CDCl3, δ ): 204.8, 165.1, 150.9, 142.9, 120.0, 118.5, 101.0, 95.4, 72.0, 70.2, 635.0, 58.5, 56.6, 54.7, 49.9, 40.4, 40.2, 39.0, 34.9, 33.2, 33.1, 30.9, 30.7, 30.2, 25.5, 25.4, 24.7, 24.7, 21.4, 20.1.HRMS [M + H]+: calcd for C30H41N2O5S, 541.2736; found, 541.2734.
(1′ S,2R,5′ S,6′ S,6a′ R,9′ S, 11b′ S,14′ R)-1′,5′,6 ′,14′-Tetrahydroxy-4′,4′-dimethyldecahydro-1′ H,7′ H-spiro[aziridine-2,8′-[6,11b]- (epoxymethano)[6a,9]methanocyclohepta[a]naphthalen]-7′ -one (35).
Compound 35 (30.1 mg) was prepared from 25 in 62% yield by a procedure similar to that used to prepare compound 24. The title compound 35 was obtained as a white amorphous solid. = −110.9 (c 0.11, MeOH); 1HNMR (300 MHz, CDCl3, δ ): 5.18 (d, J = 11.1 Hz, 1H), 4.93 (d, J = 1.3 Hz, 1H), 4.27–4.09 (m, 2H), 3.89–3.76 (m, 2H), 3.68 (d, J = 12.1 Hz, 1H), 3.46 (dd, J = 11.0, 5.9 Hz, 1H), 2.49–2.26 (m, 2H), 2.22–1.98 (m, 1H), 1.80–1.38 (m, 7H), 1.37–1.20 (m, 2H), 1.14 (m, 8H), 0.95–0.80 (m, 1H). 13C NMR (75 MHz, CDCl3, δ ): 217.9, 96.6, 75.0, 73.7, 73.6, 67.0, 63.9, 62.9, 59.8, 54.4, 49.7, 43.2, 41.3, 38.7, 33.8, 32.8, 30.0, 22.3, 21.9, 18.4. HRMS [M + H]+: calcd for C20H30NO6, 380.2073; found, 380.2063.
(2R,5′ S,6′ S,6a′ R,9′ S,11b′ S,14′ R)-5′,6 ′,14′-Trihydroxy-4 ′,4′ -dime- thyldecahydro-1′ H,7′ H-spiro[aziridine-2,8′ -[6,11b](epoxymethano)- [6a,9]methanocyclohepta[a]naphthalene]-1″,7′-dione (36).
Compound 36 (79.1 mg) was prepared from 27 in 28% yield by a procedure similar to that used to prepare compound 24. The title compound 36 was obtained as a white amorphous solid. = −100.0 (c 0.14, MeOH); 1H NMR (300 MHz, CDCl3, δ ): 5.37 (d, J = 11.1 Hz, 1H), 5.04 (s, 1H), 4.27 (dd, J = 10.4, 1.4 Hz, 1H), 4.03 (dd, J = 10.4, 1.4 Hz, 1H), 3.90–3.74 (m, 1H), 2.54–2.24 (m, 5H), 2.11 (m, 1H), 2.00–1.53 (m, 7H), 1.39–1.21 (m, 2H), 1.16 (s, 3H), 1.01 (s, 3H), 0.96–0.63 (m, 1H). 13C NMR (75 MHz, CDCl3, δ ): 217.9,211.7,97.9,73.2,72.8,64.9, 61.5, 60.1, 49.1, 49.1, 48.4, 40.5, 38.4, 35.7, 32.9, 31.5, 30.5, 26.0, 23.3, 18.6. HRMS [M + H]+: calcd for C20H28NO6, 378.1917; found, 378.1907.
(2R,5′ S,6′ S,6a′ R,9′ S,11 b′ R,14′ R)-5′,6 ′,14 ′-Trihydroxy-4 ′,4′-di- methyl-4 ′,4a′,5′,6′,9′,10 ′,11′,11a′-octahydro-3′ H,7′ H-spiro- [aziridine-2,8′-[6,11b](epoxymethano)[6a,9]methanocyclohepta- [a]naphthalene]-3′,7′-dione (37).
Compound 37 (16.6 mg) was prepared from 29 in 35% yield by a procedure similar to that used to prepare compound 24. The title compound 37 was obtained as a white amorphous solid. = −55.0 (c 0.12, MeOH); 1H NMR (300 MHz, CDCl3, δ ): 6.35 (d, J = 10.4 Hz, 1H), 6.05 (d, J = 10.4 Hz, 1H), 5.51 (s, 1H), 5.14 (s, 1H), 4.29–3.83 (m, 3H), 2.57–2.21 (m, 3H), 1.93 (m, 4h), 1.72 (m, 3h), 1.30 (m, 7H), 0.89 (m, 1H); 13C NMR (75 MHz, CDCl3, δ ): 217.7, 202.8, 142.6, 130.2, 97.5, 73.4, 72.0, 64.7, 61.8, 56.1, 50.7, 44.5, 40.5, 39.0, 31.6, 29.7, 26.0, 23.6, 22.1, 17.3; HRMS [M + H]+: calcd for C20H26NO6, 376.1760; found, 376.1750.
(2R,5′ S,6′ S,6 a′ R,9′ S,11b′ S,14′ R)-5′,6 ′,14′-Trihydroxy-4 ′,4′ -dimethyl-4 ′,4a′,5′,6′,9′,10 ′,11′,11a′-octahydro-1′H,7′ H-spiro- [aziridine-2,8′-[6,11b](epoxymethano)[6a,9]methanocyclohepta- [a]naphthalene]-1′,7′-dione (38).
Compound 38 (11.0 mg) was prepared from 30 in 61% yield by a procedure similar to that used to prepare compound 24. The title compound 38 was obtained as a white amorphous solid. = −206.9 (c 0.19, MeOH); 1H NMR (300 MHz, CDCl3, δ ): 6.80 (d, J = 10.1 Hz, 1H), 5.89 (d, J = 10.1 Hz, 1H), 5.51 (d, J = 11.5 Hz, 1H), 5.10 (d, J = 23.9 Hz, 2H), 4.31 (d, J = 10.2 Hz, 1H),4.05 (dd, J = 19.5, 10.0 Hz, 2H), 3.51 (s, 1H),2.28 (d, J = 11.7 Hz, 3h), 2.07 (d, J = 4.1 Hz, 2H), 1.90 (d, J = 9.0 Hz, 2H), 1.62 (d, J = 8.8 Hz, 3H), 1.37 (s, 2H), 1.27 (d, J = 5.5 Hz, 4H); 13C NMR (75 MHz, CDCl3, δ ): 218.3, 196.2, 161.3, 127.1, 97.8, 73.4, 72.6, 65.6, 62.0, 56.5, 49.8, 49.2, 46.2, 40.5, 35.8, 30.0, 29.7, 26.2, 24.7, 19.0; HRMS [M + H]+: calcd for C20H26NO6, 376.1760; found, 376.1750.
(9H-Fluoren-9-yl)methyl (2R,3′S,3a′R,3a1′R,6a′R,7′S,11a′ S)-7′-hy- droxy-5′,5 ′,8 ′,8′-tetramethyl-11′,14′-dioxodecahydro-2′ H-spiro- [aziridine-2,15′ -[6a,11a](epoxymethano)[3,3a1]ethanophenanthro- [1,10-de][1,3]dioxine]-1-carboxylate (39).
To a solution of 27 (203.1 mg, 0.49 mmol) and Na2CO3 (154.7 mg, 1.46 mmol) in dioxane (20 mL) was added Fmoc chloride (151.0 mg, 0.58 mmol) at 0°C. The resulting mixture was stirred at room temperature for 12 h. Next, the solution was removed, and the residue was dissolved with CH2Cl2 (50 mL). The organic layer was washed with saturated NaHCO3 solution (50 mL) and brine (50 mL), dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using silica gel column; elution with 33% EtOAc in hexane afforded the desired product 39 as a white solid (106.1 mg, 34%). = −45.9 (c 0.19, MeOH); 1H NMR (300 MHz, CDCl3, δ ): 7.83–7.73 (m, 2H), 7.65 (dd, J = 10.5,7.4 Hz, 2h), 7.37 (dtt, J = 26.4,7.6,1.4 Hz, 4H), 4.99 (d, J = 1.5 Hz, 1H), 4.65 (d, J = 11.9 Hz, 1H), 4.61–4.49 (m, 1H), 4.36–4.19 (m, 3H), 4.08 (dd, J = 10.1, 1.5 Hz, 1H), 3.96 (dd, J = 11.9, 8.8 Hz, 1H), 2.75 (d, J = 1.1 Hz, 1H), 2.56–2.32 (m, 5H), 2.15–2.07 (m, 1H), 1.99–1.54 (m, 9H), 1.44 (s, 3H), 1.22 (s, 3H), 1.08 (s, 3H). 13C NMR (75 MHz, CDCl3, δ ): 211.9, 211.2, 159.4, 143.6, 143.6, 141.2, 141.2, 127.7, 127.7, 127.1, 127.1, 125.4, 125.4, 119.9, 119.9, 101.9, 95.5, 71.8, 70.4, 68.7, 64.7, 59.6, 56.2, 54.5, 48.2, 46.9, 46.8, 38.5, 38.2, 35.8, 35.4, 33.0, 31.0, 29.8, 26.5, 25.4, 23.0, 18.5. HRMS [M + H]+: calcd for C38H42NO8, 640.2943; found, 640.2897.
(9H-Fluoren-9-yl)methyl (2R,5′S,6′S,6a′R,9′S,11b′S,14′R)- 5′,6 ′,14′-trihydroxy-4 ′,4′ -dimethyl-1′,7′-dioxodecahydro-1′ H,7′ H- spiro[aziridine-2,8′-[6,11b](epoxymethano)[6a,9]- methanocyclohepta[a]naphthalene]-1-carboxylate (40).
Compound 40 (106.1 mg) was prepared in 75% yield by a procedure similar to that used to prepare compound 24. The title compound 40 was obtained as a white solid. = −110.2 (c 0.13, MeOH); 1H NMR (300 MHz, CDCl3 and CD3OD, δ ): 7.72 (d, J = 7.4 Hz, 2H), 7.54 (dd, J = 7.1, 2.3 Hz, 2H), 7.44–7.20 (m, 4H), 5.98 (d, J = 125.4 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 4.85 (s, 1H), 4.45–4.14 (m, 4H), 4.04–3.92 (m, 1H), 3.85–3.53 (m, 2H), 2.60–2.39 (m, 2H), 2.30 (ddt, J = 15.3, 11.5, 5.8 Hz, 2H), 2.12–1.98 (m, 2H), 1.97–1.53 (m, 5h), 1.24 (dt, J = 10.7, 5.4 Hz, 2h), 1.13 (s, 3h), 0.97 (s, 3H). 13C NMR (75 MHz, CDCl3 and CD3OD, δ ): 214.2,211.6,154.8,143.8,143.7,141.2,141.2,127.6,127.6, 127.1, 127.1, 125.1, 125.1, 119.9, 119.9, 97.8,73.3,72.7,67.2,66.8,64.7, 62.7, 59.8, 49.1, 48.2, 47.0, 44.5, 44.0, 38.2, 35.7, 32.8, 30.4, 23.1, 21.1, 17.1. HRMS [M + H]+: calcd for C35H38NO8, 600.2597; found, 600.2587.
(9H-Fluoren-9-yl)methyl (2R,6′S,7′S,7a′R,10′S,12b′R,15′R)-2′-(al- lylamino)-6′,7′,15′-trihydroxy-5′,5′-dimethyl-8′-oxo-′,5a′,6 ′,7′,10 ′,11′,12 ′,12a′-octahydro-4′ H,8′ H-spiro[aziridine-2,9′ - [7,12b](epoxymethano)[7a,10]methanocyclohepta[7,8]naphtho- [1,2-d]thiazole]-1-carboxylate (42).
To a solution of 40 (70.0 mg, 0.12 mmol) in THF (4 mL) was added PyHBr3 (41.6 mg, 0.13 mmol) at room temperature. The reaction mixture was stirred at room temperature for 4 h and then poured into water and extracted with CH2Cl2 (20 mL × 3). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo to give a crude oily product 41 (65.2 mg, 82%), which was directly used in the next reaction. To a solution of 41 (65.2 mg, 0.10 mmol) in ethanol (4 mL) was added N-allylthiourea (13.4 mg, 0.12 mmol) at room temperature. The reaction mixture was heated under reflux for 6 h. After cooling and basifying with saturated NaHCO3 aqueous solution, the mixture was concentrated in vacuo to give an oily residue. The residue was purified by silica gel column; elution with 33% EtOAc in hexane afforded the desired product 42 (29.4 mg, 44%) as an amorphous gel. = −102.5 (c 0.08, MeOH); 1H NMR (300 MHz, CDCl3, δ ): 7.76 (d, J = 7.5 Hz, 2H), 7.59 (d, J = 7.4 Hz, 2H), 7.40 (t, J = 7.4 Hz, 2H), 7.33 (d, J = 7.4 Hz, 2h), 6.13 (s, 1H), 5.90 (ddt, J = 16.2, 10.7, 5.6 Hz, 1H), 5.49 (s, 1H), 5.34–5.12 (m,4H), 5.07 (s, 1H),4.53 (d, J = 10.2 Hz, 1H), 4.32 (ddd, J = 31.8, 13.6, 7.6 Hz, 3H), 3.96 (d, J = 10.2 Hz, 1H), 3.88–3.78 (m, 2H), 3.65 (d, J = 12.1 Hz, 1H), 2.63–2.46 (m, 2H), 2.45–2.28 (m, 2H), 2.05–1.88 (m, 2H), 1.84–1.74 (m, 1H), 1.61 (d, J = 9.4 Hz, 1H), 1.26 (d, J = 8.3 Hz, 6H), 0.98 (s, 3H). 13C NMR (75 MHz, CDCl3, δ ): 214.6, 166.6, 154.7, 144.0, 143.9, 142.1, 141.3, 141.3, 133.6, 133.6, 127.6, 127.6, 127.1, 127.1, 125.2, 125.2, 119.9, 119.7, 117.3, 97.6, 73.5, 73.4, 67.7, 66.7, 65.3, 64.0, 57.7, 52.8, 48.3, 47.1, 44.7, 41.0, 38.8, 35.0, 30.4, 29.7, 21.7, 20.9, 18.6. HRMS [M + H]+: calcd for C39H42N3O7S, 696.2743; found, 696.2698.
(2R,6′ S,7′S,7a′ R,10′ S,12b′ R,15′ R)-2′-(Allylamino)-6 ′,7′,15′-trihy- droxy-5,5 -dimethyl-5,5a,6,7,10,11,1212a -octahydro- 4′ H,8′ H-spiro[aziridine-2,9′-[7,12b](epoxymethano)[7a,10]- methanocyclohepta[7,8]naphtho[1,2-d]thiazol]-8′-one (43).
To a solution of 42 (28.0 mg, 0.04 mmol) in DMF (0.8 mL) was added piperidine (0.2 mL) at room temperature. The reaction mixture was stirred at room temperature for 1 h and then poured into water and extracted with CH2Cl2 (10 mL × 3). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo to give a crude oily residue. The residue was purified by silica gel column; elution with 5% MeOH in CH2Cl2 afforded the desired product 43 (15.6 mg, 82%) as an amorphous gel. = −30.8 (c 0.13, MeOH); 1H NMR (300 MHz, CDCl3, δ ): 5.90 (ddt, J = 17.2, 10.2, 5.6 Hz, 1H), 5.38–5.11 (m,3H), 5.07 (d, J = 1.3 Hz, 1H), 4.86 (d, J = 11.6 Hz, 1H), 4.39 (dd, J = 10.1,1.4 Hz, 1H),4.00(dd, J = 10.1,1.4 Hz, 1H), 3.91 (dd, J = 11.3, 8.8 Hz, 1H), 3.82 (d, J = 3.9 Hz, 2H), 3.73 (d, J = 25.0 Hz, 2H), 3.50 (s, 1H), 2.94 (d, J = 22.3 Hz, 2H), 2.58–2.25 (m, 4H), 2.23–1.97 (m, 2H), 1.97–1.62 (m, 3H), 1.62–1.54 (m, 1H), 1.27 (d, J = 2.4 Hz, 5H), 1.00 (s, 3H). 13C NMR (75 MHz, CDCl3δ ): 218.5, 165.7, 142.8, 133.8, 119.7, 117.2, 97.5, 73.3, 73.2, 65.6, 62.7, 58.2, 52.7, 49.2, 48.2, 40.8, 38.9, 35.0, 31.8, 30.5, 29.7, 26.5, 21.0, 19.9. HRMS [M + H]+: calcd for C24H32N3O5S, 474.2063; found, 474.2053.
(2R,3′ S,3a′ R,3a1′ R,6a′ R,7′ S,11′S,11a′ S)-1-Acryloyl-7′,11′-dihy- droxy-5′,5 ′,8 ′,8′-tetramethyldecahydro-2′ H-spiro[aziridine-2,15′ - [6a, 11a](epoxymethano)[3,3a1]ethanophenanthro[1,10-de][1,3]- dioxin]-14′-one (44).
To a solution of 25 (66.0 mg, 0.16 mmol), acrylic acid (12.5 mg, 0.17 mmol), and DIPEA (60.9 mg, 0.47 mmol) in CH2Cl2 (20 mL) was added HBTU (89.5 mg, 0.24 mmol) at 0°C. The resulting mixture was stirred at room temperature for 12 h. Next, the mixture was diluted with CH2Cl2 (30 mL). The organic layer was washed with saturated NaHCO3 solution (50 mL) and brine (50 mL), dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using silica gel column; elution with 33% EtOAc in hexane afforded the desired product 44 as a white solid (48.1 mg, 65%). = −25.4 (c 0.13, MeOH); 1H NMR (300 MHz, CDCl3, δ ): 6.71–5.96 (m, 2H), 5.89–5.52 (m, 1H), 4.97 (m, 2H), 4.56–3.98 (m, 2H), 3.88 (m, 1H), 3.47 (m, 1H), 2.77 (m, 4H), 2.43 (m, 3H), 1.95 (m, 3H), 1.72 (m, 4H), 1.43 (m, 4h), 1.24 (m, 2h), 1.12 (m, 6h). 13C NMR (75 MHz, CDCl3, δ ): 212.1, 175.0, 131.0, 129.0, 101.6, 94.7, 74.5, 72.9, 70.7, 62.7, 58.2, 56.8, 55.1, 51.2, 41.0, 39.0, 38.1, 34.5, 33.4, 29.9, 29.8, 27.5, 25.5, 22.7, 19.8. HRMS [M + H]+: calcd for C26H36NO7, 474.2492; found, 474.2478; [M + Na]+: calcd for C26H35NNaO7, 496.2311; found, 496.2303.
(2R,3′S,3a′ R,3a1 R,6a′ R,7 ′S,11′ S,11a′ S)-7′,11′-Dihydroxy- 5′,5 ′,8 ′,8′-tetramethyl-1-propionyldecahydro-2′ H-spiro[aziridine- 2,15′-[6a,11a](epoxymethano)[3,3a1]ethanophenanthro[1,10-de]-dioxin]-14′-one (45).
Compound 45 (35.0 mg) was prepared in 74% yield by a procedure similar to that used to prepare compound 44. The title compound 45 was obtained as a white solid. = −36.0 (c 0.10, CHCl3); 1H NMR (300 MHz, CDCl3, δ ): 5.03 (d, J = 12.0 Hz, 1H), 4.99–4.92 (m, 1H), 4.28 (d, J = 9.9 Hz, 1H), 4.05 (d, J = 9.9 Hz, 1H), 3.91 (dd, J = 12.0, 8.1 Hz, 1H), 3.48 (dd, J = 11.4, 5.5 Hz, 1H), 2.69 (d, J = 1.0 Hz, 1H), 2.55–2.28 (m, 5H), 2.03–1.88 (m, 2h), 1.79 (m, 1H), 1.73 (s, 3H), 1.71–1.57 (m, 2H), 1.57–1.45 (m, 2H), 1.43 (s, 3H), 1.35–1.22 (m, 3H), 1.15 (s, 3h), 1.13 (s, 3H),1.08 (m, 3H). 13CNMR (75 MHz, CDCl3, δ ): 213.5, 184.8, 101.6, 94.7, 74.4, 73.0, 70.8, 62.8, 58.3, 56.9, 54.9, 51.2, 40.9, 39.0, 38.2, 34.7, 33.6, 33.4, 30.3, 29.9, 29.7, 27.4, 25.5, 22.7, 19.8, 8.6. HRMS [M + H]+: calcd for C26H38NO7, 476.2648; found, 476.2632.
Measuring Compound Reactivity with GSH.
The oridonin or its derivatives (10.0 mg) were incubated with an excess of GSH (15–20 mg) in deuterated phosphate buffer at pH 7.4 (0.05 mL) and d6-DMSO (0.50 mL) at 37°C. The reaction was monitored by the change of representative hydrogen signals with 1H NMR analysis, and the adduct products were confirmed with the HRMS. The reactivities of the covalent warheads could be initially classified based on the integration change of the representative proton NMR.
In Vitro Determination of Newly Synthesized Compounds against Cancer Cell Proliferation.
Breast cancer cells (MDA-MB- 231, MDA-MB-468, SK-BR3, and MCF-7 lines) were seeded in 96-well plates at a density of 1 × 104 cells per well and treated with DMSO or testing compounds at 0.1, 0.33, 1.0, 3.3, and 10 μM concentrations for 72 h. Proliferation was measured by treating cells with 3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) in a CellTiter 96t aqueous non-radioactive cell proliferation assay kit (Promega, Madison, WI). Absorbance of all wells was determined by measuring OD at 560 nm after 1 h incubation at 37°C on a 96-well iMark microplate absorbance reader (BioRad, Hercules, CA). Each individual compound was tested in quadruplicate wells for each concentration.
Colony-Formation Assay.
Breast cancer MDA-MB-231 cells were seeded in 6-well tissue culture plates with a density of 600 cells per well and maintained in regular culture media. Similarly, breast cancer MDA- MB-468, SK-BR3, or MCF-7 cells were seeded with a density of 800 cells per well. After 24 h, the cells were treated with 44 at different concentrations (1, 5, 10, 15, and 20 μM) or DMSO as the vehicle. The culture media with the compounds were changed every 72 h. At the end of 2 weeks, the wells were washed twice with PBS buffer, and 2 mL of 0.01% crystal violet staining buffer was added to each well and incubated for 10 min. The wells were then washed with PBS for 5 min three times and allowed to dry. Photographs were then taken.
Cellular-Apoptosis Assay.
Breast cancer MDA-MB-231 cells were incubated in 6-well plates (2.5 × 105 cells per well). Cells were then treated with DMSO, oridonin, or testing compounds at different concentrations for 48 h, and then both adherent and floating cells were collected and washed once with PBS. Resuspended cells were incubated with 100 μL of PBS containing 1% BSA and 100 μL of Annexin V and dead-cell detection reagent at room temperature for 20 min. Apoptosis was measured immediately using the Muse Cell Analyzer with the Muse Apoptosis Kit (catalog no. MCH100105).
Western-Blot Analysis.
Breast cancer MDA-MB-231 cells were treated with DMSO or 44 at different concentrations. After 48 h of treatment, cells were harvested and lysed. Protein concentrations were quantified by the method of Bradford with bovine serum albumin as the standard. Equal amounts of total cellular protein extract (40 μg) was separated by electrophoresis on sodium dodecyl sulfate–polyacrylamide gels and transferred to NC membranes. After blocking with 5% nonfat milk, the membrane was incubated with the desired primary antibody overnight at the following dilution: anti-Bcl-2 (1:200), anticleaved- PARP (1:500), anti-NF-κB (1:1000), and β-actin (1:2000), and anti-Bax (1:1000). Subsequently, the membrane was incubated with appropriate secondary antibody. The immunoreactive bands were visualized by enhanced chemiluminescence, as recommended by the manufacturer.
In Vivo Antitumor Activity Determination.
All procedures including mice and in vivo experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of UT M. D.Anderson Cancer Center (MDACC). A total of31 female nude mice were obtained from MDACC and were used for orthotopic tumor studies at6to8 weeks of age. The mice were maintained in a barrier unit with 12 h of light–dark switching. FreshlyharvestedMDA-MB-231 cells (1.0 × 106cellspermouse, resuspended in 100μL of PBS)were injected into the no. 3 mammary fat pad of each mice and then randomly assigned into 4 groups. The mice were treated daily with 10 or 15 mg/kg of 44, oridonin or vehicle through intraperitoneal injection when the tumor volume reached 80–100 mm3 in volume. All agents were dissolved in DMSO for in vivo administration. Body weights and tumors volume were measured daily, and tumor volume was calculated according to the formula V = 0.5 × L × W2, where L = length (mm) and W = width (mm).
Statistical Analysis.
Statistical significance was determined using a Student t test or one-way ANOVA in vivo experiments. A single asterisk represents a p value of less than 0.05, double asterisks represent a p value of less than 0.01, and triple asterisks represent a p value of less than 0.001.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Breast Cancer Research Program (BCRP) Breakthrough Award (grant nos. BC160038 and BC160038P1) from the Department of Defense (DoD) (to J.Z. and Q.S.), grant nos. P30 DA028821 and R01 DA038446 from the National Institutes of Health (to J.Z.), Sanofi Innovation Awards (iAward) Program (to J.Z.), the Cancer Prevention Research Institute of Texas (CPRIT) award (toJ.Z.), Cancer Center Support grant no. P30 CA016672 from the United States National Institutes ofHealth (to The University of Texas MD Anderson Cancer Center), startup funds from MD Anderson Cancer Center (to Q.S.), and the Duncan Family Institute Seed Funding Research Program (to Q.S). We thank Dr. Lawrence C. Sowers at the Department of Pharmacology as well as Dr. Tianzhi Wang at the NMR core facility of UTMB for the NMR spectroscopy assistance and Dr. Xuemei Luo at UTMB mass spectrometry core with funding support from the UT system proteomics network for the HRMS analysis.
ABBREVIATION USED
- SAR
structure–activity relationship
- SRR
structure–reactivity relationship
- FDA
Food and Drug Administration
- GSH
glutathione
- NRF2
nuclear factor erythroid-derived 2-like 2
- NF-κB
nuclear factor κ-light-chain-enhancer of activated B cells
- ROS
reactive oxygen species
- p53
tumor protein p53
- p21
cyclin-dependent kinase inhibitor 1
- OTC
over-the-counter
- IC50
half maximal inhibitory concentration
- Bcl-2
B-cell lymphoma 2
- Bax
Bcl-2-associated X protein
- PARP
poly- (ADP-ribose) polymerase
- ER
estrogen receptor
- DR5
death receptor 5
- XBP1
X-box binding protein 1
- Rh2(esp)2
bis[rhodium(α,α,α′ ,α′-tetramethyl-1,3-benzenedipropionic acid)]
- RNS
reactive nitrogen species
- NMR
nuclear magnetic resonance
- t1/2
half-life; HRMS, high-resolution mass spectrometry
- EGFR
epidermal growth factor receptor
- MS
multiple sclerosis
- LD50
median lethal dose
- DPH
O-(2,4- dinitrophenyl)hydroxylamine
- Rh2(OAc)4
rhodium(II) acetate dimer
- Rh2(TPA)4
rhodium(II) triphenylacetate dimer
- Rh2(R- DOSP)4
tetrakis[1-[[4-alkyl(C11–C13)phenyl]sulfonyl]-(2R)- pyrrolidinecarboxylate]dirhodium(II)
- Rh2(S-DOSP)4
tetrakis- [1-[[4-alkyl(C11 – C13)phenyl]sulfonyl]-(2S)- pyrrolidinecarboxylate]dirhodium(II)
- PyHBr3
pyridinium tribromide
- THF
Tetrahydrofuran
- DMF
dimethylformamide
- HBTU
2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- DIPEA
N,N-diisopropylethylamine
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PI
propidium iodide
- DMSO
dimethyl sulfoxide
- PBS
phosphate-buffered saline
- BCA
bicinchoninic acid
- BSA
bovine serum albumin
- SDS
sodium dodecyl sulfate
- NC
nitrocellulose
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/acs.jmed-chem.7b01514.
Figures showing the reactivity of oridonin and its derivatives with GSH determined by the 1H NMR and HRMS, the reaction of representative aziridine compound and oridonin analogue, growth-inhibitory effects of the oridonin derivatives on MCF-7 and MDA-MB-231 cell lines, cell viability, microsomal and plasma stability, cell density, and 1H and 13C NMR spectra for the new compounds described in this paper. A table showing X-ray CIF file for compound 22. Molecular formula strings. (PDF)
Crystallographic data for a selection of experimental compounds. (CSV)
The authors declare no competing financial interest.
REFERENCES
- (1).Singh J; Petter RC; Baillie TA; Whitty A The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10, 307–317. [DOI] [PubMed] [Google Scholar]
- (2).Barf T; Kaptein A Irreversible protein kinase inhibitors: balancing the benefits and risks. J. Med. Chem 2012, 55, 6243–6262. [DOI] [PubMed] [Google Scholar]
- (3).Liu Q; Sabnis Y; Zhao Z; Zhang T; Buhrlage SJ; Jones LH; Gray NS Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Finlay MR; Anderton M; Ashton S; Ballard P; Bethel PA; Box MR; Bradbury RH; Brown SJ; Butterworth S; Campbell A; Chorley C; Colclough N; Cross DA; Currie GS; Grist M; Hassall L; Hill GB; James D; James M; Kemmitt P; Klinowska T; Lamont G; Lamont SG; Martin N; McFarland HL; Mellor MJ; Orme JP; Perkins D; Perkins P; Richmond G; Smith P; Ward RA; Waring MJ; Whittaker D; Wells S; Wrigley GL Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J.Med. Chem 2014, 57, 8249–8267. [DOI] [PubMed] [Google Scholar]
- (5).Engel J; Richters A; Getlik M; Tomassi S; Keul M; Termathe M; Lategahn J; Becker C; Mayer-Wrangowski S; Grutter C; Uhlenbrock N; Krull J; Schaumann N; Eppmann S; Kibies P; Hoffgaard F; Heil J; Menninger S; Ortiz-Cuaran S; Heuckmann JM; Tinnefeld V; Zahedi RP; Sos ML; Schultz-Fademrecht C; Thomas RK; Kast SM; Rauh D Targeting drug resistance in EGFR with covalent inhibitors: a structure-based design approach. J. Med. Chem 2015, 58, 6844–6863. [DOI] [PubMed] [Google Scholar]
- (6).Song Z; Ge Y; Wang C; Huang S; Shu X; Liu K; Zhou Y; Ma X Challenges and perspectives on the development of small- molecule EGFR inhibitors against T790M-mediated resistance in nonsmall-cell lung cancer. J. Med. Chem 2016, 59, 6580–6594. [DOI] [PubMed] [Google Scholar]
- (7).Schwartz PA; Kuzmic P; Solowiej J; Bergqvist S; Bolanos B; Almaden C; Nagata A; Ryan K; Feng J; Dalvie D; Kath JC; Xu M; Wani R; Murray BW Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Backus KM; Correia BE; Lum KM; Forli S; Horning BD; Gonzalez-Paez GE; Chatterjee S; Lanning BR; Teijaro JR; Olson AJ; Wolan DW; Cravatt BF Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Flanagan ME; Abramite JA; Anderson DP; Aulabaugh A; Dahal UP; Gilbert AM; Li C; Montgomery J; Oppenheimer SR; Ryder T; Schuff BP; Uccello DP; Walker GS; Wu Y; Brown MF; Chen JM; Hayward MM; Noe MC; Obach RS; Philippe L; Shanmugasundaram V; Shapiro MJ; Starr J; Stroh J; Che Y Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J. Med. Chem 2014, 57, 10072–10079. [DOI] [PubMed] [Google Scholar]
- (10).Cee VJ; Volak LP; Chen Y; Bartberger MD; Tegley C; Arvedson T; McCarter J; Tasker AS; Fotsch C Systematic study of the glutathione (GSH) reactivity of N-arylacrylamides: 1. effects of aryl substitution. J. Med. Chem 2015, 58, 9171–9178. [DOI] [PubMed] [Google Scholar]
- (11).Ward RA; Anderton MJ; Ashton S; Bethel PA; Box M; Butterworth S; Colclough N; Chorley CG; Chuaqui C; Cross DA; Dakin LA; Debreczeni JE; Eberlein C; Finlay MR; Hill GB; Grist M; Klinowska TC; Lane C; Martin S; Orme JP; Smith P; Wang F; Waring MJ Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J. Med. Chem 2013, 56, 7025–7048. [DOI] [PubMed] [Google Scholar]
- (12).Palkowitz MD; Tan B; Hu H; Roth K; Bauer R A. Synthesis of diverse N-acryloyl azetidines and evaluation of their enhanced thiol reactivities. Org. Lett 2017, 19, 2270–2273. [DOI] [PubMed] [Google Scholar]
- (13).Kathman SG; Xu Z; Statsyuk AV A fragment-based method to discover irreversible covalent inhibitors of cysteine proteases. J. Med. Chem 2014, 57, 4969–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wang J; Zhang CJ; Chia WN; Loh CC; Li Z; Lee YM; He Y; Yuan LX; Lim TK; Liu M; Liew CX; Lee YQ; Zhang J; Lu N; Lim CT; Hua ZC; Liu B; Shen HM; Tan KS; Lin Q Haem-activated promiscuous targeting of artemisinin in plasmodium falciparum. Nat. Commun 2015, 6, 10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Baillie TA Targeted covalent inhibitors for drug design. Angew. Chem., Int. Ed. Engl 2016, 55, 2–17. [DOI] [PubMed] [Google Scholar]
- (16).Wong MH; Bryan HK; Copple IM; Jenkins RE; Chiu PH; Bibby J; Berry NG; Kitteringham NR; Goldring CE; O’Neill PM; Park BK Design and synthesis of irreversible analogues of bardoxolone methyl for the identification of pharmacologically relevant targets and interaction sites. J. Med. Chem 2016, 59, 2396–2409. [DOI] [PubMed] [Google Scholar]
- (17).Federspiel JD; Codreanu SG; Goyal S; Albertolle ME; Lowe E; Teague J; Wong HL; Guengerich FP; Liebler DC Specificity of protein covalent modification by the electrophilic proteasome inhibitor carfilzomib in human cells. Mol. Cell. Proteomics 2016, 15, 3233–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bauer RA Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discovery Today 2015, 20, 1061–1073. [DOI] [PubMed] [Google Scholar]
- (19).Li CY; Wang EQ; Cheng Y; Bao JK Oridonin: an active diterpenoid targeting cell cycle arrest, apoptotic and autophagic pathways for cancer therapeutics. Int. J. Biochem. Cell Biol 2011, 43, 701–704. [DOI] [PubMed] [Google Scholar]
- (20).Xu J; Wold EA; Ding Y; Shen Q; Zhou J Therapeutic potential of oridonin and its analogs: from anticancer and antiinflammation to neuroprotection. Molecules 2018, 23, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ding Y; Ding C; Ye N; Liu Z; Wold EA; Chen H; Wild C; Shen Q; Zhou J Discovery and development of natural product oridonin-inspired anticancer agents. Eur. J. Med. Chem 2016, 122, 102–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lin Z; Guo Y; Gao Y; Wang S; Wang X; Xie Z; Niu H; Chang W; Liu L; Yuan H; Lou H ent-Kaurane diterpenoids from Chinese liverworts and their antitumor activities through Michael addition as detected in situ by a fluorescence probe. J. Med. Chem 2015, 58, 3944–3956. [DOI] [PubMed] [Google Scholar]
- (23).Nan FJ; Zhang YM; Liu HN; Ding J; Meng LH; Xu CH Synthesis and Derivatization of an ent-Kaurane Diterpenoid. Chinese Patent CN102584760A, January/13/2011. [Google Scholar]
- (24).Du Y; Villeneuve NF; Wang XJ; Sun Z; Chen W; Li J; Lou H; Wong PK; Zhang DD Oridonin confers protection against arsenic-induced toxicity through activation of the Nrf2-mediated defensive response. Environ. Health Persp 2008, 116, 1154–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhen T; Wu CF; Liu P; Wu HY; Zhou GB; Lu Y; Liu JX; Liang Y; Li KK; Wang YY; Xie YY; He MM; Cao HM; Zhang WN; Chen LM; Petrie K; Chen SJ; Chen Z Targeting of AML1-ETO in t(8;21) leukemia by oridonin generates a tumor suppressor-like protein. Sci. Transl. Med 2012, 4, [DOI] [PubMed] [Google Scholar]
- (26).Zhou GB; Kang H; Wang L; Gao L; Liu P; Xie J; Zhang FX; Weng XQ; Shen ZX; Chen J; Gu LJ; Yan M; Zhang DE; Chen SJ; Wang ZY; Chen Z Oridonin, a diterpenoid extracted from medicinal herbs, targets AML1-ETO fusion protein and shows potent antitumor activity with low adverse effects on t(8;21) leukemia in vitro and in vivo. Blood 2007, 109, 3441–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).De Silva C; Stevens R; Jordan KM Inflammatory aortitis controlled by the Chinese herbal remedy Donglingcao Pian. Rheumatology (Oxford, U. K.) 2008, 47, 1257–1259. [DOI] [PubMed] [Google Scholar]
- (28).Liu QQ; Wang HL; Chen K; Wang SB; Xu Y; Ye Q; Sun YW Oridonin derivative ameliorates experimental colitis by inhibiting activated T-cells and translocation of nuclear factor-kappa B. J. Dig. Dis 2016, 17, 104–112. [DOI] [PubMed] [Google Scholar]
- (29).Sun PY; Wu GL; Qiu ZJ; Chen YJ l-alanine-(14-oridonin) Ester Trifluoroacetate as well as Preparation Method and Application. Chinese Patent CN104017000A, 09/March/2014. [Google Scholar]
- (30).Bohanon FJ; Wang X; Graham BM; Ding C; Ding Y; Radhakrishnan GL; Rastellini C; Zhou J; Radhakrishnan RS Enhanced effects of novel oridonin analog CYD0682 for hepatic fibrosis. J. Surg. Res 2015, 199, 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ding C; Zhang Y; Chen H; Yang Z; Wild C; Ye N; Ester CD; Xiong A; White MA; Shen Q; Zhou J Oridonin ring A-based diverse constructions of enone functionality: identification of novel dienone analogues effective for highly aggressive breast cancer by inducing apoptosis. J. Med. Chem 2013, 56, 8814–8825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wang X; Bohanon F; Kandathiparampil A; Rastellini C; Ding C; Ding Y; Zhou J; Radhakrishnan R Suppressive effects of novel oridonin derivative CYD0625 on activated hepatic stellate cells. FASEB J 2015, 29, 6115. [Google Scholar]
- (33).Ding C; Zhang Y; Chen H; Yang Z; Wild C; Chu L; Liu H; Shen Q; Zhou J Novel nitrogen-enriched oridonin analogues with thiazole-fused A-ring: protecting group-free synthesis, enhanced anticancer profile, and improved aqueous solubility. J. Med. Chem 2013, 56, 5048–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Chen W; Zhou J; Wu K; Huang J; Ding Y; Yun EJ; Wang B; Ding C; Hernandez E; Santoyo J; Chen H; Lin H; Sagalowsky A; He D; Zhou J; Hsieh JT Targeting XBP1-mediated β-catenin expression associated with bladder cancer with newly synthetic oridonin analogues. Oncotarget 2016, 7, 56842–56854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wu J; Ding Y; Chen CH; Zhou Z; Ding C; Chen H; Zhou J; Chen C A new oridonin analog suppresses triple-negative breast cancer cells and tumor growth via the induction of death receptor 5. Cancer Lett 2016, 380, 393–402. [DOI] [PubMed] [Google Scholar]
- (36).Zang L; He H; Xu Q; Yu Y; Zheng N; Liu W; Hayashi T; Tashiro S; Onodera S; Ikejima T Reactive oxygen species H2O2 and •OH, but not O2•(–) promote oridonin-induced phagocytosis of apoptotic cells by human histocytic lymphoma U937 cells. Int. Immunopharmacol 2013, 15, 414–423. [DOI] [PubMed] [Google Scholar]
- (37).Cheng Y; Qiu F; Ye YC; Guo ZM; Tashiro S; Onodera S; Ikejima T Autophagy inhibits reactive oxygen species-mediated apoptosis via activating p38-nuclear factor-kappa B survival pathways in oridonin-treated murine fibrosarcoma L929 cells. FEBS J 2009, 276, 1291–1306. [DOI] [PubMed] [Google Scholar]
- (38).Kuo LM; Kuo CY; Lin CY; Hung MF; Shen JJ; Hwang TL Intracellular glutathione depletion by oridonin leads to apoptosis in hepatic stellate cells. Molecules 2014, 19, 3327–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Xu ZZ; Fu WB; Jin Z; Guo P; Wang WF; Li JM Reactive oxygen species mediate oridonin-induced apoptosis through DNA damage response and activation of JNK pathway in diffuse large B cell lymphoma. Leuk. Lymphoma 2016, 57, 888–898. [DOI] [PubMed] [Google Scholar]
- (40).Kubo I; Taniguchi M; Satomura Y; Kubota T Antibacterial activity and chemical structure of diterpenoids. Agric. Biol. Chem 1974, 38, 1261–1262. [Google Scholar]
- (41).Xu S; Luo S; Yao H; Cai H; Miao X; Wu F; Yang DH; Wu X; Xie W; Yao H; Chen ZS; Xu J Probing the anticancer action of oridonin with fluorescent analogues: visualizing subcellular localization to mitochondria. J. Med. Chem 2016, 59, 5022–5034. [DOI] [PubMed] [Google Scholar]
- (42).Schmidt TJ; Ak M; Mrowietz U Reactivity of dimethyl fumarate and methylhydrogen fumarate towards glutathione and N-acetyl-L-cysteine-preparation of S-substituted thiosuccinic acid esters. Bioorg. Med. Chem 2007, 15, 333–342. [DOI] [PubMed] [Google Scholar]
- (43).Wilson AJ; Kerns JK; Callahan JF; Moody CJ Keap calm, and carry on covalently. J. Med. Chem 2013, 56, 7463–7476. [DOI] [PubMed] [Google Scholar]
- (44).Couch RD; Browning RG; Honda T; Gribble GW; Wright DL; Sporn MB; Anderson AC Studies on the reactivity of CDDO, a promising new chemopreventive and chemotherapeutic agent: implications for a molecular mechanism of action. Bioorg. Med. Chem. Lett 2005, 15, 2215–2219. [DOI] [PubMed] [Google Scholar]
- (45).Fujita E; Nagao Y; Kaneko K; Nakazawa S; Kuroda H The antitumor and antibacterial activity of the Isodon diterpenoids. Chem. Pharm. Bull 1976, 24, 2118–2127. [DOI] [PubMed] [Google Scholar]
- (46).Zhu Y; Wang Q; Cornwall RG; Shi Y Organocatalytic asymmetric epoxidation and aziridination of olefins and their synthetic applications. Chem. Rev 2014, 114, 8199–8256. [DOI] [PubMed] [Google Scholar]
- (47).Degennaro L; Trinchera P; Luisi R Recent advances in the stereoselective synthesis of aziridines. Chem. Rev 2014, 114, 7881–7929. [DOI] [PubMed] [Google Scholar]
- (48).Smith DT; Njardarson JT A scalable rhodium-catalyzed intermolecular aziridination reaction. Angew. Chem., Int. Ed 2014, 53, 4278–4280. [DOI] [PubMed] [Google Scholar]
- (49).Xu J; Zhao J; Wang J; Feng N; Tan R; Liu Y Study on stability of oridonin solution. Zhongguo Zhong Yao Za Zhi 2009, 34, 47–49. [PubMed] [Google Scholar]
- (50).Jat JL; Paudyal MP; Gao H; Xu QL; Yousufuddin M; Devarajan D; Ess DH; Kürti L; Falck JR Direct stereospecific synthesis of unprotected N-H and N-Me aziridines from olefins. Science 2014, 343, 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Armstrong A; Baxter CA; Lamont SG; Pape AR; Wincewicz R Amine-promoted, organocatalytic aziridination of enones. Org. Lett 2007, 9, 351–353. [DOI] [PubMed] [Google Scholar]
- (52).Shen YM; Zhao MX; Xu J; Shi Y An amine-promoted aziridination of chalcones. Angew. Chem., Int. Ed 2006, 45, 8005–8008. [DOI] [PubMed] [Google Scholar]
- (53).Pesciaioli F; De Vincentiis F; Galzerano P; Bencivenni G; Bartoli G; Mazzanti A; Melchiorre P Organocatalytic asymmetric aziridination of enones. Angew. Chem., Int. Ed 2008, 47, 8703–8706. [DOI] [PubMed] [Google Scholar]
- (54).Hata Y; Watanabe M Reaction of aziridinecarboxylic acids with thiols in aqueous solution. the formation of β-amino acid. Tetrahedron 1987, 43, 3881–3888. [Google Scholar]
- (55).Ding C; Zhang Y; Chen H; Wild C; Wang T; White MA; Shen Q; Zhou J Overcoming synthetic challenges of oridonin A-ring structural diversification: regio- and stereoselective installation of azides and 1,2,3-triazoles at the C-1, C-2, or C-3 position. Org. Lett 2013, 15, 3718–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Ding C; Wang L; Chen H; Wild C; Ye N; Ding Y; Wang T; White MA; Shen Q; Zhou J ent-Kaurane-based regio- and stereoselective inverse electron demand hetero-Diels-Alder reactions: synthesis of dihydropyran-fused diterpenoids. Org. Biomol. Chem 2014, 12, 8442–8452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Zhang Y; Wu Y; Wu D; Tashiro S; Onodera S; Ikejima T NF-kappab facilitates oridonin-induced apoptosis and autophagy in HT1080 cells through a p53-mediated pathway. Arch. Biochem. Biophys 2009, 489, 25–33. [DOI] [PubMed] [Google Scholar]
- (58).Hsieh TC; Wijeratne EK; Liang JY; Gunatilaka AL; Wu JM Differential control of growth, cell cycle progression, and expression of NF-kappaB in human breast cancer cells MCF-7, MCF-10A, and MDA-MB-231 by ponicidin and oridonin, diterpenoids from the Chinese herb rabdosia rubescens. Biochem. Biophys. Res. Commun 2005, 337, 224–231. [DOI] [PubMed] [Google Scholar]
- (59).Chen S; Gao J; Halicka HD; Huang X; Traganos F; Darzynkiewicz Z The cytostatic and cytotoxic effects of oridonin (rubescenin), a diterpenoid from rabdosia rubescens, on tumor cells of different lineage. Int. J. Oncol 2005, 26, 579–588. [PubMed] [Google Scholar]
- (60).Zhang CL; Wu LJ; Zuo HJ; Tashiro S; Onodera S; Ikejima T Cytochrome c release from oridonin-treated apoptotic A375-S2 cells is dependent on p53 and extracellular signal-regulated kinase activation. J. Pharmacol. Sci 2004, 96, 155–163. [DOI] [PubMed] [Google Scholar]
- (61).Cheng Y; Qiu F; Ye YC; Tashiro S; Onodera S; Ikejima T Oridonin induces G2/M arrest and apoptosis via activating ERK-p53 apoptotic pathway and inhibiting PTK-Ras-Raf-JNK survival pathway in murine fibrosarcoma L929 cells. Arch. Biochem. Biophys 2009, 490, 70–75. [DOI] [PubMed] [Google Scholar]
- (62).Cui Q; Tashiro S; Onodera S; Ikejima T Augmentation of oridonin-induced apoptosis observed with reduced autophagy. J. Pharmacol. Sci 2006, 101, 230–239. [DOI] [PubMed] [Google Scholar]
- (63).Cui Q; Tashiro S; Onodera S; Minami M; Ikejima T Autophagy preceded apoptosis in oridonin-treated human breast cancer MCF-7 cells. Biol. Pharm. Bull 2007, 30, 859–864. [DOI] [PubMed] [Google Scholar]
- (64).Zhang CL; Wu LJ; Tashiro S; Onodera S; Ikejima T Oridonin induces a caspase-independent but mitochondria- and MAPK-dependent cell death in the murine fibrosarcoma cell line L929. Biol. Pharm. Bull 2004, 27, 1527–1531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.