

Abstract
Developmental transcription programs are epigenetically regulated by multi-protein complexes, including the menin- and MLL-containing trithorax (TrxG) complexes, which promote gene transcription by depositing the H3K4me3 activating mark at target gene promoters. We recently reported that in Ewing sarcoma, MLL1 (lysine methyltransferase 2A, KMT2A) and menin are overexpressed and function as oncogenes. Small molecule inhibition of the menin-MLL interaction leads to loss of menin and MLL1 protein expression and to inhibition of growth and tumorigenicity. Here, we have investigated the mechanistic basis of menin-MLL-mediated oncogenic activity in Ewing sarcoma. Bromouridine sequencing (Bru-seq) was performed to identify changes in nascent gene transcription in Ewing sarcoma cells, following exposure to the menin-MLL interaction inhibitor MI-503. Menin-MLL inhibition resulted in early and widespread reprogramming of metabolic processes. In particular, the serine biosynthetic pathway (SSP) was the pathway most significantly affected by MI-503 treatment. Baseline expression of SSP genes and proteins (PHGDH, PSAT1, and PSPH), and metabolic flux through the SSP were confirmed to be high in Ewing sarcoma. In addition, inhibition of PHGDH resulted in reduced cell proliferation, viability and tumor growth in vivo, revealing a key dependency of Ewing sarcoma on the SSP. Loss of function studies validated a mechanistic link between menin and the SSP. Specifically, inhibition of menin resulted in diminished expression of SSP genes, reduced H3K4me3 enrichment at the PHGDH promoter, and complete abrogation of de novo serine and glycine biosynthesis, as demonstrated by metabolic tracing studies with 13C-labeled glucose. These data demonstrate that the SSP is highly active in Ewing sarcoma and that its oncogenic activation is maintained, at least in part, by menin-dependent epigenetic mechanisms involving trithorax complexes.
Keywords: cancer metabolism, PHGDH, epigenetic, EWS-FLI1, Ewing sarcoma
Introduction
Ewing sarcoma, an aggressive tumor of presumed stem cell origin, is the second commonest bone and soft tissue tumor in children and young adults. Despite intensive therapy, prognosis is poor in patients with metastatic and relapsed disease [1–3]. Moreover, patients who survive face debilitating and often life-threatening health consequences as a result of the highly toxic therapy [4–7]. There is an urgent need to identify novel opportunities for therapeutic intervention. Ewing sarcomas are characterized by pathognomonic EWS-ETS family chromosomal translocations, most commonly EWS-FLI1 [3]. EWS-FLI1 promotes oncogenesis through global alteration of chromatin structure, leading to widespread epigenetic reprogramming and disruption of normal stem cell development and differentiation [8–11]. Normal development is orchestrated in part by the reciprocal activities of the multi-protein Polycomb and Trithorax (TrxG) complexes, which regulate gene expression via covalent modifications to chromatin [12]. We recently demonstrated that Ewing sarcomas are critically dependent upon the TrxG histone methyltransferase, MLL1 (lysine methyltransferase 2A, KMT2A) and the scaffolding protein menin for maintenance of their tumorigenic properties [13]. Pharmacologic inhibition of the interaction between menin and MLL1 proteins leads to loss of tumorigenicity, concomitant with down-regulation of oncogenic HOXD genes, as well as loss of menin and MLL1 protein expression [13]. Although posterior HOXD genes are clear downstream mediators of menin-MLL-mediated tumorigenicity, the additional transcriptional targets of menin-MLL in the context of Ewing sarcoma, and their oncogenic effects, are unknown. To begin to address this gap in knowledge, we have used an unbiased bromouridine sequencing (Bru-seq) approach [14] to interrogate transcriptional changes downstream of menin-MLL inhibition, revealing a link between menin and cellular metabolism, in particular serine and glycine biosynthesis.
Cells primarily obtain serine and glycine through import or de novo biosynthesis using the serine synthesis pathway (SSP) [15]. Autonomous production of serine by the SSP frequently enables cancer cells to survive and continue proliferating irrespective of the availability of exogenous serine [16,17]. De novo serine synthesis diverts the glycolytic intermediate 3-phosphoglycerate from the glycolysis pathway, and occurs in three sequential enzymatic steps, catalyzed by phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase (PSAT1), and phosphoserine phosphatase (PSPH) [15]. Serine can be further metabolized to glycine via serine hydroxymethyltransferases (SHMT1 or SHMT2) [15]. Serine and glycine serve as major sources of metabolic intermediates that are required for synthesis of nucleotides, phospholipids and ATP, as well for maintenance of redox homeostasis and regulation of DNA and histone methylation [15,18,19]. Activation of the SSP in cancer has been linked to genomic amplification of the PHGDH locus [16,20], as well as to transcriptional and epigenetic [17,21–25] mechanisms. PHGDH is not amplified in Ewing sarcoma [26–28], implicating alternative mechanisms in maintaining oncogenic activation of the SSP. Notably, it was recently reported that EWS-FLI1 itself modulates SSP gene expression [29]. In the current work we describe a novel role for the menin-MLL complex in epigenetic regulation of the SSP. Importantly, significant efforts are underway to develop inhibitors of this epigenetic regulatory complex [30]. Likewise, there is tremendous interest in targeting de novo serine biosynthesis in the context of cancer [31,32]. Thus, these findings provide new insights into the oncogenic functions of the menin-MLL complex, and identify a potential therapeutic vulnerability in Ewing sarcoma.
Materials and Methods
Cell lines
Ewing sarcoma cell lines were kindly provided by Dr. Timothy Triche at Children’s Hospital Los Angeles (CHLA), USA, 2004; Dr. Heinrich Kovar (CCRI, St. Anna Kinderkrebsforschung, Vienna, Austria) 2010; and the Children’s Oncology Group (COG) cell bank (cogcell.org), 2012. Identities were confirmed by short tandem repeat (STR) profiling. ASC52telo human mesenchymal stem cells were purchased from ATCC. Cell lines were cultured according to COG- or ATCC-recommended protocols. All cell lines were routinely tested for mycoplasma contamination using the e-Myco™ plus Mycoplasma PCR Detection Kit (Bulldog Bio, Portsmouth, NH, USA).
Bru-Seq and bioinformatic analysis
Cell line samples were prepared as described previously [14] and sequenced using Illumina HiSeq 2000, 50-bp single-end reads through the University of Michigan Sequencing Core. Reads were processed, mapped, and quantified per gene using the Bru-Seq data analysis pipeline [14]. Differential expression (DE) was calculated using DESeq v1.24 for (i) individual cell line MI503 vs. MI-NC treatments and (ii) combined cell line MI503 vs. MI-NC treatment [33]. Bru-Seq gene identifiers were mapped to HGNC gene symbols, Entrez gene identifiers, and then to Gene Ontology (GO) and KEGG pathway gene sets. GO biological process gene sets were downloaded on 30 Sep 2016 using the GO.db Bioconductor package [34–36]. KEGG gene sets were downloaded using the KEGG web API on the same date [37]. Gene set enrichment was calculated using a Fisher’s exact test to quantify the overlap of DE genes with GO and KEGG gene sets. Gene sets with ≤3 or ≥500 total genes were excluded. P-values were adjusted for multiple testing using FDR for each gene set source and only gene sets with a qValue ≤ 0.1 were considered to be significantly enriched. For further detail see supplementary material, Supplementary materials and methods.
Metabolomic analysis
Cell treated with MI-503 or vehicle were cultured in DMEM containing 10% dialyzed FBS with 25 mM uniformly labeled 13C- or 12C-glucose for 24 hours in biological triplicates. Cells were lysed with dry-ice-cold 80% methanol and then cleared by centrifugation. Metabolite fractions for each sample were normalized based on protein concentrations of a parallel sample, and then dried in a SpeedVac concentrator. Dried metabolite pellets from cells were resuspended in 35 μl 50:50 methanol:water mixture for LC–MS analysis. See supplementary material, Supplementary materials and methods for details.
Tissue microarray and immunohistochemistry
Tissue microarray, immunohistochemistry, and scoring were performed as outlined previously [13]. PHGDH antibody (3PGDH 6B2; Santa Cruz Biotechnology, sc100–317) was used at 1:100 dilution.
Gene knockdown studies
Lentivirus production and lentiviral gene transfer for knockdown of menin (MEN1), MLL1 (KMT2A), and PHGDH were conducted as previously described [38]. For PHGDH knockdown, cells were transduced with lentivirus for 48 hours, followed by selection in medium containing 1.5 µg/ml puromycin for 48 hours. Assessment of knockdown by qRT-PCR and western blot, and trypan blue assays were conducted at this time point. See supplementary material, Supplementary materials and methods for shRNA sequences.
In vitro proliferation assays
Cell proliferation was assessed using trypan blue exclusion as outlined previously [13], or using the Cell Titer Glo assay (Promega). Detailed methods can be found in the supplementary materials and methods.
Chemical synthesis of menin-MLL inhibitors and treatments
MI-503 and MI-NC were prepared using the synthetic procedures reported previously [39]. Cells were treated for 72 hours or 6 days with 3 µM MI-503 or DMSO vehicle. Fresh compound was added on day 3 of treatment.
Chromatin immunoprecipitation and PCR
Chromatin immunoprecipitation-quantitative PCR (ChIP-qPCR) and quantitative real-time PCR (qRT-PCR) were performed as outlined previously [13]. Antibodies and primer sequences are listed in the supplementary materials and methods.
Western Blot
Western blot was performed according to established protocols with the following primary antibodies: anti-PHGDH 1:1000 (3PGDH 6B2; Santa Cruz Biotechnology, sc100–317); anti-PSAT1 1:1000 (Thermo Fisher PA5–22124); anti-GAPDH 1:1000 (Cell Signaling 14C10); monoclonal anti-GAPDH 1:1000 (Thermo Fisher AM4300); anti-menin 1:1000 (Bethyl A300–105A). Membranes were probed with the appropriate secondary antibodies from LiCor Biotechnology (Lincoln, NE) at 1:10,000 dilution and imaged with a LiCor Odyssey infrared imager.
Statistics
All data in barplots are presented as the mean ± SEM, and statistical significance was determined using unpaired, 2-tailed Student’s t-tests or ANOVA. P values of less than 0.05 were considered significant. Analyses were conducted using Graphpad Prism software and the R statistical programming language (https://www.R-project.org/).
Tumor Xenograft Studies
Animal studies were performed according to approved protocols reviewed by the University of Michigan Institutional Animal Care and Use Committee (PRO#00006703). A4573 and A673 cells were transduced with shGFP or shPHGDH#2 lentiviral constructs and used for xenograft studies 48 hours after puromycin selection. 500,000 viable cells were suspended in 50% (v:v) PBS:Matrigel® (Corning, Tewksbury, MA) and injected into flanks of male NCR nude mice, 8–10 weeks of age. Tumor growth was monitored 2–3 times weekly.
Ethics Statement
This investigation has been conducted in accordance with the Declaration of Helsinki and has been approved by the University of Michigan Institutional Review Board (HUM00067293).
Results
Treatment with MI-503 leads to widespread changes in metabolic programs
To investigate the mechanistic basis of menin-MLL-mediated oncogenic activity in Ewing sarcoma, we performed Bru-seq analysis [14] in three Ewing sarcoma cell lines to evaluate changes in newly transcribed mRNAs after 72 hours’ treatment with MI-503, or the control compound, MI-NC (both at 3 µM, as per previous studies [13]; supplementary material, Figure S1). A 72 hour time-point was chosen, given that adequate modulation of menin-MLL function and expression are observed at three days, with evidence of a growth inhibitory phenotype but without overwhelming cell loss. We calculated average n-fold changes for genes commonly expressed across all three cell lines. Using an absolute average ≥2-fold change, 95 genes were identified as differentially expressed (78 down-regulated and 17 up-regulated, supplementary material, Table S1). The differentially expressed genes showed significant enrichment with 38 KEGG and GO categories and demonstrated striking, widespread changes in metabolic programs (Figure 1A). Notably, although MI-503 treatment affected multiple metabolic programs, serine biosynthesis was most significantly down-regulated (Figure 1A). The SSP converts the glycolytic intermediate, 3-phosphoglycerate, to serine via three enzymatic steps (Figure 1B). Each enzyme in the SSP, as well as the downstream mitochondrial folate pathway enzymes serine hydroxymethyltransferase (SHMT2) and methylenetetrahydrofolate dehydrogenase (MTHFD2), was down regulated (Figure 1B). Importantly, down regulation of SSP genes was not a non-specific effect of impaired proliferation, as serum starvation and exposure to the DNA methyltransferase inhibitor decitabine, caused similar reductions in proliferation but had no reproducible or coordinated impact on SSP gene expression (supplementary material, Figure S2A-D). Based on these data, we investigated the role of the SSP in Ewing sarcoma pathogenesis, and the mechanistic basis for menin-dependent pathway activation.
Figure 1.
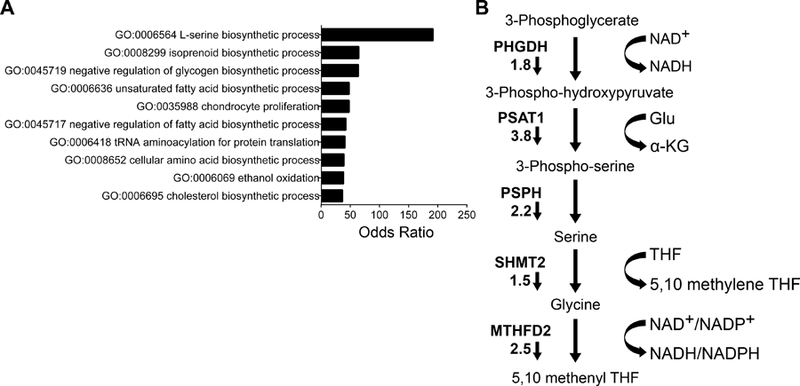
Bromouridine sequencing (Bru-seq) of MI-503 treated Ewing sarcoma cells identifies the SSP as a key oncogenic target. (A) Gene ontology analysis, showing the top ten differentially regulated pathways after MI-503 treatment. (B) Diagram of the de novo serine biosynthesis and folate pathways, showing fold changes in expression after MI-503 treatment. Small arrows indicate that fold changes in gene expression were down after MI-503 treatment.
The SSP is heterogeneously expressed in Ewing sarcoma tumors
We first investigated the baseline expression of the SSP in Ewing sarcoma cell lines and tumors. In the Cancer Cell Line Encyclopedia, PHGDH expression was higher in Ewing sarcoma than in any other cancer, an observation recently reported by others [29]. We measured expression of SSP enzymes in Ewing sarcoma cell lines using qRT-PCR and western blot, and observed robust protein expression of pathway components in all cell lines compared to mesenchymal stem cells, putative cells of origin for Ewing sarcoma (MSCs, supplementary material, Figure S3A-C). Interestingly, PHGDH transcript and protein levels were markedly higher in Ewing sarcoma cells than A549 lung carcinoma cells, which were recently reported to exhibit high PHGDH expression and to be dependent upon the SSP for their tumorigenic properties (supplementary material, Figure S3A-B and [17]). We next evaluated SSP expression in tumors. In three publicly available gene expression datasets of Ewing sarcoma tumors [40–42] expression of PHGDH, PSAT1, and PSPH was highly variable across tumor samples (Figure 2A). Notably, however, we observed strong correlations among expression of SSP genes in all three datasets, suggesting that genes in the pathway are coordinately regulated (Figure 2B). Immunostaining for PHGDH protein in 28 tumors revealed a similar pattern of heterogeneity. Seven tumors (25%) showed positive staining for PHGDH and, among positive tumors, significant intra-tumor heterogeneity was observed (Figure 2C, validation of antibody in supplementary material, Figure S4). These data demonstrate that SSP genes and proteins are highly expressed by Ewing sarcoma cell lines and that high-level expression is also evident in sub-populations of tumor cells in vivo.
Figure 2.
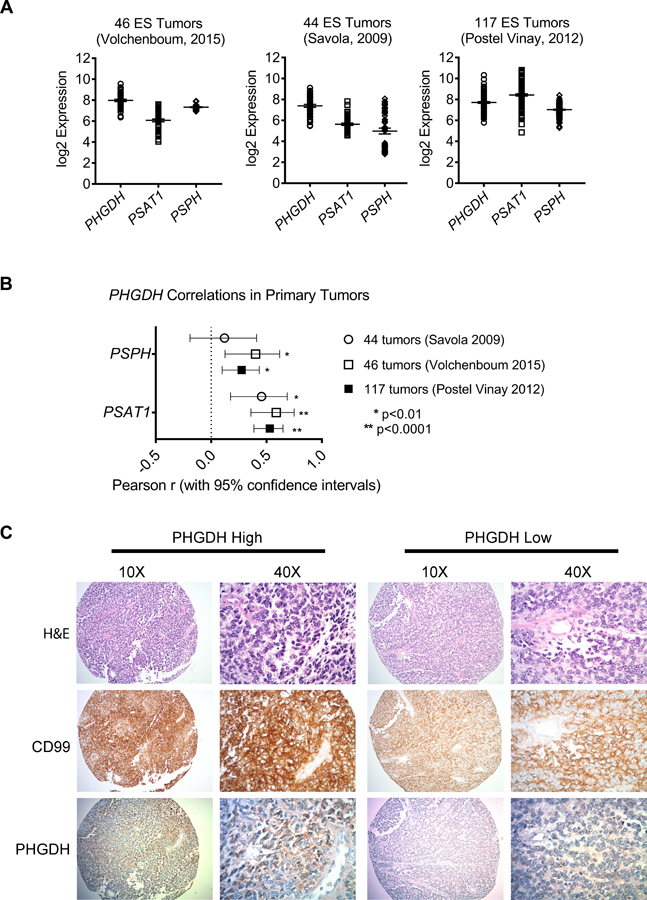
SSP expression in Ewing sarcoma primary tumors. (A, B) Data from three publicly available gene expression profiling datasets of Ewing sarcoma tumors, showing transcript expression of SSP enzymes (A) and the correlations between PHGDH and PSAT1 or PSPH (B). (C) Representative images depicting high and low immunohistochemical staining for PHGDH in a cohort of Ewing sarcoma tumors. Hematoxylin and eosin (H&E) and CD99 staining were performed in adjacent sections.
Ewing sarcoma cells depend on PHGDH for proliferation, survival and tumor growth
We next evaluated the functional importance of the SSP in Ewing sarcoma cells. De novo serine biosynthesis confers upon cells the ability to proliferate autonomously in the absence of serine and glycine [15]. Consistent with this, we found that Ewing sarcoma cells survive and proliferate in serine- and glycine-free conditions (Figure 3A). Interestingly, despite uniformly high expression of the SSP enzymes, the cell lines exhibited variable degrees of sensitivity to serine and glycine withdrawal (Figure 3A). This did not correlate with cellular proliferation rate, and the reasons for it remain to be determined. To evaluate the effect of pathway inhibition on cell survival and expansion, we knocked down expression of PHGDH, the rate-limiting step in serine biosynthesis, in four Ewing sarcoma cell lines using puromycin-selectable lentiviral shRNAs (Figure 3B). PHGDH knockdown led to cell death, and surviving PHGDH-low cells showed a marked inhibition of proliferation, despite abundant serine and glycine in the media (Figure 3C and supplementary material, Figure S5A). Moreover, PHGDH knockdown was deleterious to cells irrespective of their ability to proliferate in serine- and glycine-free conditions. As a complementary approach, we treated cells with a recently developed pharmacological inhibitor of PHGDH, CBR5884 [32]. Similar to PHGDH knockdown, pharmacological inhibition of PHGDH led to diminished viability and proliferation, even in serine- and glycine-replete conditions (supplementary material, Figure S5B-C). To evaluate the role of PHGDH in tumorigenicity, we plated equal numbers of viable control and PHGDH knockdown cells in soft agar and measured anchorage-independent growth. Knockdown of PHGDH led to reduced numbers of colonies (supplementary Figure S5D). To assess tumorigenicity in vivo, cells were injected subcutaneously into nude mice. Although tumors formed in all mice, there was a trend toward delayed engraftment and a reproducible slowing of growth in tumors established with PHGDH knockdown cells (Figure 3D). Thus, high expression of PHGDH supports a more proliferative, tumorigenic phenotype. We next assessed whether the observed effects of PHGDH knockdown on cell proliferation might be due to inhibition of 1-carbon metabolism, downstream of serine biosynthesis. To this end, we used lentiviral shRNA to knock down expression of SHMT2, which is the primary mediator of glycine production and the 1-carbon units used to make purines de novo in proliferating cells [43,44] (supplementary material, Figure S6A). SHMT2 knock-down had no appreciable effect on proliferation or cell death (supplementary material, Figure S6B-D), suggesting that compensation for SHMT2 loss by cytoplasmic SHMT1 [45] may provide the 1-carbon units and glycine needed to meet cellular demands when SHMT2 is inhibited. Additional studies are needed to determine the role of SHMT1 in Ewing sarcoma.
Figure 3.
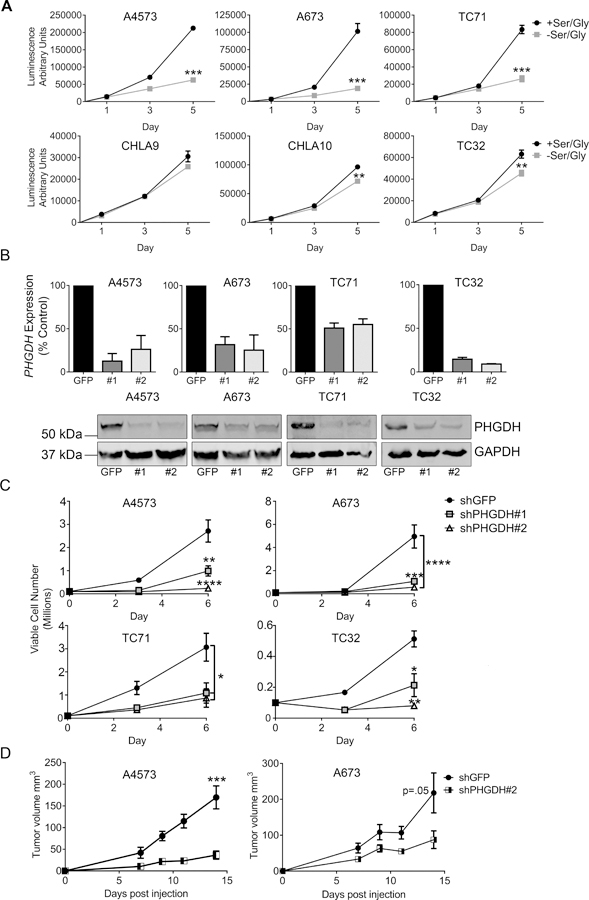
Ewing sarcoma cells depend on PHGDH for proliferation and survival. (A) Proliferation data in a panel of Ewing sarcoma cell lines, showing variable sensitivity to serine and glycine deprivation. (B) qRT-PCR data (top) and western blot (bottom) showing knockdown of PHGDH with two different shRNA constructs. (C) Proliferation is decreased following PHGDH knockdown. n=3 independent experiments. (D) Growth rates of subcutaneous tumors formed from A4573 and A673 cells harboring PHGDH knockdown or shGFP control. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 by two-tailed, unpaired t-test.
Menin inhibition leads to reduced SSP expression
We next focused on determining the mechanistic basis for menin-MLL-dependent maintenance of the SSP. We first confirmed that 72-hour treatment with MI-503 significantly down-regulated expression of PHGDH, PSAT1 and PSPH (Figure 4A). Likewise, western blot analysis demonstrated a corresponding loss of protein expression within 3 days (A4573) or 6 days (A673 and TC71) (Figure 4B-C). Given that MI-503 leads to loss of both menin and MLL1 protein expression in Ewing sarcoma [13], we next investigated the specific role of each of these proteins in SSP gene regulation. Knock-down of menin consistently led to down-regulation of the SSP enzymes in all cell lines tested (Figure 5A-B). In contrast, the impact of shRNA-mediated knockdown of MLL1 on SSP gene expression was inconsistent (Figure 5C). Finally, menin-MLL complexes function to epigenetically regulate gene expression, in large part, by promoting MLL-dependent trimethylation of H3K4 at target gene promoters [12]. Thus, we performed H3K4me3 ChIP on Ewing sarcoma cells before and after exposure to MI-503 and assessed levels of enrichment in a regulatory region of the PHGDH locus, defined based on H3K4me3 enrichment in published Ewing sarcoma cell data, and a PHGDH promoter-associated CpG island that was recently described.[46][11] H3K4me3 enrichment at the PHGDH promoter was reproducibly diminished after MI-503 treatment (Figure 5D). Together these data demonstrate that menin maintains high-level expression of SSP genes in Ewing sarcoma cells and that this is mediated, at least in part, by its function in epigenetic activation of the PHGDH locus.
Figure 4.
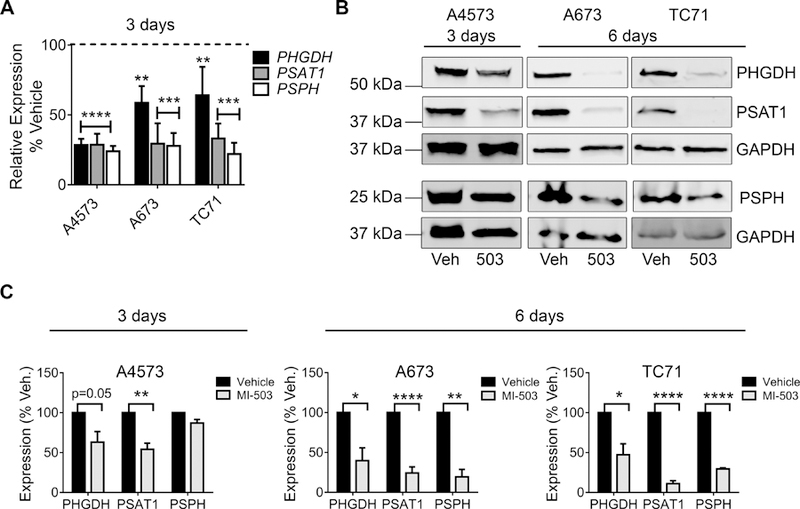
MI-503 inhibits expression of SSP enzymes. (A) qRT-PCR data showing down-regulation of SSP genes after 72-hour treatment with 3 µM MI-503 compared to DMSO vehicle. (B, C) Representative western blots (B) and densitometry (C) depicting down-regulation of SSP enzyme protein expression with MI-503 treatment for 72 hours or 6 days. n=3 independent experiments. * p < 0.05, ** p < 0.01, p < 0.001, **** p < 0.0001 by two-tailed, unpaired t-test.
Figure 5.
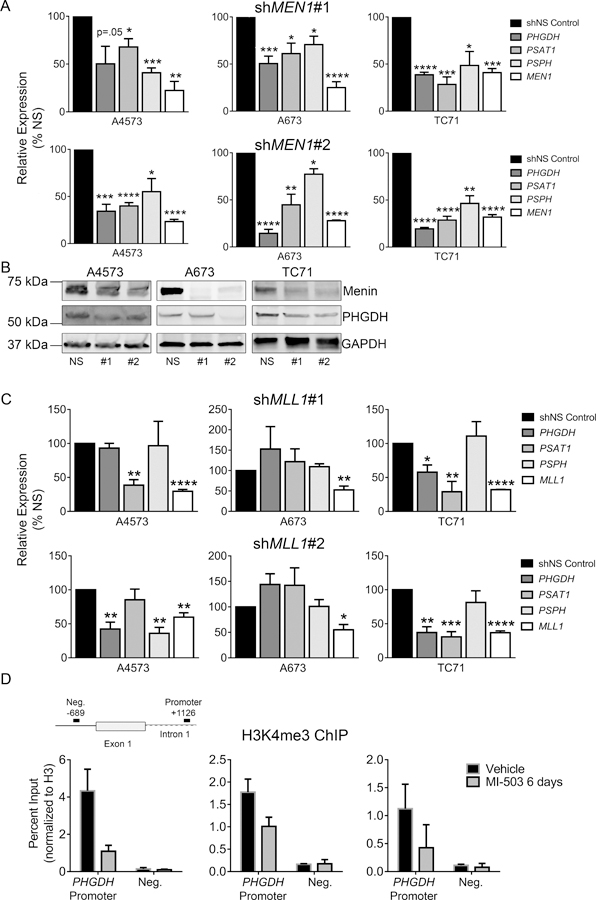
Regulation of SSP gene expression by menin. (A) qRT-PCR data showing down-regulation of SSP gene expression after menin (MEN1) knockdown with two different shRNA constructs. (B) Western blot showing that menin knockdown leads to down-regulation of PHGDH. (C) qRT-PCR data showing no consistent changes in SSP enzyme gene expression after MLL1 knockdown with two different shRNA constructs. n=3 independent experiments. (D) ChIP data showing reduced H3K4me3 enrichment at the PHGDH promoter after MI-503 treatment. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 by two-tailed, unpaired t-test. All statistical comparisons are made relative to control (shNS).
Inhibition of menin leads to reduced de novo serine and glycine biosynthesis
Having established the pattern of SSP enzyme expression and its dependence on menin, we next investigated SSP activity in Ewing cells. To achieve this, we first measured the baseline level of de novo serine and glycine biosynthesis in Ewing sarcoma cells using liquid chromatography-mass spectrometry-based metabolic tracing. This approach permits the tracing of glucose uniformly labeled with the non-radioactive carbon-13 (13C) isotope into serine and glycine (Figure 6A). Here, M represents the parent mass of the compound where each labeled carbon is +1. Notably, pathway activity was uniformly high in all cell lines, as indicated by robust incorporation of labeled carbon into serine and glycine (Figure 6B). We next assessed the effect of menin-MLL inhibition on SSP activity. Consistent with the observed reduction in expression of SSP enzymes, MI-503 treatment led to reduced incorporation of 13C-labeled glucose (M+6) into serine (M+3) and glycine (M+2) (Figure 6C). Likewise, despite potential equilibration with unlabeled serine and glycine in the growth medium [17], total cellular pools of serine and glycine were also reduced (Figure 6D). Importantly, the effects of MI-503 on the SSP were not secondary to reduced glucose uptake. Post-treatment levels of glucose in the culture media did not differ between vehicle and MI-503 treated conditions (supplementary material, Figure S7A). Moreover, levels of glycolytic intermediates in A4573 cells increased after MI-503 treatment (supplementary material, Figure S7B). The serine and glycine generated by the SSP contribute to the production of numerous downstream metabolites that play critical roles in cell growth and proliferation, including purine nucleotides [47]. Accordingly, we observed a reduction in de novo serine/glycine-derived ATP and ADP with MI-503 treatment (Figure 6E-F). In A673 cells, total level of these nucleotides also decreased (supplementary material, Figure S7C). Taken together, these data demonstrate that menin inhibition impairs de novo generation of serine/glycine and downstream metabolites in Ewing sarcoma cells.
Figure 6.
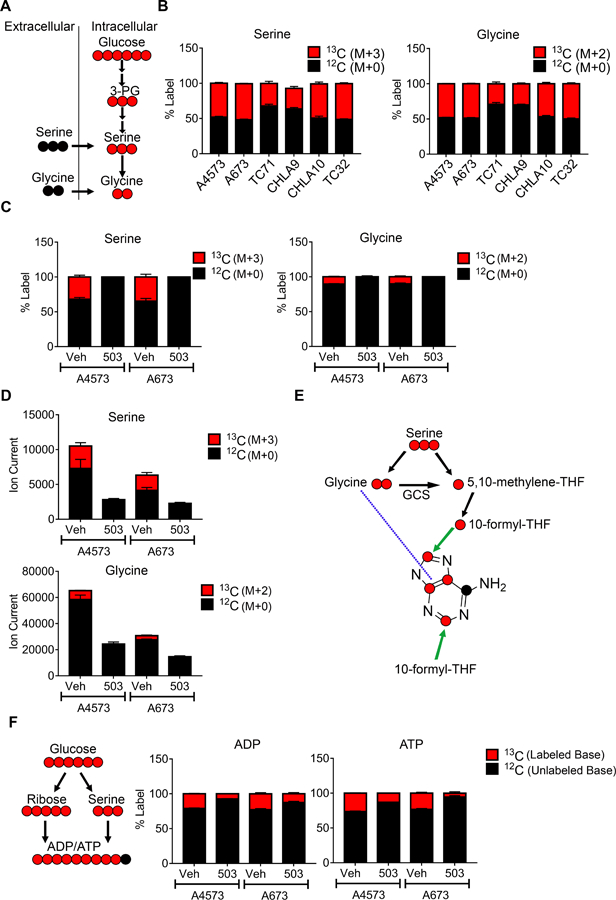
Effect of menin inhibition on de novo synthesis of serine and glycine. (A) Schematic depicting tracing of serine (M+3) and glycine (M+2) (both red) from 13C-labeled glucose via de novo synthesis. Unlabeled serine and glycine, in black, can be imported from the extracellular space. (B) Baseline fractional incorporation of 13C-labeled glucose into serine (M+3) and glycine (M+2) in a panel of Ewing sarcoma cell lines. (C) Fractional incorporation of 13C-labeled serine and glycine in vehicle- and MI-503 treated cells. (D) Total cellular pools of serine and glycine after MI-503 treatment. (E) Chemical structure of the adenine base, showing potential locations of glucose-derived, 13C-labeled serine and glycine carbon. GCS: glycine cleavage system. (F) Schematic and fractional incorporation of 13C-labeled glucose into ADP and ATP.
Discussion
The tumor-promoting functions of the menin-MLL complex are diverse and context-specific; however, a role for this complex in regulation of cancer metabolism has not yet been established. In the current work, we have discovered that, in Ewing sarcoma, maintenance of oncogenic de novo serine and glycine biosynthesis through the SSP depends on menin. Accumulating evidence from multiple cancers has identified deregulation of the SSP as a critical mechanism by which tumors sustain their proliferation, survival, and metastatic fitness [15,47,48]. High expression of SSP genes is associated with refractory disease, chemoresistance, and poor prognosis in a number of human cancers [17,49–51] and a recent paper also linked high SSP gene expression to poor outcomes in Ewing sarcoma [29]. Thus, elucidation of the mechanisms by which the SSP can be hyperactivated is of profound importance to cancer pathogenesis broadly.
Oncogenic activation of the SSP is often a consequence of genomic amplification of the PHGDH locus [16,20] and, in tumors that do not harbor this genomic lesion, transcriptional [17,21–23] as well as epigenetic mechanisms [24,25] have been identified. To date, epigenetic mechanisms linked to SSP activation include promoter activation by the histone methyltransferase G9a [25] and the histone demethylase KDM4C [24]. To our knowledge, links between SSP genes and MLL-containing Compass-like complexes, including the menin-containing trithorax complexes, have not been described. Our data show that pharmacologic inhibition of the menin-MLL interaction leads to loss of SSP gene expression, diminished H3K4me3 enrichment at the PHGDH promoter, and abrogation of de novo SSP-derived serine and glycine. Interestingly, loss of SSP gene expression was also observed in MI-503-treated MLL-rearranged leukemia cells [30], demonstrating that menin-MLL-dependent activation of the SSP extends beyond Ewing sarcoma. In addition, genetic knockdown of menin in Ewing sarcoma reproducibly resulted in loss of SSP gene expression, while loss of MLL1 did not. Thus, it is likely that menin’s impact on trithorax-dependent activation of the PHGDH promoter is due to its interaction with MLL2 rather than MLL1, given that MI-503 effectively inhibits both interactions [39]. Alternatively, menin-dependent regulation of SSP gene expression may be mediated by non-trithorax-dependent mechanisms that involve other menin protein-binding partners. Menin has numerous MLL complex-independent functions [52–56]. Of particular relevance to this study, menin was shown to cooperate with cMYC in the regulation of metabolism-associated genes, including PSAT1, in an MLL-independent fashion [53]. Moreover, the EWS-FLI1 oncoprotein itself contributes to maintenance of SSP gene expression in Ewing sarcoma [29]. The extent to which MYC, EWS-FLI1, or other transcriptional regulators cooperate with menin to hijack and maintain oncogenic activation of the SSP in Ewing sarcoma remains to be determined.
It is noteworthy that, despite robust and relatively homogenous over-expression of SSP genes in Ewing sarcoma cell lines, we observed a high degree of inter- and intra-tumor heterogeneity in SSP expression in patient tumors. PHGDH-high tumor cells were detected in only a quarter of biopsies by immunohistochemistry and high levels of PHGDH were uniformly correlated with high levels of PSAT1 and PSPH in three independent expression profiling studies. This inter-tumor heterogeneity and prevalence of high PHGDH-expressing cells bears striking similarity to what was recently observed in lung adenocarcinomas, where PHGDH expression was also heterogeneous and correlated with cell proliferation and increased DNA replication and repair [50]. We hypothesize that SSP activation is a feature of the most aggressive cells, which are selected for during establishment of cell lines, a hypothesis that is supported by the finding that PHGDH expression was higher among high-risk patients [29,42,50]. The importance of tumor heterogeneity to Ewing sarcoma metastasis and aggressiveness has recently become evident [57–59] and the current findings implicate heterogeneity of SSP activation as a potentially important player in mediating aggressive cell phenotypes. Further studies are now required to elucidate how and why the SSP contributes to maintenance and progression of Ewing sarcoma. Finally, the observation that Ewing sarcoma cells exhibit variable sensitivity to serine and glycine withdrawal, despite uniformly robust activity of the pathway, is intriguing. This finding contrasts with previous work showing that the ability of cancer cells to proliferate in the absence of serine and glycine is correlated with expression of PHGDH and flux through the SSP [16]. However, additional studies have demonstrated that cells continue to consume high levels of extracellular serine despite high SSP activity [60]. These data suggest that Ewing sarcoma cells may rely simultaneously on de novo serine biosynthesis, as well as import of serine from the extracellular environment. In support of this, the presence of abundant exogenous serine in culture media was unable to rescue cells from PHGDH loss of function. Inhibition of PHGDH was recently shown to impair the incorporation of one-carbon units from endogenous and exogenous serine into nucleotides [31]. It is plausible, therefore, that Ewing sarcoma cells similarly utilize the SSP to coordinate the fate of endogenous and exogenous one-carbon units. Another possibility is that the SSP may be used to generate components other than serine and glycine that are critical for survival. In addition to serine, the SSP also yields equimolar quantities of α-ketoglutarate (α-KG), which can promote anaplerosis into the TCA cycle to support anabolic metabolism, cell proliferation and epigenetic reprogramming [16,61,62]. Metabolic tracing studies in MI-503 treated cells demonstrated that Ewing sarcoma cells utilize the SSP for the generation of purine nucleotides; however, the precise functions of these metabolites in supporting the oncogenic phenotype are currently unknown. The mechanism(s) underlying the oncogenic function of the SSP in Ewing sarcoma remain an active area of investigation. In summary, identification of novel and actionable biological targets is urgently needed in Ewing sarcoma, particularly for patients with aggressive disease. Here, we show that targeting the menin-MLL complex in Ewing sarcoma leads to loss of oncogenic serine biosynthesis, highlighting a novel role for menin in cancer. Given the critical dependence of Ewing sarcomas cells on the SSP, and the efforts underway to develop inhibitors of the menin-MLL complex and the serine biosynthetic pathway in cancer, our findings are likely to have important biological and translational implications.
Supplementary Material
Table S1. List of changes in BruSeq-measured gene expression on MI-503 treatment
Acknowledgements
We would like to thank the members of the Lawlor and Lyssiotis labs for their intellectual input, as well as Michelle Paulsen for preparation of the Bru-seq samples. We would also like to acknowledge the University of Michigan Comprehensive Cancer Center Vector and Sequencing Cores. We gratefully acknowledge the COG Cell Bank for cell lines, as well as Drs. Lewis Cantley and Edouard Mullarky for the PHGDH shRNAs. Support for this work was provided by the following sources: SARC Sarcoma SPORE 1U54-CA168512, Hyundai Hope on Wheels and Russell G. Adderley Endowment (ERL); 1R01-CA160467, 1R01-CA-200660 and Leukemia and Lymphoma Society Scholar grant 1215-14 (JG). C.A.L. was supported by a Dale F. Frey Award for Breakthrough Scientists from the Damon Runyon Cancer Research Foundation (DFS-09-14); Junior Scholar Award from The V Foundation for Cancer Research (V2016-009); Kimmel Scholar Award from the Sidney Kimmel Foundation for Cancer Research (SKF-16-005); and a 2017 AACR NextGen Grant for Transformative Cancer Research (17-20-01-LYSS). Metabolomics studies were supported by NIH grant DK097153. Shared resources from the UMCCC Core Grant (P30 CA046592) are acknowledged: bioinformatics, tissue and molecular pathology. Additional funding was provided by: Hartwell Foundation Postdoctoral Fellowship (LKS), Charles Woodson Research Fund (ERL), Alex’s Lemonade Stand Pediatric Oncology Student Training Program (AZ), and Cancer Biology training grant T32CA009676 and Michigan Institute for Translational & Clinical Research Postdoctoral Translational Scholars Program (UL1TR000433) (CJH).
Footnotes
Conflict of interest statement
Drs. Grembecka and Cierpicki receive research support from Kura Oncology, Inc. They have also an equity ownership in the company.
References:
- 1.Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol 2010; 11: 184–192. [DOI] [PubMed] [Google Scholar]
- 2.Arnaldez FI, Helman LJ. New strategies in ewing sarcoma: lost in translation? Clin Cancer Res 2014; 20: 3050–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawlor ER, Sorensen PH. Twenty Years on: What Do We Really Know about Ewing Sarcoma and What Is the Path Forward? Crit Rev Oncog 2015; 20: 155–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ginsberg JP, Goodman P, Leisenring W, et al. Long-term survivors of childhood Ewing sarcoma: report from the childhood cancer survivor study. J Natl Cancer Inst 2010; 102: 1272–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armstrong GT, Kawashima T, Leisenring W, et al. Aging and risk of severe, disabling, life-threatening, and fatal events in the childhood cancer survivor study. J Clin Oncol 2014; 32: 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Youn P, Milano MT, Constine LS, et al. Long-term cause-specific mortality in survivors of adolescent and young adult bone and soft tissue sarcoma: a population-based study of 28,844 patients. Cancer 2014; 120: 2334–2342. [DOI] [PubMed] [Google Scholar]
- 7.Teepen JC, van Leeuwen FE, Tissing WJ, et al. Long-Term Risk of Subsequent Malignant Neoplasms After Treatment of Childhood Cancer in the DCOG LATER Study Cohort: Role of Chemotherapy. J Clin Oncol 2017; 35: 2288–2298. [DOI] [PubMed] [Google Scholar]
- 8.Boulay G, Sandoval GJ, Riggi N, et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017. [DOI] [PMC free article] [PubMed]
- 9.Riggi N, Knoechel B, Gillespie SM, et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014; 26: 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riggi N, Suva ML, Suva D, et al. EWS-FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res 2008; 68: 2176–2185. [DOI] [PubMed] [Google Scholar]
- 11.Tomazou EM, Sheffield NC, Schmidl C, et al. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep 2015; 10: 1082–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuettengruber B, Bourbon HM, Di Croce L, et al. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017; 171: 34–57. [DOI] [PubMed] [Google Scholar]
- 13.Svoboda LK, Bailey N, Van Noord RA, et al. Tumorigenicity of Ewing sarcoma is critically dependent on the trithorax proteins MLL1 and menin. Oncotarget 2017; 8: 458–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paulsen MT, Veloso A, Prasad J, et al. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods 2014; 67: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer 2016; 16: 650–662. [DOI] [PubMed] [Google Scholar]
- 16.Possemato R, Marks KM, Shaul YD, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011; 476: 346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeNicola GM, Chen PH, Mullarky E, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 2015; 47: 1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maddocks OD, Labuschagne CF, Adams PD, et al. Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol Cell 2016; 61: 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong CC, Qian Y, Yu J. Interplay between epigenetics and metabolism in oncogenesis: mechanisms and therapeutic approaches. Oncogene 2017; 36: 3359–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Locasale JW, Grassian AR, Melman T, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 2011; 43: 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu M, Xia Y, Ding J, et al. Transcriptional Profiling Reveals a Common Metabolic Program in High-Risk Human Neuroblastoma and Mouse Neuroblastoma Sphere-Forming Cells. Cell Rep 2016; 17: 609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bao XR, Ong SE, Goldberger O, et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife 2016; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye J, Mancuso A, Tong X, et al. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci U S A 2012; 109: 6904–6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao E, Ding J, Xia Y, et al. KDM4C and ATF4 Cooperate in Transcriptional Control of Amino Acid Metabolism. Cell Rep 2016; 14: 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding J, Li T, Wang X, et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab 2013; 18: 896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tirode F, Surdez D, Ma X, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 2014; 4: 1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 2014; 4: 1326–1341. [DOI] [PubMed] [Google Scholar]
- 28.Brohl AS, Solomon DA, Chang W, et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 2014; 10: e1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanner JM, Bensard C, Wei P, et al. EWS/FLI is a Master Regulator of Metabolic Reprogramming in Ewing Sarcoma. Mol Cancer Res 2017. [DOI] [PMC free article] [PubMed]
- 30.Borkin D, He S, Miao H, et al. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015; 27: 589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pacold ME, Brimacombe KR, Chan SH, et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol 2016; 12: 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mullarky E, Lucki NC, Beheshti Zavareh R, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci U S A 2016; 113: 1778–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010; 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gene Ontology C. Gene Ontology Consortium: going forward. Nucleic Acids Res 2015; 43: D1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carlson MR, Pages H, Arora S, et al. Genomic Annotation Resources in R/Bioconductor. Methods Mol Biol 2016; 1418: 67–90. [DOI] [PubMed] [Google Scholar]
- 37.Kanehisa M, Sato Y, Kawashima M, et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016; 44: D457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.von Levetzow C, Jiang X, Gwye Y, et al. Modeling initiation of Ewing sarcoma in human neural crest cells. PLoS One 2011; 6: e19305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grembecka J, He S, Shi A, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol 2012; 8: 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Postel-Vinay S, Veron AS, Tirode F, et al. Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet 2012; 44: 323–327. [DOI] [PubMed] [Google Scholar]
- 41.Volchenboum SL, Andrade J, Huang L, et al. Gene Expression Profiling of Ewing Sarcoma Tumors Reveals the Prognostic Importance of Tumor-Stromal Interactions: A Report from the Children’s Oncology Group. J Pathol Clin Res 2015; 1: 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Savola S, Klami A, Tripathi A, et al. Combined use of expression and CGH arrays pinpoints novel candidate genes in Ewing sarcoma family of tumors. BMC Cancer 2009; 9: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jain M, Nilsson R, Sharma S, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012; 336: 1040–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Narkewicz MR, Sauls SD, Tjoa SS, et al. Evidence for intracellular partitioning of serine and glycine metabolism in Chinese hamster ovary cells. Biochem J 1996; 313 (Pt 3): 991–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ducker GS, Chen L, Morscher RJ, et al. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab 2016; 24: 640–641. [DOI] [PubMed] [Google Scholar]
- 46.Truong V, Huang S, Dennis J, et al. Blood triglyceride levels are associated with DNA methylation at the serine metabolism gene PHGDH. Sci Rep 2017; 7: 11207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mattaini KR, Sullivan MR, Vander Heiden MG. The importance of serine metabolism in cancer. J Cell Biol 2016; 214: 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amelio I, Cutruzzola F, Antonov A, et al. Serine and glycine metabolism in cancer. Trends Biochem Sci 2014; 39: 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ross KC, Andrews AJ, Marion CD, et al. Identification of the Serine Biosynthesis Pathway as a Critical Component of BRAF Inhibitor Resistance of Melanoma, Pancreatic, and Non-Small Cell Lung Cancer Cells. Mol Cancer Ther 2017; 16: 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang B, Zheng A, Hydbring P, et al. PHGDH Defines a Metabolic Subtype in Lung Adenocarcinomas with Poor Prognosis. Cell Rep 2017; 19: 2289–2303. [DOI] [PubMed] [Google Scholar]
- 51.Pollari S, Kakonen SM, Edgren H, et al. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res Treat 2011; 125: 421–430. [DOI] [PubMed] [Google Scholar]
- 52.Li BE, Gan T, Meyerson M, et al. Distinct pathways regulated by menin and by MLL1 in hematopoietic stem cells and developing B cells. Blood 2013; 122: 2039–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu G, Yuan M, Shen S, et al. Menin enhances c-Myc-mediated transcription to promote cancer progression. Nat Commun 2017; 8: 15278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sowa H, Kaji H, Hendy GN, et al. Menin is required for bone morphogenetic protein 2- and transforming growth factor beta-regulated osteoblastic differentiation through interaction with Smads and Runx2. J Biol Chem 2004; 279: 40267–40275. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y, Ozawa A, Zaman S, et al. The tumor suppressor protein menin inhibits AKT activation by regulating its cellular localization. Cancer Res 2011; 71: 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Getz AM, Visser F, Bell EM, et al. Two proteolytic fragments of menin coordinate the nuclear transcription and postsynaptic clustering of neurotransmitter receptors during synaptogenesis between Lymnaea neurons. Sci Rep 2016; 6: 31779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pedersen EA, Menon R, Bailey KM, et al. Activation of Wnt/beta-Catenin in Ewing Sarcoma Cells Antagonizes EWS/ETS Function and Promotes Phenotypic Transition to More Metastatic Cell States. Cancer Res 2016; 76: 5040–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krook MA, Nicholls LA, Scannell CA, et al. Stress-induced CXCR4 promotes migration and invasion of ewing sarcoma. Mol Cancer Res 2014; 12: 953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Franzetti GA, Laud-Duval K, van der Ent W, et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 2017; 36: 3505–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hosios AM, Hecht VC, Danai LV, et al. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev Cell 2016; 36: 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang IY, Kwak S, Lee S, et al. Psat1-Dependent Fluctuations in alpha-Ketoglutarate Affect the Timing of ESC Differentiation. Cell Metab 2016; 24: 494–501. [DOI] [PubMed] [Google Scholar]
- 62.Carey BW, Finley LW, Cross JR, et al. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015; 518: 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of changes in BruSeq-measured gene expression on MI-503 treatment