

Abstract
Background
Kabuki syndrome (KS) is a rare pediatric congenital disorder with multiple congenital anomalies and intellectual disabilities, which is inherited in an autosomal dominant manner. Mutations in KMT2D and KDM6A have been proven to be the primary cause in most cases of KS.
Case presentation
Here we report two Chinese boys with clinical features of KS referred to our hospital for clinical diagnosis. Next-generation sequencing was performed on MiSeq to analyze the genetic mutations in both patients. In both, two novel de novo mutations in KMT2D gene (c.5235delA, p.(A1746Lfs*39) and c.7048G > A, p.(Q2350*)) were detected, both of which were subsequently confirmed by the two-generation pedigree analysis based on Sanger sequencing. A systematic literature review of previously reported mutational spectrum of KMT2D was also conducted.
Conclusions
Two novel de novo mutations in KMT2D gene were identified and considered to be pathogenic in both of KS patients. Our data adds information to the growing knowledge on the mutational spectrum of KS.
Electronic supplementary material
The online version of this article (10.1186/s12881-018-0545-5) contains supplementary material, which is available to authorized users.
Keywords: Kabuki syndrome, KMT2D, Novel, Mutation
Background
Kabuki syndrome (KS) (OMIM#147920), also previously known as Kabuki makeup syndrome, or Niikawa-Kuroki syndrome, first reported by Japanese researchers Kuroki [1] and Niikawa [2], is a congenital disorder with multiple congenital anomalies and intellectual disabilities [3]. The cardinal diagnostic manifestations of KS include distinctive facial features, mild-to-moderate intellectual disability, skeletal anomalies, dermatoglyphic abnormalities, and postnatal growth deficiencies [4]. The prevalence of this syndrome is estimated to be 1/86,000–1/32,000 [4].
It has been proved that KS is an autosomal dominant disorder and can be mainly caused by loss-of-function in two different genes-KMT2D and KDM6A. In 2010, KMT2D (NM_003482.3, formerly known as MLL2) was identified as the first causative gene in KS patients using whole-exome sequencing [5], which locates on chromosome 12q13. In 2012, KDM6A (NM_021140.3) was identified as the second causative gene in three KMT2D mutation-negative KS patients [6], which locates on chromosome Xp11.23. These two genes belong to a family of genes called chromatin-modifying enzymes, and act together in the epigenetic control of regulating a diverse set of gene transcriptions involved in embryogenesis and development [7, 8].
Cases of KS patients have been extensively reported from different parts all over the world during past years. However, there are only few cases from China. To the best of our knowledge, only 25 sporadic KS patients have been reported in China [9–18], in which 15 cases performed genetic analysis, including 14 cases with KMT2D mutation [15, 17] and 1 case with KDM6A mutation [16]. Here, our present study reported two novel de novo mutations of KMT2D in two young Chinese boys with KS.
Case presentation
This study conformed to the Tenets of the Declaration of Helsinki and was approved by the Ethics Board of the First Affiliated Hospital of Dalian Medical University. The CARE guidelines were followed in reporting our cases. Written informed consents from both patients and their parents were obtained before collecting blood samples.
Patient 1 was the second child (the first one was spontaneous abortion) of healthy, non-consanguineous Chinese parents. There was no family history of genetic disorder. He was born at 36 weeks of gestation due to his mother’s irregular vaginal bleeding. His birth parameters were as follows: weight, 2850 g (25-50th percentile); length, 47 cm (10-25th percentile); and head circumference, 31.5 cm (5-10th percentile). He was admitted to a neonatal care unit for 11 days because of neonatal septicemia and neonatal jaundice. Feeding difficulty and recurrent respiratory tract infection were observed. He showed motor delay and could not turn over until 11 months of age. At 26 months of age, he visited our hospital due to motor and intelligence developmental delay. His height was only 86 cm (3rd-10th percentile). He could not walk alone and his speech was delayed. Facial examination showed long palpebral fissures with lateral eversion of the lower eyelids, arched eyebrows with laterally thinning, depressed nasal tip, lower lip concave, and large ears (Fig. 1a). Cardiac auscultation and ultrasound were normal. He had brachydactyly and prominent finger pads especially for the fifth finger (Fig. 1c). He had sacral dimpling (Fig. 1e). His cognitive disability was mild with an intelligence quotient of 45. In addition, he had abnormal dentitions and abnormal genitourinary system. Urological ultrasound indicated that his right side had ectopic kidney, and left side was narrow and had cyst, and the demarcation between cortex and medulla of both kidneys was not clear. Magnetic resonance imaging of brain and vertebra demonstrated no obvious abnormalities except for the undetectable coccygeal vertebra. Blood routine, urine routine, renal function, thyroid function, electroencephalogram, auditory evoked potential, cardiac ultrasound, electrocardiogram, insulin and levodopa in growth hormone releasing were normal.
Fig. 1.
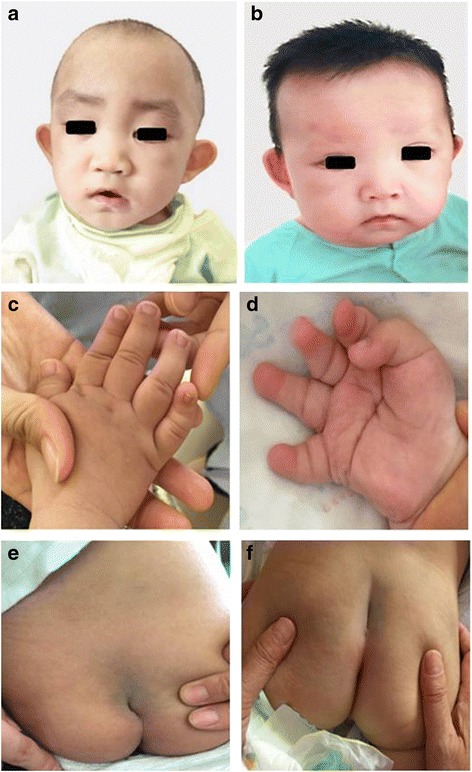
Clinical features of the patients. a and b showed makeup appearance. c and d showed short fingers especially for the fifth finger. e and f showed sacral dimpling
Patient 2 was the second child of healthy, non-consanguineous Chinese parents. There was also no family history of genetic disorder. He was born 40 weeks of gestation by spontaneous vaginal delivery and diagnosed as neonatal hypoglycemia and intrauterine infection. At 6 months of age, he visited our hospital due to motor developmental delay. He could not stand up and sit alone. He had distinctive facial appearance, long palpebral fissures with lateral eversion of the lower eyelids, arched eyebrows with laterally thinning eyebrows, depressed nasal tip, large ears, low hairline, and lower lip concave (Fig. 1b). He also had brachydactyly and prominent finger pads especially for the fifth finger and palm with a straight line across it, and sacral dimpling (Fig. 1d and f). He had joint hypermobility and hypotonia. The examination in blood routine, urine routine and abdominal ultrasound showed no apparent abnormality. Additionally, magnetic resonance imaging of brain suggested cerebellar vermis dysplasia, and urological ultrasound indicated incomplete cryptorchidism on the left side.
As our patients had typical facial features, skeletal anomalies and postnatal growth deficiency, we diagnosed both of them as KS by clinical findings. The basic situation and clinical features were summarized in Table 1. Then we performed molecular genetic testing for the KMT2D gene.
Table 1.
Summary of clinical and genotypic features of 2 Chinese children with KS
| Case ID | Patient 1 | Patient 2 |
|---|---|---|
| Gender | M | M |
| Age at diagnosis (months) | 26 | 5 |
| Elongated palpebral fissures | + | + |
| Eversion of the lateral third of the lower eyelid | + | + |
| Laterally sparse eyebrows | + | + |
| Depressed nasal tip | + | + |
| Micrognathia | + | + |
| Abnormal dentition | + | - |
| Spinal column abnormalities | + | + |
| Joint hypermobility/dislocation | - | + |
| Single palmar crease | + | + |
| Clinodactyly of fifth digits | + | + |
| Postnatal growth deficiency | + | + |
| Genitourinary anomalies | + | + |
| Cerebellar vermis dysplasia | - | + |
| Hypotonia | + | + |
| Motor delay | + | + |
| Feeding problem | + | - |
| Recurrent infection | + | - |
| Causing gene | KMT2D | KMT2D |
| Exon | 22 | 31 |
| Mutation | c.5235delA | c.7048G>A |
| Amino-acid change | p.(A1746Lfs*39) | p.(Q2350*) |
| Mutation type | Frameshift | Nonsense |
| Novelty | Novel | Novel |
Abbreviations: M, male; +, present; -, absent
Peripheral blood sample was collected from both patients and genomic DNA was then extracted from blood with a QIAamp DNA Mini Kit (catalog no. 51304, QIAGEN). Library was constructed using TruSight One Sequencing Panel (Illumina, San Diego, CA, USA, which is the largest sequencing panel available and includes 4813 clinically relevant genes), and sequenced on MiSeq platform (Illumina). The sequence was analyzed using ANNOVAR [19], PolyPhen2 [20], Mutation-Taster [21], and various databases, including ClinVar, dbSNP, 1000 genomes, Exome Sequencing Project 6500, and HGMD (Human Gene Mutation Data), were used in our study for screening and annotation of gene variants in accordance with the American college of medical genetics and genomics guidelines [22]. Furthermore, confirmation of the variants found in both patients and analysis of their parental samples were done by Sanger sequencing using standard procedures.
Mutational analysis identified two novel variants in our patients (Table 1). Each patient had one unique individual mutation, and both variants were not detected in their parents. These two de novo mutations included one small deletion leading to a frameshift and premature stop codon (c.5235delA heterozygous mutation, p.(A1746Lfs*39)) from patient 1 (Fig. 2a-c), and one nonsense mutation (c.7048G > A heterozygous mutation, p.(Q2350*)) from patient 2 (Fig. 2d-f).
Fig. 2.
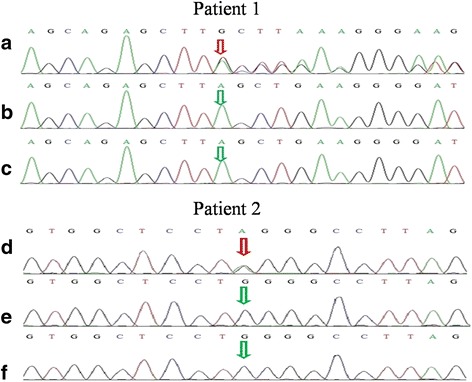
Sanger sequencing results for patients and their parents. a-c were for patient 1 and his parents, which demonstrated the A deletion (c.5235delA heterozygous mutation, p.(A1746Lfs*39); red arrow) in patient 1, and no deletion at c.5235A (green arrows) in his parents; d-f were for patient 2 and his parents, which demonstrated the presence of nonsense mutation (c.7948G > A heterozygous mutation, p.(Q2350*); red arrow) in patient 2, and the absence of mutation at c.7948G (green arrows) in his parents
Discussion & conclusions
Kabuki syndrome is a rare congenital disease that is characterized by five cardinal manifestations. Its incidence rate is approximately 1 in 32,000 individuals; however, the rate may be underestimated because of misdiagnosis and missed diagnosis. Diagnosis of KS is mainly clinical, based on a combination of distinctive dysmorphic face, intellectual disability, and multiple congenital abnormalities. Our present study reports a new case of two young Chinese boys with KS, both of which were characterized by the most striking facial appearances, skeletal anomalies, visceral anomalies and postnatal growth deficiency. More specifically in detail for both patients, we observed the typical facial appearances, including long palpebral fissures with lateral eversion of the lower eyelids, arched eyebrows with laterally thinning, depressed nasal tip, lower lip concave, and large ears (Fig. 1a-b), and skeletal anomalies, referring to brachydactyly and prominent finger pads especially for the fifth finger and sacral dimpling (Fig. 1c-f), and visceral anomalies, especially for the urogenital system, one with ectopic kidney, and the other with incomplete cryptorchidism. Specially, patient 1 showed feeding difficulty and recurrent respiratory tract infection, and his motor delay was obviously backward. In addition, he had abnormal genitourinary system, urological ultrasound indicating his ectopic kidney, and abnormal dentitions, which are less frequently observed according to previous studies. Patient 2 had cerebellar vermis dysplasia and motor delay, and his urological ultrasound indicated incomplete cryptorchidism on the left side.
Causative genes for KS were identified in 2010 and 2012 for KMT2D and KMD6A, respectively. Mutations in KMT2D were found to be the most common cause and present in 55–80% of KS patients subjected to genetic analysis, and a number of different KMT2D mutations have been reported to date. We searched the HGMD database for mutations in KMT2D, and performed a detail search for further mutations described in original articles in NCBI PubMed using the key words of “Kabuki syndrome”, “KMT2D mutation”, and “MLL2 mutation” in different combinations, without language restriction. Only articles that were fully available online were used in our analysis (see Additional file 1: Table S1). Then we manually reviewed the obtained articles, including mutation screening studies and molecularly proven case reports, to get the available clinical and molecular information of KS and KMT2D mutations. The KMT2D mutations were summarized and categorized as “frameshift”, “nonsense”, “missense”, “splice-site”, “deletion”, “duplication” or “indel”. Taking all the reviewed studies together, we found that 589 variants in KMT2D gene have been reported so far, including 216 frameshift variants, 178 nonsense mutations, 123 missense mutations, 53 splice-site variants, 10 deletions, 8 duplications and 1 indel (Fig. 3a), in which the majority mutations were frameshift, nonsense and missense, accounting for 36.67%, 30.05% and 20.88%, respectively. We also observed that although the mutations were distributed throughout nearly all exons, there seemed to be some exons in which mutations were mainly located such as exons 39 (23.31%), 48 (13.16%), 31 (9.96%) and 34 (8.27%) (Fig. 3b), which was similar with previous studies [23, 24]. However, we did not see the mutations enrichment phenomenon in the above exons when the length of each exon was taken into consideration (Fig. 3c).
Fig. 3.
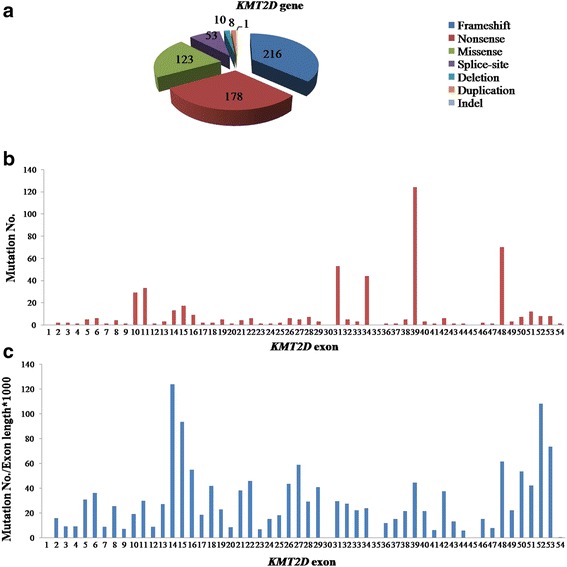
Overview of mutation type and exon distribution of published KMT2D mutations. a: Mutation types of previously published mutations in KMT2D. b: Mutation number distribution in KMT2D exons. c: Mutation distribution in KMT2D exons (with exon length taking into account)
In our present study, one frameshift mutation in exon 22 (c.5235delA heterozygous mutation, p.(A1746Lfs*39)) and one nonsense mutation in exon 31 (c.7048G > A heterozygous mutation, p.(Q2350*)) were identified in KMT2D using next-generation sequencing. Considering that all the patients’ parents were not found to carry KMT2D mutations, so both the patients harbored two de novo genetic mutations. In summary, we identified two novel de novo KMT2D mutations in two Chinese boys with KS, which adds information to the growing knowledge on the mutational spectrum of KS.
Additional file
Table S1. Summary of published mutations in KMT2D. (DOCX 64 kb)
Acknowledgements
We thank all the patients and their families for their collaboration in the study.
Funding
This study was funded by the National Nature Science Foundation of China (Grant No. 81471308) and National Health and Family Planning Commission of China and China Food and Drug Administration (Registration No. CMR-20161129-1003).
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
CX and CW contributed equally as co-first authors. JL was the corresponding author. CX, CW and YW performed the mutational analysis and literature review. JZ and LW drafted the figures and revised the manuscript. YW, RL and JL participated in the clinical diagnosis. CX, CW and JL wrote the manuscript. All authors read and approved the finial manuscript.
Ethics approval and consent to participate
This study conformed to the Tenets of the Declaration of Helsinki and was approved by the Ethics Board of the First Affiliated Hospital of Dalian Medical University (FB-KY-2016-47). Informed consent form from the patients and their parents was obtained before collecting blood samples.
Consent for publication
Written informed consent was obtained from all patients and their parents for publication of this case report and any accompanying images.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chengqi Xin, Email: chengqixin@126.com.
Chun Wang, Email: wangchun36@126.com.
Yachen Wang, Email: wangyachen100@163.com.
Jingyuan Zhao, Email: zhaojingyuan3344@sina.cn.
Liang Wang, Email: wangliang2000@aliyun.com.
Runjie Li, Email: lrj.62@163.com.
Jing Liu, Email: liujing@dmu.edu.cn.
References
- 1.Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr. 1981;99(4):570–573. doi: 10.1016/S0022-3476(81)80256-9. [DOI] [PubMed] [Google Scholar]
- 2.Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr. 1981;99(4):565–569. doi: 10.1016/S0022-3476(81)80255-7. [DOI] [PubMed] [Google Scholar]
- 3.Verhagen JM, Oostdijk W, Terwisscha van Scheltinga CE, Schalij-Delfos NE, van Bever Y. An unusual presentation of kabuki syndrome: clinical overlap with CHARGE syndrome. Eur J Med Genet. 2014;57(9):510–512. doi: 10.1016/j.ejmg.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Niikawa N, Kuroki Y, Kajii T, Matsuura N, Ishikiriyama S, Tonoki H, Ishikawa N, Yamada Y, Fujita M, Umemoto H, et al. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet. 1988;31(3):565–589. doi: 10.1002/ajmg.1320310312. [DOI] [PubMed] [Google Scholar]
- 5.Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, et al. Exome sequencing identifies MLL2 mutations as a cause of kabuki syndrome. Nat Genet. 2010;42(9):790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lederer D, Grisart B, Digilio MC, Benoit V, Crespin M, Ghariani SC, Maystadt I, Dallapiccola B, Verellen-Dumoulin C. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with kabuki syndrome. Am J Hum Genet. 2012;90(1):119–124. doi: 10.1016/j.ajhg.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.FitzGerald KT, Diaz MO. MLL2: A new mammalian member of the trx/MLL family of genes. Genomics. 1999;59(2):187–192. doi: 10.1006/geno.1999.5860. [DOI] [PubMed] [Google Scholar]
- 8.Aziz A, Liu QC, Dilworth FJ. Regulating a master regulator: establishing tissue-specific gene expression in skeletal muscle. Epigenetics. 2010;5(8):691–695. doi: 10.4161/epi.5.8.13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma KH, Chow SN, Yau FT. Isolated adrenocorticotropin deficiency in a child with kabuki syndrome. J Pediatr Endocrinol Metab. 2005;18(6):607–609. doi: 10.1515/JPEM.2005.18.6.607. [DOI] [PubMed] [Google Scholar]
- 10.Jiang YQ, Cao YH, Sun JF. Kabuki syndrome one case report. Chin J Dermatol. 2000;33(2):132. [Google Scholar]
- 11.Atar M, Lee W, O'Donnell D. Kabuki syndrome: oral and general features seen in a 2-year-old Chinese boy. Int J Paediatr Dent. 2006;16(3):222–226. doi: 10.1111/j.1365-263X.2006.00699.x. [DOI] [PubMed] [Google Scholar]
- 12.Yang XJ, Zeng Y, Xiong F. Kabuki syndrome case report. Zhonghua Er Ke Za Zhi. 2010;48(11):868–870. [PubMed] [Google Scholar]
- 13.Huang HQ, Gong GS. Four cases of kabuki make-up syndrome. Chin J Clinicians( Electronic Edition) 2013;7(2):857–858. [Google Scholar]
- 14.Lu Y, Cao KL. A case report: hearing disorder in kabuki make-up (Niikawa-Kuroki) syndrome in China. Journal of Otology. 2014;9:136–140. doi: 10.1016/j.joto.2014.12.002. [DOI] [Google Scholar]
- 15.Liu S, Hong X, Shen C, Shi Q, Wang J, Xiong F, Qiu Z. Kabuki syndrome: a Chinese case series and systematic review of the spectrum of mutations. BMC Med Genet. 2015;16:26. doi: 10.1186/s12881-015-0171-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang P, Tan H, Xia Y, Yu Q, Wei X, Guo R, Peng Y, Chen C, Li H, Mei L, et al. De novo exonic deletion of KDM6A in a Chinese girl with kabuki syndrome: a case report and brief literature review. Am J Med Genet A. 2016;170(6):1613–1621. doi: 10.1002/ajmg.a.37634. [DOI] [PubMed] [Google Scholar]
- 17.Wu BB, Su YJ, Wang HJ, Zhang P, Li L, Zhou WH. Report of 6 kabuki syndrome cases caused by KMT2D gene mutation and literature review. Chin J Evid Based Pediatr. 2017;12(2):135–139. [Google Scholar]
- 18.Wang LC, Chiu I, Wang PJ, Wu MH, Wang JK, Hung Y. Kabuki make-up syndrome associated with congenital heart disease. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi. 1994;35(1):63–69. [PubMed] [Google Scholar]
- 19.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 22.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banka S, Veeramachaneni R, Reardon W, Howard E, Bunstone S, Ragge N, Parker MJ, Crow YJ, Kerr B, Kingston H, et al. How genetically heterogeneous is kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet. 2012;20(4):381–388. doi: 10.1038/ejhg.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bogershausen N, Gatinois V, Riehmer V, Kayserili H, Becker J, Thoenes M, Simsek-Kiper PO, Barat-Houari M, Elcioglu NH, Wieczorek D, et al. Mutation update for kabuki syndrome genes KMT2D and KDM6A and further delineation of X-linked kabuki syndrome subtype 2. Hum Mutat. 2016;37(9):847–864. doi: 10.1002/humu.23026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of published mutations in KMT2D. (DOCX 64 kb)
Data Availability Statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.