

ABSTRACT
Blood vessel epicardial substance (BVES) is a tight-junction associated protein that was originally discovered from a cDNA screen of the developing heart. Research over the last decade has shown that not only is BVES is expressed in cardiac and skeletal tissue, but BVES is also is expressed throughout the gastrointestinal epithelium. Mice lacking BVES sustain worse intestinal injury and inflammation. Furthermore, BVES is suppressed in gastrointestinal cancers, and mouse modeling has shown that loss of BVES promotes tumor formation. Recent work from multiple laboratories has revealed that BVES can regulate several molecular pathways, including cAMP, WNT, and promoting the degradation of the oncogene, c-Myc. This review will summarize our current understanding of how BVES regulates the intestinal epithelium and discuss how BVES functions at the molecular level to preserve epithelial phenotypes and suppress tumorigenesis.
Keywords: BVES, colitis-associated cancer, POPDC1, POPEYE
Introduction to the intestinal epithelium
The intestine is divided into two anatomical compartments: the small intestine, where food is absorbed and digested, and the large intestine, or colon, where water and electrolytes are absorbed.1 At the histological level, the small intestine consists of villi, or finger-like projections, which protrude into the lumen providing maximal surface area to absorb food. At the base of each villus is a crypt, or invagination, where cells divide and migrate upward to repopulate the mature epithelium. Whereas the small intestine contains “crypt-villus units,” the colon consists of only crypts. A single layer of cells lines these crypts and villi and is collectively referred to as “the intestinal epithelium.”
As the intestine is constantly enduring mechanical and chemical stress, as well as infectious challenges, the epithelium regenerates entirely every 7 days. This regeneration is initiated by stem cells residing in the crypt base. These Lgr5+ stem cells asymmetrically divide yielding a daughter stem cell and a committed cell.2–4 The committed cells are collectively referred to as the “the transit amplifying population” and migrate upward along the crypt-villus axis, differentiating as they replace sloughed cells.2 Broadly, these migrating progenitors can differentiate into absorptive cells (enterocytes) or secretory cells (goblet, enteroendocrine, and Paneth cells). Whether an intestinal cell commits to an absorptive or secretory fate is largely determined by balancing inputs from the Wnt, Notch, and BMP signaling pathways. Understanding this complex dynamic – stem cells giving rise to various lineages, tasked with absorption of nutrients and partitioning luminal microbial populations all while enduring mechanical and chemical stress in the setting of pathogens, carcinogens, and physical injury – has been the subject of intense investigation over the past several decades.
The intestine as a barrier and junctional constituents
The intestinal epithelium is a selectively permeable barrier allowing for the absorption of nutrients, water, and electrolytes while simultaneously preventing penetration of intestinal pathogens and other antigenic materials. Junctional complexes – such as tight junctions, adherens junctions, hemidesmosomes, and gap junctions – within these epithelial cells are critical for linkage of the cytoskeleton, cell-cell signaling, sub-cellular partitioning of membrane proteins, and for regulating para-cellular permeability that ultimately allows the intestinal epithelium to perform its function. Hemidesmosomes couple the basal membrane of the epithelium to the underlying basal lamina and coordinate with the more laterally localized desmosomes to help nucleate intermediate filament networks of the cytoskeleton.5 Gap junctions connect neighboring cells and allow for direct cell-cell passage of ions, metabolites, and second messengers through hexamers consisting mainly of connexin proteins.6 Adherens junctions initiate and maintain cell-cell contacts by coupling the transmembrane glycoprotein E-cadherin and cytoplasmic constituents p120-catenin, β-catenin, and α-catenin with the actin cytoskeleton.7 Maintaining the integrity of adherens complex is critical both in regulating barrier function and oncogenesis. For example, mice expressing a dominant-negative form of N-cadherin, which disrupts the adherens junction, develop spontaneous colitis,8 and E-cadherin reductions in colorectal cancer are associated with increased invasive potential.9 Interestingly, disruption of E-cadherin in the epidermis leads to alterations in the tight junction, highlighting the interconnectivity of the junctional barrier network.10
Indeed, tight junctions are often in close association with adherens junctions near the apical-lateral membrane and help to form a selective, semipermeable barrier restricting the paracellular transport of solutes, ions, and water. Furthermore, tight junctions allow for the sub-cellular partitioning of apical and baso-lateral proteins, assisting in defining cellular polarity. Principal constituents of tight junctions include the transmembrane proteins of the claudin family, occludins, tricellulin, junctional adhesion molecules (JAMs), and blood vessel epicardial substance (BVES). Multiple adaptor proteins such as the zonula occludens (ZO-1–3) and cingulin as well as the RHOGEF, RHOA, and ZONAB signaling molecules are also located cytoplasmically in association with the tight junction. Given the complexity of tight junction signaling, this review will focus on the role of one particular tight junction protein, BVES, and explore its role in maintaining epithelial integrity while also discussing a role for BVES in non-epithelial tissue. We summarize work over the last 15 years on BVES – a protein initially discovered via a cDNA screen of the developing heart11,12 – and discuss its emerging and key role in governing intestinal epithelial biology, from tumorigenesis to barrier function to intestinal injury responses.
BVES structure, expression, and localization
BVES is the founding member of the POPDC family, which also consists of POPDC2 and POPDC3. POPDC2 and POPDC3 share 50% homology while BVES is only 25% homologous to either family member, suggesting evolutionary divergence.13,14 BVES, the most well-studied family member, is a three-pass transmembrane protein, and in humans, the protein consists of 360 residues with three alpha-helical membrane spanning, hydrophobic domains, two extracellular N-linked glycosylation sites,15 and a large intracellular domain, where many protein-protein interactions occur (Figure 1). The extracellular amino terminus of BVES extends from amino acid 1–42, and the two N-glycosylation sites potentially protect BVES from proteolysis or help traffic it to the cellular membrane.23,24 Within the intracellular carboxy-terminus (a.a. 114–360) is the POPEYE domain (a.a.172–266), which is highly conserved (80%) across the family throughout different vertebrates.14,16 Kawaguchi et al. showed that BVES exists as a dimer via interactions near the carboxy end of the POPEYE domain and that this homotypic interaction requires two lysine residues (K272 and K273).18
Figure 1.
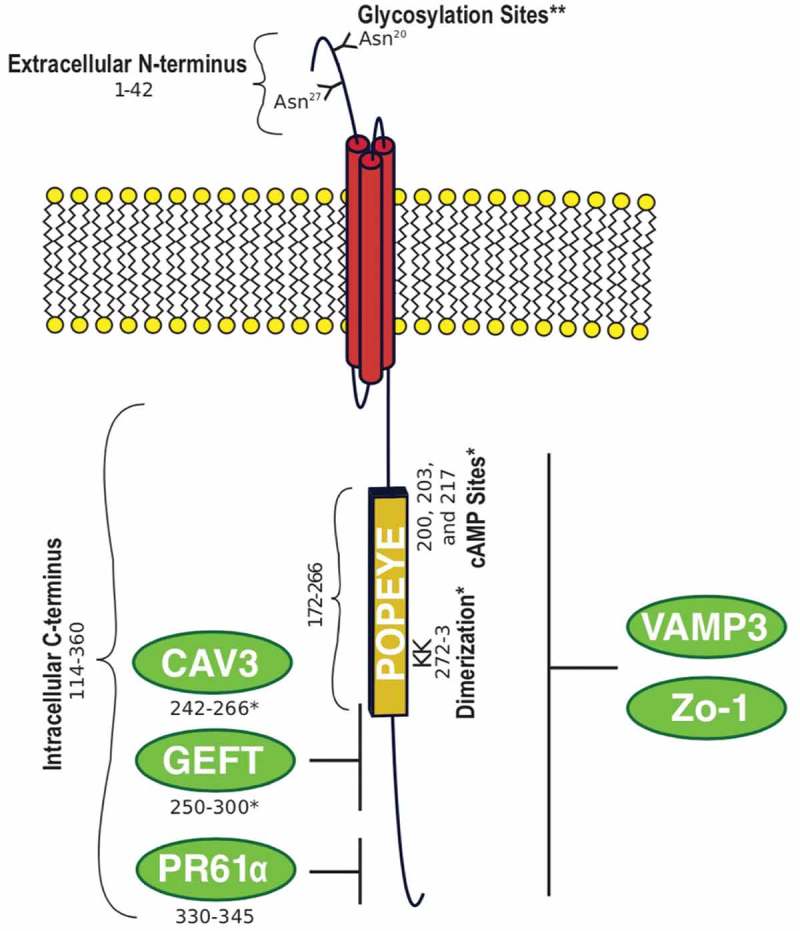
BVES structure. Human BVES protein is 360 amino acids with an extracellular N-terminus, a three-pass transmembrane domain, and a large intracellular C-terminus containing the highly conserved POPEYE domain.*mapped to murine BVES amino acids. ** mapped to chick BVES amino acids. Glycosylation;15 POPEYE;16 cAMP;17 Dimerization;18 CAV3;19 GEFT;20 PR61α;21 VAMP3;22 Zo-1.13
Given its location at the membrane and that it possesses a three-pass transmembrane domain, it was postulated that BVES contributes to cell adherence, but interestingly, BVES lacks any motifs or domains commonly observed in known classes of adhesion molecules. And to date, little is known about its protein folding, translocation to the membrane, or how and if it is regulated at the post-translational level. Furthermore, sequence alignments analyses do not predict enzymatic activity. The novelty of the POPDC family structure, implicates a unique cellular function.
While the structure of BVES does not provide immediate clues to its function, its expression pattern suggests some tissue specific capabilities. BVES is expressed in muscle and epithelial tissues in a wide-variety of organs: heart,11,12 smooth and skeletal muscle,25 retina,26 intestine,27,28 lung,29 and breast.27 One of the common properties of BVES-expressing tissue is cell adherence. Indeed, further work by Osler et al. identified that BVES contributes to cell-cell adhesion. When cells are subconfluent, BVES is primarily localized to the cytoplasm (Figure 2). But when cells establish contact, BVES rapidly traffics to the cellular membrane(Figure 2),13,30 in a poorly understood process. BVES movement to the membrane localizes to points of cell-cell contact and co-localizes with tight junction constituents ZO−1 and occludin, but not with adherens junction-associated proteins such as β-catenin and E-cadherin or desmosomal associated proteins. Overall, based on BVES localization and its association with junctional complexes, it was hypothesized that BVES could be an important regulator of epithelial states.
Figure 2.
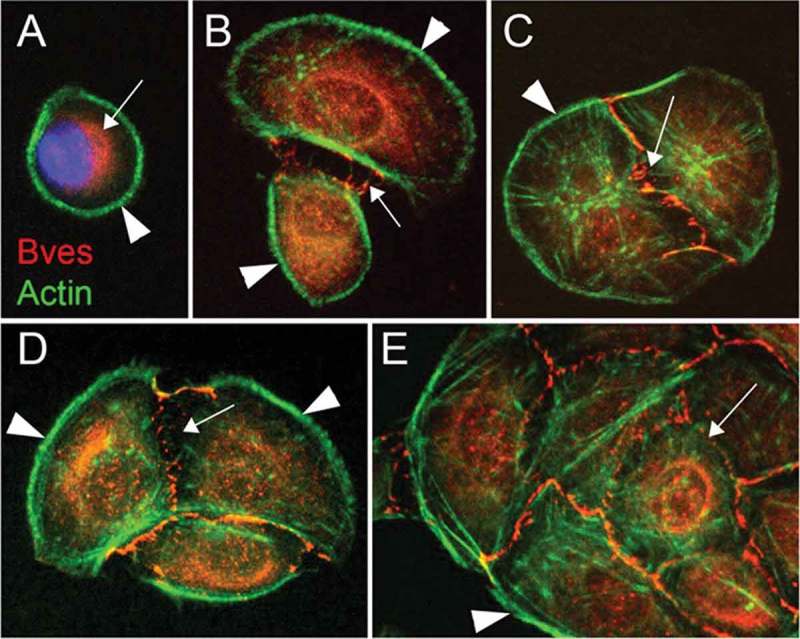
BVES localization is dynamic. BVES subcellular localization changes depending on cell-cell contact. BVES (red) is primarily confined to the cytoplasm when cells (Epicardial mesothelial cells) are subconfluent (A-B), but as cells make contact, BVES localizes to the plasma membrane (C-E). This figure has been reproduced.13 © The Company of Biologists Limited. Reproduced by permission of he Company of Biologists Limited. Permission to reuse must be obtained from the rightsholder.
Loss of BVES is associated with mesenchymal-like phenotype
A hallmark of epithelial cells is their ability to aggregate through cell-cell adhesion into an organized epithelium functioning, collectively, as a tissue.31 Within the epithelium, there is coordinated cell motility, proliferation, and differentiation. The dynamic nature of the epithelium is apparent when there is loss of cell-cell contact, leading to individual epithelial cells assuming a fibroblast-like or mesenchymal-like morphology.32 This phenotypic change is termed epithelial-mesenchymal transition (EMT); in many tissues, upon regaining cell-cell contacts, cells undergo reciprocal mesenchymal to epithelial transition (MET). Cellular adhesion complexes, including tight and adherens junctions are key regulators of these transitions, providing a means for cellular signaling in addition to mechanical adhesion. For example, the role of the adherens junction as a modulator of canonical WNT signaling through sequestration of β-catenin at the cell membrane is well established.33 Similarly, tight junctions play a fundamental role in outside-in signaling cascades. RhoA downregulation following epithelial cell confluency leads to reductions in proliferation, which is due in part to an interaction with Guanine nucleotide exchange factor H1 (GEFH) and the tight junction protein Cingulin. In a similar fashion, ZO-1 can modify gene expression programs and cell growth by sequestering ZONAB (a Y-box transcription factor) at the tight junction, leading to coordinated regulation of cytoskeletal and transcriptional programs and providing a link between junctional integrity and cell signaling programs.34,35
Because of the dynamic subcellular localization of BVES, and its impact on epithelial junctions, it was postulated that BVES contributed to maintaining epithelial states similar to the regulatory role of E-cadherin. Using the human corneal epithelial cell line (HCE), Osler et al. showed that knocking down BVES impairs tight junction and adherens junction formation as evidenced by disrupted membrane localization of β-catenin, E-cadherin, ZO-1, and occludin.13 Furthermore, loss of junctional complexes were functionally significant as transepithelial resistance (TER), a readout for junctional integrity and barrier competency of an epithelium, was reduced in the setting of BVES loss.13 Conversely, overexpression of BVES in HCE cells increased TER. Moreover, overexpression of BVES with mutations at lysines 272 and 273, which prevents endogenous BVES from trafficking to the cellular membrane, converts HCE cells to a fibroblast morphology. Concomitant increase in vimentin expression, a mesenchymal cell marker, reduction in cytokeratin, and enhanced mobility and invasion18 were also observed, all indicating a transition from an epithelial to a more mesenchymal state. This was further supported by the in vivo observation that Bves-/- mice have impaired skeletal muscle recovery after injury.25 Recovery from injury requires the orchestration of cellular migration and re-establishment of cell polarization – both of which were abnormal in Bves-/- muscle. Thus, initial reports about BVES suggested that BVES promoted epithelial states.
Pivotal studies began to elucidate the cellular mechanisms by which BVES regulates epithelial states. Smith et al. conducted a yeast-two-hybrid screen and identified that murine BVES interacted with Guanine nucleotide exchange factor T (GEFT) through BVES amino acids 250–300.20 Broadly, GEFs functional principally to stimulate the exchange of GDP for GTP and can activate Rho GTPases.7 GEFT activates Rac1 and Cdc42 to induce lamelipodia and filopodia formation during cellular migration. Importantly, Rho GTPases contribute to tight junction composition and conversely, tight junctions are implicated in the regulation of Rho GTPases. For example, the RhoA activator GEF-H1 can be sequestered through an interaction with ZO-1 and the adaptor protein cingulin to reduce RhoA activation.7 Alternatively, expression of a constitutively active RhoA leads to alterations in tight junction localization with redistribution of occludin and ZO-1.36 It was shown that overexpression of BVES in NIH 3T3 cells resulted in less motile and more round cells,20 and this correlated with less active forms of Rac1 and Cdc42. However, the report was not conclusive as to how BVES regulates RhoA signaling. In a follow up study, Russ et al. provided evidence that BVES acts to reduce RhoA signaling as well as maintain junctional integrity in epithelial cells. They demonstrated that overexpression of BVES reduced RhoA activity and overexpression of a truncated-BVES mutant that disrupted endogenous BVES localization increased RhoA activity.37 Moreover, they observed that in addition to co-localizing with ZO-1, BVES immunoprecipitated with ZO-1. When BVES was overexpressed, ZO-1 and occludin were confined to the cellular membrane. But when the mutant BVES was expressed, ZO-1 and occludin were primarily intracellular. This suggested that BVES was required for junctional composition.
More recently, Hager et al. identified Vesicle-associated membrane protein 3 (VAMP3), a SNARE protein family member involved in vesicular transport, as a BVES interacting protein via a yeast-two-hybrid approach.22 VAMP3 is widely expressed and interacts with syntaxin-4 to tether vesicular cargo to the cellular membrane38 and is required for cellular migration and movement through aiding in the trafficking of β-1-integrins. Studies have shown that interfering with VAMP3 function, such as silencing via siRNA or using a tetanus toxin that inhibits VAMP3, reduces cellular migration and cell adhesion with associated impairment of β-1-integrins recycling,39,40 suggesting that VAMP3-integrin trafficking is critical in coordinating cellular migration and adhesion. Hager et al. showed that silencing BVES disrupted β-1-integrin recycling during cellular migration. In addition to the role for BVES in modulating GEF activity, the identification of VAMP3 as a BVES binding partner implicates BVES in multiple pathways controlling cellular migration.
Non-epithelial roles of BVES
In 2012, Froese et al. discovered that BVES has an important regulatory role in cardiac myocytes.17 In their work, they identified that Bves-/- mice had impaired stress-induced bradycardia, suggesting a sinus node defect. They identified a high-affinity cAMP binding domain within the POPEYE domain and biochemical experiments showed that BVES directly bound to cAMP. Mutating single amino acids (D200, E203, and V217) modulated the affinity of the interaction. Moreover, BVES interacted with the 2-pore domain potassium channel TREK-1, and this was sensitive to cAMP stimulation. It was postulated that BVES could recruit TREK-1 to the membrane to enhance current, and that this recruitment was modulated by levels of cAMP.17 This report expanded the known regulatory roles of BVES by showing its importance in cardiac pacemaking and a role in determining membrane localization of other proteins. Moreover, the report showed a firm in vivo functional relevance to BVES. A subsequent report in 2016 by Schindler et al. identified a BVES missense variant (S201F) by whole-exome sequencing in a family of four with cardiac arrhythmias and limb-girdle muscular dystrophy (LGMD).41 Skeletal muscle biopsies from the affected patients showed impaired BVES membrane localization. The variant, which was within the cAMP binding domain, showed 50% reduction in cAMP affinity in vitro and impaired TREK-1 recruitment to the membrane. Furthermore, this BVES mutant, expressed in zebrafish, phenocopied the heart and skeletal muscle defects observed in the family carrying the homozygous allele. Thus, these studies revealed the powerful clinical implications of disrupting BVES function and continued to expand its widening regulatory role in cardiac myocytes and skeletal muscle tissue.
The role and functional impact of a BVES:cAMP interaction in normal epithelium is unknown; however, Amunjela, et al have recently demonstrated that BVES can immunoprecipitate with cAMP in breast cancer lines. Further, cAMP increases BVES protein levels and may influence cell migration.42 Importantly, cAMP is known to play a key role in junctional integrity. Treatment of primary HUVEC cells with cAMP derivatives improves tight junction continuity and decreases paracellular permeability.43 In ovarian cancer cells, the cAMP-dependent protein kinase (PKA) can phosphorylate claudin-3. A mutation in claudin 3 at the PKA phosphorylation site which mimics constitutive phosphorylation impairs junctional assembly as reflected by reductions in TER.44 Additional studies defining the BVES:cAMP and its contribution to junctional integrity in the epithelium will be required.
BVES as a tumor suppressor
Epithelial state changes, such as EMT, occur during differing stages of tumor development and growth. Indeed, tumor cells at the invasive front tend to possess mesenchymal traits (hypermigratory, poorly differentiated, and hyperproliferative).45 They also tend to be incapable of cell-cell contact mediated growth arrest.45 Loss of adhesion junction molecules has been associated with increased tumor invasiveness,46,47 and colorectal cancers that retain E-cadherin expression are associated with decreased invasiveness.48 Similarly, tight junction proteins are implicated in tumor biology with claudin family members being overexpressed in ovarian, CRC, and gastric cancers.49,50 However, claudin-1 has also demonstrated tumor suppressive effects in gastric tumors,21 suggesting context and tissue dependence or tumor type-specific function. Moreover, high expression of ZO-1 is a good prognostic indicator in non-small cell lung cancer51 and decreased expression of ZO-1 in breast and colon cancer are correlated with progression.52,53 Thus, although our understanding is incomplete, dysregulation of adherens and tight junction constituents is a feature of malignancies.
Expanding on the role of BVES in cancer, Williams et al. showed that BVES was underexpressed in several epithelial malignancies, including breast and colorectal cancer.27 Restoration of BVES expression in CRC cell lines induced an epithelial-like morphology (Figure 3) and decreased cellular proliferation, invasion, growth as subcutaneous xenografts in athymic mice and reduction in splenic metastasis in mouse experiments.27 Conversely, disrupting BVES in non-malignant human corneal epithelial cells induced a mesenchymal-like morphology and increased cellular proliferation and migration. In sum, BVES acted as a regulator of EMT in CRC.
Figure 3.
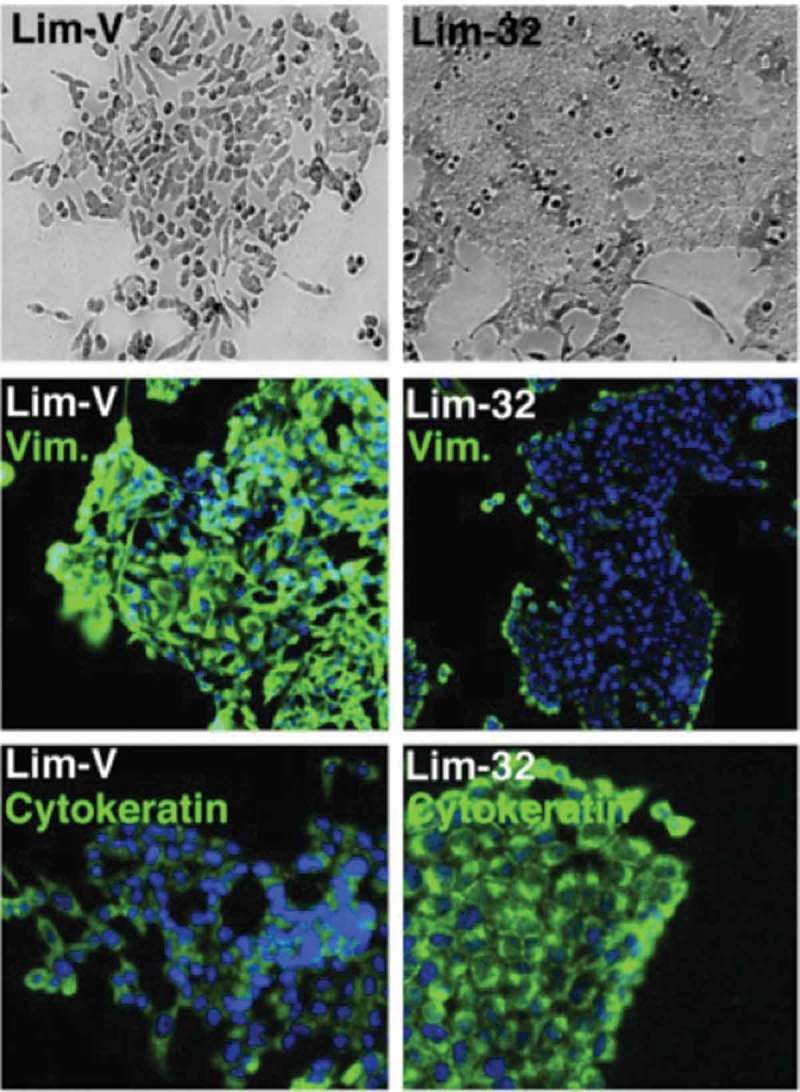
BVES expression in CRC cell lines induces epithelial-like phenotype. Bright field microscopy and immunofluorescent staining for vimentin and cytokeratin in Lim2405 cells (Lim-V) and Lim2405 cells that express BVES (Lim-32). This figure has been reproduced.27 © American Society for Clinical Investigation. Reproduced by permission of American Society for Clinical Investigation. Permission to reuse must be obtained from the rightsholder.
The study concluded that this regulation was in part due to BVES-induced regulation of RhoA signaling. But how BVES reduced cellular proliferation, migration, and invasion remained only partially explained. Given the increasing number of identified BVES interacting proteins, it was reasonable to predict that other unknown interacting proteins could contribute to the tumor suppressive mechanisms of BVES. Moreover, whether BVES had an in vivo role in suppressing tumorigenesis was not established.
Parang et al. conducted the first in vivo study testing BVES as a tumor modifier.54 Using the well-established azoxymethane repeated cycles of dextran sodium sulfate (AOM/DSS)55–58 model of inflammatory carcinogenesis, they showed that Bves-/- mice developed worse inflammatory injury, greater tumor multiplicity, and a higher degree of intratumoral dysplasia. The authors observed increased intratumoral Wnt signaling at the protein level. Analysis of RNA sequencing of Bves-/- tumors revealed activation of c-Myc signaling networks and subsequent immunohistochemistry showed increased c-Myc protein. The authors posited that BVES may regulate cellular c-Myc levels. Indeed, overexpressing BVES in vitro decreased c-Myc protein with associated increase in ubiquitylated c-Myc levels. Furthermore, knocking down BVES by siRNA increased c-Myc protein. Co-immunoprecipitation and proximity ligation assays revealed that BVES associates with c-Myc. Given that BVES modified c-Myc post-translationally, they hypothesized that BVES interacted with PPP2R5A (PR61α), a regulatory subunit of the serine-threonine protein phosphatase 2A (PP2A) heterotrimeric complex that dephosphorylates c-Myc and promotes c-Myc ubiquitylation. By yeast-two-hybrid, co-immunoprecipitation, and proximity ligation assay, they showed that BVES interacted with PR61α and identified that residues 330–345 in human BVES were required for the BVES:PR61α interaction. Deletion of those 15 amino acids uncoupled the interaction, and expression of that BVES mutant had no effect on c-Myc levels, suggesting that BVES likely requires PR61α to promote c-Myc degradation. This was a key study illustrating that BVES can regulate oncogenic signaling pathways. Interestingly, in inflammatory bowel disease, c-Myc has been postulated to contribute to colitis-associated cancer progression.59–61 Together, this study continued to broaden the known regulatory roles of BVES, from cardiac and skeletal muscle maintenance to tumor progression. As PP2A dephosphorylates a number of other targets, many of which are implicated in tumorigenesis,62,63 determining whether BVES directs additional PP2A activity may elucidate novel regulatory roles. For instance, PP2A has been shown to dephosphorylate tight junction proteins such as ZO-1, occludin, and claudin-1 leading to increased paracellular permeability.64
While several studies have demonstrated that BVES has an important role in intestinal malignancy, additional studies implicate BVES in hepatocellular and breast cancer.65–67 The tissue-specific functions of BVES in regulating tumorigenesis remain unclear, especially in vivo, but the recent literature strongly suggests BVES acts as a tumor suppressor in a variety of epithelial tissues, likely, at least in part, due to its role in promoting c-Myc degradation. The majority of phenotypes associated with BVES loss require cellular or physiologic stress, suggesting that under homeostasis, BVES loss has minimal deleterious effects. This may be due, in part, to physiologic redundancy via compensation by other POPEYE domain containing proteins such as POPDC2 and POPDC3.
BVES promoter methylation as a biomarker in malignancy
Hypermethylation of CpG islands in proximal promoters of tumor suppressor genes is a common mechanism if silencing their expression in malignancy.68 In a study of non-small cell lung cancer patients, Feng et al. detected that the BVES promoter was hypermethylated in tumors compared to non-malignant tissue.29 Williams et al. subsequently identified a large CpG island from nucleotides −997 to + 394 in the BVES proximal promoter and demonstrated that BVES was suppressed via hypermethylation of this island. Treatment of CRC cell lines with 5-aza-2ʹ-deoxycytidine, a demethylating agent, restored BVES expression.27 Similarly, others have shown that the BVES promoter is hypermethylated in gastric malignancies compared to normal tissue.28,29
Aside from sporadic cancers, Parang et al. analyzed patients with ulcerative colitis and colitis-associated cancer. Similar to prior studies, they determined that the BVES promoter was hypermethylated in colitis-associated cancers compared to patients with ulcerative colitis who did not have colitis-associated cancer. Interestingly, they showed that the BVES promoter was also hypermethylated in distant, non-malignant mucosa of patients who also had colitis-associated cancer, suggesting that BVES promoter methylation status could be used a biomarker of distant cancer.54
The current method of cancer screening in inflammatory bowel disease patients, who are at a significantly elevated risk of developing colitis-associated cancer,69 is surveillance colonoscopy, in which the colon is biopsied with the hope that cancer can detected at an early and therefore treatable stage. However, the screening for malignancy in the colon of inflammatory bowel disease patients can be challenging because the lesions are frequently flat, making it difficult to identify, especially in the setting of inflammatory changes. Parang et. al’s report suggest that BVES promoter methylation may be a useful biomarker for the presence of colitis-associated cancer, or even dysplasia, and that measuring BVES promoter methylation status could serve as a clinically useful tool to identify patients at risk for colon dysplasia or cancer.
BVES and its stem cell role
A growing number of studies are supporting the hypothesis that BVES regulates multiples intracellular signaling pathways and its expression is key in maintaining an epithelial phenotype. In support of this, Bves-/- mice have increased intestinal proliferation at baseline.70 Lgr5 is a transmembrane receptor for R-spondin that amplifies Wnt tone and is a marker of intestinal stem cells,71,72 Bves-/- mice crossed with an Lgr5-eGFP-IRES-creERT2 intestinal stem cell reporter mouse have an expanded stem cell compartment compared to wildtype mice. Consistent with loss of BVES promoting “stemness,” enteroid cultures derived from Bves-/- mice had a higher plating efficiency and increased stem-cell markers such as Axin2, CD44, and Cyclin-D1 by qPCR.70 The precise molecular mechanism underlying why Bves-/- mice have an expanded stem cell compartment is unclear, but the authors posit that it could be due to dysregulation of the numerous pathways BVES is known to coordinate, such as the Wnt pathway.
Conclusions: BVES as a sensor
Since 1999, when BVES was initially discovered, our understanding of BVES, from its structure to its function, has expanded substantially. This three-transmembrane domain protein, with its unique molecular domains, is key in maintaining epithelial states. In a variety of tissues, it has been repeatedly shown that disruption of the BVES protein results in epithelial cells acquiring a more mesenchymal phenotype, becoming more proliferative, migratory, and less-adherent with impaired adherens and tight-junction formation. The number of intracellular signaling pathways that BVES regulates is growing with each new study. An emerging picture is that BVES acts as a regulator of epithelial state in large part due to its intracellular carboxy terminus acting as a docking partner or scaffold for a variety of protein-protein interactions, such as c-Myc and PP2A. Our proposed model, building from the work of others, is that BVES acts as a sensor of cell-cell contact to arrest cellular mechanisms such as proliferation and migration. When cells make contact with each other, BVES, through recruitment of protein-protein interactions at the cell membrane, transduces “outside” contact into intracellular signaling that induces an epithelial phenotype. Thus, when BVES function is lost, such as through mutation or promoter hypermethylation, there is loss of epithelial regulation and a higher risk of malignancy.
Future studies
Future studies of BVES require further investigation into other binding partners that would help us understand how it regulates such a variety of intracellular signaling pathways. As previous studies have demonstrated an interaction between BVES and cAMP, the importance of this interaction needs evaluation specifically in intestinal epithelial cells. Moreover, although recent studies have shed some light into the in vivo function of BVES, it remains to be seen how BVES functions within other tissues such as lung or breast. Generation of tissue-specific BVES knockout mice will be key in revealing more about its function.
As much as we know about what pathways BVES can regulate, our understanding of BVES itself at the transcriptional, translational, and post-translational level is very limited. We do not have a strong grasp of what transcription factors regulate BVES expression, and we know little of the kinetics of BVES protein turnover or its posttranslational modifications. Uncovering how BVES is regulated will be key in continuing to understand the function of this regulator of epithelial states.
Funding Statement
This work was supported by NIH grants R01DK099204 (CSW), P30DK058404 (Vanderbilt Digestive Disease Research Center), and UL1TR000445 (Vanderbilt CTSA), Merit Review Grants from the Office of Medical Research, Department of Veterans Affairs 1I01BX001426 (CSW), T32GM007347 (BP, JJT), F30 DK096718 (BP), and F30 600 DK111107 (JJT).This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases [DK111107];National Institute of Diabetes and Digestive and Kidney Diseases [DK096718];National Institutes of Health [DK080221]. The funders had no role in the decision to publish, or in preparation of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Noah TK, Donahue B, Shroyer NF.. Intestinal development and differentiation. Exp Cell Res. 2011;317:2702–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Santa Barbara P, van den Brink GR, Roberts DJ. Development and differentiation of the intestinal epithelium. Cell Mol Life Sci. 2003;60:1322–1332. doi: 10.1007/s00018-003-2289-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crosnier C, Stamataki D, Lewis J. Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet. 2006;7:349–359. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt GH, Winton DJ, Ponder BA. Development of the Pattern of Cell Renewal in the Crypt-Villus Unit of Chimaeric Mouse Small Intestine. Development. 1988;103:785–790. [DOI] [PubMed] [Google Scholar]
- 5.Walko G, Castañón MJ, Wiche G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015;360:363–378. doi: 10.1007/s00441-014-2061-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans WH, Martin PEM. Gap junctions: structure and function (Review). Mol Membr Biol. 2002;19:121–136. doi: 10.1080/09687680210139839. [DOI] [PubMed] [Google Scholar]
- 7.Balda MS, Matter K. Tight junctions and the regulation of gene expression. Biochim Biophys Acta. 2009;1788:761–767. doi: 10.1016/j.bbamem.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 8.Hermiston ML, Gordon JI. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science. 1995;270:1203–1207. [DOI] [PubMed] [Google Scholar]
- 9.Dorudi S, Sheffield JP, Poulsom R, Northover JM, Hart IR. E-cadherin expression in colorectal cancer. Am J Pathol. 1993;142:981–986. [PMC free article] [PubMed] [Google Scholar]
- 10.Tunggal JA, Helfrich I, Schmitz A, Schwarz H, Günzel D, Fromm M, Kemler R, Krieg T, Niessen CM. E-cadherin is essential for in vivo epidermal barrier function by regulating tight junctions. EMBO J. 2005;24:1146–1156. doi: 10.1038/sj.emboj.7600605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reese DE, Zavaljevski M, Streiff NL, Bader D. BVES: A novel gene expressed during coronary blood vessel development. Dev Biol. 1999;209:159–171. doi: 10.1006/dbio.1999.9246. [DOI] [PubMed] [Google Scholar]
- 12.Andrée B, Hillemann T, Kessler-Icekson G, Schmitt-John T, Jockusch H, Arnold HH, Brand T. Isolation and characterization of the novel popeye gene family expressed in skeletal muscle and heart. Dev Biol. 2000;223:371–382. doi: 10.1006/dbio.2000.9725. [DOI] [PubMed] [Google Scholar]
- 13.Osler ME, Chang MS, Bader DM. Bves modulates epithelial integrity through an interaction at the tight junction. J Cell Sci. 2005;118:4667–4678. doi: 10.1242/jcs.02588. [DOI] [PubMed] [Google Scholar]
- 14.The BT. Popeye domain-containing gene family. Cell Biochem Biophys. 2005;43:95–103. doi: 10.1385/CBB:43:1:095. [DOI] [PubMed] [Google Scholar]
- 15.Knight RF, Bader DM, Backstrom JR. Membrane topology of Bves/Pop1A, a cell adhesion molecule that displays dynamic changes in cellular distribution during development. J Biol Chem. 2003;278:32872–32879. doi: 10.1074/jbc.M301961200. [DOI] [PubMed] [Google Scholar]
- 16.Osler ME, Smith TK, Bader DM. Bves, a member of the Popeye domain-containing gene family. Dev Dyn. 2006;235:586–593. doi: 10.1002/(ISSN)1097-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Froese A, Breher SS, Waldeyer C, Schindler RFR, Nikolaev VO, Rinné S, Wischmeyer E, Schlueter J, Becher J, Simrick S, et al. Popeye domain containin proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J Clin Invest. 2012;122:1119–1130. doi: 10.1172/JCI59410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawaguchi M, Hager HA, Wada A, Koyama T, Chang MS, Bader DM. Identification of a novel intracellular interaction domain essential for Bves function. PLoS One. 2008;3:e2261. doi: 10.1371/journal.pone.0002261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alcalay Y, Hochhauser E, Kliminski V, Dick J, Muayad ZA, Parnes D, Schlesinger H, Abassi Z, Shainberg A, Schindler RF, et al. Popeye domain containing 1 (Popdc1/Bves) is a caveolae-associated protein involved in Ischemia Tolerance. PLoS ONE. 2013;8(9):e71100. doi: 10.1371/journal.pone.0071100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith TK, Hager H, Francis R, Dm K, Cw L, Dm B. Bves directly interacts with GEFT, and controls cell shape and movement through regulation of Rac1/Cdc42 activity. Proc Natl Acad Sci U S A. 2008;105:8298–8303. doi: 10.1073/pnas.0802345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang TL, Ito K, Ko TK, Liu Q, Salto-Tellez M, Yeoh KG, Fukamachi H, Ito Y. has tumor suppressive activity and is a direct target of RUNX3 in gastric epithelial cells: Gastroenterology. Vol. 138P Claudin-1; 2010. p. 255–265. doi: 10.1053/j.gastro.2009.08.044. [DOI] [PubMed] [Google Scholar]
- 22.Hager H, Rj R, Ee C, Proux-Gillardeaux V, Dm B. Identification of a novel Bves function: regulation of vesicular transport. EMBO J. 2010;29:532–545. doi: 10.1038/emboj.2009.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hager HA, Bader DM. Bves: ten years after. Histol Histopathol. 2009;24:777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kukuruzinska MA, Lennon K. Protein N-glycosylation: molecular genetics and functional significance. Crit Rev Oral Biol Med. 1998;415–448. doi: 10.1177/10454411980090040301. [DOI] [PubMed] [Google Scholar]
- 25.Andrée B, Fleige A, Arnold H-H BT. Mouse Pop1 is required for muscle regeneration in adult skeletal muscle. Mol Cell Biol. 2002;22:1504–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu YC, Liu CY, Chen YH, Chen RF, Huang CJ, Wang IJ. Blood vessel epicardial substance (Bves) regulates epidermal tight junction integrity through atypical protein kinase C. J Biol Chem. 2012;287:39887–39897. doi: 10.1074/jbc.M112.372078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams CS, Zhang B, Smith JJ, Jayagopal A, Barrett CW, Pino C, Russ P, Presley SH, Peng D, Rosenblatt DO, et al. BVES regulates EMT in human corneal and colon cancer cells and is silenced via promoter methylation in human colorectal carcinoma. J Clin Invest. 2011;121:4056–4069. doi: 10.1172/JCI57873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim M, Jang HR, Haam K, Kang TW, Kim JH, Kim SY, Noh SM, Song KS, Cho JS, Jeong HY, et al. Frequent silencing of popeye domain-containing genes, BVES and POPDC3, is associated with promoter hypermethylation in gastric cancer. Carcinogenesis. 2010;31:1685–1693. doi: 10.1093/carcin/bgq144. [DOI] [PubMed] [Google Scholar]
- 29.Feng Q, Hawes SE, Stern JE, Wiens L, Lu H, Dong ZM, Jordan CD, Kiviat NB, Vesselle H. DNA methylation in tumor and matched normal tissues from non-small cell lung cancer patients. Cancer Epidemiol Biomark Prev. 2008;17:645–654. doi: 10.1158/1055-9965.EPI-07-2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith TK, Bader DM. Characterization of Bves expression during mouse development using newly generated immunoreagents. Dev Dyn. 2006;235:1701–1708. doi: 10.1002/(ISSN)1097-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guillot C, Lecuit T. Mechanics of epithelial tissue homeostasis and morphogenesis. Science. 2013;340:1185–1189. doi: 10.1126/science.1235249. [DOI] [PubMed] [Google Scholar]
- 32.Ragkousi K, Gibson MC. Cell division and the maintenance of epithelial order. J Cell Biol. 2014;207:181–188. doi: 10.1083/jcb.201408044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orsulic S, Huber O, Aberle H, Arnold S, Kemler R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J Cell Sci. 1999;112:1237–1245. [DOI] [PubMed] [Google Scholar]
- 34.Jayagopal A, Yang JL, Haselton FR, Chang MS. Tight junction-associated signaling pathways modulate cell proliferation in uveal melanoma. Investig Ophthalmol Vis Sci. 2011;52:588–593. doi: 10.1167/iovs.10-5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kavanagh E, Buchert M, Tsapara A, Choquet A, Balda MS, Hollande F, Matter K. Functional interaction between the ZO-1-interacting transcription factor ZONAB/DbpA and the RNA processing factor symplekin. J Cell Sci. 2006;119:5098–5105. doi: 10.1242/jcs.02791. [DOI] [PubMed] [Google Scholar]
- 36.Jou TS, Schneeberger EE, Nelson WJ. Structural and functional regulation of tight junctions by RhoA and Rac1 small GTPases. J Cell Biol. 1998;142:101–115. doi: 10.1083/jcb.142.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Russ PK, Pino CJ, Williams CS, Bader DM, Haselton FR, Chang MS. Bves modulates tight junction associated signaling. PLoS One. 2011;6:e14563. doi: 10.1371/journal.pone.0014563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fields IC, Shteyn E, Pypaert M, Proux-Gillardeaux V, Kang RS, Galli T, Fölsch H. v-SNARE cellubrevin is required for basolateral sorting of AP-1B-dependent cargo in polarized epithelial cells. J Cell Biol. 2007;177:477–488. doi: 10.1083/jcb.200610047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tayeb MA, Skalski M, Cha MC, Kean MJ, Scaife M, Coppolino MG. Inhibition of SNARE-mediated membrane traffic impairs cell migration. Exp Cell Res. 2005;305:63–73. doi: 10.1016/j.yexcr.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Luftman K, Hasan N, Day P, Hardee D, Hu C. Silencing of VAMP3 inhibits cell migration and integrin-mediated adhesion. Biochem Biophys Res Commun. 2009;380:65–70. doi: 10.1016/j.bbrc.2009.01.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schindler RFR, Scotton C, Zhang J, Passarelli C, Ortiz-Bonnin B, Simrick S, Schwerte T, Poon KL, Fang M, Rinné S, et al. POPDC1S201F causes muscular dystrophy and arrhythmia by affecting protein trafficking. J Clin Invest. 2016;126:239–253. doi: 10.1172/JCI79562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tucker SJ, Amunjela JN. Dysregulation of POPDC1 promotes breast cancer cell migration and proliferation. Biosci Rep. 2017 Dec 22; 37(6):1–15, BSR20171039.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beese M, Wyss K, Haubitz M, Kirsch T. Effect of cAMP derivates on assembly and maintenance of tight junctions in human umbilical vein endothelial cells. BMC Cell Biol. 2010;11:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.D’Souza T, Agarwal R, Morin PJ. Phosphorylation of Claudin-3 at threonine 192 by cAMP-dependent protein kinase regulates tight junction barrier function in ovarian cancer cells. J Biol Chem. 2005;280:26233–26240. [DOI] [PubMed] [Google Scholar]
- 45.Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells — an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–749. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- 46.Thoreson M, Ab R. Altered expression of the catenin p120 in human cancer: implications for tumor progression. Differentiation. 2002;70:583–589. doi: 10.1046/j.1432-0436.2002.700607.x. [DOI] [PubMed] [Google Scholar]
- 47.Yap A. The morphogenetic role of cadherin cell adhesion molecules in human cancer: a thematic review. Cancer Invest. 1998;16:252–261. doi: 10.3109/07357909809039774. [DOI] [PubMed] [Google Scholar]
- 48.van Aken J, Cuvelier CA, de Wever N, Roels J, Gao Y, Mareel MM. Immunohistochemical analysis of E-cadherin expression in human colorectal tumours. Pathol Res Pract. 1993;18:975–978. doi: 10.1016/S0344-0338(11)80667-9. [DOI] [PubMed] [Google Scholar]
- 49.Dhawan P, Singh AB, Deane NG, No YR, Shiou SR, Schmidt C, Neff J, Washington MK, Beauchamp RD. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J Clin Invest. 2005;115:1765–1767. doi: 10.1172/JCI24543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hewitt K, Agarwal R, Morin P. The claudin gene family: expression in normal and neoplastic tissues. BMC Cancer. 2006;6:186. doi: 10.1186/1471-2407-6-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ni S, Xu L, Huang J, Feng J, Zhu H, Wang G, Increased WX. ZO-1 expression predicts valuable prognosis in non-small cell lung cancer. Int J Clin Exp Pathol. 2013;6:2887–2895. [PMC free article] [PubMed] [Google Scholar]
- 52.Martin TA, Watkins G, Mansel RE, Jiang WG. Loss of tight junction plaque molecules in breast cancer tissues is associated with a poor prognosis in patients with breast cancer. Eur J Cancer. 2004;40:2717–2725. doi: 10.1016/j.ejca.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 53.Kaihara T, Kusaka T, Nishi M, Kawamata H, Imura J, Kitajima K, Itoh-Minami R, Aoyama N, Kasuga M, Oda Y, et al. Dedifferentiation and decreased expression of adhesion molecules, E-cadherin and ZO-1, in colorectal cancer are closely related to liver metastasis. J Exp Clin Cancer Res. 2003;22:117–123. [PubMed] [Google Scholar]
- 54.Parang B, Kaz AM, Barrett CW, Short SP, Ning W, Keating CE, Mittal MK, Naik RD, Washington MK, Revetta FL, et al. BVES regulates c-Myc stability via PP2A and suppresses colitis-induced tumourigenesis. Gut. 2017;66:852–862. doi: 10.1136/gutjnl-2015-310255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Snider AJ, Bialkowska AB, Ghaleb AM, Yang VW, Obeid LM, Hannun YA. Murine model for colitis-associated cancer of the colon. Methods Mol Biol. 2016;1438:245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Waldner MJ, Neurath MF. Mechanisms of immune signaling in colitis-associated cancer. CMGH. 2015;1:6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, Signori E, Fazio VM. The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J Carcinog. 2011. October;29(10):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parang B, Barrett CW, Williams CS. AOM/DSS model of colitis-associated cancer. Methods Mol Biol. 2016;1422:297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ciclitira PJ, Macartney JC, Evan G. Expression of c-myc in non-malignant and pre-malignant gastrointestinal disorders. J Pathol. 1987;151:293–296. doi: 10.1002/(ISSN)1096-9896. [DOI] [PubMed] [Google Scholar]
- 60.Brentnall TA, Pan S, Mp B, Crispin DA, Mirzaei H, Cooke K, Tamura Y, Nikolskaya T, JeBailey L, Dr G, et al. Proteins that underlie neoplastic progression of ulcerative colitis. Proteomics Clin Appl. 2009;3:1326–1337. doi: 10.1002/prca.200900061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suzuki R, Miyamoto S, Yasui Y, Sugie S, Tanaka T. Global gene expression analysis of the mouse colonic mucosa treated with azoxymethane and dextran sodium sulfate. BMC Cancer. 2007;7:84. doi: 10.1186/1471-2407-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arnold HK, Sears RC. A tumor suppressor role for PP2A-B56 through negative regulation of c-Myc and other key oncoproteins. Cancer Metastasis Rev. 2008;27:147–158. doi: 10.1007/s10555-008-9128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thompson J, Williams C. Protein Phosphatase 2A in the regulation of Wnt signaling, stem cells, and cancer. Genes. 2018;9:121. doi: 10.3390/genes9030127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nunbhakdi-Craig V, Machleidt T, Ogris E, Bellotto D, White CL, Sontag E. Protein phosphatase 2A associates with and regulates atypical PKC and the epithelial tight junction complex. J Cell Biol. 2002;158:967–978. doi: 10.1083/jcb.200206094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Han P, Fu Y, Liu J, Wang Y, He J, Gong J, Li M, Tan Q, Li D. Netrin-1 promotes cell migration and invasion by down-regulation of BVES expression in human hepatocellular carcinoma. Am J Cancer Res. 2015;5:1396–1409. [PMC free article] [PubMed] [Google Scholar]
- 66.Han P, Fu Y, Luo M, He J, Liu J, Liao J, Tian D, Yan W. BVES inhibition triggers epithelial-mesenchymal transition in human hepatocellular carcinoma. Dig Dis Sci. 2014;59:992–1000. doi: 10.1007/s10620-013-2992-3. [DOI] [PubMed] [Google Scholar]
- 67.Amunjela J, Tucker S. POPDC1 is suppressed in human breast cancer tissues and is negatively regulated by EGFR in breast cancer cell lines. Cancer Lett. 2017;406:81–92. doi: 10.1016/j.canlet.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 68.Esteller M. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene. 2002;21(35):5427–5440. [DOI] [PubMed] [Google Scholar]
- 69.Mantovani A, Mantovani A, Allavena P, Allavena P, Sica A, Sica A, Balkwill F, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 70.Reddy VK, Short SP, Barrett CW, Mittal MK, Keating CE, Thompson JJ, Harris EI, Revetta F, Bader DM, Brand T, et al. BVES regulates intestinal stem cell programs and intestinal crypt viability after radiation. Stem Cells. 2016;34:1626–1636. doi: 10.1002/stem.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 72.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]