

Abstract
Background
Avian influenza A H5N1 virus can cause lethal disease in humans. The virus can trigger severe pneumonia and lead to acute respiratory distress syndrome. Data from clinical, in vitro and in vivo suggest that virus-induced cytokine dysregulation could be a contributory factor to the pathogenesis of human H5N1 disease. However, the precise mechanism of H5N1 infection eliciting the unique host response are still not well understood.
Methods
To obtain a better understanding of the molecular events at the earliest time points, we used RNA-Seq to quantify and compare the host mRNA and miRNA transcriptomes induced by the highly pathogenic influenza A H5N1 (A/Vietnam/3212/04) or low virulent H1N1 (A/Hong Kong/54/98) viruses in human monocyte-derived macrophages at 1-, 3-, and 6-h post infection.
Results
Our data reveals that two macrophage populations corresponding to M1 (classically activated) and M2 (alternatively activated) macrophage subtypes respond distinctly to H5N1 virus infection when compared to H1N1 virus or mock infection, a distinction that could not be made from previous microarray studies. When this confounding variable is considered in our statistical model, a clear set of dysregulated genes and pathways emerges specifically in H5N1 virus-infected macrophages at 6-h post infection, whilst was not found with H1N1 virus infection. Furthermore, altered expression of genes in these pathways, which have been previously implicated in viral host response, occurs specifically in the M1 subtype. We observe a significant up-regulation of genes in the RIG-I-like receptor signaling pathway. In particular, interferons, and interferon-stimulated genes are broadly affected. The negative regulators of interferon signaling, the suppressors of cytokine signaling, SOCS-1 and SOCS-3, were found to be markedly up-regulated in the initial round of H5N1 virus replication. Elevated levels of these suppressors could lead to the eventual suppression of cellular antiviral genes, contributing to pathophysiology of H5N1 virus infection.
Conclusions
Our study provides important mechanistic insights into the understanding of H5N1 viral pathogenesis and the multi-faceted host immune responses. The dysregulated genes could be potential candidates as therapeutic targets for treating H5N1 disease.
Electronic supplementary material
The online version of this article (10.1186/s12920-018-0335-0) contains supplementary material, which is available to authorized users.
Keywords: Influenza A virus, H5N1, Macrophage polarization, Transcriptomics, RNA-Seq
Background
H5N1 infection could trigger severe pneumonia and lead to acute respiratory distress syndrome, which could be lethal in humans. According to the World Health Organization, there were 856 human reported cases of influenza A H5N1 virus infection resulting in 452 deaths from January 2003 to May 2017; the fatality rate was over 50%. Contributory factors such as high viral load in the infected lungs, tropism for the lower respiratory tract as well as dysregulated host response after infection have been proposed to explain the unusual virulence of this virus [1, 2]. In humans, lungs of H5N1 virus infected patients have markedly upregulation of cytokines [3]. Mice, ferrets and macaques experimentally infected by H5N1 virus, also showed dysregulated cytokine expression [4–6]. We previously reported that H5N1 virus infection triggers high pro-inflammatory cytokine and chemokine expression in primary human macrophages compared to that by seasonal H1N1 virus infection. Although these data suggest that dysregulated host response contributes to the pathogenesis of H5N1 virus infection, the precise mechanisms as well as the host response profile has not been well studied.
We and others have used microarray or real time quantitative polymerase chain reaction (RT-qPCR) to measure systematically the mRNA expression profile in influenza A virus infected cells. Results indicate that highly pathogenic avian H5N1 virus triggers dysregulated pro-inflammatory cytokine expression in vitro and in vivo [7–10], which may be relevant to the unusual severity of H5N1 disease in humans.
Macrophages are key immune cells that produce cytokines in host defense, and could be one of the primary targets during H5N1 virus infection. Building on these studies, here we use RNA sequencing (RNA-Seq) to characterize the host mRNA and microRNA (miRNA) transcriptomes in human macrophages after infection by the highly pathogenic H5N1 or low pathogenic H1N1 influenza virus to identify the initial molecular events relating to viral pathogenesis at the early stages of infection. The sensitivity and specificity of RNA-Seq enabled us to identify a strong confounding effect introduced by M1 and M2 populations. Statistically quantifying and correcting this effect, a process that cannot be adequately performed with previous microarray data, enabled us to refine the set of H5N1 virus induced differentially expressed genes specific to the M1 subtype. In particular, we observe significant up-regulation of genes in the RIG-I-like receptor signaling pathway early in H5N1 virus infection which lead to expression of suppressors for interferon (IFN) signaling and in turn cause suppression of host antiviral mechanism at later post-infection time. Results here provide new mechanistic insights and explanations for the severity of H5N1 infections in humans.
Methods
Viruses
Viruses used in this study include A/Vietnam/3212/04 (H5N1), isolated from a fatal human case in Vietnam, 2004; and A/Hong Kong/54/98 (H1N1), a human influenza A H1N1 virus. Isolation and propagation of viruses were performed as previously described [10].
Primary human macrophage culture
Peripheral blood monocytic cells were isolated from blood packs of healthy donors, obtained from the Hong Kong Red Cross Blood Transfusion Service, by centrifugation with Ficoll-Paque PLUS density gradient media (GE Healthcare, Uppsala, Sweden). Monocytes were further purified by plastic adherence and let to differentiate in RPMI-1640 medium (Life Technologies, Grand Island, NY, USA) supplemented with 5% heat-inactivated autologous plasma. Differentiated cells were seeded onto multi-well tissue culture plates for subsequent experiments.
Influenza virus infection of macrophage
Influenza A H1N1 and H5N1 viruses were used at a multiplicity of infection (MOI) of 2 for infection of differentiated monocyte-derived macrophages. Viruses were allowed to adsorb to cells for 30 min before the inoculum was removed, followed by a wash with pre-warmed culturing medium. Infected macrophages were then incubated with serum free medium (SFM) (Life Technologies) supplemented with penicillin and streptomycin for indicated period of time. Mock-infected cells were included as controls.
Total RNA isolation
Total RNA was extracted from cells with mirVana™ miRNA Isolation Kit (Ambion, Foster City, CA, USA) at 1-, 3-, and 6-h after infection per manufacturer’s instructions. Quality of RNA was assessed by an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). As an indication of high RNA quality, all samples isolated attained a RNA Integrity Number (RIN) of at least 9.0.
Illumina mRNA library preparation
Ribosomal RNAs (rRNAs) were removed from 1 μg total RNA using the RiboMinus™ Transcriptome Isolation kit (Human/Mouse) (Invitrogen) according to the manufacturer’s instructions. The remaining RNA was heat fragmented at 94 °C for 5 min in 5 × Array Fragmentation Buffer (Ambion). The fragmented RNA was reverse transcribed to cDNA using SuperScript Double Stranded cDNA synthesis kit (Invitrogen, Carlsbad, CA, USA). The resulting cDNA was purified using QiaQuick PCR column (Qiagen, Valencia, CA, USA), and the ends were ligated with adapters using the Quick Ligation™ Kit (New England Biolabs, Ipswich, MA, USA).
Illumina small RNA library preparation
The rRNA-depleted RNA described as above was used for preparation of small RNA libraries. The RNA was ligated with the 3′ and 5′ adapter using T4 RNA Ligase 1 and Ligase 2, respectively (New England Biolabs). The following sequences were used as the 3′ and 5′ adapter, respectively: 5’-rAppAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG/3ddC/− 3′, and 5’-rArCrArCrUrCrUrUrUrCrCrCrUrArCrArCrGrArCrGrCrUrCrUrUrCrCrGrArUrCrU-3′. The ligation product was reverse transcribed to cDNA using SuperScript II Reverse Transcriptase (Invitrogen) and an oligonucleotide primer (5’-CTCGGCATTCCTGCTGAACCGCTC–3′).
Illumina sequencing and data generation
DNA sequencing was performed on Illumina GAIIx platform per manufacturer’s instructions (Illumina, San Diego, CA, USA). Single end sequencing reads of 38-bp were generated for mRNA samples and paired-end sequencing reads of 76-bp were generated for miRNA samples. Each library sample was sequenced on three or more lanes of the Illumina flow-cell. Base calling was performed using Off-line Base Caller (OLB) v1.6.0 software.
Sequencing data analysis of mRNAs
The mRNA sequence reads were trimmed of adapter, homopolymer, and residue rRNA sequences, and the resulting reads were mapped to the Genome Reference Consortium Human Build 37 (GRCh37). Only manually curated genes with an annotated protein product as described in the SWISSPROT/UniProt database [11] were used for analysis.
Sequencing data analysis of miRNAs
The small RNA sequence reads were trimmed of low-quality sequences and adapter sequences. The trimmed reads were compared to the miRBase database (http://www.mirbase.org/). Due to imperfect Dicer processing, a 5-bp overhang on the 5′- and 3′-end of mature/mature* miRNAs and 2 mismatches within the mature/mature* miRNAs were allowed in the mapping. The expression level of mature/mature* miRNAs was calculated using the RPM normalization model (Reads mapped Per Million mappable reads). The differential expression of miRNAs was evaluated by comparing RPM values derived from influenza A H1N1 or H5N1 virus-infected cells against the values derived from mock control, with the significance assessed with Z-score using Z-test. The remaining reads were mapped to additional RNA databases, including miRNA precursors, rRNA (18S, 28S, and 5.8S), tRNA (GtRNAdb) [12], snoRNA (snoRNABase) [13], and mRNA (NCBI RefSeq database) with a minimum similarity of 92%. An average of 25.6% of the raw reads was usable for miRNA quantification.
Clustering analysis
Multidimensional scaling (MDS) analysis was used to identify broad library-wise trends. Although the first dimension indicated a slight dissimilarity at 6-h post-infection in all three time-series, no library-wise confounding factors were identified. The Bioconductor package edgeR was used to perform MDS analysis [14]. The k-means clustering was performed using an implementation designed for count-based data [15]. An elbow plot was used to select k = 3 as the appropriate number of clusters.
Macrophage subtype population estimation
Non-negative matrix factorization to estimate the contribution of the two subtypes used Kullback-Leibler divergence as a cost function [16]. Bootstrap estimation was used to determine the 95% confidence intervals.
Statistical model to compute mRNA differential expression
The Bioconductor package edgeR was used to fit a negative binomial model to the expression of individual genes [14]. Experiment-wide and gene-wise estimates of dispersion were used as recommended. The estimated macrophage subtype percentage was always included as a covariate (xsubtype). The covariate (xclass) consists of a binary label that identifies membership in one of two possible groups: sample or control. Statistical significance was determined using a likelihood ratio test (the likelihood of the full model over the model missing xclass). Multiple testing corrections were performed using the Benjamini-Hochberg FDR method. The genes with likelihood ratio ≥ 5 and p-value ≤0.03 were considered to be statistically significant.
Pathway analysis
GSEA (Gene Set Enrichment Analysis) pathway analysis was performed using pre-ranked gene lists (using the negative binomial model significance and the sign of model coefficient βclass as the ranking metric), the GSEA software package and the MSigDB gene sets C2:CP (canonical pathways) and C5 (Gene Ontology gene sets) [17]. Results with a false discovery rate less than 5% were considered significant.
Results
Macrophage heterogeneity and plasticity in response to H1N1 and H5N1 virus infection
The mRNAs isolated from primary human macrophages after infection by H5N1, H1N1 viruses, or mock infection at 1-, 3-, and 6-h post infection were analyzed by RNA-Seq. At least 20 million filtered reads were generated for each sample time-point (Additional file 1: Table S1). MDS analysis was used to identify broad sample-wise trends. The overall orientation of the samples did not correspond strongly to any biologically meaningful pattern (Additional file 2: Figure S1). K-means clustering analysis was subsequently performed for the gene expression profiles across time-points (Additional file 2: Figure S2). We identified three strong clusters: two of which are oppositional clusters indicative of differential gene expression originating from at least two distinct cell types, and a third cluster corresponding to genes that are expressed at a similar level across all cell types (Fig. 1a). Inspection of the genes present in each of the two oppositional clusters revealed a strong association to markers of M1 or M2 macrophages (Table 1). Using non-negative matrix factorization with Kullback-Leibler divergence as the cost function [16], we estimated the proportion of the two presumptive macrophage subtypes from the RNA-Seq reads. This analysis revealed a markedly varied population of macrophage subtypes along infection and across time, with a distinct pattern in subtype ratios observed in response to H5N1 virus infection when compared to H1N1 virus or mock infection (Fig. 1b). We then estimated expression of macrophage subtype marker genes [18] across samples while considering the estimated proportions of macrophage subtypes as a confounding factor. The expression of genes known to be restricted to one of the two macrophage subtypes was fit to a negative binomial linear model with the estimated division between subtypes included as a covariate. Of the genes with sufficient expression level for the model to be fitted, 12/12 (100%) M1 markers fitted correctly (βsubtype > 0) to one of the estimated subpopulations (7 at p < 0.05), while 16/20 (80%) M2 markers fitted correctly (βsubtype < 0) to the alternative subpopulation (9 at p < 0.05) (Table 1). Thus, the macrophage subtype marker genes accurately fitted a model of their expression based on estimated macrophage proportions. This phenomenon was retrospectively investigated using previously published microarray data generated under identical experimental conditions [10]. We observed that the average rank order of genes across donor samples, with the most positive and negative βsubtype as calculated from the RNA-Seq data showed a similar oppositional trend in the previous microarray data. However, the qualitative nature of the microarray data did not predict the presence of distinct macrophage subtypes within the statistical threshold of our model (Fig. 1c).
Fig. 1.
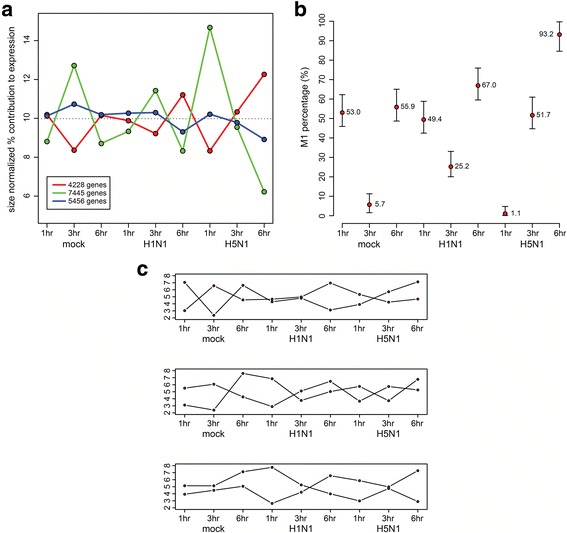
Differential gene expression profile reflecting macrophage polarization in response to influenza A virus infection. a k-means clustering analysis of RNA-Seq counts using a Poisson-based distance metric. The final representative values in each cluster were normalized to the total library size for the purpose of visualization. b The estimated percentage of the M1 macrophage subtype in each sample as estimated by non-negative matrix factorization. 95% confidence intervals are denoted by the whiskers displayed around each point. c The relative pattern of expression of genes identified as strong M1 and M2 subtype markers based on RNA-Seq data. Each point represents the mean sample-wise rank of the top 1000 putative M1-associated genes (red) and top 1000 putative M2-associated genes (blue) based on the cell subtype coefficient estimated from the linear model applied to the RNA-Seq dataset
Table 1.
Fitted expression of macrophage subtype-specific markers
| Gene | Subtype | β0a | βsubtype | p-value | Gene | Subtype | β0 | βsubtype | p-value |
|---|---|---|---|---|---|---|---|---|---|
| CCL2 | M1 | − 11.57 | 1.73 | 0.005 | CCL17 | M2a | − 12.04 | − 1.04 | < 0.001 |
| CCL3 | M1 | − 9.66 | 1.18 | 0.009 | CCL18 | M2a | − 13.42 | − 0.459 | 0.219 |
| CCL5 | M1 | − 11.82 | 2.03 | 0.034 | CCL22 | M2a | − 7.24 | 0.0164 | 0.904 |
| CXCL10 | M1 | expressed in H5N1 6-h only | CCL24 | M2a | − 13.58 | − 1.19 | 0.066 | ||
| CXCL11 | M1 | − 24.95 | 13.07 | 0.001 | CXCR1 | M2a | not expressed | ||
| CXCL16 | M1 | − 8.47 | 0.0759 | 0.443 | CXCR2 | M2a | not expressed | ||
| CXCL9 | M1 | − 13.76 | 2.25 | 0.027 | IL1R2 | M2a | − 11.25 | − 0.300 | 0.027 |
| IL12A | M1 | − 13.98 | 0.576 | 0.150 | CD163 | M2a | − 9.96 | 0.238 | 0.176 |
| IL23A | M1 | − 13.64 | 0.835 | 0.148 | FCER2 | M2a | − 12.21 | − 1.28 | 0.003 |
| IL8 | M1 | − 8.87 | 0.881 | 0.100 | IL10 | M2a/b/c | − 11.86 | 0.379 | 0.035 |
| CCR7 | M1 | − 11.97 | 0.367 | 0.351 | ARG1 | M2a/b/c | − 14.54 | − 0.289 | 0.652 |
| TLR2 | M1 | − 9.74 | 0.288 | 0.028 | IL1RN | M2a/c | − 7.34 | 0.677 | 0.002 |
| TLR4 | M1 | − 8.28 | 0.488 | < 0.001 | MRC1 | M2a/c | − 12.23 | − 0.869 | 0.182 |
| MRC2 | M2a/c | − 9.79 | − 0.455 | 0.032 | |||||
| CCL1 | M2b | − 13.94 | − 0.895 | 0.098 | |||||
| CCL16 | M2c | − 13.39 | − 0.814 | 0.067 | |||||
| CCL18 | M2c | − 13.42 | − 0.459 | 0.219 | |||||
| CXCL13 | M2c | − 10.44 | − 0.739 | < 0.001 | |||||
| TGFB1 | M2c | − 9.45 | − 0.508 | 0.047 | |||||
| CCR2 | M2c | − 12.94 | − 0.343 | 0.378 | |||||
| CD14 | M2c | − 8.83 | − 0.592 | 0.003 | |||||
| SLAMF1 | M2c | − 11.22 | − 0.166 | 0.251 | |||||
aβ0 indicates the intercept for the fitted linear model
Together, our analyses reveal that the activation state of the macrophage population can vary in response to virus infection and across time, suggesting that macrophage subtype switching is a highly plastic process. Macrophage subtype composition is thus an important factor that must be considered when investigating the impact of viral infections on host gene expression.
Differentially regulated gene expression in response to H1N1 and H5N1 virus infection
To estimate global differential expression, we extended the above-described negative binomial statistical model to all genes. The estimated M1 subtype percentage was again included as a model covariate (see Materials and Methods). The significance of differential expression was evaluated using a likelihood ratio test against a null model. To identify differential expression in an unbiased manner and identify the most informative grouping of samples, we considered all 256 possible combinations of the nine RNA-Seq samples using a two-class (sample vs. control) experimental comparison design (e.g. one sample vs. the rest, all samples at one time-point vs. all samples at other time points). The total likelihood of all combinations was scaled to one. Most configurations never contribute a satisfactory model of differential expression for any gene. However, a large number of genes produced models that are highly consistent with differential expression: 1) at 6-h post-infection in all three time-series, and 2) at 6-h post-infection with H5N1 virus (Fig. 2). The resulting list of differentially expressed genes, considering M1 and M2 macrophage subtypes, is more sensitive and accurate compared to previous microarray studies (Additional file 3). It allows us to apply stringent criteria (likelihood ratio ≥ 5 and p-value ≤0.03) to select the candidate genes with significant differential expression for downstream pathway analysis.
Fig. 2.
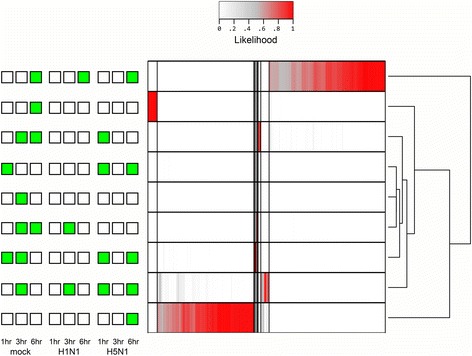
Heatmap of the scaled likelihood for each gene representing a specific pattern of differential expression. Of the 256 possible groupings, only the nine shown here have significant likelihood of differential expression for at least one gene. The top (6 h-specific differential expression) and bottom (H5N1, 6 h-specific differential expression) patterns are the most prominent
H5N1 virus infection induces markedly up-regulated expression of RLRs and leads to the significant induction of IFN signaling
Pathway analysis using curated pathways and Gene Ontology terms [17] revealed that H5N1 virus induced gene expression at 6-h post-infection is predominately associated with host defense pathways, which share several common regulators (Additional file 1: Table S2 and Table 2). The majority of genes identified having altered expression in the M1 subpopulation, are associated with these pathways. In agreement with our previous microarray data [10], RIG-I-like receptor (RLR) signaling pathway is strongly up-regulated specifically in response to H5N1 virus infection. Such genes include: DDX58, IFIH1, DHX58, and TRIM25, which detect viral nucleic acids; MAVS, TRAF2, AKT1, RELA, and NFKBIA, which initiate or propagate a signaling cascade upon detection to regulate the transcription of target response genes; as well as cytokine genes such as CCL2, CCL5, CXCL10, and IL12B (Fig. 3). In addition, we observed significant up-regulation of many other genes related to IFN signaling pathways, such as IFIT1, IFIT2, IFIT3, IFIT5, RSAD2, OASL, GSP1, HERC5, IFI44, IFI44L, CD274, GBP2, GBP4, OAS2, DDX60, EIF2AK2, IRF1, BST2, and MNDA. All of these data clearly indicate that at the early stages of infection, influenza A H5N1 virus strongly activates a more prominent IFN signaling through RLRs, when compared with H1N1 virus infection.
Table 2.
Significantly enriched pathways in response to influenza H5N1 virus infection at 6-h post-infection
| Pathway Name | Gene Count | Enrichment | FDR p-value | |
|---|---|---|---|---|
| upregulated | NOD-like receptor signaling pathway [KEGG] | 58 | 10 | 0.012 |
| RIG-I-like receptor signaling pathway [KEGG] | 66 | 18 | 0.006 | |
| Chemokine receptors bind chemokines [Reactome] | 51 | 12 | 0.020 | |
| Cytosolic DNA sensing pathway [KEGG] | 55 | 17 | 0.019 | |
| downregulated | Packaging of telomere ends [Reactome] | 43 | 20 | 0.000 |
| RNA polymerase I promoter clearance [Reactome] | 69 | 30 | 0.005 | |
| Viral mRNA translation [Reactome] | 82 | 39 | 0.009 | |
| RNA polymerase I promoter opening [Reactome] | 48 | 26 | 0.009 | |
| Ribosome [KEGG] | 84 | 42 | 0.011 | |
| Insulin synthesis and secretion [Reactome] | 126 | 50 | 0.011 | |
| Peptide chain elongation [Reactome] | 82 | 38 | 0.039 |
Fig. 3.
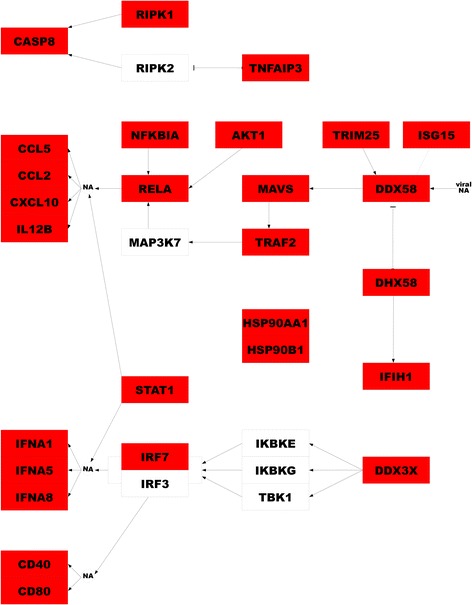
The genes and their functional relationships within the RLR signaling pathway. Genes highlighted in red show strong M1 subtype specific differential expression in response to H5N1 virus infection at 6-h post-infection. “NA” indicates nucleic acids
RNA-Seq data was confirmed using real-time PCR on selected genes in the pathway. Relative expression levels of these selected genes peaked 6-h post-infection with both viruses, with induced levels markedly higher after infection by the H5N1 virus when compared with the H1N1 virus (Additional file 2: Figure S3).
Positive and negative regulation of antiviral responses in H5N1 virus-infected macrophages
To get a better understanding of how innate immune response regulated after influenza A virus infection, we compared the expression level of the suppressors of cytokine signaling (SOCS) in H5N1 and H1N1 virus infected cells. The SOCS family, consisting of eight members, SOCS1–7 and cytokine inducible SH2 containing protein (CISH), are negative regulators for cytokine responses. As illustrated in Fig. 4, expression of SOCS1 and SOCS3 were at least 2.5-fold higher in H5N1 virus-infected macrophages than in mock-infected cells at 6-h post infection. By contrast, SOCS genes were not significantly up-regulated following H1N1 virus infection. These observations are in agreement with our previous microarray data. SOCS1 and SOCS3 are also known to act as negative regulators of influenza A H3N2 virus triggered innate immune response by suppression of type I IFN expression and signaling [19].
Fig. 4.
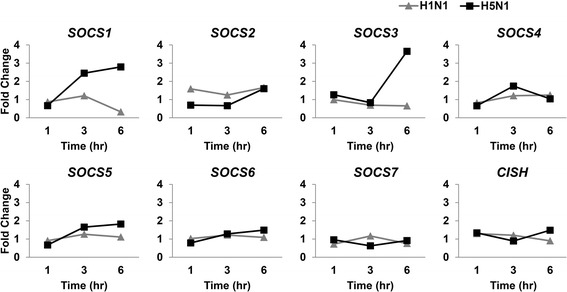
Expression patterns of the SOCS family genes at 1-, 3-, and 6-h post-infection compared to mock infection. SOCS1 and SOCS3 were expressed with at least 2.5-fold higher in H5N1-infected macrophages than in mock-infected control cells at 6-h post-infection. No genes were significantly up-regulated in H1N1 virus-infected cells
To assess if up-regulation of SOCS at early stages of H5N1 virus infection would correlate with decreased expression of antiviral IFNs later in infection, we extended our investigation to later time points. While IFN-β was found to be hyper-induced in H5N1- compared to H1N1 virus-infected macrophages at 6-h post infection, the expression of IFN-β started to fall in H5N1 virus-infected cells at 8-h post infection. Expression of IFN-β in H5N1 virus-infected cells continued to fall at 12 and 24-h post infection, while increased expression of IFN-β was observed in H1N1 virus-infected cells during the same period (Fig. 5a).
Fig. 5.
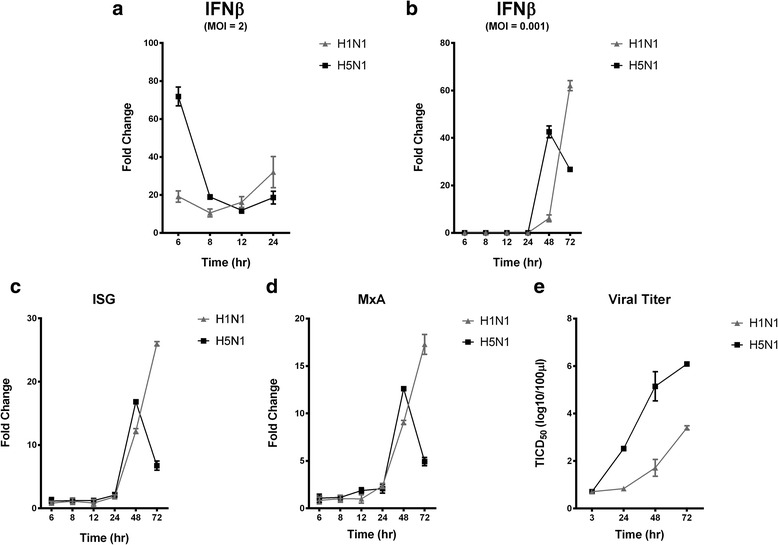
Kinetics of antiviral gene expression and virus replication in influenza A virus infected human macrophages. Expression of IFN-β in human macrophages after (a) single-round or (b) multiple-rounds of influenza A virus replication. Expression kinetics of antiviral genes, (c) ISG15 and (d) MxA in response to influenza A virus infection. e Influenza A viral replication kinetics in human macrophages. Data of mean ± S.D. of three independent experiments are shown
As SOCS1 and SOCS3 were induced by H5N1 virus in the initial round of viral replication at 6-h post infection, we next investigated if this cellular event could affect the expression of IFN-β after multiple rounds of influenza virus replication by infecting the cells using MOI of 0.001. IFN-β expression was found to be hyper-induced after H5N1 virus infection compared that with H1N1 virus infection at 48-h post infection. Interestingly, at later post infection time, IFN-β was significantly decreased in H5N1 virus-infected cells, while there was a further increase in response to H1N1 virus infection (Fig. 5b). Similar expression pattern of interferon-stimulated gene (ISG) (Fig. 5c) and myxovirus resistance protein 1 (MxA) (Fig. 5d) was observed as well. Viral replication kinetics correlated well with the expression kinetics of these antiviral genes of which H5N1 virus infection results in a much higher viral load compared to H1N1 virus infection in macrophages, especially at later post-infection time (Fig. 5e). This data highlights the importance of SOCS1 and SOCS3 in H5N1 virus-induced innate immune response, suggesting a possible relationship between hyper-induction of these negative regulators of IFN signaling in the initial rounds of virus replication, with a concomitant decreased expression in antiviral genes leading to an increase in viral load at the later stage of infection that could contribute to the pathogenesis of H5N1 virus infection.
Distinct cellular miRNA expression patterns in response to influenza A H1N1 and H5N1 virus infection
MiRNA are 21–23 nt RNA molecules that negatively regulate the transcription or translation of specific target mRNA genes by binding to partially complementary sites at the 3’ UTR of the target mRNAs. We identified the expression patterns of the miRNAs in H1N1 and H5N1 virus-infected macrophages at 1-, 3-, and 6-h post-infection and assessed their differential expression levels relative to mock-infected cells (Fig. 6). A total of 105,842,096 filtered high-quality reads, representing 2,854,995 unique miRNA species, were generated for miRNAs for H5N1 or H1N1 virus-infected macrophages at 1-, 3-, and 6-h post-infection. A total of 361 mature miRNAs and 113 mature* miRNAs were identified from the nine samples (Additional file 1: Table S3). Due to insufficient read counts (Fig. 6a) to support a macrophage subtype-specific statistical model for miRNA expression data, we used a combined macrophage statistical method to characterize the differential expression. We calculated the miRNA expression fold change in influenza A virus-infected cells compared to mock-infected control cells and assessed the significance of the differential expression for each miRNA using Z-statistics (Additional file 4). A total of 102 mature miRNAs were found to be significant based on the criteria that fold-change ≥1.2 and Z-score ≥ 3 (Fig. 6b, c).
Fig. 6.
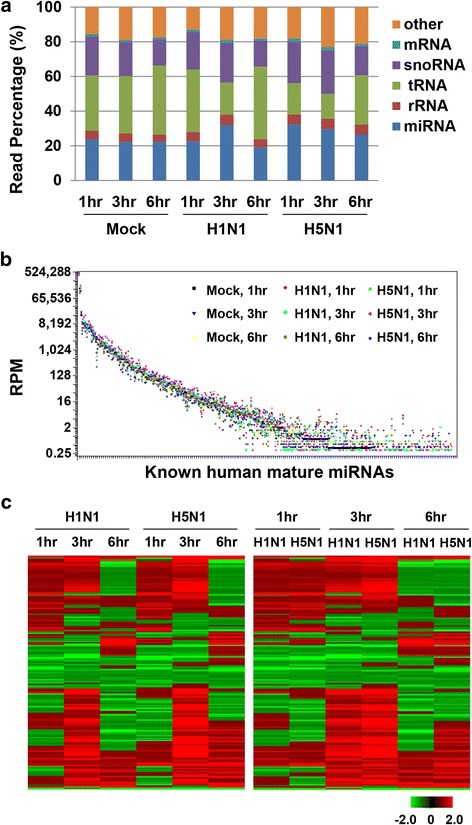
Small RNA expression profiles of macrophages in response to H5N1 virus- and H1N1 virus-infection. a Composition of human macrophage small RNA libraries at different post-infection time points. b Expression patterns for the identified human mature miRNAs ordered by descending RPM in mock-infected macrophage at 1-h post-infection. RPM represents number of reads per million mappable reads. c Heatmaps of the known mature miRNA differential expression profiles in influenza A H5N1 virus- and H1N1 virus-infected macrophages compared with mock-infected cells at different post-infection time points. Co-regulated genes were clustered
Top ranked differentially expressed miRNAs regulate innate immunity pathways
In order to identify the distinct cellular response to H5N1 virus and H1N1 virus-infection underlying the differentially expressed miRNAs, we seek to make correlations between the differential expressions of miRNAs with that of the mRNAs quantified above by computationally predicting the miRNA target genes. MiRNAs with significant differential expression (fold-change ≥1.2 and Z-score ≥ 3) were used for target prediction at each time point using DIANA-microT [20]. We tabulated a set of potential target genes based on an inversely correlated expression pattern between the target mRNA and miRNAs, e.g. up-regulated miRNA and down-regulated target mRNAs or vice versa, and performed pathway enrichment analysis for the target genes inversely regulated compared to the regulating miRNAs (Additional file 5). Overall, many pathways are enriched with genes targeted by differentially expressed miRNAs, which is expected due to the large number of interactions between miRNAs and their target mRNAs. Specifically, the RIG-I like receptor signaling pathway is significantly activated in response to H5N1 virus at multiple time points in response to infection. (Additional file 1: Table S4). The enrichment of this pathway is most significant at 6-h post-infection in H5N1 virus-infected cells, with at least 10 genes targeted by 10 different down-regulated miRNAs (Additional file 2: Figure S4). Our results imply the specific regulatory roles of miRNAs in response to H5N1 influenza virus infection.
Discussion
Studies have indicated that human macrophages are one of the target cells of influenza A H5N1 virus infection in lung, and that the pathogenicity of this virus may be due to the dysregulation of the host immune response [10, 21–24]. In an effort to identify specific mechanisms underlying the pathogenesis of the highly pathogenic avian influenza A H5N1 virus in human, we compared the host transcriptomes of human macrophages infected with either highly pathogenic A/Vietnam/3212/04 (H5N1) virus or the low pathogenic A/Hong Kong/54/98 (H1N1) virus at 1-, 3-, and 6-h post-infection using RNA-Seq. The high sensitivity of the RNA-Seq platform enables identification of macrophage cell heterogeneity and plasticity in response to influenza A virus infection. Here, we demonstrate for the first time that macrophage subtype could be an important confounding factor that should be taken into account to interpret differential gene expression in this experimental system. When accounted for using a statistical model, the RNA-Seq data reveals the quantitative differences of thousands of genes during H5N1 and H1N1 virus infection.
Our present study, together with recent microarray and RNA-Seq analysis of H5N1 virus-infected human airway epithelial and endothelial cells [25, 26] showed a common pattern of gene expression. Notwithstanding cell type differences used in these studies, genes involved in IFN signaling, as well as a number of cytokine and chemokine genes (CCL5 and CXCL10) were differentially up-regulated in response to H5N1 virus infection. These expression profiles are in agreement with previous reports, implicating the importance of macrophages, epithelial cells and endothelial cells in disease pathogenesis of H5N1 virus infection. [22, 23, 27, 28]. In vivo experiments employing mice [29], ferrets [7], or ducks [30] to investigate the host gene expression profiles after H5N1 virus infection, have revealed a general phenomenon, whereby IFN and chemokine pathways are significantly up-regulated in lungs after H5N1 virus infection. Despite a pronounced similarity in host response across species following infection by highly pathogenic H5N1 virus, there were notable fine differences in certain aspects of the observed response. In duck, for example, induced expression of antiviral gene IFIT5 was observed only following infection with low pathogenic virus, whereas highly pathogenic H5N1 virus strain did not induce expression of this gene [30]. By contrast, our present study with human macrophages observed a significant up-regulation of IFIT5 in response to highly pathogenic H5N1 virus infection. Of note, there was no significant up-regulation of immune-related genes in H5N1 virus-infected mouse lung at early post-infection time. At later post-infection time, the expression of immune-related genes increased dramatically in mice [29] which is similar to our present observation in human macrophage that most immune-related genes were found to be significantly up-regulated at later post-infection time at 6-h but not at earlier post-infection time at 1- or 3-h. Innate immune cells differentially recognize the invasion of pathogens through members of the pathogen recognition receptors (PRRs) family, consisting of Toll-like receptors (TLRs), RLRs, and nucleotide oligomerization domain (NOD)-like receptors (NLRs). TLRs recognize bacteria, viruses, fungi, and protozoa, whereas RLRs are activated by viral infection. Both TLRs and RLRs play an important role in the production of type I IFNs and various cytokines. NLRs detect bacteria and virus and then regulate interleukin-1β (IL-1β) maturation through activation of caspase-1. Our RNA-Seq data identifies marked up-regulation of RLR family members, RIG-I, IFIH1, and DHX58 in response to H5N1 virus infection compared with H1N1 virus infection. Among these members, RIG-I and MDA5 contain one DExD/H-box helicase domain and two CARD domains. The helicase domain of RLRs recognizes specific viral RNA patterns, and the CARD domains interact with MAVS for triggering the activation of kinase such as TANK-binding kinase 1 (TBK1), involved in NF-κB activation. Transcription factors, including IRF3, IRF7, and NF-κB, are then activated and translocated into the nucleus, where they activate the transcription of the IFNs. DHX58 lack a CARD domain, which was suggested to play a regulatory role in the RLR signaling pathway.
In our case, significant up-regulation of IFN was specific to H5N1 virus at 6-h post-infection (βH5N1,6h = 2.62; corrected p = 0.0001). At this time point, we observed concomitant negative regulation of the innate immunity pathway through SOCS1 and SOCS3 induced by H5N1 virus infection and subsequently the down-regulation of IFN and ISGs in the later infection time was associated with the strong activation of RLRs at the early stage of infection. We also observed reduced expression of antiviral cytokine IFN-β, ISG15 and MxA after multiple rounds of viral replication in H5N1 compared to H1N1 virus-infected cells at the late stage of infection. Taken together, hyper-induction of the negative regulators of IFN signaling, and the reduced expression of antiviral cytokines are the likely contributors to the higher viral load observed in H5N1 virus infection. Previous reports have revealed that TLRs and NLRs are involved in inflammatory disorder. Here we suggest that H5N1 virus-induced RLR activation elicits an early hyper-inflammatory response and fails to mount proper antiviral responses at the late stage of infection that contributes to the H5N1 virus pathogenesis.
Previous work has reported the miRNA profiling in an alveolar epithelial cell line, A549 cells, after infection by influenza A viruses, including the H5N1 virus (A/Thailand/NK165/2005) [31]. Here, we investigated the miRNA expression profiles in primary human cells, human monocyte-derived macrophages. We revealed that 10 targeted genes are regulated inversely with their targeting miRNAs, although the differential regulation of these genes is not significant based on our stringent criteria. Interestingly, we found that many of the targeting miRNAs were previously reported to associate with signal transduction or inflammatory cascades. For instance, we found that the expression pattern of the hsa-let-7 family of tumor suppressor miRNAs inversely correlates with that of multiple target mRNA genes in the RIG-I like receptor signaling pathway (Additional file 1: Table S4). This family of miRNAs has also been reported to be part of the feedback mechanism of MAPK signaling. Another candidate, miR-146a, was predicted to target an important signal transducer, TRAF6 in RIG-I signaling. MiR-146a is significantly suppressed in response to H5N1 virus infection (Z-score = − 12.1) with a differential expression in H5N1- compared to H1N1 virus-infected cells (1.5 fold down in H5N1 vs. H1N1). Recent studies suggested that miR-146a plays an important role in innate immunity and inflammation. MiR-146a is significantly reduced in inflammatory diseases, such as sepsis [32], osteoclastogenesis in arthritis [33], subclinical inflammation in type 2 diabetes [34] and chronic obstructive pulmonary disease [35]. It was suggested that miR-146a is induced via TLR-2, − 4 and − 5 ligands, but not responsive to TLR-3, − 7 and − 9 activation [36]. Expression of this miRNA, however, could be stimulated in response to the challenge by pro-inflammatory cytokines such as TNF-α [24, 37]. A recent report has demonstrated that expression of miR-146a is correlated with cytokine production and inversely correlated to TNF-α production [38]. MiR-146a is thought to be an important negative feedback regulator in controlling pro-inflammatory signaling in innate immune responses [39]. The suppression of miR-146a in response to H5N1 virus infection might be an important factor associated with the cytokine dysregulation observed in H5N1 disease [1, 40] and functional study of this miRNA, especially in vivo examination, may therefore be important in understanding H5N1 pathogenesis. This correlation indicates the regulatory roles of miRNAs in cellular response to influenza A virus infection, while further investigation should be performed to study the differences in expression levels of miRNAs that contribute to viral pathogenesis.
Conclusions
Data here reveals that two macrophage populations, M1 and M2 respond distinctly to H5N1 virus infection. Genes in significantly enriched pathways in response to H5N1 virus infection was specifically correlated in M1 subtype, whilst RIG-I-like receptor signaling pathway, in particular IFN and ISGs are broadly affected. The negative regulators of IFN signaling, the suppressors of cytokine signaling, SOCS1 and SOCS3, were found to be markedly up-regulated in the initial round of H5N1 virus replication. Elevated levels of these suppressors could lead to the eventual suppression of antiviral gene expression that may contribute to the pathogenesis of H5N1 virus infection. Taken together, this study provides important mechanistic insights into the understanding of H5N1 viral pathogenesis and highlights possible candidates as therapeutic targets in treating H5N1 diseases for further study.
Additional files
Table S1. Sequencing statistics of mRNA transcriptomes. Table S2. Significantly enriched Gene Ontology terms in response to H5N1 virus infection. Table S3. Sequencing statistics of miRNA transcriptomes. Table S4. MiRNAs and their target mRNAs in RIG-I like receptor signaling pathway. (DOCX 41 kb)
Figure S1. Multidimensional scaling (MDS) analysis of broad library-wise trends. No major trends were observed related to infection type or time post-infection. Figure S2. Elbow plot used to define appropriate number of clusters for k-means clustering. Figure S3. Expression of selected genes in the RIG-I-like receptor signaling pathway analyzed by real-time PCR. Figure S4. The interaction network between inversely regulated miRNAs and mRNAs enriched in RIG-I like receptor signaling pathway at 6-h post-infection in H5N1 virus-infected cells. (DOCX 3058 kb)
Differential expression of the genes in response to H5N1 virus infection at 6-h post-infection. (XLS 8081 kb)
Differential expression of mature miRNAs with a minimum fold-change of 1.2. (XLS 137 kb)
Pathway enrichment of the mRNA genes inversely regulated with the targeting miRNAs for H1N1 and H5N1 virus-infected macrophage cells at 1-, 3-, and 6-h post-infection. (XLS 154 kb)
Acknowledgements
We thank Yip Tsz Fung for revising the manuscript.
Funding
This work was supported by RFCID grant (no. 08070532), UGC Theme-based Research Scheme (no. T11–705/14-N) and U.S. NIH grant U41 HG006623.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the NCBI Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) with GEO platform accession GPL9115, series accession GSE99079, and sample accession GSM2609096-GSM2609104 and GSM2631402-GSM2631410.
Abbreviations
- AKT1
AKT serine/threonine kinase 1
- BST2
Bone marrow stromal cell antigen 2
- CCL2
C-C motif chemokine ligand 2
- CCL5
C-C motif chemokine ligand 5
- CD274
CD274 molecule encoding an immune inhibitory receptor ligand
- CISH
SH2 containing protein
- CXCL10
C-X-C motif chemokine ligand 10
- DDX60
DExD/H-box helicase 60
- DHX58
DExH-box helicase 58
- EIF2AK2
Eukaryotic translation initiation factor 2 alpha kinase 2
- GBP2
Guanylate binding protein 2
- GSP1
member RAS oncogene family
- HERC5
HECT and RLD domain containing E3 ubiquitin protein ligase 5
- IFI44
Interferon induced protein 44
- IFI44L
Interferon induced protein 44 like
- IFIH1
Interferon induced with helicase C domain 1
- IFIT1
Interferon induced protein with tetratricopeptide repeats 1
- IFN
Interferon
- IL12B
Interleukin 12B
- IL-1β
Interleukin-1β
- IRF
Interferon regulatory factor
- ISG
Interferon-stimulated genes
- MAVS
Mitochondrial antiviral signaling protein
- MDS
Multidimensional scaling
- miRNA
microRNA
- MNDA
Myeloid cell nuclear differentiation antigen
- MOI
Multiplicity of infection
- MxA
Myxovirus resistance protein 1
- NFKBIA
NF-κB inhibitor alpha
- NF-κB
Nuclear factor kappa B
- NLR
NOD-like receptors
- OAS2
2′-5′-oligoadenylate synthetase 2
- OASL
2′-5′-oligoadenylate synthetase like
- PRR
Pathogen recognition receptors
- RELA
RELA proto-oncogene NF-κB subunit
- RLR
RIG-I-like receptor
- RNA-Seq
RNA sequencing
- rRNA
ribosomal RNA
- RSAD2
Radical S-adenosyl methionine domain containing 2
- RT-qPCR
Real time quantitative polymerase chain reaction
- SFM
Serum free medium
- SOCS
Suppressors of cytokine signaling
- TBK
TANK-binding kinase 1
- TLR
Toll-like receptors
- TRAF2
TNF receptor associated factor 2
- TRIM25
Tripartite motif containing 25
Authors’ contributions
NZ and AHYT performed the experiments. YJB, GDB and SZ analyzed the data. JSMP, SL and SMYL conceived the project and supervised the study. NZ, YJB, GDB, SL and SMYL wrote the paper. All authors revised and approved the paper.
Ethics approval and consent to participate
Consent from blood donors was obtained by Hong Kong Red Cross to use blood components for research experiments. The use of human blood samples in this project has been reviewed and obtained human ethics approval (ref no. UW 08–073) issued by Institutional Review Board of the University of Hong Kong.
Viruses were obtained from specimens collected from patients as part of their routine clinical care. Thus, there is no necessity to obtain specific consent from the patients. Virus isolates obtained in this way are routinely used for research and for public health purposes. There is no requirement to obtain specific consent from each patient for the use of virus isolates coming from their clinical specimens.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Na Zhang, Email: zngoluk@hotmail.com.
Yun-Juan Bao, Email: ybao2@nd.edu.
Amy Hin-Yan Tong, Email: amyhytong2@yahoo.ca.
Scott Zuyderduyn, Email: scott.zuyderduyn@utoronto.ca.
Gary D. Bader, Email: gary.bader@utoronto.ca
J. S. Malik Peiris, Email: malik@hku.hk
Si Lok, Email: si.lok@sickkids.ca.
Suki Man-Yan Lee, Email: suki@hku.hk, Email: myleesuki01@gmail.com.
References
- 1.de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, et al. Fatal outcome of human influenza a (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12(10):1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salomon R, Hoffmann E, Webster RG. Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc Natl Acad Sci U S A. 2007;104(30):12479–12481. doi: 10.1073/pnas.0705289104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peiris JS, Yu WC, Leung CW, Cheung CY, Ng WF, Nicholls JM, et al. Re-emergence of fatal human influenza a subtype H5N1 disease. Lancet. 2004;363(9409):617–619. doi: 10.1016/S0140-6736(04)15595-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baskin CR, Bielefeldt-Ohmann H, Tumpey TM, Sabourin PJ, Long JP, García-Sastre A, et al. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc Natl Acad Sci U S A. 2009;106(9):3455–3460. doi: 10.1073/pnas.0813234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008;4(8):e1000115. doi: 10.1371/journal.ppat.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zitzow LA, Rowe T, Morken T, Shieh WJ, Zaki S, Katz JM. Pathogenesis of avian influenza a (H5N1) viruses in ferrets. J Virol. 2002;76(9):4420–4429. doi: 10.1128/JVI.76.9.4420-4429.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cameron CM, Cameron MJ, Bermejo-Martin JF, Ran L, Xu L, Turner PV, et al. Gene expression analysis of host innate immune responses during lethal H5N1 infection in ferrets. J Virol. 2008;82(22):11308–11317. doi: 10.1128/JVI.00691-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakrabarti AK, Vipat VC, Mukherjee S, Singh R, Pawar SD, Mishra AC. Host gene expression profiling in influenza a virus-infected lung epithelial (A549) cells: a comparative analysis between highly pathogenic and modified H5N1 viruses. Virol J. 2010;7:219. doi: 10.1186/1743-422X-7-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cilloniz C, Shinya K, Peng X, Korth MJ, Proll SC, Aicher LD, et al. Lethal influenza virus infection in macaques is associated with early dysregulation of inflammatory related genes. PLoS Pathog. 2009;5(10):e1000604. doi: 10.1371/journal.ppat.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SM, Gardy JL, Cheung CY, Cheung TK, Hui KP, Ip NY, et al. Systems-level comparison of host-responses elicited by avian H5N1 and seasonal H1N1 influenza viruses in primary human macrophages. PLoS One. 2009;4(12):e8072. doi: 10.1371/journal.pone.0008072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Magrane M, Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database. 2011;2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan PP, Lowe TM. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37(Database issue):D93–DD7. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie J, Zhang M, Zhou T, Hua X, Tang L, Wu W. Sno/scaRNAbase: a curated database for small nucleolar RNAs and cajal body-specific RNAs. Nucleic Acids Res. 2007;35(Database):D183–D187. doi: 10.1093/nar/gkl873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai L, Huang H, Blackshaw S, Liu JS, Cepko C, Wong WH. Clustering analysis of SAGE data using a Poisson approach. Genome Biol. 2004;5(7):R51. doi: 10.1186/gb-2004-5-7-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee DD, Seung HS. Algorithms for non-negative matrix factorization. NIPS Proceedings. 2000;13:556–562. [Google Scholar]
- 17.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benoit M, Desnues B, Mege J-L. Macrophage Polarization in Bacterial Infections. J Immunol. 2008;181(6):3733. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 19.Pauli EK, Schmolke M, Wolff T, Viemann D, Roth J, Bode JG, et al. Influenza a virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008;4(11):e1000196. doi: 10.1371/journal.ppat.1000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maragkakis M, Reczko M, Simossis VA, Alexiou P, Papadopoulos GL, Dalamagas T, et al. DIANA-microT web server: elucidating microRNA functions through target prediction. Nucleic Acids Res. 2009;37(suppl 2):W273–W2W6. doi: 10.1093/nar/gkp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicholls JM, Chan MC, Chan WY, Wong HK, Cheung CY, Kwong DL, et al. Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat Med. 2007;13(2):147–149. doi: 10.1038/nm1529. [DOI] [PubMed] [Google Scholar]
- 22.Cheung CY, Poon LL, Lau AS, Luk W, Lau YL, Shortridge KF, et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet. 2002;360(9348):1831–1837. doi: 10.1016/S0140-6736(02)11772-7. [DOI] [PubMed] [Google Scholar]
- 23.Lee SMY, Cheung C-Y, Nicholls JM, Hui KPY, Leung CYH, Uiprasertkul M, et al. Hyperinduction of Cyclooxygenase-2-mediated Proinflammatory Cascade: a mechanism for the pathogenesis of avian influenza H5N1 infection. J Infect Dis. 2008;198(4):525–535. doi: 10.1086/590499. [DOI] [PubMed] [Google Scholar]
- 24.Hui KP, Lee SM, Cheung CY, Ng IH, Poon LL, Guan Y, et al. Induction of proinflammatory cytokines in primary human macrophages by influenza A virus (H5N1) is selectively regulated by IFN regulatory factor 3 and p38 MAPK. J Immunol. 2009;182(2):1088–1098. doi: 10.4049/jimmunol.182.2.1088. [DOI] [PubMed] [Google Scholar]
- 25.Josset L, Zeng H, Kelly SM, Tumpey TM, Katze MG. Transcriptomic characterization of the novel avian-origin Influenza A (H7N9) virus: specific host response and responses intermediate between Avian (H5N1 and H7N7) and Human (H3N2) viruses and implications for treatment options. MBio. 2014;5(1):e01102–e01113. doi: 10.1128/mBio.01102-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeng H, Belser JA, Goldsmith CS, Gustin KM, Veguilla V, Katz JM, et al. A(H7N9) virus results in early induction of Proinflammatory cytokine responses in both human lung epithelial and endothelial cells and shows increased human adaptation compared with Avian H5N1 virus. J Virol. 2015;89(8):4655–4667. doi: 10.1128/JVI.03095-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan MC, Cheung CY, Chui WH, Tsao SW, Nicholls JM, Chan YO, et al. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir Res. 2005;6:135. doi: 10.1186/1465-9921-6-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tundup S, Kandasamy M, Perez JT, Mena N, Steel J, Nagy T, et al. Endothelial cell tropism is a determinant of H5N1 pathogenesis in mammalian species. PLoS Pathog. 2017;13(3):e1006270. doi: 10.1371/journal.ppat.1006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park SJ, Kumar M, Kwon HI, Seong RK, Han K, Song JM, et al. Dynamic changes in host gene expression associated with H5N8 avian influenza virus infection in mice. Sci Rep. 2015;5:16512. doi: 10.1038/srep16512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar A, Vijayakumar P, Gandhale PN, Ranaware PB, Kumar H, Kulkarni DD, et al. Genome-wide gene expression pattern underlying differential host response to high or low pathogenic H5N1 avian influenza virus in ducks. Acta Virol. 2017;61(1):66–76. doi: 10.4149/av_2017_01_66. [DOI] [PubMed] [Google Scholar]
- 31.Makkoch J, Poomipak W, Saengchoowong S, Khongnomnan K, Praianantathavorn K, Jinato T, et al. Human microRNAs profiling in response to influenza A viruses (subtypes pH1N1, H3N2, and H5N1) Exp Biol Med (Maywood) 2016;241(4):409–420. doi: 10.1177/1535370215611764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Brahmakshatriya V, Zhu H, Lupiani B, Reddy SM, Yoon BJ, et al. Identification of differentially expressed miRNAs in chicken lung and trachea with avian influenza virus infection by a deep sequencing approach. BMC Genomics. 2009;10:512. doi: 10.1186/1471-2164-10-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakasa T, Shibuya H, Nagata Y, Niimoto T, Ochi M. The inhibitory effect of microRNA-146a expression on bone destruction in collagen-induced arthritis. Arthritis Rheum. 2011;63(6):1582–1590. doi: 10.1002/art.30321. [DOI] [PubMed] [Google Scholar]
- 34.Balasubramanyam M, Aravind S, Gokulakrishnan K, Prabu P, Sathishkumar C, Ranjani H, et al. Impaired miR-146a expression links subclinical inflammation and insulin resistance in type 2 diabetes. Mol Cell Biochem. 2011;351(1):197–205. doi: 10.1007/s11010-011-0727-3. [DOI] [PubMed] [Google Scholar]
- 35.Sato T, Liu X, Nelson A, Nakanishi M, Kanaji N, Wang X, et al. Reduced miR-146a increases prostaglandin E(2) in chronic obstructive pulmonary disease fibroblasts. Am J Respir Crit Care Med. 2010;182(8):1020–1029. doi: 10.1164/rccm.201001-0055OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams AE, Perry MM, Moschos SA, Larner-Svensson HM, Lindsay MA. Role of miRNA-146a in the regulation of the innate immune response and cancer. Biochem Soc Trans. 2008;36(Pt 6):1211–1215. doi: 10.1042/BST0361211. [DOI] [PubMed] [Google Scholar]
- 37.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103(33):12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nahid MA, Satoh M, Chan EK. Mechanistic role of microRNA-146a in endotoxin-induced differential cross-regulation of TLR signaling. J Immunol. 2011;186(3):1723–1734. doi: 10.4049/jimmunol.1002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roy S, Sen CK. MiRNA in innate immune responses: novel players in wound inflammation. Physiol Genomics. 2011;43(10):557–565. doi: 10.1152/physiolgenomics.00160.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peiris JS, de Jong MD, Guan Y. Avian influenza virus (H5N1): a threat to human health. Clin Microbiol Rev. 2007;20(2):243–267. doi: 10.1128/CMR.00037-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequencing statistics of mRNA transcriptomes. Table S2. Significantly enriched Gene Ontology terms in response to H5N1 virus infection. Table S3. Sequencing statistics of miRNA transcriptomes. Table S4. MiRNAs and their target mRNAs in RIG-I like receptor signaling pathway. (DOCX 41 kb)
Figure S1. Multidimensional scaling (MDS) analysis of broad library-wise trends. No major trends were observed related to infection type or time post-infection. Figure S2. Elbow plot used to define appropriate number of clusters for k-means clustering. Figure S3. Expression of selected genes in the RIG-I-like receptor signaling pathway analyzed by real-time PCR. Figure S4. The interaction network between inversely regulated miRNAs and mRNAs enriched in RIG-I like receptor signaling pathway at 6-h post-infection in H5N1 virus-infected cells. (DOCX 3058 kb)
Differential expression of the genes in response to H5N1 virus infection at 6-h post-infection. (XLS 8081 kb)
Differential expression of mature miRNAs with a minimum fold-change of 1.2. (XLS 137 kb)
Pathway enrichment of the mRNA genes inversely regulated with the targeting miRNAs for H1N1 and H5N1 virus-infected macrophage cells at 1-, 3-, and 6-h post-infection. (XLS 154 kb)
Data Availability Statement
The datasets supporting the conclusions of this article are available in the NCBI Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) with GEO platform accession GPL9115, series accession GSE99079, and sample accession GSM2609096-GSM2609104 and GSM2631402-GSM2631410.