

Abstract
Background
Complete mitochondrial (mt) genomes have been used extensively to test hypotheses about microevolution and to study population structure, phylogeography, and phylogenetic relationships of Anura at various taxonomic levels. Large-scale mt genomic reorganizations have been observed among many fork-tongued frogs (family Dicroglossidae). The relationships among Dicroglossidae and validation of the genus Feirana are still problematic. Hence, we sequenced the complete mt genomes of Nanorana taihangnica (=F. taihangnica) and N. yunnanensis as well as partial mt genomes of six Quasipaa species (dicroglossid taxa), two Odorrana and two Amolops species (Ranidae), and one Rhacophorus species (Rhacophoridae) in order to identify unknown mt gene rearrangements, to investigate the validity of the genus Feirana, and to test the phylogenetic relationship of Dicroglossidae.
Results
In the mt genome of N. taihangnica two trnM genes, two trnP genes and two control regions were found. In addition, the trnA, trnN, trnC, and trnQ genes were translocated from their typical positions. In the mt genome of N. yunnanensis, three control regions were found and eight genes (ND6, trnP, trnQ, trnA, trnN, trnC, trnY and trnS genes) in the L-stand were translocated from their typical position and grouped together. We also found intraspecific rearrangement of the mitochondrial genomes in N. taihangnica and Quasipaa boulengeri. In phylogenetic trees, the genus Feirana nested deeply within the clade of genus Nanorana, indicating that the genus Feirana may be a synonym to Nanorana. Ranidae as a sister clade to Dicroglossidae and the clade of (Ranidae + Dicroglossidae) as a sister clade to (Mantellidae + Rhacophoridae) were well supported in BI analysis but low bootstrap in ML analysis.
Conclusions
We found that the gene arrangements of N. taihangnica and N. yunnanensis differed from other published dicroglossid mt genomes. The gene arrangements in N. taihangnica and N. yunnanensis could be explained by the Tandem Duplication and Random Loss (TDRL) and the Dimer-Mitogenome and Non-Random Loss (DMNR) models, respectively. The invalidation of the genus Feirana is supported in this study.
Keywords: Dicroglossidae, Feirana, Mitochondrial genome, Gene rearrangement, Phylogeny
Background
Vertebrate mitochondrial (mt) genomes are closed circular molecules that generally have lengths varying from 15 to 27 kb [1]. They typically encode 37 genes including two ribosomal RNAs (12S and 16S rRNAs), 22 transfer RNAs (tRNAs), 13 protein–coding genes, and one long non-coding region (NCR) called the control region (CR; also referred to as the D–loop region) [2, 3]. The mt genome has several valuable characteristics including small size, rapid evolutionary rate, relatively conserved gene content and organization, maternal inheritance, and limited recombination [4]. Complete mt genomes have been extensively used to test hypotheses about microevolution, to study population structure, phylogeography, and phylogenetic relationships at various taxonomic levels, and to identify cryptic species [2, 5, 6]. The mitochondrial DNA (mtDNA) of many neobatrachian anurans shows gene rearrangement of the relative position of NADH dehydrogenase subunit 5 (ND5); this has been reported in Ranidae, Dicroglossidae, Mantellidae and Rhacophoridae [7–11]. Rearrangements of two transfer ribonucleic acid M (trnM) genes were also reported in dicroglossid and mantellid mt genomes [5, 8–10, 12–17]. Other mt genomic rearrangements can also be found in some species of neobatrachians. For example, Rhacophorus schlegelii [11], Mantella madagascariensis [9], and Rana kunyuensis [17] possessed duplicated control regions. By contrast, Nanorana taihangnica [13] lost the trnT gene and Polypedates megacephalus [18] lost the ATPase subunit 8 (ATP8) and ND5 genes. Gene rearrangements in the mitochondrial genome can be mainly explained by six available models: the recombination model [8, 19], the Tandem Duplication and Random Loss model (TDRL) [20], the Tandem Duplication and Non-Random Loss model (TDNL) [21], the tRNA miss-priming model [22], the Dimer-Mitogenome and Non-Random Loss model (DMNR) [23] and/or the Double Replications and Random Loss model (DRRL) [24].
Within the Neobatrachia, the monophyly of the combined Mantellidae and Rhacophoridae has been generally accepted, but the relationships of Ranidae, Dicroglossidae and (Mantellidae + Rhacophoridae) have been in controversy. The relationship of ((Dicroglossidae + (Ranidae + (Rhacophoridae + Mantellidae)) was supported by Frost et al. [25], Kakehashi et al. [1], Kurabayashi and Sumida [26], Kurabayashi et al. [27], Li et al. [7], Xia et al. [28], Pyron and Wiens [29] and Yuan et al. [30]. However, Chen et al. [31], Ren et al. [32], Zhang et al. [33] and Zhou et al. [12] supported the relationship of ((Ranidae + Dicroglossidae) + (Rhacophoridae + Mantellidae)). Furthermore, the relationships within Dicroglossidae are extremely problematic, and have received much attention. The dicroglossids are divided into two subfamilies and four tribes: Dicroglossinae (Dicroglossini, Limnonectini, and Paini) and Occidozyginae (Occidozygini) with the classification of spiny frogs and non-spiny frogs belonging to the tribe Paini (Dicroglossidae) remaining obscure [25, 34–36]. The taxonomy of this group has been revised numerous times [37–40]. The genus Feirana of tribe Paini including three species (F. taihangnica, F. quadranus and F. kangxianensis) is widely distributed in China [36, 41, 42] and was considered to be a synonym to Nanorana by Frost et al. [25, 43] and Che et al. [35, 44]. Although Dubois transferred Feirana species to the subgenus Rana (Paa) [38], Fei et al. [39] assigned them to the newly created subgenus Paa (Quadrana). Dubois [38] and Fei et al. [40] placed the subgenus Quadrana as genus Feirana. So, the validity of genus Feirana is still unknown.
Large-scale mt genomic rearrangements in many Dicroglossidae species have been observed. However, complete information on Nanorana and Quasipaa mt genomes is still lacking except for Nanorana parkeri [15], Nanorana pleskei [31], Quasipaa boulengeri [16], Quasipaa spinosa [12], Yerana yei [14] and Nanorana taihangnica [13]. Compared with neobatrachian families, the dicroglossid mt genomes investigated thus far feature differences in gene arrangements, which gave us more chances to discuss the potential reasons for gene rearrangements in the mitochondrial genome.
In the present study, we determined the complete mt genomes of N. taihangnica and N. yunnanensis as well as the partial mt genomes of six Quasipaa species (Dicroglossidae), two Odorrana and two Amolops species (Ranidae), and one Rhacophorus species (Rhacophoridae). In this paper, we follow the system of anuran taxonomy published by Fei et al. [42] and Frost et al. [43] to prevent unnecessary confusion in taxonomy. The data was used to determine unknown mt gene rearrangements, to investigate the validity of the genus Feirana, and to test the phylogenetic relationships of Ranidae and Dicroglossidae.
Methods
Ethical statement
The thirteen species studied (N. taihangnica, N. yunnanensis, Q. boulengeri, Q. exilispinosa, Q. jiulongensis, Q. robertingeri, Q. shini, Q. verrucospinosa, Odorrana livida, O. schmackeri, Amolops hongkongensis, A. wuyiensis, Rhacophorus dennysi) are not protected by the provisions of the laws of People’s Republic of China on the protection of wildlife. Thus, the experiments in this study were performed with toe-clip tissue samples collected from all frog specimens and stored in 100% ethanol. Sample acquisition was reviewed, approved and carried out in accordance with the relevant guidelines of the Committee of Animal Research Ethics of Zhejiang Normal University.
Sample collection
Specimens included two species of Nanorana (N. taihangnica = F. taihangnica, N. yunnanensis), seven samples belonging to six species of Quasipaa (Q. boulengeri, Q. exilispinosa, Q. jiulongensis, Q. robertingeri, Q. shini, Q. verrucospinosa) (Dicroglossidae) including two Q. boulengeri samples from two different sites, two species of Odorrana (O. livida, O. schmackeri) (Ranidae), two species of Amolops (A. hongkongensis, A. wuyiensis) (Ranidae), and R. dennysi (Rhacophoridae). Information on all of the sequenced samples is shown in Table 1. We were unable to successfully sequence the displacement loop (D-loop) region of these samples except for N. taihangnica and N. yunnanensis because of highly repetitive regions in the D-loop or other unknown reasons despite many optimization efforts; this is similar to the report of Zhang et al. [45].
Table 1.
Information on the samples used in this study. Specimen sources and GenBank accession numbers are also shown
| Species | Collection Locality | Collection Date | Specimen No. | Accession No. |
|---|---|---|---|---|
| N. taihangnica | Luanchuan, Henan | 17-July-2010 | LGW-LC-001 | KF199146 |
| N. yunnanensis | Luoping, Yunnan | 22-Oct-2010 | STJW-LP-001 | KF199150 |
| Q. boulengeri | Tongshan, Hubei | 7-Jun-2009 | JFW-TS-002 | KF199152 |
| Q. exilispinosa | Wuyishan, Fujian | 8-Jun-2010 | XJW-WYS-001 | KF199151 |
| Q. jiulongensis | Wuyishan, Fujian | 8-Jun-2010 | JLJW-WYS-002 | KF199149 |
| Q. shini | Longsheng, Guangxi | 10-Oct-2011 | XJW-LS-001 | KF199148 |
| Q. verrucospinosa | Pingbian, Yunnan | 7-July-2007 | DYJW-PB-003 | KF199147 |
| Q. boulengeri | Luoping, Yunnan | 22-Oct-2010 | JFW-LPS-002 | KX233867 |
| Q. robertingeri | Hejiang, Sichuan | 7-July-2012 | HJJW-HJ-001 | KX233868 |
| O. livida | Wenzhou, Zhejiang | 7-Aug-2013 | DLW-WZ-004 | KX233865 |
| O. schmackeri | Wenzhou, Zhejiang | 7-Aug-2013 | HCW-WZ-001 | KX233866 |
| A. hongkongensis | Wuyishan, Fujian | 13-July-2013 | DYTW-WYS-001 | KX233864 |
| A. wuyiensis | Wenzhou, Zhejiang | 7-Aug-2013 | WYTW-WZ-001 | KM282625 |
| R. dennysi | Yangjiang, Guangdong | 1-Sept-2011 | DFSW-YJ-002 | KX233869 |
PCR and sequencing
Total DNA was extracted from the clipped toe of each frog specimen using a DNeasy Tissue Kit (Qiagen, Germany). We amplified overlapping fragments that covered the entire mt genome of N. taihangnica and N. yunnanensis by normal PCR and long-and-accurate polymerase chain reaction (LA PCR) methods slightly modified from Yu et al. [5, 46] and Zhang et al. [45]. All PCR procedures were performed using a MyCycler Thermal Cycler (Bio-Rad, Hercules, CA, USA). TaKaRa Ex-Taq and LA-Taq kits (Takara Biomedical, Dalian, China) were used for the normal and LA-PCR reactions. The resulting PCR fragments were electrophoresed on 1% agarose gels, and all target DNAs were purified from excised pieces of gel using a SanPrep DNA Gel Extraction Kit (Sangon Biotech, Shanghai, China) prior to sequencing. The sequences for each fragment were obtained in an automated DNA sequencer (ABI 3730) from both strands. The long fragments were sequenced using specific primer walking of both strands.
Sequence assembly and analysis
Sequences were checked and assembled using SeqMan (Lasergene version 5.0) [47]. The locations of the 13 protein coding genes and two rRNA genes were determined by comparison with the available RefSeq sequences of closely related anurans downloaded from GenBank using ClustalW in Mega 5.0 [48, 49]. All tRNA genes were identified by their cloverleaf secondary structure using tRNA-scan SE 1.21 [50] or determined by comparison with the homologous sequences of other anurans. The mt genomes (see Fig. 1) of all taxa were analyzed to determine the corresponding mt gene arrangements. The resultant sequences were deposited in GenBank with accession numbers KF199146-KF199152, KX233864-KX233869 and KM282625 (see Table 1).
Fig. 1.

Mitochondrial map of thirteen species of tribe Paini used in this study. The tRNAs are labeled according to the three-letter amino acid codes. The gene name above the median indicates the direction of transcription is from left to right, whereas the gene name below the median indicates right to left. Genes with red letters indicates that the direction of transcription is from right to left. The red square frame shows the region of WANOLCY or modified WANOLCY
Molecular phylogenetic analysis
With the recently increased number of mitochondrial genomes available for Anura, phylogenetic analyses were performed with 83 anurans for which complete or partial mt genomes were available including 14 samples of the 13 species from this study. In total this included the ingroup of 33 species from Ranidae [1, 27, 28, 45, 46, 51–63], 28 species from Dicroglossidae [5, 10, 12–14, 16, 17, 31–33, 64], 13 species from Rhacophoridae [11, 18, 65], one species from Mantellidae [9], one species from Petropedetidae [45], one species from Pyxicephalidae [45], one species from Phrynobatrachidae [45], one species from Ptychadenidae [45], one species from Brevicipitidae (outgroup) [26], one species from Hyperoliidae (outgroup) [26] and two species of Microhylidae (outgroups) [66, 67]. In order to discuss the phylogenetic relationship of Anura, we used the amino acid data and the nucleotides data to compare the identical topology or not according to the methods of Zhang et al. [6] and Zhou et al. [12]. The amino acid sequences of 10 mt protein-coding genes were separately aligned in Mega 5.0 [48] excluding the ATP8, ND5 and ND6 genes for the following reasons: (a) the ATP8 sequence was too short in length and had too little good information (only 18 nucleotides or < 0.5% of the total nucleotides of combined PCGs) after G-Block analysis, (b) the loss of the ND5 gene in some species [18], and (c) the heterogeneous base composition and poor phylogenetic performance for ND6 which failed to support the consistency analysis with other PCGs [45]. The alignments were revised using Gblocks 0.91b software with the default parameters [68] to select conserved regions of the putative amino acids. We concatenated the alignments of the 10 other mitochondrial protein-coding genes and got an alignment consisting of 2497 amino acid residues as 10Paa dataset. An alignment of 7491 nucleotides sites with 4919 variable informative sites was converted from 2497 amino acids data directly using the amino acid alignment as the backbone. Saturation analysis was performed for subsets with first, second, and third codon positions using DAMBE 4.2.13 [69]. The results showed that the third codon positions were saturated. Thus, we excluded the third codon positions from further phylogenetic analyses and obtained a dataset called 10P consisting of 4994 nucleotide sites from the 1st and 2nd codon positions of the 10 protein-coding genes according to the methods of Cameron et al. [70], Zhang et al. [6] and Zhou et al. [12].
The phylogeny was analyzed using the combined datasets 10P (nucleotides dataset) and 10Paa (amino acid dataset) by the maximum likelihood (ML) and Bayesian inference (BI) methods. To improve the fit of the substitution model to the datasets of 10P and 10Paa, we compared data partitioning schemes according to the Akaike Information Criterion (AIC) and Bayesian Information Criterion (BIC) using the program PartitionFinder v1.0 and PartitionFinderProtein [71]. We set the 10 coding-genes as 20 partitions in dataset 10P and 10 partitions in dataset 10Paa, respectively. For the dataset 10P, twenty-partitions were optimal: 1) first codon positions of the 10 protein-coding genes; 2) second codon positions of the 10 protein-coding genes. The best substitution model of twenty-partitions in ten different genes of dataset 10P is always GTR + I + G. For the dataset 10Paa, ten-partitions were optimal: 10 protein-coding genes with MTMAM. So, the optimal model for 10P with twenty partitions and the optimal model for 10Paa with ten partitions was chosen for ML by the RaxML program [72] and Bayesian analyses by MrBayes3.1.2 [73–75], respectively. ML and BI analyses for datasets of 10P and 10Paa were separately performed using the RaxML program [72] with 1000 bootstrap replications and a modified version of MrBayes3.1.2 [73]. During BI analysis, the following settings were applied: number of Markov chain Monte Carlo (MCMC) generations = 10 million; sampling frequency = 1000; burn-in = 1000. The burn–in size was determined by checking convergences of -log likelihood (−ln L). The robustness of the resulting ML tree was evaluated using bootstrap percentages calculated from nonparametric bootstrap analyses, and statistical support of the resulting BI trees was determined based on Bayesian posterior probability (BPP).
Results
Genome organization of mtDNA
The N. taihangnica mt genome is 21,322 base pairs (bp) in length and contains 13 protein coding genes, two rRNA genes, 24 tRNA genes (including extra trnM and trnP genes), and 10 NCRs including two control regions (CRs). The two CRs were located between the cytochrome b (Cyt b) and trnL genes (CR1 2014 bp) and between the trnC and trnT genes (CR2 2698 bp). Remarkably, CR1 and CR2 have nearly identical nucleotide sequences (99.9% similarity with only 1 substitution in 2014 alignment sites) excluding the extra 5′-635 bases and 3′-49 bases in CR2. Tandem duplication of the trnM gene and an additional trnP gene were found (Fig. 1). The trnT-trnP-trnF tRNA cluster moved from the typical neobatrachian LTPF tRNA cluster to a position between the CR1 and NADH dehydrogenase subunit 1 (ND1) genes. The typical LTPF tRNA cluster was replaced by a trnL-trnP-pseudo trnF tRNA cluster. The pseudo-trnF showed 89.9% nucleotide similarity with the corresponding trnF gene in the trnT-trnP-trnF tRNA cluster. This pseudo-trnF contained the same anticodon nucleotides (Fig. 2) compared to trnF. The trnA, trnN, trnC, and trnQ genes were translocated from their typical positions and replaced by a 40–138 bp NCR (Fig. 1). The trnQ gene moved from the typical dicroglossid IQMM tRNA cluster to a location between a 209 bp NCR and a 208 bp NCR (Fig. 1) and within the former IQMM tRNA cluster the trnQ gene was replaced by a 40 bp NCR between the trnI and tandem trnM genes. The trnA, trnN, and trnC genes also moved from the WANCY tRNA cluster to a position between a 208 bp NCR and CR1 (Fig. 1). The positions of the Light-strand replication origin (OL) are located between a 138 bp NCR (non-coding region) and a 52 bp NCR for the translocations of trnA, trnN, and trnC genes, and a W-NCR (138 bp)-OL-NCR (52 bp)-Y gene cluster was formed in the position of the typical WANOLCY gene cluster. Furthermore, a new cluster consisting of a L-NCR (209 bp)-Q-NCR (208 bp)-A-N-C gene arrangement was observed (Fig. 1). The two trnP genes contained the same anticodon nucleotides (Fig. 2). The trnF pseudogene contained the same anticodon nucleotides as in trnF whereas trnA and trnN contained different anticodon nucleotides (Fig. 2).
Fig. 2.
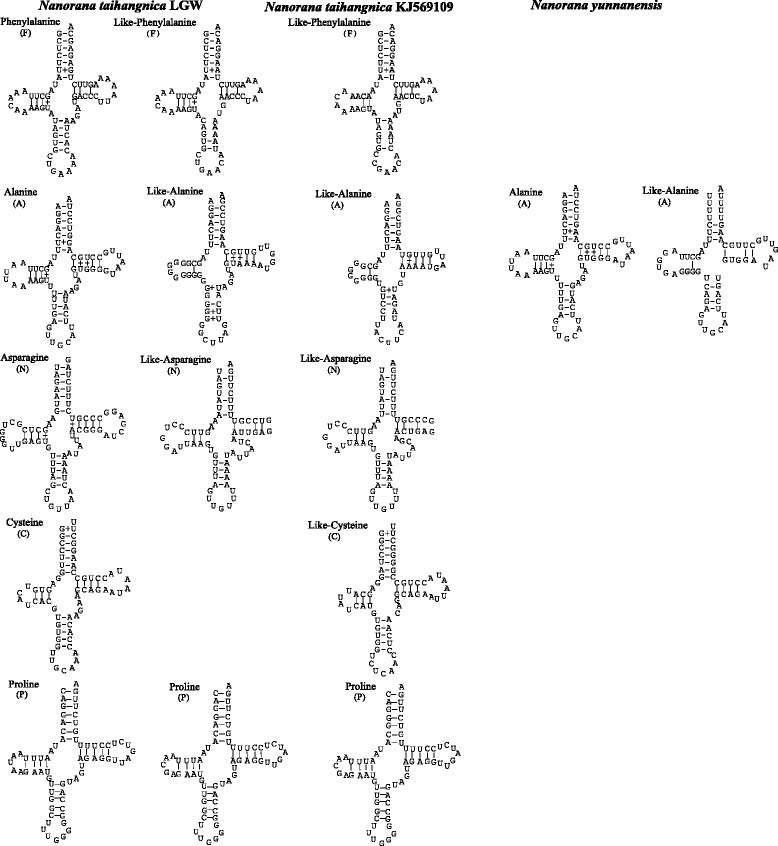
Predicted secondary structures for the tRNA genes and the corresponding pseudo-tRNA genes of N. taihangnica and N. yunnanensis. Dashes (−) indicate Watson–Crick base pairing and plus (+) indicate G + U base pairing. Arms of the tRNAs (clockwise from the top) are the amino acid acceptor (AA) arm, the TyC (T) arm, the anticodon (AC) arm, and the dihydrouridine (DHU) arm
The N. yunnanensis mt genome is 23,685 bp in length and contains 13 protein coding genes, two rRNA genes, 23 tRNA genes (including an extra trnM gene), and nine non-coding regions (including three control regions) (Fig. 1). Eight genes (ND6, trnP, trnQ, trnA, trnN, trnC, trnY and trnS genes) in the L-stand were translocated from the typical position to CR regions or near to CR regions and grouped together. CR1, CR2 and CR3 with lengths of 1635 bp, 1581 bp and 1560 bp, respectively, were found between Cyt b and trnQ, between ND6 and trnP, and between trnS and trnL, respectively (Fig. 1). The three CRs have a similar sequence with a length of 1372 bp. The typical WAN(OL)CY tRNA cluster was replaced by a modified W-NCR (139 bp) -OL-NCR (84 bp) arrangement. Through trnP translocation, the LTF tRNA cluster replaced the LTPF tRNA cluster. Through trnQ translocation, the I-NCR (40 bp)-MM tRNA cluster replaced the IQMM tRNA cluster. Through ND6 gene translocation, a 231 bp NCR replaced the ND6 gene in the original region. Through translocation of the trnS gene to between cytochrome c oxidase subunit I (COI) and trnD, the 51 bp NCR replaced trnS. A 47 bp NCR was found between the trnS and ND5 genes.
The detailed gene rearrangements of other known dicroglossids, ranids and rhacophorids in this study are described below.
Quasipaa boulengeri, Q. jiulongensis, Q. verrucospinosa
The typical WANOLCY tRNA cluster was replaced by a W-A-NCR-OL-NCR-N-NCR-C-Y tRNA cluster, the NCR of which ranged from 20 bp to 201 bp. The typical IQMM tRNA cluster was found. A 41–56 bp NCR was also found between the trnS and ND5 genes.
Quasipaa robertingeri
The typical WANOLCY tRNA cluster was replaced by a W-A-N-310 bp NCR-OL-C-Y tRNA cluster. The typical IQMM tRNA cluster was found. A 46 bp NCR was also found between the trnS and ND5 genes.
Quasipaa exilispinosa, Q. shini
The typical WANOLCY and IQMM tRNA clusters were also found in Q. spinosa and Q. yei. A 32–48 bp NCR was found between the trnS and ND5 genes.
Odorrana livida
The typical WANOLCY tRNA cluster and a 52 bp NCR between the ND5 and ND6 gene were found.
Odorrana schmackeri
The typical WANOLCY tRNA cluster was replaced by a W-A-NCR-OL-NCR-C-Y tRNA cluster. A 296 bp NCR with tandem sequence between the ND5 and ND6 genes was found.
Amolops hongkongensis, A. wuyiensis
The typical WANOLCY tRNA cluster and no NCR between the ND5 and ND6 gene were found.
Rhacophorus dennysi
The typical WANOLCY tRNA cluster was found and the ND5 gene between trnS and ND6 was translocated to the region between CR and trnT.
Phylogenetic analysis
All BI and ML phylogenetic analyses performed in this study showed similar topologies (Figs. 3 and 4). In the phylogeny of Dicroglossidae, Ranidae, Mantellidae and Rhacophoridae, the monophyly of Dicroglossidae, Ranidae and Rhacophoridae are well supported. Dicroglossidae is a sister clade of Ranidae (1.00 in posterior probability for nucleotides and amino acids datasets; 64% and 69% bootstrap frequencies for nucleotides and amino acids, respectively), and Mantellidae is a sister clade of Rhacophoridae (1.00 in posterior probability for nucleotides and amino acids datasets; 58% and 75% bootstrap frequencies for nucleotides and amino acids, respectively). Then the clade of (Ranidae + Dicroglossidae) is a sister clade of (Mantellidae + Rhacophoridae) (1.00 in posterior probability for nucleotides and amino acids datasets; 46% and 59% bootstrap frequencies for nucleotides and amino acids, respectively).
Fig. 3.
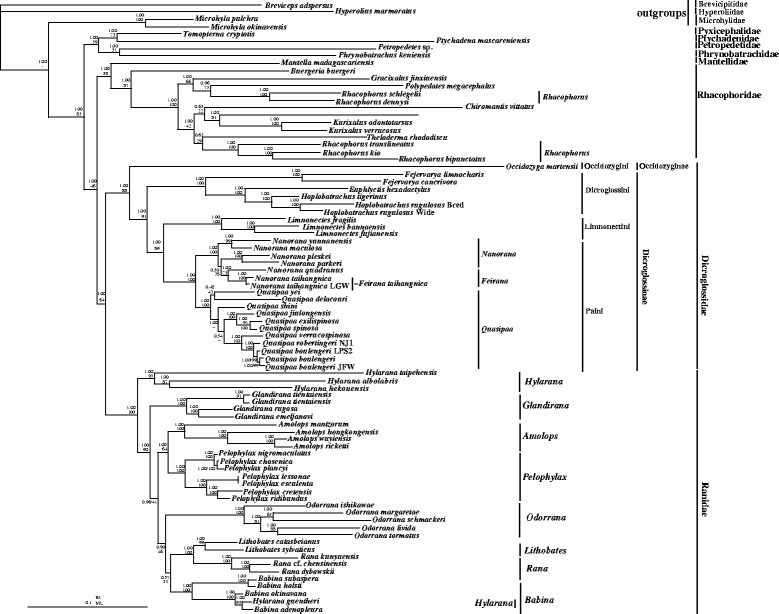
Phylogenetic relationships of Dicroglossidae, Ranidae, Mantellidae and Rhacophoridae based on 10 protein-coding genes using nucleotide datasets. Phylogenetic analyses using nucleotide datasets were carried out for the 83 frogs based on all 10 protein-coding genes from their respective mt genomes. Branch lengths and topology are from the BI analysis. The tree was rooted with four out-groups (Microhyla pulchra (NC_024547), M. okinavensis (NC_010233), Breviceps adspersus (NC_023379) and Hyperolius marmoratus (NC_023381)). Numbers above the nodes are the bootstrap values of ML in the bottom and the posterior probabilities of BI in the top
Fig. 4.
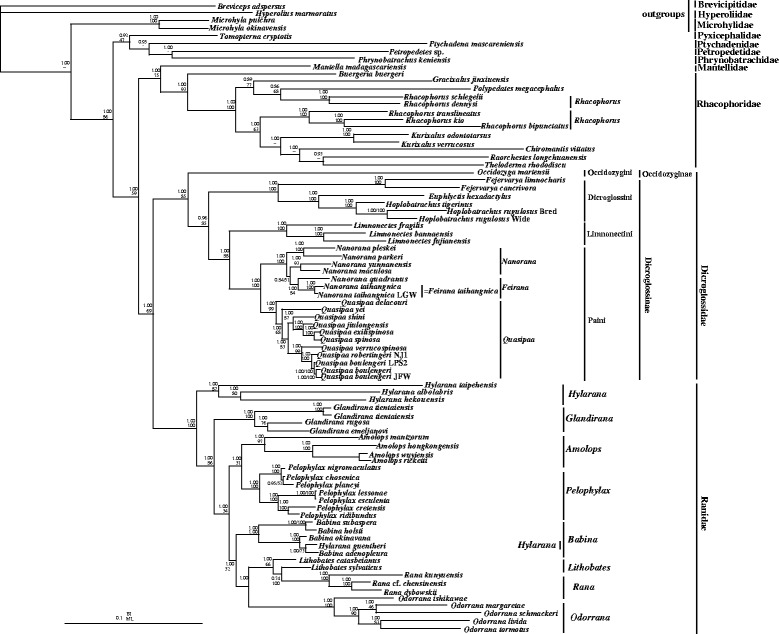
Phylogenetic relationships of Dicroglossidae, Ranidae, Mantellidae and Rhacophoridae based on 10 protein-coding genes using amino acid datasets. Phylogenetic analyses using amino acid datasets were carried out for the 83 frogs based on all 10 protein-coding genes from their respective mt genomes. Branch lengths and topology are from the BI analysis. The tree was rooted with four out-groups (Microhyla pulchra, M. okinavensis, Breviceps adspersus and Hyperolius marmoratus). Numbers above the nodes are the bootstrap values of ML in the bottom and the posterior probabilities of BI in the top
In the dicroglossid clade, Dicroglossidae was divided into two clades: Occidozyginae and Dicroglossinae (Figs. 3 and 4). Among the dicroglossid frogs in this study, O. martensii (Occidozyginae: Occidozygini) occupied the basal phylogenetic position (1.00 in BI of both datasets; 88% in ML of nucleotide and 85% in ML of amino acids). The monophyly of Quasipaa and Feirana was supported but the monophyly of Nanorana was not supported because the clade of (N. taihangnica (=F. taihangnica) + N. quadranus (=F. quadranus)) was supported within the clade of Nanorana (Figs. 3 and 4). In Quasipaa, the relationship of Q. delacouri + (Q. yei + ((((Q. spinosaa + Q. exilispinosa) + Q. jiulongensis) + Q. shini) + ((Q. boulengeri + Q. robertingeri) + Q. verrucospinosa))) was supported in BI and ML of nucleotide datasets (most nodes: 1.00 posterior probability, > 68% bootstrap frequencies). In the ranid clade, the monophyly of the Genera Odorrana, Pelophylax, Amolops and Glandirana was well supported, but the monophyly of Babina and Hylarana was not supported because Hylarana guentheri (KM035413) clustered into the clade of Genus Babina. Using the Blast function in NCBI, we found that the mt genome of Hylarana guentheri (KM035413) [57] was the most similar to Babina adenopleura (DQ283117) [46] with 98% identity, which suggests that Hylarana guentheri (KM035413) was misidentified and possibly corresponds to Babina adeopleura..
Discussion
The mtDNA arrangement
In Dicroglossidae, a 32–85 bp NCR between the trnS (AGY) and ND5 genes was observed in Paini and Limnonectini, while a 7–39 bp NCR between the trnS (AGY) and ND6 genes was observed in Dicroglossini and Occidozygini for the translocation of the ND5 gene (Fig. 1). The presence of short non-coding sequences among the rearranged genes has also been observed in previous studies [5]. In this study, both N. yunnanensis and N. taihangnica have a short apomorphic NCR between trnI and trnM; this was also reported in N. quadranus by Zhang et al. [76].
Interspecies tRNA gene rearrangements are well known [11–16], but few were found in the current work. Comparing the tRNA gene rearrangements of mt genomes in all known frogs, we found that the LTPF tRNA cluster, the IQMM tRNA cluster and the WANCY tRNA cluster can easily undergo gene rearrangement, the phenomenon appearing not only in interspecies but also intraspecies comparisons (eg. N. taihangnica and Q. boulengeri). Comparing two mt genomes of N. taihangnica (=F. taihangnica) between this study and a previously sequenced N. taihangnica [13], we found that different gene rearrangements of the trnP, trnF, trnQ, trnA, trnN and trnC genes, the IQMM tRNA cluster and the WANCY tRNA cluster existed. The trnT gene of the LTPF tRNA cluster was lost in the previously sequenced N. taihangnica [13], whereas the trnT gene between CR1 and trnP gene was found in N. taihangnica of this study. The L-NCR (35 bp) -PF tRNA cluster in the previously sequenced N. taihangnica [13] was also found in N. taihangnica of this study but an extra TPF tRNA cluster between CR1 and a 289 bp NCR occurred in N. taihangnica of this study. The IQMM tRNA cluster was found in previously sequenced N. taihangnica whereas the I-NCR (40 bp)-MM tRNA cluster was found in N. taihangnica of this study because the trnQ gene was translocated. The WANOLCY tRNA cluster was found in previously sequenced N. taihangnica while W- NCR (138 bp)-OL-NCR (52 bp)-Y was found in N. taihangnica of this study because of the translocation of trnA, trnN and trnC. Comparing mt genomes of Q. boulengeri between this study and other known sequences [16, 30], we found different gene rearrangements in the WANOLCY tRNA cluster as also found by Xia et al. [77]. In species of the Genus Nanorana and Quasipaa, two types of tRNA clusters (I-NCR-MM or IQMM, WANOLCY or WAOLNCY) were found. Even in the same species, N. taihangnica and Q. boulengeri, different tRNA clusters were found, which may motivate future discussions on mitochondrial gene arrangements among Nanorana and Quasipaa species. This suggests that more mt genomes of Nanorana and Quasipaa species need to be sequenced to further determine how these different gene arrangements formed.
In N. yunnanensis, seven tRNA genes (trnQ, trnA, trnC, trnY, trnS, trnN and trnP) and the ND6 gene on the L-stand were translocated into or near to control regions and grouped together. We did not find any other species of Anura where these gene arrangements existed, but in a fish Crossorhombus azureus (Pleuronectiformes: Bothidae) [23] seven tRNA genes (trnQ, trnA, trnC, trnY, trnS1, trnE, trnP) and the ND6 gene encoded by the Light-strand (L-strand) were translocated to a position between trnT and trnF, which is very similar to our results.
Possible gene rearrangement mechanisms
In N. taihangnica, we observed several gene rearrangements (extra CR, trnP and trnM genes as well as translocation of trnQ, trnA, trnN, and trnC) in the region between CR and the cox1 gene. We propose that the gene rearrangements may be explained by the TDRL model. Although long tandem duplication is a very rare event in mtDNA duplication, the duplication can happen between the origin for H-strand replication (OH) in the CR and the origin for L-strand replication (OL) in the WANCY tRNA cluster, which is a distance of about two-thirds of the genomic length. The mechanism for duplication between the CR and the WANOLCY tRNA genes in N. taihangnica could be caused by the OH and OL structures and be explained by the TDRL model [20], which is similar to the research of Shi et al. [24]. The hypothesized intermediate steps are as follows. Firstly, the above-mentioned OH and OL structures initiated DNA synthesis twice during mitochondrial replication, causing tandem duplication of the genes located between the CR and the WANCY region in the ancestral mitogenome (Fig. 5). Secondly, one of each of the duplicated gene pairs was randomly deleted completely or partially and then lost its function or became a pseudogene (Fig. 5).
Fig. 5.
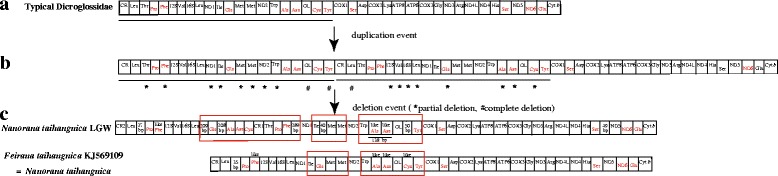
Proposed mechanism of gene rearrangements in N. taihangnica under a model of tandem duplication of gene regions and subsequent gene deletions. a Typical Dicroglossidae gene order. b Tandem duplication in the area from the control region (CR) to trnY (Tyr) and subsequent deletions of partial genes or complete genes resulting in the derived gene order. c State in N. taihangnica. * means that the tandem replicated genes were partially deleted and # means that the tandem replicated genes were completely deleted
In N. yunnanensis the genes of the mitogenome are extensively rearranged with clustering of eight genes on the L-strand in the same polarity and three control regions in an unexpected gene order. These special features of eight genes in the same polarity on the L-strand and two noncoding regions were reported in Crossorhombus azureus which proposed a new mechanism for gene rearrangement [23]. We can use this gene rearrangement mechanism to explain the polarity of gene rearrangement in N. yunnanensis. The hypothesized intermediate steps are as follows. Firstly, the inferred “dimer-mitogenome” intermediate of the N. yunnanensis mtDNA (Fig. 6) could be formed by two entire mitogenomes if the two mt genomes were linked by the head-to-tail method. Secondly, some duplicated genes were non-randomly deleted completely except that all ten genes on the L-strand of one mt monomer were retained; some duplicated genes were also non-randomly deleted completely or partially from the other mt monomer (Fig. 6). Thirdly, the region of CR-trnP-trnQ-trnA-trnN-trnC-trnY-trnS-trnS-ND6-trnE was duplicated. Fourthly, some duplicated genes were randomly deleted completely. So the Dimer-Mitogenome and Non-Random Loss model (DMNR) [23] and the TDRL model [20] may be more appropriate to explain the gene arrangements in N. yunnanensis. But we have no suitable model to explain the four non-coding region (256 bp between trnQ and trnY, 722 bp between trnY and ND6, 1317 bp between trnP and trnA, 394 bp between trnC and trnS).
Fig. 6.
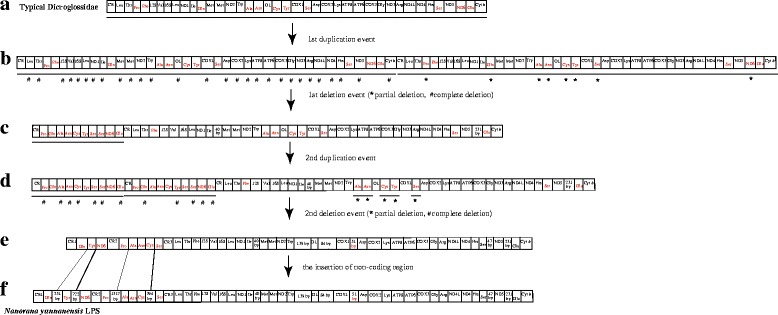
Proposed mechanism of gene rearrangements in the N. yunnanensis mitogenome under a model of tandem duplication of gene regions and subsequent gene deletions. a Typical Dicroglossidae gene order. b The dimeric molecule with two monomers linked head-to-tail and subsequent first deletions of partial genes or complete genes resulting in the derived gene order. c State in N. yunnanensis after the first duplication and deletion. d Tandem duplication in the area from the control region (CR) to trnE (Glu) and subsequent second deletions of partial genes or complete genes resulting in the derived gene order. e State in N. yunnanensis after the second duplication and deletion. f Tandem duplication in the region of CR1-trnQ (Gln), CR2-trnP (Pro) and trnS (Ser)-CR3, respectively, and subsequent third deletions of partial genes or complete genes resulting in the derived gene order. g State in N. yunnanensis after the third duplication and deletion. * means that the tandem replicated genes were partially deleted and # means that the tandem replicated genes were completely deleted
Phylogenetic analyses of Dicroglossidae
The evolutionary relationships of dicroglossid taxa indicated by the phylogenetic trees were mostly similar to previously reported molecular phylogeny [5]. Roelants et al. [78] suggested that Occidozygini is a sister clade to ((Dicroglossini + Paini) + Limnonectini), whereas van der Meijden et al. [79] found that Occidozyga (Occidozygini) is located within Dicroglossinae. Dubois [37] returned Occidozygini to Dicroglossinae as a tribe based on the strength of evidence produced by van der Meijden et al. [79]. In the present study, Occidozygini was found to be a sister clade to (Dicroglossini + (Paini + Limnonectini)), and Occidozygini (Occidozyginae) was observed to be a basal clade to Dicroglossinae.
In phylogenetic trees, the clade of (N. quadranus + ((N. taihangnica + N. taihangnica (KJ569109)) was clustered into the Nanorana. Although N. taihangnica and N. quadranus belong to the genus Feirana according to Fei et al. [42], we draw the conclusion that genus Feirana is not valid according to the phylogenetic relationship of Nanorana and Feirana, which was also supported by Frost et al. [25, 43] and Che et al. [35, 44]
Invalidation of Q. robertingeri as a species
The validity of Quasipaa robertingeri is also heatedly debated. Che et al. [35] found that Quasipaa robertingeri nested deeply within Q. boulengeri and suggested that Q. robertingeri should be synonymous with Q. boulengeri, which is supported by Frost et al. [25]. However, Fei et al. [36, 42] insisted on the validity of Q. robertingeri as a species. The data of Pyron and Wiens [29] supported the proposal that Q. robertingeri was a sister clade to Q. shini, not to Q. boulengeri. To compare the genetic divergence we analyzed the complete mt genomes and 16S RNA gene of Q. boulengeri and Q. robertingeri in Mega 5.0 with the parameter p-distance model. The average genetic distance between Q. boulengeri and Q. robertingeri using mt genomes and 16S RNA was determined to be 4.3% and 1.1%, respectively, which is lower than the lowest interspecies mt genomes between Q. spinosa and Q. exilispinosa (6.8%) and 16S RNA diversity as a species threshold (3%) [80], respectively. Although Q. boulengeri is as a sister clade to Q. robertingeri in phylogenetic relationship, the genetic distance between Q. boulengeri and Q. robertingeri is lower than the genetic distance between interspecies of Quasipaa. The different gene arrangement of Q. boulengeri and Q. robertingeri cannot be used as a species delimitation method because the gene rearrangement can also happened within intraspecies. So we deduce that Q. robertingeri may not be a valid species.
Conclusion
The characteristics of mt genomes and gene arrangements provide novel insights into the phylogenetic relationships among several major lineages of Dicroglossidae. The phylogenetic relationship of ((Ranidae + Dicroglossidae) + (Mantellidae + Rhacophoridae)) is supported in BI analyses. Feirana is not a valid genus according to the phylogenetic relationship with Nanorana. Quasipaa robertingeri may be an invalid species according to genetic divergence. The gene arrangements of N. taihangnica and N. yunnanensis differed from those of other published dicroglossid mt genomes. The mt genomes are promising markers for discussing the reasons for intraspecies gene rearrangements, and the current results broadens our knowledge of the evolution of anuran mt genomes.
Acknowledgements
Computational experiments have been performed with Dell server in the lab of Prof. Bao Yi-Xin. We are grateful to Master Yu-Bo Lin and Kun-Zheng Deng for their help in the study.
Funding
This research was supported by the Natural Science Foundation of Zhejiang Province (LQ16C030001), the National Natural Science Foundation of China (Nos. 31472015 and 31370042) and the Science Technology Commission of Zhejiang Province (2014C32068)) for the study design, data collection and analyses..
Availability of data and materials
The sequences of frogs in this study were deposited in GenBank with accession numbers KF199146-KF199152, KX233864-KX233869 and KM282625. https://www.ncbi.nlm.nih.gov/nuccore. The alignment data used for the phylogenetic analyses are available from the corresponding authors upon request on researchgate.
Abbreviations
- AIC
Akaike Information Criterion
- ATP6/8
ATPase subunit 6/8
- BI
Bayesian inference
- BIC
Bayesian Information Criterion
- BPP
Bayesian posterior probability
- CR
Control region
- Cyt b
cytochrome b
- DMNR
The Dimer-Mitogenome and Non-Random Loss model
- DRRL
Double Replications and Random Loss model
- MCMC
Markov chain Monte Carlo
- NCR
Non-coding region
- ND1–6
NADH dehydrogenase subunit 1–6
- rRNAs
Ribosomal ribonucleic acid
- TDRL
The Tandem Duplication and Random Loss model
- tRNAX
Transfer RNA acid X
Authors’ contributions
Conceived and designed the experiments: YDN ZJY ZLP. Performed the experiments: YDN ZJY ZLP. Analyzed the data: ZJY YDN. Contributed reagents/materials/analysis tools: ZJY YDN ZRQ. Wrote the paper: YDN ZJY KBS ZLP ZRQ. All the authors read and approved the final version of the manuscript.
Ethics approval
All the samples of frogs were collected on public land and all frog samples were permitted under the scientific research in China. We have the permission of our university to collect the frogs.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kakehashi R, Kurabayashi A, Oumi S, Katsuren S, Hoso M, Sumida M. Mitochondrial genomes of Japanese Babina frogs (Ranidae, Anura): unique gene arrangements and the phylogenetic positionof genus Babina. Genes Genet Syst. 2013;88(1):59–67. doi: 10.1266/ggs.88.59. [DOI] [PubMed] [Google Scholar]
- 2.Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27(8):1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boore JL. In: The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. In: comparative genomics: empirical and analytical approaches to gene order dynamics, map alignment and the evolution of gene families. Sankoff D, Nadeau JH, editors. Dordrecht: Kluwer Academic Publishers; 2000. pp. 133–216. [Google Scholar]
- 4.Avise JC. Molecular markers, natural history and evolution. In. New York: Champman and Hall; 1994. [Google Scholar]
- 5.Yu D, Zhang J, Li P, Zheng R, Shao C. Do cryptic species exist in Hoplobatrachus rugulosus? An examination using four nuclear genes, the Cyt b gene and the complete MT genome. PLoS One. 2015;10(4):e0124825. doi: 10.1371/journal.pone.0124825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang JY, Zhou CF, Gai YH, Song DX, Zhou KY. The complete mitochondrial genome of Parafronurus youi (Insecta: Ephemeroptera) and phylogenetic position of the Ephemeroptera. Gene. 2008;424(1–2):18–24. doi: 10.1016/j.gene.2008.07.037. [DOI] [PubMed] [Google Scholar]
- 7.Li E, Li X, Wu X, Feng G, Zhang M, Shi H, Wang L, Jiang J. Complete nucleotide sequence and gene rearrangement of the mitochondrial genome of Occidozyga martensii. J Genet. 2014;93(3):631–641. doi: 10.1007/s12041-014-0418-4. [DOI] [PubMed] [Google Scholar]
- 8.Kurabayashi A, Sumida M, Yonekawa H, Glaw F, Vences M, Hasegawa M. Phylogeny, recombination, and mechanisms of stepwise mitochondrial genome reorganization in mantellid frogs from Madagascar. Mol Biol Evol. 2008;25(5):874–891. doi: 10.1093/molbev/msn031. [DOI] [PubMed] [Google Scholar]
- 9.Kurabayashi A, Usuki C, Mikami N, Fujii T, Yonekawa H, Sumida M, Hasegawa M. Complete nucleotide sequence of the mitochondrial genome of a Malagasy poison frog Mantella madagascariensis: evolutionary implications on mitochondrial genomes of higher anuran groups. Mol Phylogenet Evol. 2006;39(1):223–236. doi: 10.1016/j.ympev.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 10.Liu ZQ, Wang YQ, Su B. The mitochondrial genome organization of the rice frog, Fejervarya limnocharis (Amphibia: Anura): a new gene order in the vertebrate mtDNA. Gene. 2005;346:145–151. doi: 10.1016/j.gene.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 11.Sano N, Kurabayashi A, Fujii T, Yonekawa H, Sumida M. Complete nucleotide sequence of the mitochondrial genome of Schlegeli's tree frog Rhacophorus schlegelii (family Rhacophoridae): duplicated control regions and gene rearrangements. Genes Genet Syst. 2005;80(3):213–224. doi: 10.1266/ggs.80.213. [DOI] [PubMed] [Google Scholar]
- 12.Zhou Y, Zhang JY, Zheng RQ, Yu BG, Yang G. Complete nucleotide sequence and gene organization of the mitochondrial genome of Paa spinosa (Anura: Ranoidae) Gene. 2009;447(2):86–96. doi: 10.1016/j.gene.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 13.Chen Z, Zhai X, Zhang J, Chen X. The complete mitochondrial genome of Feirana taihangnica (Anura: Dicroglossidae) Mitochondrial DNA. 2015;26(3):485–486. doi: 10.3109/19401736.2014.908362. [DOI] [PubMed] [Google Scholar]
- 14.Chen Z, Zhai X, Zhu Y, Chen X. Complete mitochondrial genome of the Ye’s spiny-vented frog Yerana yei (Anura: Dicroglossidae) Mitochondrial DNA. 2015;26(3):489–490. doi: 10.3109/19401736.2014.926542. [DOI] [PubMed] [Google Scholar]
- 15.Jiang L, Ruan Q, Chen W. The complete mitochondrial genome sequence of the Xizang plateau frog, Nanorana parkeri (Anura: Dicroglossidae) Mitochondrial DNA Part A. 2016;27(5):3184–3185. doi: 10.3109/19401736.2015.1074206. [DOI] [PubMed] [Google Scholar]
- 16.Shan X, Xia Y, Zheng YC, Zou FD, Zeng XM. The complete mitochondrial genome of Quasipaa boulengeri (Anura: Dicroglossidae) Mitochondrial DNA. 2014;25(2):83–84. doi: 10.3109/19401736.2013.782023. [DOI] [PubMed] [Google Scholar]
- 17.Yu DN, Zhang JY, Zheng RQ, Shao C. The complete mitochondrial genome of Hoplobatrachus rugulosus (Anura: Dicroglossidae) Mitochondrial DNA. 2012;23(5):336–337. doi: 10.3109/19401736.2012.690748. [DOI] [PubMed] [Google Scholar]
- 18.Zhang P, Zhou H, Liang D, Liu YF, Chen YQ, Qu LH. The complete mitochondrial genome of a tree frog, Polypedates megacephalus (Amphibia : Anura : Rhacophoridae), and a novel gene organization in living amphibians. Gene. 2005;346:133–143. doi: 10.1016/j.gene.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Lunt DH, Hyman BC. Animal mitochondrial DNA recombination. Nature. 1997;387(6630):247. doi: 10.1038/387247a0. [DOI] [PubMed] [Google Scholar]
- 20.Arndt A, Smith M. Mitochondrial gene rearrangement in the sea cucumber genus Cucumaria. Mol Biol Evol. 1998;15(8):1009–1016. doi: 10.1093/oxfordjournals.molbev.a025999. [DOI] [PubMed] [Google Scholar]
- 21.Lavrov DV, Boore JL, Brown WM. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: duplication and nonrandom loss. Mol Biol Evol. 2002;19(2):163–169. doi: 10.1093/oxfordjournals.molbev.a004068. [DOI] [PubMed] [Google Scholar]
- 22.Cantatore P, Gadaleta M, Roberti M, Saccone C, Wilson A. Duplication and remoulding of tRNA genes during the evolutionary rearrangement of mitochondrial genomes. Nature. 1987;329:853–855. doi: 10.1038/329853a0. [DOI] [PubMed] [Google Scholar]
- 23.Shi W, Dong X, Wang Z, Miao X, Wang S, Kong X. Complete mitogenome sequences of four flatfishes (Pleuronectiformes) reveal a novel gene arrangement of L-strand coding genes. BMC Evol Biol. 2013;3:173. doi: 10.1186/1471-2148-13-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi W, Miao XG, Kong XY. A novel model of double replications and random loss accounts for rearrangements in the Mitogenome of Samariscus latus (Teleostei: Pleuronectiformes) BMC Genomics. 2014;15:352. doi: 10.1186/1471-2164-15-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frost DR, Grant T, Faivovich J, Bain RH, Haas A, Haddad CFB, De Sa RO, Channing A, Wilkinson M, Donnellan SC. The amphibian tree of life. Bull Am Mus Nat Hist. 2006;297:1–291. doi: 10.1206/0003-0090(2006)297[0001:TATOL]2.0.CO;2. [DOI] [Google Scholar]
- 26.Kurabayashi A, Sumida M. Afrobatrachian mitochondrial genomes: genome reorganization, gene rearrangement mechanisms, and evolutionary trends of duplicated and rearranged genes. BMC Genomics. 2013;14(1):633. doi: 10.1186/1471-2164-14-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurabayashi A, Yoshikawa N, Sato N, Hayashi Y, Oumi S, Fujii T, Sumida M. Complete mitochondrial DNA sequence of the endangered frog Odorrana ishikawae (family Ranidae) and unexpected diversity of mt gene arrangements in ranids. Mol Phylogenet Evol. 2010;56:543–553. doi: 10.1016/j.ympev.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 28.Xia Y, Zheng YC, Miura I, Wong PB, Murphy RW, Zeng X. The evolution of mitochondrial genomes in modern frogs (Neobatrachia): nonadaptive evolution of mitochondrial genome reorganization. BMC Genomics. 2014;15(1):691. doi: 10.1186/1471-2164-15-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pyron RA, Wiens JJ. A large-scale phylogeny of Amphibia including over 2800 species, and a revised classification of extant frogs, salamanders, and caecilians. Mol Phylogen Evol. 2011;61(2):543–583. doi: 10.1016/j.ympev.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 30.Yuan S, Xia Y, Zheng Y, Zeng X. Next-generation sequencing of mixed genomic DNA allows efficient assembly of rearranged mitochondrial genomes in Amolops chunganensis and Quasipaa boulengeri. Peer J. 2016;4:e2786. doi: 10.7717/peerj.2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen G, Wang B, Liu J, Xie F, Jiang JP. Complete mitochondrial genome of Nanorana pleskei (Amphibia: Anura: Dicroglossidae) and evolutionary characteristics of the amphibian mitochondrial genomes. Curr Zool. 2011;57(6):785–805. doi: 10.1093/czoolo/57.6.785. [DOI] [Google Scholar]
- 32.Ren Z, Zhu B, Ma E, Wen J, Tu T, Cao Y, Hasegawa M, Zhong Y. Complete nucleotide sequence and gene arrangement of the mitochondrial genome of the crab-eating frog Fejervarya cancrivora and evolutionary implications. Gene. 2009;441(1–2):148–155. doi: 10.1016/j.gene.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 33.Zhang JF, Nie LW, Wang Y, Hu LL. The complete mitochondrial genome of the large-headed frog, Limnonectes bannaensis (Amphibia: Anura), and a novel gene organization in the vertebrate mtDNA. Gene. 2009;442(1–2):119–127. doi: 10.1016/j.gene.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 34.Jiang JP, Zhou KY. Phylogenetic relationships among Chinese ranids inferred from sequence data set of 12S and 16S rDNA. Herpetol J. 2005;15(1):1–8. [Google Scholar]
- 35.Che J, Hu J, Zhou W, Murphy RW, Papenfuss TJ, Chen M, Rao D, Li P, Zhang YP. Phylogeny of the Asian spiny frog tribe Paini (family Dicroglossidae) sensu Dubois. Mol Phylogenet Evol. 2009;50(1):59–73. doi: 10.1016/j.ympev.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 36.Fei L, Hu S, Ye C, Huang Y. Fauna Sinica. Amphibia. Vol. 3. Anura Ranidae. Beijing: Chinese Academy of Science. Science Press; 2009. [Google Scholar]
- 37.Dubois A. Amphibia Mundi. 1.1. An ergotaxonomy of recent amphibians. Alytes. 2005;23(1–2):1–24. [Google Scholar]
- 38.Dubois A. Notes sur la classification des Ranidae (Amphibiens Anoures) Bull Soc Linn Lyon. 1992;61:305–352. [Google Scholar]
- 39.Fei L, Ye C, Huang Y. Atlas of amphibians of China. Chongqing: Chongqing Branch, Science and Techonology Literature Publishing House (In Chinese); 1990. [Google Scholar]
- 40.Fei L, Ye C, Huang Y, Jiang J, Xie F. An illustrated key to Chinese amphibians. Chongqing: Sichun Publishing House of Science and Technology (In Chinese); 2005. [Google Scholar]
- 41.Yang X, Wang B, Hu J, Jiang J. A new species of the genus Feirana (Amphibia: Anura: Dicroglossidae) from the western Qinling Mountains of China. Asian Herpetol Res. 2011;2(2):72–86. doi: 10.3724/SP.J.1245.2011.00072. [DOI] [Google Scholar]
- 42.Fei L, Ye CY, Jiang JP. Colored atlas of Chinese amphibians (in Chinese) Chengdu, Sichuan: Sichuan Publishing House of Science and Technology; 2010. [Google Scholar]
- 43.Amphibian Species of the World: an Online Reference. Version 6.0 (1 Dec. 2016). Electronic Database accessible at http://research.amnh.org/herpetology/amphibia/index.html. American Museum of Natural History, New York, USA.
- 44.Che J, Pang J, Zhao H, Wu G, Zhao E, Zhang Y. Molecular phylogeny of the Chinese ranids inferred from nuclear and mitochondrial DNA sequences. Biochem Syst Ecol. 2007;35(1):29–39. doi: 10.1016/j.bse.2006.09.003. [DOI] [Google Scholar]
- 45.Zhang P, Liang D, Mao RL, Hillis DM, Wake DB, Cannatella DC. Efficient sequencing of anuran mtDNAs and a mitogenomic exploration of the phylogeny and evolution of frogs. Mol Biol Evol. 2013;30(8):1899–1915. doi: 10.1093/molbev/mst091. [DOI] [PubMed] [Google Scholar]
- 46.Yu D, Zhang J, Zheng R. The complete mitochondrial genome of Babina adenopleura (Anura: Ranidae) Mitochondrial DNA. 2012;23(6):423–425. doi: 10.3109/19401736.2012.710214. [DOI] [PubMed] [Google Scholar]
- 47.Burland TG. DNASTAR's Lasergene sequence analysis software. Methods Mol Biol. 2000;132:71–91. doi: 10.1385/1-59259-192-2:71. [DOI] [PubMed] [Google Scholar]
- 48.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yan L, Geng ZZ, Yan P, Wu XB. The complete mitochondrial genome of Glandirana tientaiensis (Ranidae, Anura) Mitochondrial DNA Part A. 2016;27(2):1154–1155. doi: 10.3109/19401736.2014.936415. [DOI] [PubMed] [Google Scholar]
- 52.Shan X, Xia Y, Kakehashi R, Kurabayashi A, Zou FD, Zeng XM. Complete mitochondrial genome of Amolops mantzorum (Anura: Ranidae) Mitochondrial DNA. 2016;27(1):705–707. doi: 10.3109/19401736.2014.913152. [DOI] [PubMed] [Google Scholar]
- 53.Li Y, Wu X, Zhang H, Yan P, Xue H, Wu X. The complete mitochondrial genome of Amolops ricketti (Amphidia, Anura, Ranidae) Mitochondrial DNA Part A. 2016;27(1):242–243. doi: 10.3109/19401736.2014.883606. [DOI] [PubMed] [Google Scholar]
- 54.Sumida M, Kanamori Y, Kaneda H, Kato Y, Nishioka M, Hasegawa M, Yonekawa H. Complete nucleotide sequence and gene rearrangement of the mitochondrial genome of the Japanese pond frog Rana Nigromaculata. Genes Genet Syst. 2001;76(5):311–325. doi: 10.1266/ggs.76.311. [DOI] [PubMed] [Google Scholar]
- 55.Ryu SH, Hwang UW. Complete mitochondrial genome of the Seoul frog Rana chosenica (Amphibia, Ranidae): comparison of R. Chosenica and R. Plancyi. Mitochondrial DNA. 2011;22(3):53–54. doi: 10.3109/19401736.2011.603313. [DOI] [PubMed] [Google Scholar]
- 56.Hofman S, Pabijan M, Dziewulska-Szwajkowska D, Szymura JM. Mitochondrial genome organization and divergence in hybridizing central European waterfrogs of the Pelophylax Esculentus Complex (Anura, Ranidae) Gene. 2012;491(1):71–80. doi: 10.1016/j.gene.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 57.Wu X, Li Y, Zhang H, Jiang Z, Xue H, Yan P, Wu X. The complete mitochondrial genome of Hylarana guentheri (Amphidia, Anura, Ranidae) Mitochondrial DNA Part A. 2016;27(2):1223–1224. doi: 10.3109/19401736.2014.945528. [DOI] [PubMed] [Google Scholar]
- 58.Lin Y, Tao B, Fang X, Wang T, Zhang J. The complete mitochondrial genome of Lithobates catesbeianus (Anura: Ranidae) Mitochondrial DNA. 2014;25(6):447–448. doi: 10.3109/19401736.2013.814115. [DOI] [PubMed] [Google Scholar]
- 59.Ni N, Yu D, Storey KB, Zheng R, Zhang J. The complete mitochondrial genome of Lithobates sylvaticus (Anura: Ranidae) Mitochondrial DNA Part A. 2016;27(4):2460–2461. doi: 10.3109/19401736.2015.1033697. [DOI] [PubMed] [Google Scholar]
- 60.Li J, Yin W, Xia R, Lei G, Fu C. Complete mitochondrial genome of a brown frog, Rana kunyuensis (Anura: Ranidae) Mitochondrial DNA Part A. 2016;27(1):34–35. doi: 10.3109/19401736.2013.869681. [DOI] [PubMed] [Google Scholar]
- 61.Li J, Lei G, Fu C. Complete mitochondrial genomes of two brown frogs, Rana dybowskii and Rana Cf. chensinensis (Anura: Ranidae) Mitochondrial DNA Part A. 2016;27(1):155–156. doi: 10.3109/19401736.2013.878921. [DOI] [PubMed] [Google Scholar]
- 62.Chen Z, Zhang J, Zhai X, Zhu Y, Chen X. Complete mitochondrial genome of the green odorous frog Odorrana Margaretae (Anura: Ranidae) Mitochondrial DNA. 2015;26(3):487–488. doi: 10.3109/19401736.2014.926533. [DOI] [PubMed] [Google Scholar]
- 63.Su X, Wu XB, Yan P, Cao SY, Hu YL. Rearrangement of a mitochondrial tRNA gene of the concave-eared torrent frog, Amolops tormotus. Gene. 2007;394(1):25–34. doi: 10.1016/j.gene.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 64.Alam MS, Kurabayashi A, Hayashi Y, Sano N, Khan MMR, Fujii T, Sumida M. Complete mitochondrial genomes and novel gene rearrangements in two dicroglossid frogs, Hoplobatrachus tigerinus and Euphlyctis hexadactylus, from Bangladesh. Genes Genet Syst. 2010;85(3):219–232. doi: 10.1266/ggs.85.219. [DOI] [PubMed] [Google Scholar]
- 65.Sano N, Kurabayashi A, Fujii T, Yonekawa H, Sumida M. Complete nucleotide sequence and gene rearrangement of the mitochondrial genome of the bell-ring frog, Buergeria buergeri (family Rhacophoridae) Genes Genet Syst. 2004;79(3):151–163. doi: 10.1266/ggs.79.151. [DOI] [PubMed] [Google Scholar]
- 66.Wu X, Li Y, Zhang H, Yan L, Wu X-B. The complete mitochondrial genome of Microhyla pulchra (Amphidia, Anura, Microhylidae) Mitochondrial DNA Part A. 2016;27(1):40–41. doi: 10.3109/19401736.2013.869685. [DOI] [PubMed] [Google Scholar]
- 67.Igawa T, Kurabayashi A, Usuki C, Fujii T, Sumida M. Complete mitochondrial genomes of three neobatrachian anurans: a case study of divergence time estimation using different data and calibration settings. Gene. 2008;407(1):116–129. doi: 10.1016/j.gene.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 68.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 69.Xia X, Xie Z. DAMBE: software package for data analysis in molecular biology and evolution. J Hered. 2001;92(4):371–373. doi: 10.1093/jhered/92.4.371. [DOI] [PubMed] [Google Scholar]
- 70.Cameron SL, Miller KB, D'Haese CA, Whiting MF, Barker SC. Mitochondrial genome data alone are not enough to unambiguously resolve the relationships of Entognatha, Insecta and Crustacea Sensu Lato (Arthropoda) Cladistics. 2004;20(6):534–557. doi: 10.1111/j.1096-0031.2004.00040.x. [DOI] [PubMed] [Google Scholar]
- 71.Lanfear R, Calcott B, Ho SY, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29(6):1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- 72.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 73.Huelsenbeck JP, Ronquist F. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 74.Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 75.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang DR, Chen MY, Murphy RW, Che J, Pang J, Hu JS, Luo J, Wu S, Ye H, Zhang YP. Genealogy and palaeodrainage basins in Yunnan Province: phylogeography of the Yunnan spiny frog, Nanorana yunnanensis (Dicroglossidae) Mol Ecol. 2010;19(16):3406–3420. doi: 10.1111/j.1365-294X.2010.04747.x. [DOI] [PubMed] [Google Scholar]
- 77.Xia Y, Zheng Y, Murphy RW, Zeng X. Intraspecific rearrangement of mitochondrial genome suggests the prevalence of the tandem duplication-random loss (TDLR) mechanism in Quasipaa boulengeri. BMC Genomics. 2016;17(1):965. doi: 10.1186/s12864-016-3309-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roelants K, Jiang J, Bossuyt F. Endemic ranid (Amphibia: Anura) genera in southern mountain ranges of the Indian subcontinent represent ancient frog lineages: evidence from molecular data. Mol Phylogenet Evol. 2004;31(2):730–740. doi: 10.1016/j.ympev.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 79.van der Meijden A, Vences M, Hoegg S, Meyer A. A previously unrecognized radiation of ranid frogs in southern Africa revealed by nuclear and mitochondrial DNA sequences. Mol Phylogenet Evol. 2005;37(3):674–685. doi: 10.1016/j.ympev.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 80.Vences M, Thomas M, Van der Meijden A, Chiari Y, Vieites DR. Comparative performance of the 16S rRNA gene in DNA barcoding of amphibians. Front Zool. 2005;2(1):5. doi: 10.1186/1742-9994-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sequences of frogs in this study were deposited in GenBank with accession numbers KF199146-KF199152, KX233864-KX233869 and KM282625. https://www.ncbi.nlm.nih.gov/nuccore. The alignment data used for the phylogenetic analyses are available from the corresponding authors upon request on researchgate.