

Abstract
Changes in gene regulation have long been thought to play an important role in primate evolution. However, although a number of studies have compared genome-wide gene expression patterns across primate species, fewer have investigated the gene regulatory mechanisms that underlie such patterns, or the relative contribution of drift versus selection. Here, we profiled genome-scale DNA methylation levels in blood samples from five of the six extant species of the baboon genus Papio (4–14 individuals per species). This radiation presents the opportunity to investigate DNA methylation divergence at both shallow and deeper timescales (0.380–1.4 My). In contrast to studies in human populations, but similar to studies in great apes, DNA methylation profiles clearly mirror genetic and geographic structure. Divergence in DNA methylation proceeds fastest in unannotated regions of the genome and slowest in regions of the genome that are likely more constrained at the sequence level (e.g., gene exons). Both heuristic approaches and Ornstein–Uhlenbeck models suggest that DNA methylation levels at a small set of sites have been affected by positive selection, and that this class is enriched in functionally relevant contexts, including promoters, enhancers, and CpG islands. Our results thus indicate that the rate and distribution of DNA methylation changes across the genome largely mirror genetic structure. However, at some CpG sites, DNA methylation levels themselves may have been a target of positive selection, pointing to loci that could be important in connecting sequence variation to fitness-related traits.
Keywords: DNA methylation, neutral evolution, genetic divergence, molecular evolution, baboons, gene regulation
Introduction
Changes in gene regulation have long been hypothesized to play an important role in trait evolution (Britten and Davidson 1971; King and Wilson 1975; Jacob 1977; Wray 2007; Stern and Orgogozo 2008). Regulatory changes have the potential to be more modular, and hence more specific to the individual tissues, environmental conditions, or developmental time points targeted by selection, than protein-coding changes (Stern 2000). In addition, regulatory regions are believed to have larger mutational target sizes, increasing the rate at which they may evolve (Landry et al. 2007). In support of the importance of regulatory evolution, a number of studies have identified regulatory changes that contribute to species-specific adaptations. For example, noncoding variants that regulate the ectodysplasin and pitx1 genes underlie morphological changes that separate saltwater threespine sticklebacks (Gasterosteus aculeatus) from their close freshwater relatives (Colosimo et al. 2004, 2005; Shapiro et al. 2004). Similarly, wing pattern mimicry in Heliconius butterflies has been repeatedly shaped by regulatory evolution near the optix gene, in which convergent changes at different cis-regulatory variants have produced similar patterns of wing coloration (Reed et al. 2011; Heliconius Genome Consortium 2012). Together, these and other case studies (e.g., Abzhanov et al. 2004; Prud’Homme et al. 2006; Manceau et al. 2011; Jones et al. 2012; Poelstra et al. 2014) provide compelling examples of the importance of regulatory sequence changes to adaptive evolution.
However, evaluating the role of gene regulation in adaptive trait evolution also requires understanding the genome-wide distribution of selectively relevant regulatory variants. To address this question, two approaches have commonly been employed: sequence-based tests for selection and comparative analyses of gene expression phenotypes themselves. The first approach has identified signatures of natural selection in regulatory regions both within and between species (e.g., Pollard et al. 2006; Prabhakar et al. 2006; Kosiol et al. 2008). In primates, for example, genes associated with developmental or neuronal functions have been argued to contain more signatures of positive selection in noncoding regions than in their coding sequences (Haygood et al. 2010). Relative to other genetic variants, loci that affect gene expression in humans also have larger integrated haplotype scores, providing evidence for recent positive selection (Nédélec et al. 2016; Kim-Hellmuth et al. 2017). Consistent with these findings, variants associated with disease risk, fecundity, and other selectively relevant traits are often found within noncoding regions, and likely affect gene expression levels (Nicolae et al. 2010; Wray 2013).
The second approach investigates patterns of gene expression across species to search for cases consistent with adaptive evolution. Several patterns have emerged from this work. First, overall differences in gene expression accumulate over evolutionary time, such that more closely related species have more similar gene expression profiles. Global clustering approaches from the same tissue thus tend to faithfully reproduce the species phylogeny (Brawand et al. 2011; Sudmant et al. 2015), and exceptions to this pattern suggest possible cases of natural selection. For example, gene expression levels in testis, but not in other tissues, group humans and gorillas to the exclusion of chimpanzees and bonobos (Brawand et al. 2011). This pattern is consistent with elevated sexual selection on male reproductive physiology in chimpanzees and bonobos, which are characterized by unusually large testis to body size ratios relative to other primates (Schultz 1938). Second, stabilizing selection appears to constrain most gene expression levels. Comparative analyses of gene expression have found that most genes are characterized by low levels of intra- and interspecific divergence, a pattern consistent with stabilizing selection (Rifkin et al. 2003; Gilad, Oshlack, Smyth, et al. 2006; Khaitovich et al. 2006; Blekhman et al. 2008; Coolon et al. 2014; Hodgins-Davis et al. 2015). Furthermore, within species, regulatory variants of large effect tend to have low allele frequencies, suggesting that they are typically selected against (Battle et al. 2014; Hernandez et al. 2017; Schoech et al. 2017). In support of this argument, experimental mutation accumulation lines exhibit an excess of gene expression variation compared with that observed in natural populations. They also accumulate differences in gene expression at a faster rate than observed in between-species comparisons (Denver et al. 2005; Rifkin et al. 2005).
Thus, both sequence-based studies and comparative studies of gene expression support a central role for selection on gene expression evolution, dominated by stabilizing selection but with an additional contribution made by positive selection (Signor and Nuzhdin 2018). However, gene expression patterns themselves are a product of multiple underlying regulatory mechanisms, which govern chromatin accessibility, transcription factor binding, and mRNA processing, splicing, and stability. These mechanisms link genetic variation in DNA sequence to selectively relevant gene expression phenotypes (Gallego Romero et al. 2012; Pai and Gilad 2014). For example, in humans, genetic variants associated with chromatin accessibility and DNA methylation are often also associated with gene expression, suggesting that these mechanisms functionally link DNA sequence variation to gene expression (Degner et al. 2012; Banovich et al. 2014; Gate et al. 2018). Between species, however, we know considerably less about how gene regulatory mechanisms evolve, including their relative contributions to lineage-specific shifts in gene expression levels (Pai and Gilad 2014).
Comparative studies to date have focused most intensively on DNA methylation, an epigenetic regulatory mechanism that refers to the covalent addition of a methyl group to a cytosine base and that can affect transcription factor binding, chromatin accessibility, and gene expression (Klose and Bird 2006; Weber et al. 2007; Jones 2012; but see also Shibata et al. 2012; Zhou et al. 2014; Villar et al. 2015; Berthelot et al. 2018 for work on other mechanisms). In primates, comparisons between humans, chimpanzees, and rhesus macaques suggest that divergence in DNA methylation is associated with changes in gene expression (Zeng et al. 2012; Heyn et al. 2013), explaining 15–21% of expression differences between species (Pai et al. 2011). Like gene expression, divergence in DNA methylation also increases with genetic distance (Hernando-Herraez et al. 2013). However, comparisons among human populations suggest that DNA methylation evolves in a more clock-like fashion than gene expression, possibly because gene expression phenotypes evolve under greater functional constraint (Carja et al. 2017). Unlike for gene expression levels (Rifkin et al. 2003; Gilad, Oshlack, Smyth, et al. 2006; Khaitovich et al. 2006; Whitehead and Crawford 2006; Blekhman et al. 2010; Brawand et al. 2011; Rohlfs and Nielsen 2014), the relative contribution of genetic drift and natural selection to DNA methylation evolution across species has not been investigated.
Here, we address this gap by investigating the evolution of genome-wide DNA methylation levels in the baboon genus Papio. Baboons radiated in sub-Saharan Africa over the past 1.4 My to include six currently recognized extant species: anubis baboons (P. anubis, also called the olive baboon), hamadryas baboons (P. hamadryas), and Guinea baboons (P. papio) in the northern half of Africa and the Arabian peninsula; and yellow baboons (P. cynocephalus), chacma baboons (P. ursinus), and Kinda baboons (P. kindae) in central and southern Africa (fig. 1A: Jolly 1993; Rogers et al. forthcoming). Studying DNA methylation divergence in this species complex thus provides additional resolution on the rate of DNA methylation evolution in primates, as previous studies have concentrated either on deeply diverged great apes (5–15 My of divergence) or on closely related human populations (Pai et al. 2011; Hernando-Herraez et al. 2013, 2015; Heyn et al. 2013; Mendizabal et al. 2016; Carja et al. 2017). Genetic evidence indicates that branching events leading to the extant baboon species occurred on an intermediate timescale, between 0.380 and 1.4–2.0 Ma (Zinner et al. 2013; Rogers et al. forthcoming). Further, because baboon genetic diversity is unusually well-characterized (Wall et al. 2016; Leffler 2017), focusing on baboons also allowed us to investigate the relationship between DNA methylation and patterns of genetic variation across the genome.
Fig. 1.

Geographic and genetic structure in baboon DNA methylation patterns. (A) The first two principal components from a PCA of baboon DNA methylation profiles (subsampled to n = 4 individuals per species) projected onto the geographic distribution of baboon species in Africa. Northern clade (cool colors) and southern clade (warm colors) baboons separate along the first PC. The distribution of the six commonly recognized baboon species in Africa and the Arabian peninsula is based on Zinner et al. (2013) and modified from a map created by Kenneth Chiou (CC BY 3.0 license); note that points reflect coordinates for DNA methylation data in PC space, not sampling location. Phylogenetic relationships between the five species included in this data set, with rhesus macaque as an outgroup, are shown in the inset (divergence dates within baboons from Rogers et al. [forthcoming] and between baboons and macaques from Perelman et al. [2011]). (B) Procrustes transformation of PC1 and PC2 of the DNA methylation data (empty squares) conforms with PC1 and PC2 of genotype data (solid circles) from the same samples (Procrustes t0 = 0.89, P < 10−6). PVE values on the x- and y-axis are provided for the genotype data.
To do so, we generated genome-scale bisulfite sequencing data from blood samples obtained from 4 to 14 members of each of five of the extant species (all but chacma baboons). We asked: 1) to what degree does phylogenetic divergence between baboon species predict evolutionary change in DNA methylation levels? 2) how are clade- and species-specific shifts in DNA methylation distributed across the baboon genome? and 3) what are the relative contributions of natural selection and genetic drift to patterns of DNA methylation across species? Our results show that divergence in DNA methylation is closely linked to genetic divergence in baboons. Additionally, heterogeneity in DNA methylation divergence is explained by a combination of functional context, mean methylation level, and differences in selective constraint. At a subset of sites, these differences are consistent with spatially clustered, lineage-specific selective shifts, suggesting candidate loci for which interspecific changes in gene expression may be explained by selection on DNA methylation.
Results
Genome-Wide Variation in DNA Methylation Reflects Geography and Phylogenetic Structure
We generated DNA methylation profiles from blood samples for 39 baboons and 5 rhesus macaques (Macaca mulatta) (supplementary table S1, Supplementary Material online) using reduced representation bisulfite sequencing (RRBS: Gu et al. 2011; Boyle et al. 2012). After filtering for CpG sites where at least half of our study subjects were sequenced to at least 5× coverage, the data set included DNA methylation estimates for 2,450,153 CpG sites throughout the genome. As expected for RRBS data, these sites were strongly enriched in or near CpG dense regions of the genome, including CpG islands, CpG shores, gene bodies, and promoters (supplementary fig. S1, Supplementary Material online). At least one CpG site in the promoter or gene body was included for 75.2% of Ensembl-annotated protein-coding genes in the reference anubis baboon genome (Panu2.0; supplementary fig. S1, Supplementary Material online). To investigate patterns of DNA methylation variation across Papio, we subsequently focused on the subset of 756,262 CpG sites that were not constitutively hyper- or hypomethylated (mean methylation level across all study subjects). Two of the species we sampled (hamadryas baboons and anubis baboons) included individuals from multiple source populations (supplementary table S1, Supplementary Material online). However, because source population was not significantly associated with variation in DNA methylation within species (supplementary Methods, Supplementary Material online), we grouped all samples from the same species together for subsequent analysis.
To investigate the relationship between DNA methylation levels and genetic divergence, we first performed principal component analysis (PCA) on the DNA methylation data. With rhesus macaques included, the first principal component explained 14% of the overall variance in the data and separated all baboons from all rhesus macaques (supplementary fig. S2, Supplementary Material online). Subsequent PCs captured variation within Papio and were highly correlated with the top PCs when considering baboon samples only (r2 > 0.96 between PCs 2–5 including macaques and PCs 1–4 excluding macaques). To investigate species differences within Papio, we subsampled the baboon data to four individuals for each species (based on the smallest sample size per species, for Kinda baboons) and analyzed the baboon samples alone. In most subsets (79.6%), PC1 and PC2 mirror the phylogenetic history of the baboon species we sampled (fig. 1A). They first separated baboons from the northern clade from baboons from the southern clade (PC1), and then separated hamadryas baboons from all other taxa (PC2). To explicitly compare structure in the DNA methylation data with baboon genetic structure, we used Procrustes analyses on the DNA methylation data set and genotype data collected from the same RRBS data (n = 49,607 single nucleotide polymorphisms; supplementary Methods, Supplementary Material online). The first two PCs of the genotype data were significantly concordant with the first two PCs of the DNA methylation data (fig. 1B; Procrustes t0 = 0.89, P < 10−6), indicating that divergence in CpG methylation levels is closely tied to genetic divergence (near-identical results were obtained when including additional PCs, up to PC6).
Consistent with a close link between DNA methylation and genetic divergence, pairwise genetic covariance between samples strongly predicted pairwise covariance in DNA methylation levels. Across all CpG sites, a sample-wise covariance matrix based on RRBS-derived genotype data was significantly correlated with a sample-wise covariance matrix based on DNA methylation levels (n = 756,262 CpG sites; Mantel test r [95% CI] = 0.680 [0.651–0.721], P < 10−6), especially when considering the baboon samples alone (r = 0.818 [0.794–0.856], P < 10−6). However, the strength of the correlation varied systematically across genomic contexts (lowest in CpG islands and exons and highest in intergenic, unannotated regions, controlling for mean methylation: fig. 2A and supplementary Methods, Supplementary Material online) and across mean methylation levels. Specifically, in all contexts, the strongest relationship between genetic variation and DNA methylation levels was observed for intermediately methylated CpG sites, which also tend to exhibit the most variation in DNA methylation across individuals (fig. 2A). Notably, regions of the genome that support a nonconsensus phylogeny (i.e., those most likely to be affected by incomplete lineage sorting or admixture, which is common in baboons: Zinner et al. 2009, 2013; Tung and Barreiro 2017; Rogers et al. forthcoming; see Materials and Methods) exhibited a weaker association between the DNA methylation and genotype matrices than those that fit the consensus phylogeny (Mantel test r = 0.716 [0.649–0.760], n = 211,852 sites compared with 0.815 [0.766–0.858] for regions that matched the consensus phylogeny, n = 542,509 sites).
Fig. 2.

Concordance between DNA methylation variation and genetic variation depends on genomic context. (A) Correlation between pairwise genetic covariance between species and pairwise covariance in DNA methylation levels, for CpG sites stratified by genomic context and mean DNA methylation level. Each point represents n = 2,658–604,775 CpG sites. (B) Estimated mean rate of change in DNA methylation levels per million years, stratified by genomic context. Error bars represent the standard error for each estimate.
Thus, both the PCA results and the correlation between DNA methylation and genetic structure indicate that species differences in DNA methylation are associated with genetic divergence. To investigate how this relationship scales, we estimated the correlation between divergence time (0.380–1.4 My within Papio, and 8.1 My between baboons and macaques: Perelman et al. 2011; Rogers et al. forthcoming) and DNA methylation divergence per site. For this analysis, we limited the data set to CpG sites that were measured in at least three individuals of each species (n = 438,713 CpG sites). When both macaques and baboons were included in the analysis, pairwise divergence time was strongly positively correlated with pairwise DNA methylation divergence (Mantel test r = 0.970, P = 0.011), with an estimated rate of change for the average CpG site of 1.14% per million years. This estimate is similar to that obtained from baboons alone (1.27% per million years), although the baboon results are noisier and not statistically significant (Mantel test r = 0.377, P = 0.067). Divergence in DNA methylation is fastest in functionally unannotated regions of the genome and slowest in gene exons, CpG islands, promoters, and enhancers (fig. 2B and supplementary fig. S3, Supplementary Material online).
EvolutionaryShifts in DNAMethylation Levels Within Papio
We next investigated the frequency and distribution of CpG sites that exhibit 1) genus-level differences in DNA methylation between baboons and macaques, 2) clade-level differences in DNA methylation between northern and southern clade baboons, and/or 3) species-level shifts in DNA methylation levels that differentiate one baboon species from all other baboons. To do so, we first used ANOVA to identify 130,358 (17.3% of those tested), 12,791 (1.70%), and 9,999 (1.33%) CpG sites for which genus, clade (within genus), or species (within clade) membership explained significant variance in DNA methylation levels, respectively (fig. 3A; 10% false discovery rate [FDR] based on the q-value approach of Storey and Tibshirani [2003], supplementary Methods, Supplementary Material online). These sets of taxonomically structured CpG sites overlapped more than expected by chance (Fisher’s exact test log2(OR) > 0.95 and P < 10−16 for all three pairwise comparisons). CpG sites located in functionally unannotated regions, gene introns, and untranslated regions were more likely to exhibit taxonomically structured variation in DNA methylation than CpG sites in other genomic contexts (fig. 3B and supplementary table S2, Supplementary Material online). Conversely, such variation was depleted for CpG sites in gene exons. This dependency on genomic context was generally consistent between sites that exhibited significant genus-, clade-, or species-level variation. However, species-level changes were more strongly enriched in unannotated regions and more clearly depleted for other functional contexts (supplementary fig. S4 and table S3, Supplementary Material online), consistent with faster divergence in regions where genetic variation is more likely to be selectively neutral. Taxonomically structured variation was also more common for sites with more nearby (within 1 kb) CpG-disrupting genetic variants, even though the focal CpG sites themselves were not disrupted (logistic regression: β = 0.0246, P = 3.33 × 10−133; supplementary fig. S5 and supplementary Methods, Supplementary Material online). Therefore, changes in local CpG site density may contribute to some taxonomically structured variation in DNA methylation (Fukuda et al. 2013).
Fig. 3.

Interspecific differences in DNA methylation levels. (A) The number of CpG sites that exhibit significant taxonomic structure at successive levels of the phylogeny. Sites significantly overlap between genus and N–S clade (log2(OR) = 0.990, P = 2.82 × 10−240), genus and species (log2(OR) = 1.176, P = 2.760 × 10−276), and N–S clade and species (log2(OR) = 5.580, P < 100 × 10−300). (B) Enrichment by genomic context for 1) CpG sites in which DNA methylation levels show significant taxonomic structure by clade or species (green dots; background set is the full set of n = 756,262 CpG sites analyzed), and 2) CpG sites in which OU models and heuristic analyses (for clade-specific shifts) or heuristic analyses (for species-specific shifts) indicate a likely history of positive selection (blue dots; background set is n = 20,360 taxonomically structured CpG sites). Functional elements that are depleted for significant taxonomic structure overall are nevertheless enriched for a signature of selection among those sites that do exhibit taxonomic structure. (C, D) Examples of large DMRs. Dashes along the x-axis show the location of each measured CpG site in the region and lines show the smoothed mean DNA methylation level (BSmooth: Hansen et al. 2012). Thin dashed lines represent individual samples, and bold lines represent mean methylation levels per species. A Guinea baboon-specific DMR associated with the Leucine Rich Repeat and Ig Domain Containing 3 (LINGO3) gene is shown in (C) and a hamadryas baboon-specific DMR associated with the taperin (TPRN) and transmembrane protein 203 (TMEM203) genes is shown in (D).
To identify shifts in DNA methylation associated with specific baboon taxa, we focused on the set of 20,360 sites that were taxonomically structured by clade or species membership. For these sites, we then applied a binomial mixed effects model (Lea et al. 2015) to identify differential methylation 1) between each target species and all other baboons, and 2) between clades (10% FDR threshold). We required a minimum 10% difference in mean DNA methylation levels between the focal species and all other baboon species to call a species-specific shift, and a minimum 10% difference between all between-clade species pairs, as well as rhesus macaque, to call a clade-level shift. Based on these criteria, we identified 1,230–4,916 species-specific shifts per species (13,098 unique sites in all). The number of shifts per species was not a function of sample size or independent evolutionary time (linear model, P = 0.791 and P = 0.793, respectively) and we observed no consistent bias toward increased or decreased methylation in any species (controlling for mean methylation level: supplementary fig. S6, Supplementary Material online). We also identified 6,098 CpG sites with evidence for a clade-specific shift: 1,590 sites where DNA methylation in the northern clade was different from the southern clade species and macaques, 3,196 sites where DNA methylation in the southern clade was different from the northern clade species and macaques, and 1,315 sites where methylation differed between the two clades and both clades were also different from macaques. Sites that exhibited species- or clade-level shifts were more spatially clustered than expected by chance (P < 10−6, z-score = 13.70 compared with a permutation-based null; supplementary Methods, Supplementary Material online). We subsequently defined clusters of ≥3 such sites within a 2-kb window as differentially methylated regions (DMRs) as clusters of this size are highly unlikely to arise by chance (supplementary Methods, Supplementary Material online).
Overall, we identified 724 DMRs, which as a whole were enriched near genes involved in catabolic processes. DMRs specific to the southern clade were enriched near genes involved in the cellular response to stress, and DMRs specific to Guinea, hamadryas, and yellow baboons were enriched near genes involved in RNA processing, the response to organic substances, and metabolism (10% FDR threshold). We also identified nine large DMRs (≥15 CpG sites: fig. 3C and D). Four of these DMRs occur in the hamadryas lineage, three in the Guinea lineage, one is specific to the northern clade, and one is specific to the southern clade. Almost all (eight of nine) of the large DMRs overlapped with a CpG island and were within 10 kb of at least one gene. Large DMR-associated genes included single immunoglobulin domain-containing IL1R-related protein (SIGIRR), which is involved in innate immune defense, regulation of inflammation, and natural killer cell maturation; taperin (TPRN), which is implicated in hearing and sensory phenotypes; and transmembrane protein 203 (TMEM203), which is required for spermatogenesis. These loci represent candidate regions in which differences in DNA methylation may be important in translating genetic variation to phenotypic differences between baboon taxa.
Selection on DNA Methylation Patterns in Baboons
Our results indicate that DNA methylation in functionally important regions of the genome evolves more slowly than DNA methylation in unannotated regions, consistent with stabilizing selection on gene regulation and neutral evolution for functionally silent CpG sites. However, lineage-specific shifts in DNA methylation point to a possible contribution of positive selection. To investigate the relative contribution of these different selective regimes, we performed site-specific analyses using two complementary methods: 1) a heuristic approach based on comparisons between intra- and interspecific variation (Rifkin et al. 2003; Nuzhdin et al. 2004; Gilad, Oshlack, Smyth, et al. 2006; Whitehead and Crawford 2006; Gallego Romero et al. 2012), and 2) Ornstein–Uhlenbeck (OU) models of phenotypic evolution, which have recently been extended to model gene expression phenotypes and to incorporate intraspecific variation (Lande 1976; Butler and King 2004; Bedford and Hartl 2009; Rohlfs and Nielsen 2014).
The heuristic approach is based on the logic that phenotypes that evolve under positive selection will harbor less intraspecific variation than phenotypes that evolve under genetic drift (Gallego Romero et al. 2012). Therefore, CpG sites where mean methylation differs between species but variation is low within species are the most likely to have experienced a history of positive selection. To identify such sites, we focused on those in the lowest decile of within-species variance (controlling for average methylation, see Materials and Methods) that also displayed significant species or clade-specific methylation. These criteria yielded a set of 875 and 2,625 CpG sites that are candidates for positive selection to differentiate baboon clades or species, respectively. We note that this approach is likely to retain false positives (and also miss false negatives, which is common in tests for selection): Thus, this set should be treated as enriched for a likely history of positive selection, rather than as a definitive list of positively selected sites.
In the second approach, we fit Brownian motion and OU models of phenotypic evolution, which include explicit parameters for the strength of selection toward a phenotypic optimum or optima (Butler and King 2004). We used a modified approach that takes into account intraspecific phenotypic variance (following Bedford and Hartl 2009; Rohlfs and Nielsen 2014), with modifications to accommodate our data type. Simulations indicated that, in the baboon phylogeny, these models are underpowered to identify species-specific episodes of selection, but are reasonably well-powered to detect positive selection on multispecies lineages (see supplementary Methods, Supplementary Material online). As for the heuristic approach, we treat our results as enriched for specific evolutionary histories, as opposed to definitive. For each taxonomically structured site (n = 20,360 sites), we fit five models, which captured i) genetic drift across the baboon phylogeny, ii) stabilizing selection toward a single optimum, iii) positive selection toward a different phenotypic optimum in the southern baboon clade (yellow and Kinda), iv) positive selection toward a different phenotypic optimum in the northern baboon clade (anubis, Guinea, and hamadryas), and v) positive selection toward a different phenotypic optimum in a northern baboon subclade, the anubis–Guinea lineage. We defined the best model (for each site) as the one with the lowest Akaike information criterion value (Akaike 1974). Models iii–v, which include positive selection somewhere in the tree, were chosen as the best model for 12,700 CpG sites (1.68% of the initial set of sites tested, n = 756,262, and 62.4% of the 20,360 sites that exhibited significant clade- or species-level shifts).
The heuristic approach and the OU model approach produced highly overlapping sets of putative positively selected sites (Fisher’s exact test log2(OR) = 1.59, P = 1.06 × 10−41). We detected 724 CpG sites with evidence for positive clade-level selection in both methods (supplementary Methods, Supplementary Material online), which we treat as our highest-confidence set. Compared with the background set of sites with taxonomic structure in DNA methylation, which tend to occur most often in gene introns, untranslated regions, and functionally unannotated regions, sites with evidence for positive selection are strongly enriched for gene exons and functional regulatory elements, including promoters, enhancers, CpG islands, and CpG island shores (fig. 3B and supplementary table S2, Supplementary Material online). This pattern is consistent for all sets of candidate positively selected sites (supplementary fig. S4 and table S4, Supplementary Material online) and, similar to the larger set of taxonomically structured sites, candidate positively selected sites were more spatially clustered than expected by chance (z-score > 11 against a permutation-based null, P < 10−6). Seventy DMRs were associated with species-specific selection (2–26 per species) and 25 with clade-specific selection (11 assigned to the northern clade and 14 assigned to the southern clade). Consistent with our results for species-specific shifts above, candidate positively selected DMRs were enriched overall for association with genes involved in metabolic processes (10% FDR threshold). However, they were no more likely to be located near disrupted CpG sites than other, taxonomically structured sites (logistic regression: β = 1.93 × 10−3, P = 0.471; supplementary fig. S5, Supplementary Material online).
If regulatory divergence in DNA methylation levels is a consequence of genetic divergence, the genetic sequence surrounding positively selected CpG sites should also show signatures of positive selection, including reduced levels of local genetic variation. To test this prediction, we calculated nucleotide diversity (π: Nei and Li 1979) for the 1 kb centered on each taxonomically structured CpG site for each baboon species, based on data from the Baboon Genome Project Diversity Panel (2–4 individuals sequenced at 30× coverage per species; see supplementary Methods, Supplementary Material online). Averaged across all baboon lineages, nucleotide diversity around CpG sites for which we inferred a history of positive selection somewhere in the tree (mean π ± SD = 0.00233 ± 0.00307) was slightly reduced relative to nucleotide diversity around CpG sites with no evidence for positive selection (0.00243 ± 0.00214, Tukey’s Honestly Significant Difference test P = 0.0.0441; fig. 4 ). More strikingly, nucleotide diversity was significantly lower for site-lineage combinations in which positive selection was specifically inferred than for either other lineages at the same site (0.00213 ± 0.00341 vs. 0.00254 ± 0.00264, P = 5.00 × 10−9) or near sites with no evidence for positive selection (0.00213 ± 0.00341 vs. 0.00243 ± 0.00214, P = 1.75 × 10−8). For “nonselected” lineages, local nucleotide diversity did not differ from nucleotide diversity at sites with no evidence for positive selection (P = 0.138).
Fig. 4.
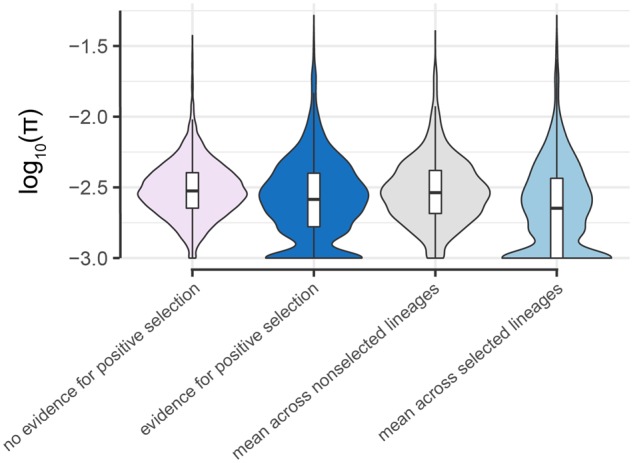
Nucleotide diversity near CpG sites is lower in lineages where positive selection has been inferred. Log10(π) (with an offset of 0.001 to avoid undefined values) for the 1-kb window surrounding CpG sites where DNA methylation levels are taxonomically structured, and 1) we inferred no evidence for positive selection (pink: n = 16,907 sites) or 2) where positive selection was inferred on any baboon lineage (dark blue: n = 3,043 sites, based on the intersection set of the heuristic and OU approaches for multispecies lineages and results from the heuristic approach for single lineages). Log10(π) for sites in dark blue are replotted in gray for lineages unaffected by putative positive selection and in light blue for lineages putatively affected by positive selection. Lineage-site combinations linked to positive selection (light blue) exhibit lower local nucleotide diversity than all other classes (Tukey’s Honestly Significant Difference test: P = 1.75 × 10−8 compared with sites with no evidence of positive selection [pink]; P = 0.0160 against the same sites, but with π averaged across all lineages [dark blue]; P = 5.00 × 10−9 against the same sites, but with π averaged across nonselected lineages only [gray]). π was calculated separately for each species prior to averaging, and is log transformed here for visualization purposes only. Box plots show median (black bar) and interquartile range (whiskers).
Discussion
Together, our findings provide novel insight into the rate and determinants of DNA methylation divergence in primates. In contrast to comparative studies of human populations (Fraser et al. 2012; Heyn et al. 2013; Carja et al. 2017), but like studies across the more deeply diverged great apes (Hernando-Herraez et al. 2013, 2015), global divergence in DNA methylation patterns in baboons is clearly apparent, even among species that diverged relatively recently (e.g., anubis and Guinea baboons: diverged ∼ 0.38 Ma). Roughly speaking, our results suggest that primate taxa can become clearly distinguishable based on DNA methylation data after ∼35,000 generations (assuming a generation time for baboons of 11 years: Swedell 2011; Rogers et al. forthcoming), although this rate varies by genomic context. Notably, although yellow baboons and Kinda baboons diverged earlier than anubis and Guinea baboons (∼0.6 Ma, closer to when hamadryas baboons diverged from the anubis–Guinea lineage), global patterns of DNA methylation separate these two southern clade species less clearly than any of the northern clade species. This difference may reflect recent admixture in the southern part of the yellow baboon range (Zinner et al. 2009; Keller et al. 2010), or smaller long-term effective population sizes in the northern clade species (Rogers et al. forthcoming). Among the northern clade species, anubis baboons fall closest to southern clade baboons, which may also be a consequence of hybridization: anubis baboons and yellow baboons hybridize in Kenya today, and have likely done so in the past as well (Alberts and Altmann 2001; Charpentier et al. 2012; Wall et al. 2016; Rogers et al. forthcoming).
Our results are in line with emerging evidence that, in comparisons involving clearly divergent lineages, variation in DNA methylation levels is largely tied to variation in nearby genetic sequence (Hernando-Herraez et al. 2013, 2015). Specifically, DNA methylation patterns in baboons recapitulate phylogenetic structure, and local genomic context predicts both the rate at which DNA methylation evolves and the probability of a past history of selection. These observations are consistent with analyses in great apes, which revealed that interspecific differences in DNA methylation tend to occur at loci that also contain high levels of species-specific mutations (Hernando-Herraez et al. 2015). Similarly, in Arabidopsis lines, interaccession differences can largely be explained by cis-acting methylation quantitative trait loci (Dubin et al. 2015). Thus, while environmental variation may be important for explaining variation in DNA methylation within populations (Jirtle and Skinner 2007; Feil and Fraga 2012), including baboons (Lea et al. 2016), genetic effects are likely to dominate in between-population and between-species comparisons. Indeed, in our data set, hamadryas baboon and anubis baboon samples were obtained from multiple populations, representing both captive and natural settings. However, despite exposure to different diets and housing conditions, population differences explained very little variance in the overall data set (supplementary Methods, Supplementary Material online).
Our data set also facilitates initial comparisons of DNA methylation evolution against gene expression data sets. Although our findings resemble those of cross-species gene expression analyses in that they globally reproduce the species phylogeny, they also suggest that the evolution of DNA methylation is less constrained on average. While CpG sites are enriched in gene bodies, promoters, and CpG islands, the majority of CpG sites in primate genomes fall in functionally unannotated regions. Our analyses show that DNA methylation levels in unannotated regions are both faster evolving, and, compared with all rapidly evolving sites, underrepresented for signatures of positive selection (fig. 3B). Thus, while several lines of evidence indicate that gene expression levels for most genes are constrained by stabilizing selection, the same pattern probably does not hold for most CpG sites. This difference may explain why the evolution of DNA methylation levels looks more clock-like than for gene expression (Carja et al. 2017), a pattern now observed in human populations, Arabidopsis accessions, and here, in baboons (Becker et al. 2011; Schmitz et al. 2011; van der Graaf et al. 2015; Carja et al. 2017). It is also consistent with experimental studies showing that DNA methylation levels influence gene regulation at only a subset of CpG sites (Maeder et al. 2013; Ford et al. 2017; Lea et al. 2018).
Nevertheless, we do find support for positive selection on DNA methylation levels for a small fraction of the CpG sites we profiled. Tests for selection on phenotypic variation have important limitations (e.g., unknown mutational variance, the assumption of relatively simple evolutionary scenarios: Butler and King 2004; Gilad, Oshlack, Rifkin, et al. 2006; Rohlfs and Nielsen 2014). However, they are still likely to enrich for true cases of positive selection (Blekhman et al. 2008; Rohlfs and Nielsen 2014). Here, the strong enrichment of putatively selected sites within genes and gene regulatory elements, the overlap between two different methods for identifying selected sites, and the identification of coherent DMRs associated with candidate selected sites all indicate that we have captured a set of CpG sites of interest for baboon evolutionary history. Additionally, we identified a loss of local nucleotide diversity—a purely DNA sequence-based analysis—specifically near sites and in lineages inferred to be affected by positive selection, in an analysis based only on DNA methylation phenotypes.
Recent evidence shows that changes in DNA methylation can play an important role in phenotypic evolution. For example, loss of sight in cave-dwelling tetra fish (Astyanax mexicanus) is due to DNA methylation-mediated repression of genes involved in eye development (Gore et al. 2018). Our results suggest that comparative studies of DNA methylation in recent radiations can help identify other loci of interest, and could potentially be combined with outlier scans based on other types of data (e.g., Bergey et al. 2016 in baboons). Notably, in baboons, we found several large DMRs linked to genes involved in immunity, sensory perception, and spermatogenesis, three categories previously identified in sequence-based scans for selection in primates (Kosiol et al. 2008). These examples suggest that, at least in some instances, natural selection on gene regulation has been directed toward changes in DNA methylation phenotypes. If so, variation in DNA methylation at candidate selected sites should functionally affect gene expression. To the best of our knowledge, no data sets on expression variation across baboon species currently exist to test this prediction. However, new data sets could complement the types of analyses reported here, or experimental approaches could be leveraged to connect DNA methylation variation with gene expression in vitro (Liu et al. 2016; Lea et al. 2018). We anticipate that such combinations of comparative, genetic, and experimental approaches will ultimately help resolve the much-debated role of epigenetic marks in adaptive evolution (Laland et al. 2014; Verhoeven et al. 2016).
Materials and Methods
RRBS Data Generation, Processing, and Quality Control
DNA methylation data were generated for 39 baboons across five of the six recognized extant species (9 anubis, 6 yellow, 14 hamadryas, 6 Guinea, and 4 Kinda baboons; supplementary table S1, Supplementary Material online). We also generated RRBS data for five rhesus macaques as an outgroup. For P. anubis samples from the Washington National Primate Research Center (WaNPRC), P. papio from the Brookfield Zoo, and P. hamadryas from the North Carolina Zoo, we extracted genomic DNA using the QIAGEN DNeasy Blood & Tissue Kit, following the manufacturer’s recommendations. Other samples were obtained as previously extracted DNA (see supplementary table S1, Supplementary Material online). All DNA samples were extracted from whole blood with the exception of 2 P. cynocephalus, 1 P. anubis, 2 P. kindae, and 1 P. hamadryas for whom samples were obtained from banked white blood cells. Differences in source tissue (whole blood vs. banked white blood cells) do not contribute to any of the first ten principal components of variation in DNA methylation within this sample (t-test, all P-values > 0.20). Differences in cell type composition also appear unlikely to drive species-specific methylation levels (supplementary Methods, Supplementary Material online).
RRBS libraries for each sample were prepared following Boyle et al. (2012). Briefly, Illumina TruSeq barcoded libraries were constructed using 180 ng of genomic DNA per sample. Libraries were pooled together in sets of 10–12 samples, subjected to sodium bisulfite conversion using the EpiTect Bisulfite Conversion kit (QIAGEN), and then polymerase chain reaction amplified for 16 cycles prior to sequencing on the Illumina HiSeq 2500 platform. Each pooled set of libraries was sequenced in a single lane to 17.2 million reads per sample (SD = 12.8 million reads: supplementary table S1, Supplementary Material online). To assess the efficiency of the bisulfite conversion, 1 ng of unmethylated lambda phage DNA (Sigma Aldrich) was added to each sample prior to library construction.
Sequences were trimmed for adapter contamination, RRBS end repair, and base quality using Trim Galore! (Babraham Bioinformatics) before being mapped to the anubis baboon reference genome (Panu2.0) using BSMAP (Xi and Li 2009). We removed sites that overlapped genetic variants in which one allele abolishes a CpG site found in the reference genome. Combined with BSMAP’s three-nucleotide mapping option, this step eliminates most heterospecific mapping biases within Papio (supplementary Methods and fig. S7, Supplementary Material online). The DNA methylation level at each CpG site was calculated as the proportion of reads with unconverted (i.e., methylated) cytosine bases to total reads covering that site. Based on reads mapped to the lambda phage genome, all samples had a bisulfite conversion efficiency >98.5%, with no significant contribution of species identity to variance in conversion efficiency (ANOVA F = 1.303, P = 0.27; supplementary table S1, Supplementary Material online).
After excluding sites for which data were missing for ≥50% of our study subjects or for which mean coverage was <5×, we retained 2,450,153 CpG sites for downstream analysis. As expected for RRBS data sets, these sites were enriched in functionally important regions of the genome and displayed typical mammalian patterns of CpG DNA methylation (supplementary fig. S1, Supplementary Material online). To focus on the sites most likely to exhibit biologically meaningful variation, we further excluded constitutively hypermethylated (mean DNA methylation level >0.90) and constitutively hypomethylated (mean DNA methylation level <0.10) sites and those that were near-invariant (SD < 0.05), resulting in a final analysis set of 756,262 CpG sites.
Where possible, we modeled DNA methylation levels as count data (the number of methylated reads and total reads for each site), which retains information about the uncertainty in each estimate due to variation in read coverage (Dolzhenko and Smith 2014; Sun et al. 2014; Lea et al. 2015; Lea et al. 2017). However, because some of our analyses (e.g., PCA, OU models) required continuous data, we also estimated DNA methylation levels as the ratio of methylated reads to total reads within each individual for each CpG site. Because variation in sequencing coverage can systematically bias DNA methylation estimates, for these analyses we used the residuals of the raw ratios after regressing out site-specific total read coverage for each individual.
Functional Element Annotations and Enrichment Analysis
We used gene body and CpG island annotations for Panu2.0 obtained from Ensembl (Cunningham et al. 2015) and the UCSC Genome Browser (Karolchik et al. 2014), respectively. Gene promoters were defined as the 2-kb region upstream of the 5ʹ-most annotated gene transcription start site (following Deng et al. 2009; Shulha et al. 2013; Lea et al. 2015), and CpG island shores were defined as the 2-kb regions flanking either side of a CpG island (Irizarry et al. 2009). Because baboon enhancer annotations are not available, we defined putative baboon enhancers by projecting coordinates from ENCODE H3K4me1 ChIP-seq of human peripheral blood mononuclear cells (Dunham et al. 2012) onto the Panu2.0 genome using the UCSC Genome Browser liftover tool (Hinrichs et al. 2006).
Gene ontology (GO) enrichment analyses were performed using the Cytoscape module ClueGO (Bindea et al. 2009). To link differentially methylated sites to genes, we first identified clusters of CpG sites with similar patterns of differential methylation (DMRs). We called DMRs when ≥3 CpG sites within a 2-kb window exhibited the same type of lineage-specific change (e.g., hypomethylation in hamadryas baboons), and bounded the DMR by the first and last CpG site that exhibited lineage-specific methylation. We then collapsed overlapping DMRs. We assigned a DMR to a gene when a CpG site within the DMR fell within 10 kb of the gene body. To test for gene set enrichment, we analyzed GO Biological Processes that fell between levels 3 and 8 of the GO tree, included at least four genes in our data set, and for which at least 5% of genes assigned to the term were present in the test set. We also collapsed GO parent–child terms with at least 50% overlap. Enrichment analyses were corrected for multiple hypothesis testing using the Benjamini–Hochberg method (Benjamini and Hochberg 1995). Gene set enrichment analyses for DNA methylation data can be biased if some gene sets are systematically associated with larger numbers of CpG sites than others (Geeleher et al. 2013). However, in our data set, genes associated with differentially methylated sites were not associated with more tested sites than other genes (logistic regression: z = 0.032, P = 0.983).
Covariance between Genetic Structure and DNA Methylation Patterns
To assess the relationship between phylogenetic structure and DNA methylation patterns in our data set, we conducted PCA in R (version 3.2.5; R Core Team 2016) on the scaled variance–covariance matrix of the DNA methylation level data. We ran the PCA both including and excluding the rhesus macaque samples, and in baboons after subsampling to the same number of individuals per species (n = 4; fig. 1A and B, and supplementary fig. S2, Supplementary Material online).
To test the correlation between DNA methylation levels and pairwise genetic distance between samples, we used Mantel tests. We called genotypes from RRBS data for 49,607 biallelic single nucleotide polymorphisms (see supplementary Methods, Supplementary Material online) and calculated the pairwise genetic covariance. We then compared a genotype-based covariance matrix with the pairwise covariance of DNA methylation profiles using the R package vegan (Oksanen et al. 2016), stratified by both functional compartment (gene, enhancer, CpG island, CpG shore, promoter, unannotated) and mean methylation level (fig. 2A). We also tested whether windows of the genome where genetic structure followed an alternate phylogeny (a consequence of incomplete lineage sorting or admixture) exhibited a lower correlation between genetic and DNA methylation covariance (see supplementary Methods, Supplementary Material online).
Finally, to investigate the relationship between DNA methylation divergence and genetic divergence between species, we retained CpG sites for which each species was represented by at least three individuals and a total (across individuals) of at least ten reads (n = 438,713 CpG sites). We calculated the mean DNA methylation level per species for each retained CpG site and the difference in mean methylation between each species pair. We then tested whether divergence time (based on Rogers et al. forthcoming for baboons and Perelman et al. 2011 for baboon–macaque) predicted the Euclidean distance between species using a Mantel test.
Lineage-Specific Changes in DNA Methylation
For sites in which clade or species significantly contributed to variance in DNA methylation levels (n = 20,360 taxonomically structured sites, identified using ANOVA and a 10% FDR threshold), we tested for lineage-specific shifts using the beta-binomial model implemented in the program MACAU (Lea et al. 2015). We tested each species for differences in DNA methylation level when compared with all other baboons and we also tested whether southern clade baboons had different methylation levels than northern clade baboons. For each comparison and CpG site, we considered the model:
| (1) |
where ri is the total read count for ith individual, yi is the methylated read count for that individual, and πi is an unknown parameter that represents the true methylation level for that individual at the site of interest. MACAU then uses a logit link to model πi as a function of the predictor variable of interest (here, species or clade membership):
| (2) |
where wi is a vector of fixed effect covariates including an intercept and the sample-specific bisulfite conversation rate; α is a vector of coefficients for wi; xi represents species or clade membership coded as 1 (for the taxon of interest) or 0 (for any other taxa) and β is the coefficient for the effect of taxonomic membership; is an n-vector of independent residual error with variance σ2; and I is an n-by-n identity matrix. We did not model genetic nonindependence in this analysis; thus, the K matrix input to MACAU was an identity matrix.
In addition to a 10% FDR threshold (q-value: Storey and Tibshirani 2003, here based on a comparison to a permutation-based empirical null), we required a minimum difference of 10% in mean methylation between either 1) the focal species compared with all other species, for species-level shifts, or 2) for all pairwise comparisons between northern clade and southern clade species, for clade-level shifts. We assigned clade-level shifts to one of the two lineages based on post hoc comparison to rhesus macaques. For example, we assigned a shift to the northern clade when there was a mean difference in DNA methylation of ≥10% between northern clade baboons and macaques, but not between southern clade baboons and macaques.
Identification of Candidate Directionally Selected Sites
To test for positive selection using the heuristic approach, we first calculated the intraspecific variance for each of the 756,262 CpG sites in our primary data set, after mean-centering DNA methylation levels for each species. We then binned the CpG sites into 5% quantiles based on mean methylation level, and retained sites with intraspecific variance in the lowest 10% quantile for each bin. We intersected these low-variance sites with the set of sites that exhibited species- or clade-specific methylation, based on the criteria outlined for identifying taxonomic structure with ANOVA followed by binomial mixed modeling. This intersection set is likely to be enriched for a history of positive selection.
As an alternative approach, we fit OU models of the evolutionary process, based on the phylogenetic tree for baboons (Rogers et al. forthcoming). In OU models, trait evolution is modeled as the sum of stochastic and deterministic forces, with parameters for the strength of selection, the strength of genetic drift, and the trait optimum. In addition, because these models assume that phenotypes have a continuous distribution, we transformed DNA methylation levels using a logit link function. A basic OU model has the form:
| (3) |
where dx captures the continuous rate of change in the trait value x, represents the pull toward the optimum trait value , is the rate of neutral drift, and W is distributed normally with variance corresponding to the amount of independent evolutionary time, dt. For multiple species m, the OU process can be written as a multivariate normal distribution:
| (4) |
where is an m-by-1 vector of , the optimum trait values for species j. captures the covariance between species and is determined by the phylogenetic covariance, , and such that the covariance between species j and k, , is given by . To incorporate intraspecific variance into the OU process, which increases the power to identify true instances of positive selection (Rohlfs and Nielsen 2014), the vector is expanded to an n-by-1 vector where each element,, is equal to for the species j to which individual i belongs. The covariance matrix is replaced by the n-by-n covariance matrix between individuals, with a new parameter added to the diagonal of the covariance matrix to take into account within-species variance.
Different evolutionary regimes correspond to different OU process parameter values. Values of at or near 0 correspond to genetic drift (no pull toward an optimum trait value), while nonzero values of indicate a history of selection. If and is constant across lineages, the trait has evolved under stabilizing selection. If and varies between lineages, the trait has evolved under directional (positive) selection on at least part of the phylogenetic tree. We therefore used Akaike information criterion to compare five OU models for each CpG site in which species or clade membership significantly contributed to DNA methylation variation based on ANOVA (see Results: Selection on DNA Methylation Patterns in Baboons and supplementary Methods, Supplementary Material online, for simulation results on power to detect selective shifts).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We are grateful to Justin O’Riain, Laurel Serieys, Christian Roos, Julia Fischer, Dietmar Zinner, Jane Phillips-Conroy, Mark Wilson, and the North Carolina Zoo for contributing samples. We thank Amanda Lea for her assistance in generating and analyzing the DNA methylation data used in this study, Rori Rohlfs for valuable aid in using OU models, and members of the Tung lab for helpful comments and discussion. The work reported in this article also benefited from the activities and discussions within the Baboon Genome Analysis Consortium, for which the full list of participants is presented by Rogers et al. (forthcoming). Finally, we thank the Baylor College of Medicine Human Genome Sequencing Center for access to the baboon genome assembly (Panu 2.0). RRBS data are deposited in NCBI’s Short Read Archive (Project Accession SRP156478). Code for fitting OU models is available at https://github.com/TaurVil/PapioMethylation, last accessed January 2, 2019. This work was supported by the National Science Foundation (Grant Number BCS-1751783 to J.T. and T.P.V.), the National Institutes of Health (R01-GM102526 to J.T. and P51-OD011132 in support of Yerkes National Primate Research Center), a pilot award from the National Center for Advancing Translational Sciences (Grant Number UL1TR001117), and high-performance computing resources supported by the North Carolina Biotechnology Center (Grant Number 2016-IDG-1013).
References
- Abzhanov A, Protas M, Grant BR, Grant PR, Tabin CJ. 2004. Bmp4 and morphological variation of beaks in Darwin’s finches. Science 3055689: 1462–1465. [DOI] [PubMed] [Google Scholar]
- Akaike H. 1974. A new look at the statistical model identification. IEEE Trans Automat Contr. 196: 716–723. [Google Scholar]
- Alberts SC, Altmann J. 2001. Immigration and hybridization patterns of yellow and anubis baboons in and around Amboseli, Kenya. Am J Primatol. 534: 139–154. [DOI] [PubMed] [Google Scholar]
- Banovich NE, Lan X, McVicker G, Van de Geijn B, Degner JF, Blischak JD, Roux J, Pritchard JK, Gilad Y. 2014. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet. 109: e1004663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle A, Mostafavi S, Zhu X, Potash JB, Weissman MM, McCormick C, Haudenschild CD, Beckman KB, Shi J, Mei R. 2014. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 241: 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C, Hagmann J, Müller J, Koenig D, Stegle O, Borgwardt K, Weigel D. 2011. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 4807376: 245. [DOI] [PubMed] [Google Scholar]
- Bedford T, Hartl DL. 2009. Optimization of gene expression by natural selection. Proc Natl Acad Sci U S A. 1064: 1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Methodol. 57:289–300. [Google Scholar]
- Bergey CM, Phillips-Conroy JE, Disotell TR, Jolly CJ. 2016. Dopamine pathway is highly diverged in primate species that differ markedly in social behavior. Proc Natl Acad Sci U S A. 11322: 6178–6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthelot C, Villar D, Horvath JE, Odom DT, Flicek P. 2018. Complexity and conservation of regulatory landscapes underlie evolutionary resilience of mammalian gene expression. Nat Ecol Evol. 21: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman W-H, Pagès F, Trajanoski Z, Galon J et al. . 2009. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 258: 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blekhman R, Marioni JC, Zumbo P, Stephens M, Gilad Y. 2010. Sex-specific and lineage-specific alternative splicing in primates. Genome Res. 202: 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blekhman R, Oshlack A, Chabot AE, Smyth GK, Gilad Y. 2008. Gene regulation in primates evolves under tissue-specific selection pressures. PLoS Genet. 411: e1000271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle P, Clement K, Gu H, Smith ZD, Ziller M, Fostel JL, Holmes L, Meldrim J, Kelley F, Gnirke A et al. . 2012. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol. 1310: R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csárdi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M et al. . 2011. The evolution of gene expression levels in mammalian organs. Nature 4787369: 343–348. [DOI] [PubMed] [Google Scholar]
- Britten RJ, Davidson EH. 1971. Repetitive and non-repetitive DNA sequences and a speculation on the origins of evolutionary novelty. Q Rev Biol. 462: 111–138. [DOI] [PubMed] [Google Scholar]
- Butler MA, King AA. 2004. Phylogenetic comparative analysis: a modeling approach for adaptive evolution. Am Nat. 1646: 683–695. [DOI] [PubMed] [Google Scholar]
- Carja O, MacIsaac JL, Mah SM, Henn BM, Kobor MS, Feldman MW, Fraser HB. 2017. Worldwide patterns of human epigenetic variation. Nat Ecol Evol. 110: 1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier MJ, Fontaine MC, Cherel E, Renoult JP, Jenkins T, Benoit L, Barthes N, Alberts SC, Tung J. 2012. Genetic structure in a dynamic baboon hybrid zone corroborates behavioural observations in a hybrid population. Mol Ecol. 213: 715–731. [DOI] [PubMed] [Google Scholar]
- Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G, Dickson M, Grimwood J, Schmutz J, Myers RM, Schluter D, Kingsley DM. 2005. Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science 3075717: 1928–1933. [DOI] [PubMed] [Google Scholar]
- Colosimo PF, Peichel CL, Nereng K, Blackman BK, Shapiro MD, Schluter D, Kingsley DM. 2004. The genetic architecture of parallel armor plate reduction in threespine sticklebacks. PLoS Biol. 25: e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coolon JD, McManus CJ, Stevenson KR, Graveley BR, Wittkopp PJ. 2014. Tempo and mode of regulatory evolution in Drosophila. Genome Res. 245: 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgeral S. 2015. Ensembl (2015). Nucleic Acids Res. 43(D1): D662–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Pai AA, Pique-Regi R, Veyrieras J-B, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE et al. . 2012. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 4827385: 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Shoemaker R, Xie B. 2009. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 27:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, Morris K, Streelman JT, Kim SK, Lynch M, Thomas WK. 2005. The transcriptional consequences of mutation and natural selection in Caenorhabditis elegans. Nat Genet. 375: 544. [DOI] [PubMed] [Google Scholar]
- Dolzhenko E, Smith AD. 2014. Using beta-binomial regression for high-precision differential methylation analysis in multifactor whole-genome bisulfite sequencing experiments. BMC Bioinformatics 15:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis C, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature, 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin MJ, Zhang P, Meng D, Remigereau M-S, Osborne EJ, Casale FP, Drewe P, Kahles A, Jean G, Vilhjálmsson B et al. . 2015. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 4:e05255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, Fraga MF. 2012. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 132: 97–109. [DOI] [PubMed] [Google Scholar]
- Ford EE, Grimmer MR, Stolzenburg S, Bogdanovic O, de Mendoza A, Franham PJ, Blancafort P, Lister R. 2017. Frequent lack of repressive capacity of promoter DNA methylation identified through genome-wide epigenomic manipulation. bioRxiv: 170506. https://doi.org/10.1101/170506 [Google Scholar]
- Fraser HB, Lam LL, Neumann SM, Kobor MS. 2012. Population-specificity of human DNA methylation. Genome Biol. 132: R8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, Ichiyanagi K, Yamada Y, Go Y, Udono T, Wada S, Maeda T, Soejima H, Saitou N, Ito T et al. . 2013. Regional DNA methylation differences between humans and chimpanzees are associated with genetic changes, transcriptional divergence and disease genes. J Hum Genet. 587: 446. [DOI] [PubMed] [Google Scholar]
- Gallego Romero I, Ruvinsky I, Gilad Y. 2012. Comparative studies of gene expression and the evolution of gene regulation. Nat Rev Genet. 137: 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gate RE, Cheng CS, Aiden AP, Siba A, Tabaka M, Lituiev D, Machol I, Godon MG, Subramaniam M, Shamim M et al. . 2018. Genetic determinants of co-accessible chromatin regions in activated T cells across humans. Nat Genet 50(8): 1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeleher P, Hartnett L, Egan LJ, Golden A, Raja Ali RA, Seoighe C. 2013. Gene-set enrichment is severely biased when applied to genome-wide methylation data. Bioinformatics 2915: 1851–1857. [DOI] [PubMed] [Google Scholar]
- Gilad Y, Oshlack A, Rifkin SA. 2006. Natural selection on gene expression. Trends Genet. 228: 456–461. [DOI] [PubMed] [Google Scholar]
- Gilad Y, Oshlack A, Smyth GK, Speed TP, White KP. 2006. Expression profiling in primates reveals a rapid evolution of human transcription factors. Nature 4407081: 242–245. [DOI] [PubMed] [Google Scholar]
- Gore AV, Tomins KA, Iben J, Ma L, Castranova D, Davis AE, Parkhurst A, Jeffery WR, Weinstein BM. 2018. An epigenetic mechanism for cavefish eye degeneration. Nat Ecol Evol 2:1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. 2011. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 64: 468–481. [DOI] [PubMed] [Google Scholar]
- Hansen KD, Langmead B, Irizarry RA. 2012. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 1310: R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haygood R, Babbitt CC, Fedrigo O, Wray GA. 2010. Contrasts between adaptive coding and noncoding changes during human evolution. Proc Natl Acad Sci U S A. 10717: 7853–7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heliconius Genome Consortium. 2012. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez RD, Uricchio LH, Hartman K, Ye J, Dahl A, Zaitlen N. 2017. Singleton variants dominate the genetic architecture of human gene expression. bioRxiv: 219238. https://doi.org/10.1101/219238 [Google Scholar]
- Hernando-Herraez I, Heyn H, Fernandez-Callejo M, Vidal E, Fernandez-Bellon H, Prado-Martinez J, Sharp AJ, Esteller M, Marques-Bonet T. 2015. The interplay between DNA methylation and sequence divergence in recent human evolution. Nucleic Acids Res. 4317: 8204–8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando-Herraez I, Prado-Martinez J, Garg P, Fernandez-Callejo M, Heyn H, Hvilsom C, Navarro A, Esteller M, Sharp AJ, Marques-Bonet T. 2013. Dynamics of DNA methylation in recent human and great ape evolution. PLoS Genet. 99: e1003763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, Moran S, Hernando-Herraez I, Sayols S, Gomez A, Sandoval J, Monk D, Hata K, Marques-Bonet T, Wang L et al. . 2013. DNA methylation contributes to natural human variation. Genome Res. 239: 1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs A, Karolchik D, Baertsch R, Barber G, Bejerano G, Clawson H. 2006. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 34: D590–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgins-Davis A, Rice DP, Townsend JP. 2015. Gene expression evolves under a house-of-cards model of stabilizing selection. Mol Biol Evol. 328: 2130–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M et al. . 2009. The human colon cancer methylome shows similar hypo-and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 412: 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F. 1977. Evolution and tinkering. Science 1964295: 1161–1166. [DOI] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK. 2007. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 84: 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly CJ. 1993. Species, subspecies, and baboon systematics. In Species, species concepts and primate evolution. Boston: Springer; p. 67–107. [Google Scholar]
- Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, Swofford R, Pirun M, Zody MC, White S et al. . 2012. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 4847392: 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA. 2012. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 137: 484–492. [DOI] [PubMed] [Google Scholar]
- Karolchik D, Barber GP, Casper J, Clawson H, Cline MS, Diekhans M et al. . 2014. The UCSC Genome Browser database: (2014) update. Nucleic Acids Res. 42:764–770.24157835 [Google Scholar]
- Keller C, Roos C, Groeneveld L, Fischer J, Zinner D. 2010. Introgressive hybridization in southern African baboons shapes patterns of mtDNA variation. Am J Phys Anthropol. 1421: 125–136. [DOI] [PubMed] [Google Scholar]
- Khaitovich P, Tang K, Franz H, Kelso J, Hellmann I, Enard W, Lachmann M, Pääbo S. 2006. Positive selection on gene expression in the human brain. Curr Biol. 1610: R356–R358. [DOI] [PubMed] [Google Scholar]
- Kim-Hellmuth S, Bechheim M, Puetz B, Mohammadi P, Nedelec Y, Giangreco N, Becker J, Kaiser V, Fricker N, Beier E et al. . 2017. Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations. Nat Commun. 8:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M, Wilson A. 1975. Evolution at two levels in humans and chimpanzees. Science 1884184: 107–116. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. 2006. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 312: 89–97. [DOI] [PubMed] [Google Scholar]
- Kosiol C, Vinar T, da Fonseca RR, Hubisz MJ, Bustamante CD, Nielsen R, Siepel A. 2008. Patterns of positive selection in six mammalian genomes. PLoS Genet. 48: e1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laland K, Uller T, Feldman M, Sterelny K, Müller GB, Moczek A, Jablonka E, Odling-Smee J, Wray GA, Hoekstra HE et al. . 2014. Does evolutionary theory need a rethink? Nature 5147521: 161–164. [DOI] [PubMed] [Google Scholar]
- Lande R. 1976. Natural selection and random genetic drift in phenotypic evolution. Evolution 302: 314–334. [DOI] [PubMed] [Google Scholar]
- Landry C, Lemos B, Rifkin SA, Dickinson W, Hartl DL. 2007. Genetic properties influencing the evolvability of gene expression. Science 3175834: 118–121. [DOI] [PubMed] [Google Scholar]
- Lea AJ, Altmann J, Alberts SC, Tung J. 2016. Resource base influences genome‐wide DNA methylation levels in wild baboons (Papio cynocephalus). Mol Ecol. 258: 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea AJ, Tung J, Zhou X. 2015. A flexible, efficient binomial mixed model for identifying differential DNA methylation in bisulfite sequencing data. PLoS Genet. 1111: e1005650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea AJ, Vilgalys TP, Durst PAP, Tung J. 2017. Maximizing ecological and evolutionary insight from bisulfite sequencing data sets. Nat Ecol Evol. 18: 1074–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea AJ, Vockley CM, Johnston RA, Del Carpio CA, Barreiro LB, Reddy TE, Tung J. 2018. Genome-wide quantification of the effects of DNA methylation on human gene regulation. eLife 7: e37513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler EM. 2017. Evolutionary insights from wild vervet genomes. Nat Genet. 4912: 1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, Jaenisch R. 2016. Editing DNA methylation in the mammalian genome. Cell 1671: 233–247.e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, Ho QH, Sander JD, Reyon D, Bernstein BE et al. . 2013. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. 3112: 1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manceau M, Domingues VS, Mallarino R, Hoekstra HE. 2011. The developmental role of Agouti in color pattern evolution. Science 3316020: 1062–1065. [DOI] [PubMed] [Google Scholar]
- Mendizabal I, Shi L, Keller TE, Konopka G, Preuss TM, Hsieh T-F, Hu E, Zhang Z, Su B, Yi SV. 2016. Comparative methylome analyses identify epigenetic regulatory loci of human brain evolution. Mol Biol Evol. 3311: 2947–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nédélec Y, Sanz J, Baharian G, Szpiech ZA, Pacis A, Dumaine A, Grenier J-C, Freiman A, Sams AJ, Hebert S et al. . 2016. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell 1673: 657–669.e621. [DOI] [PubMed] [Google Scholar]
- Nei M, Li W-H. 1979. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci U S A. 7610: 5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. 2010. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 64: e1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuzhdin SV, Wayne ML, Harmon KL, McIntyre LM. 2004. Common pattern of evolution of gene expression level and protein sequence in Drosophila. Mol Biol Evol. 217: 1308–1317. [DOI] [PubMed] [Google Scholar]
- Oksanen JF, Blanchet G, Kindt R, Legendre P, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2016. vegan: Community Ecology Package. R package version 2.3–5. [Google Scholar]
- Pai AA, Bell JT, Marioni JC, Pritchard JK, Gilad Y. 2011. A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet. 72: e1001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai AA, Gilad Y. 2014. Comparative studies of gene regulatory mechanisms. Curr Opin Genet Dev. 29:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelman P, Johnson WE, Roos C, Seuánez HN, Horvath JE, Moreira MA, Kessing B, Pontius J, Roelke M, Rumpler Y et al. . 2011. A molecular phylogeny of living primates. PLoS Genet. 73: e1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelstra JW, Vijay N, Bossu CM, Lantz H, Ryll B, Müller I, Baglione V, Unneberg P, Wikelski M, Grabherr MG et al. . 2014. The genomic landscape underlying phenotypic integrity in the face of gene flow in crows. Science 3446190: 1410–1414. [DOI] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, King B, Kern AD, Dreszer T, Katzman S, Siepel A, Pedersen JS, Bejerano G, Baertsch R et al. . 2006. Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 210: e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar S, Noonan JP, Pääbo S, Rubin EM. 2006. Accelerated evolution of conserved noncoding sequences in humans. Science 3145800: 786. [DOI] [PubMed] [Google Scholar]
- Prud’Homme B, Gompel N, Rokas A, Kassner VA, Williams TM, Yeh S-D, True JR, Carroll SB. 2006. Repeated morphological evolution through cis-regulatory changes in a pleiotropic gene. Nature 440:1050. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2016. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/. [Google Scholar]
- Reed RD, Papa R, Martin A, Hines HM, Counterman BA, Pardo-Diaz C, Jiggins CD, Chamberlain NL, Kronforst MR, Chen R et al. . 2011. Optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science 3336046: 1137–1141. [DOI] [PubMed] [Google Scholar]
- Rifkin SA, Houle D, Kim J, White KP. 2005. A mutation accumulation assay reveals a broad capacity for rapid evolution of gene expression. Nature 4387065: 220. [DOI] [PubMed] [Google Scholar]
- Rifkin SA, Kim J, White KP. 2003. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat Genet. 332: 138–144. [DOI] [PubMed] [Google Scholar]
- Rogers J, Raveendran M, Harris RA, Mailund T, Leppala K, Athanasiadis G, Schierup MH, Cheng J, Munch K, Walker JA et al. . Forthcoming. The comparative genomics and complex population history of Papio baboons. Sci Adv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlfs RV, Nielsen R. 2015. Phylogenetic ANOVA: The expression variance and evolution model for quantitative trait evolution. Systematic biology 64(5):695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RJ, Schultz MD, Lewsey MG, O’Malley RC, Urich MA, Libiger O, Schork NJ, Ecker JR. 2011. Transgenerational epigenetic instability is a source of novel methylation variants. Science 3346054: 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoech A, Jordan D, Loh P-R, Gazal S, O’Connor L, Balick DJ, Palamara PF, Finucane H, Sunyaev SR, Price AL. 2017. Quantification of frequency-dependent genetic architectures and action of negative selection in 25 UK Biobank traits. bioRxiv: 188086. https://doi.org/10.1101/188086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulha HP, Cheung I, Guo Y, Akbarian S, Weng Z. 2013. Coordinated cell type-specific epigenetic remodeling in prefrontal cortex begins before birth and continues into early adulthood. PLoS Genet 9: e1003433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz AH. 1938. The relative weight of the testes in primates. Anat Rec. 723: 387–394. [Google Scholar]
- Shapiro MD, Marks ME, Peichel CL, Blackman BK, Nereng KS, Jónsson B, Schluter D, Kingsley DM. 2004. Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature 4286984: 717–723. [DOI] [PubMed] [Google Scholar]
- Shibata Y, Sheffield NC, Fedrigo O, Babbitt CC, Wortham M, Tewari AK, London D, Song L, Lee B-K, Iyer VR et al. . 2012. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet. 86: e1002789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signor SA, Nuzhdin SV. 2018. The evolution of gene expression in cis and trans. Trends Genet 34(7): 532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DL. 2000. Perspective: evolutionary developmental biology and the problem of variation. Evolution 544: 1079–1091. [DOI] [PubMed] [Google Scholar]
- Stern DL, Orgogozo V. 2008. The loci of evolution: how predictictable is genetic evolution? Evolution 629: 2155–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 10016: 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Alexis MS, Burge CB. 2015. Meta-analysis of RNA-seq expression data across species, tissues and studies. Genome Biol. 16:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Xi Y, Rodriguez B, Park HJ, Tong P, Meong M, Goodell MA, Li W. 2014. MOABS: model based analysis of bisulfite sequencing data. Genome Biol. 152: R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedell L. 2011. Baboons, mandrills, and mangabeys. In: Campbell CJ, Fuentes A, MacKinnon KC, Panger M, Bearder SK, editors. Primates in perspective. New York (NY): Oxford University Press, p. 241–277. [Google Scholar]
- Tung J, Barreiro LB. 2017. The contribution of admixture to primate evolution. Curr Opin Genet Dev. 47:61. [DOI] [PubMed] [Google Scholar]
- van der Graaf A, Wardenaar R, Neumann DA, Taudt A, Shaw RG, Jansen RC, Schmitz RJ, Colomé-Tatché M, Johannes F. 2015. Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc Natl Acad Sci U S A. 11221: 6676–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven KJ, Vonholdt BM, Sork VL. 2016. Epigenetics in ecology and evolution: what we know and what we need to know. Mol Ecol. 258: 1631–1638. [DOI] [PubMed] [Google Scholar]
- Villar D, Berthelot C, Aldridge S, Rayner TF, Lukk M, Pignatelli M, Park TJ, Deaville R, Erichsen JT, Jasinska AJ et al. . 2015. Enhancer evolution across 20 mammalian species. Cell 1603: 554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD, Schlebusch SA, Alberts SC, Cox LA, Snyder‐Mackler N, Nevonen K, Carbone L, Tung J. 2016. Genome‐wide ancestry and divergence patterns from low‐coverage sequencing data reveal a complex history of admixture in wild baboons. Mol Ecol. 2514: 3469–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D. 2007. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 394: 457. [DOI] [PubMed] [Google Scholar]
- Whitehead A, Crawford DL. 2006. Variation within and among species in gene expression: raw material for evolution. Mol Ecol. 155: 1197–1211. [DOI] [PubMed] [Google Scholar]
- Wray GA. 2007. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 83: 206–216. [DOI] [PubMed] [Google Scholar]
- Wray GA. 2013. Genomics and the evolution of phenotypic traits. Annu Rev Ecol Evol Syst. 441: 51–72. [Google Scholar]
- Xi Y, Li W. 2009. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 10(1):232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, Konopka G, Hunt BG, Preuss TM, Geschwind D, Yi SV. 2012. Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution. Am J Hum Genet. 913: 455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Cain CE, Myrthil M, Lewellen N, Michelini K, Davenport ER, Stephens M, Pritchard JK, Gilad Y. 2014. Epigenetic modifications are associated with inter-species gene expression variation in primates. Genome Biol. 1512: 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinner D, Groeneveld LF, Keller C, Roos C. 2009. Mitochondrial phylogeography of baboons (Papio spp.): indication for introgressive hybridization? BMC Evol Biol. 9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinner D, Wertheimer J, Liedigk R, Groeneveld LF, Roos C. 2013. Baboon phylogeny as inferred from complete mitochondrial genomes. Am J Phys Anthropol. 1501: 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.