

Abstract
Mitochondrial diseases represent a significant clinical challenge. Substantial efforts have been devoted to identifying therapeutic strategies for mitochondrial disorders, but effective interventions have remained elusive. Recently, we reported attenuation of disease in a mouse model of the human mitochondrial disease Leigh syndrome through pharmacological inhibition of the mechanistic target of rapamycin (mTOR). The human mitochondrial disorder MELAS/MIDD (Mitochondrial Encephalopathy with Lactic Acidosis and Stroke-like Episodes/Maternally Inherited Diabetes and Deafness) shares many phenotypic characteristics with Leigh syndrome. MELAS/MIDD often leads to organ failure and transplantation and there are currently no effective treatments. To examine the therapeutic potential of mTOR inhibition in human mitochondrial disease, four kidney transplant recipients with MELAS/MIDD were switched from calcineurin inhibitors to mTOR inhibitors for immunosuppression. Primary fibroblast lines were generated from patient dermal biopsies and the impact of rapamycin was studied using cell-based end points. Metabolomic profiles of the four patients were obtained before and after the switch. pS6, a measure of mTOR signaling, was significantly increased in MELAS/MIDD cells compared to controls in the absence of treatment, demonstrating mTOR overactivation. Rapamycin rescued multiple deficits in cultured cells including mitochondrial morphology, mitochondrial membrane potential, and replicative capacity. Clinical measures of health and mitochondrial disease progression were improved in all four patients following the switch to an mTOR inhibitor. Metabolomic analysis was consistent with mitochondrial function improvement in all patients.
Keywords: chronic kidney disease, mitochondria
INTRODUCTION
Mitochondrial diseases are a clinically and genetically heterogeneous group of disorders that arise as a result of mitochondrial dysfunction.1 Inherited mitochondrial diseases can be caused by mutations of mitochondrial DNA (mtDNA) or of nuclear genes that encode mitochondrial proteins, with an overall prevalence estimated at approximately 1 in 5000.1 The clinical phenotypes of patients affected by mitochondrial disorders are considerably heterogeneous.2
Individuals with mitochondrial disorders resulting from mutation of mtDNA may harbor a mixture of mutated and wild-type mtDNA within each cell. The percentage of mutant mtDNA varies between individuals, as well as among organs and tissues within the same individual, contributing to the varied clinical phenotype.3
Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome is a genetic disorder that results from mutations affecting mtDNA encoding reduced nicotinamide adenine dinucleotide dehydrogenase or transfer RNAs.4 The most frequent mutation in patients with MELAS syndrome is a 3243A>G mtDNA point mutation, but more than a dozen distinct causal genetic variants have been linked to the disease. Patients with MELAS syndrome typically undergo normal development but develop bilateral deafness in late childhood; diabetes in early adulthood; progressive neurologic disabilities from the third decade of life with migraines, seizures, stroke-like episodes, and encephalopathy; heart failure; and progressive kidney failure.4 Not all MELAS patients will follow this clinical pattern, with some showing a limited phenotype, such as maternally inherited diabetes and deafness (MIDD).5
Currently, there is no effective treatment for mitochondrial diseases of any etiology and management mainly is supportive. In small uncontrolled studies, vitamins C and K, thiamine, riboflavin, and ubiquinone have shown varying degrees of benefit in individual cases.6
Recent experimental studies have shown that reduced mTOR signaling through caloric restriction or genetic manipulation rescues the lifespan of yeast harboring mutations in genes encoding mitochondrial proteins.7 Further-more, we recently reported a robust attenuation of disease in a mouse model of the human mitochondrial disease Leigh syndrome (Ndufs4−/− mice) using pharmacologic inhibition of mTOR.8,9 We observed that the Ndufs4−/− mice show increased tissue mTOR activation in affected tissue (brain) associated with metabolic defects and progressive neurologic disease. Importantly, we found that rapamycin, a specific inhibitor of mTOR, substantially delayed the onset of neurologic symptoms and lesions and extended the lifespan of the Ndufs4−/− mice.8 These data then were confirmed in induced pluripotent stem cells from patients with Leigh syndrome.10
Rapamycin is an immunosuppressive drug used after solid organ transplantation to prevent allograft rejection.11,12 Thus, transplant recipients receiving rapamycin or derivatives present a unique opportunity to study the effects of mTOR inhibition on human mitochondrial diseases. In this study, we identified 4 kidney transplant recipients with MELAS/MIDD syndrome. The 4 patients had stable allografts with an immunosuppressive regimen based on calcineurin inhibitors but their global health status was declining rapidly. Based on the findings in the mouse model we examined activation of the mTOR pathway in patient-derived fibroblasts and switched the immunosuppression regimen from calcineurin inhibitors to mTOR inhibitors accordingly.
RESULTS
Patients
Patient 1 was a man born in 1962 and was diagnosed with MELAS syndrome in 1996 (m.3243A>G mutation) (Table 1). Heteroplasmy was determined in 3 different samples: blood leukocytes (10%–15% mutant mtDNA), urine epithelial cells (5%–10% mutant mtDNA), and cheek cells (40%–45% mutant mtDNA). At the time of diagnosis, the patient had severe hearing impairment, diabetes, and kidney dysfunction with proteinuria. The kidney biopsy performed in 1996 showed focal and segmental glomerulosclerosis. The patient had his first stroke-like episode in 2001. He reached end-stage renal disease in 2004 and began hemodialysis. In 2005, he received a kidney from a deceased donor. The patient had no evidence of cardiac dysfunction but had muscle weakness. His immunosuppressive regimen was based on steroids, mycophenolate mofetil, and tacrolimus. His Karnofsky and Eastern Cooperative Oncology Group (ECOG) scores were 90% and 1, respectively, at the time of transplant. After transplantation, the patient did not experience any complications related to immunosuppression and the estimated glomerular filtration rate remained stable at approximately 72 ml/min during subsequent years. However, his general condition was found to be declining dramatically with considerable weight loss (−18 kg; Δ, −30%), anorexia, and excessive fatigability. Biological markers, such as serum albumin and prealbumin, showed severe malnutrition. We excluded cancer, chronic infection, inflammation, and depression as a cause of malnutrition. The patient’s Karnofsky and ECOG scores decreased to 30% and 3, respectively. We concluded that the cachexia was related to progression of MELAS syndrome.
Table 1|.
Demographic and clinical characteristics of the patients
| Characteristics | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| Age at diagnosis, yr | 34 | 42 | 33 | 44 |
| Sex | Male | Female | Male | Male |
| Sensorineural hearing loss | Yes | Yes | Yes | Yes |
| Diabetes | Yes | Yes | Yes | Yes |
| Heart dysfunction | No | No | Yes | No |
| Muscle weakness | Yes | Yes | Yes | Yes |
| Peripheral neuropathy | Yes | Yes | No | No |
| Age at transplantation, yr | 44 | 52 | 51 | 59 |
| Immunosuppressive regimen before switch | Steroids/MMF/tacrolimus | Steroids/Aza/cyclosporine | Steroids/MMF/tacrolimus | Steroids/MMF/tacrolimus |
| Switch to mTOR inhibitors after transplantation, mo | 89 | 96 | 40 | 3 |
| mTOR inhibitor after switch | Rapamycin | Everolimus | Rapamycin | Everolimus |
| Mean mTOR inhibitors trough levels, ng/ml | 8 ± 3 | 7 ± 3 | 7 ± 3 | 8 ± 2 |
| Karnofsky, %/ECOG scores before switch | 30/3 | 30/3 | 40/2 | 90/1 |
| eGFR before the switch, ml/min | 72 | 78 | 56 | 74 |
| eGFR after the switch, ml/min | 74 | 79 | 60 | 74 |
| HbA1c before the switch, % | 7.1 | 6.9 | 7.2 | 6.3 |
| HbA1c after the switch, % | 6.9 | 7.0 | 7.1 | 6.5 |
| Cholesterol level before the switch, mg/dl | 275 | 221 | 195 | 179 |
| Cholesterol level after the switch, mg/dl | 332 | 296 | 251 | 227 |
| Triglyceride level before the switch, mg/dl | 195 | 204 | 177 | 135 |
| Triglyceride level after the switch, mg/dl | 231 | 267 | 243 | 195 |
Data are means ± SD.
Aza, azathioprine; ECOG, Eastern Cooperative Oncology Group; eGFR, estimated glomerular filtration rate; Hb, hemoglobin; MMF, mycophenolate mofetil; mTOR, mechanistic target of rapamycin.
Patient 2 was the sister of patient 1. She was born in 1954 and MELAS syndrome was diagnosed at the same time as her brother’s diagnosis in 1996 (m.3243A>G mutation) (Table 1). Heteroplasmy was determined in 3 different samples: blood leukocytes (<5% mutant mtDNA), urine epithelial cells (15%–20% mutant mtDNA), and cheek cells (25%–30% mutant mtDNA). She had hearing loss, diabetes, and severe renal impairment with proteinuria. A kidney biopsy showed focal and segmental glomerulosclerosis with severe interstitial fibrosis. She had her first stroke-like episode in 1999. She reached end-stage renal disease in 2001 and received her first kidney transplantation in 2003. In 2010, she had her second stroke-like episode. The patient had no evidence for heart dysfunction but had muscle weakness. She reached end-stage renal failure and received a kidney transplant. Her immunosuppressive regimen included steroids, azathioprine, and cyclosporine. The Karnofsky and ECOG scores at the time of transplantation were 90% and 1, respectively. After transplantation, she did not experience any complication related to immunosuppression and the estimated glomerular filtration rate remained stable at approximately 78 ml/min for several years. However, similar to her brother, we observed a progressive anorexia with severe body weight loss (−30 kg; Δ, −50%), a dramatic decrease in nutrition markers, and an increase in fatigability. We excluded cancer, infection, or depression as a cause of cachexia. The Karnofsky and ECOG scores decreased to 30% and 3, respectively. We therefore concluded that the phenotype was related to MELAS syndrome progression.
Patient 3 was a man born in 1965 who was diagnosed with a mitochondrial disorder in 1998 (m.3243A>G mutation) (Table 1). Heteroplasmy was determined in 2 different tissues: blood leukocytes (15%–20% mutant mtDNA) and cheek cells (30%—35% mutant mtDNA). At the time of diagnosis, the patient had severe hearing impairment, chronic kidney disease with proteinuria, and diabetes. A kidney biopsy performed in 2001 showed a focal and segmental glomerulosclerosis lesion. He underwent hemodialysis in 2008 and received his first kidney transplantation from a deceased donor 2 years later. Progressively, he developed severe and refractory systolic heart dysfunction related to mitochondrial disease. He had never had a stroke-like episode and presented with muscle weakness. Given the lack of stroke-like episodes he was classified as having MIDD syndrome according to criteria.5 He underwent hemodialysis and received a kidney transplantation from a deceased donor. The Karnofsky and ECOG scores at the time of transplantation were 90% and 1, respectively. The immunosuppressive regimen included steroids, mycophenolate mofetil, and tacrolimus. After the transplantation, the patient did not experience any complication related to immunosuppression and the estimated glomerular filtration rate remained stable at approximately 56 ml/min. However, his systolic heart function continued to worsen, leading to severe congestive heart failure. His systolic ejection fraction decreased progressively from 42% to 30% and was resistant to standard treatment. In addition to weight gain related to edema secondary to heart failure, the general condition of the patient was dramatically worsening with excessive fatigability and severe markers of malnutrition. His Karnofsky score and ECOG scores decreased to 40% and 2, respectively. After multiple examinations, we concluded that his symptoms were related to progression of MELAS syndrome.
Patient 4 was a man born in 1955 who was diagnosed with MELAS in 1999 (m.3243A>G mutation) (Table 1). Heteroplasmy was determined in 2 different samples: blood leukocytes (10%–15%, mutant mtDNA) and cheek cells (40%–45% mutant mtDNA). At the time of diagnosis the patient presented with hearing impairment, kidney dysfunction with proteinuria, and diabetes. A kidney biopsy was performed in 2002 and showed a focal and segmental glomerulosclerosis lesion. He had a stroke-like episode in 2008. He underwent hemodialysis in 2014 and received his first kidney transplantation from a deceased donor 1 year later. The immune-suppressive regimen included steroids, mycophenolate mofetil, and tacrolimus. The Karnofsky and ECOG scores at the time of transplantation were 90% and 1, respectively.
Cellular effects of blocking mTOR overactivation in primary cultured fibroblasts with m.3243A>G mutation
Hyperactive mTOR signaling in primary fibroblasts from patients with m.3243A>G mutation.
In our previous work we found mTOR to be hyperactivated in whole brain lysates of the Leigh syndrome mouse model.8 To examine if the mTOR pathway also was hyperactive in primary MELAS/MIDD cells we cultured primary fibroblasts from patients and control individuals and collected protein lysates for Western blot.Importantly, we observed that the MELAS/MIDD cells show a significant increase in phosphorylation of ribosomal protein S6 (rpS6) compared with control fibroblasts, indicative of hyperactivated mTOR signaling (Figure 1a; dose-response in Supplementary Figure S1 and additional targets in Supplementary Figure S2).
Figure 1|. Mechanistic target of rapamycin (mTOR) activation and mitochondrial defects in primary mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/maternally inherited diabetes and deafness (MELAS/MIDD) fibroblast lines.
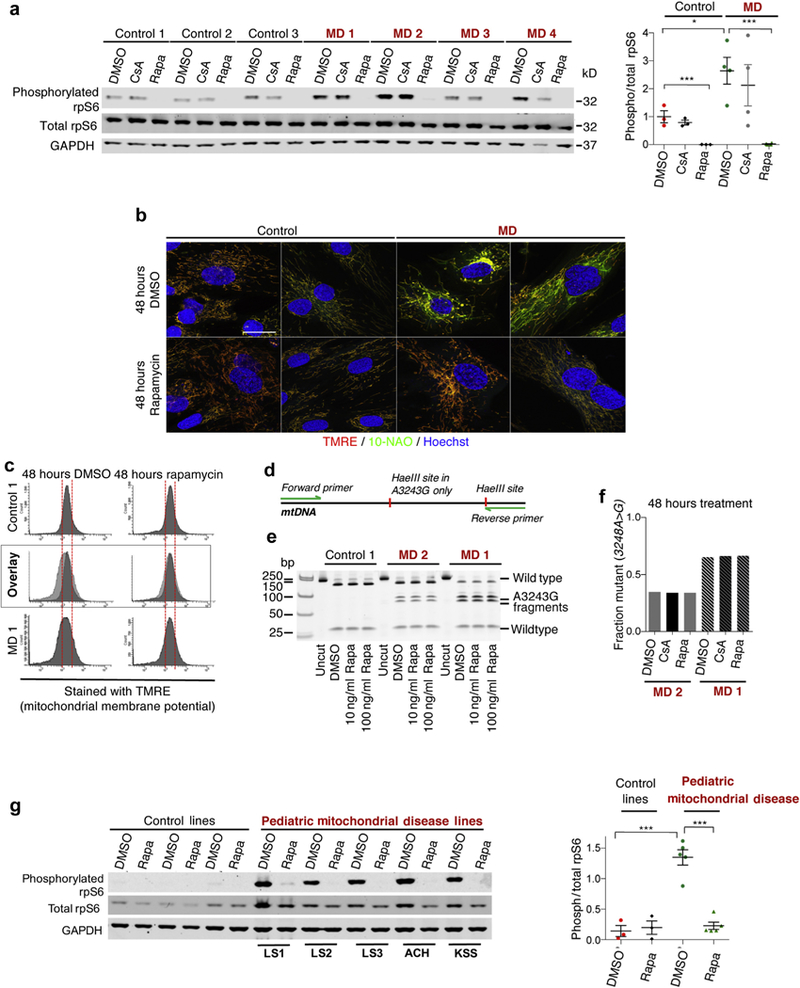
(a) Western blot of lysates from primary MELAS/MIDD patient and control fibroblasts. Cells were treated for 48 hours with dimethylsulfoxide (DMSO) (vehicle-solvent control), rapamycin (Rapa), or cyclosporin A (CsA), and lysates were probed for phosphorylated ribosomal protein S6, total rpS6, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and quantification. (b) Representative confocal microscopy images of primary fibroblast lines treated for 48 hours with DMSO or rapamycin and stained for mitochondrial membrane potential (tetramethylrhodamine ethyl ester [TMRE], red), mitochondrial mass (10-N-nonyl acridine orange [10-NAO], green), and DNA (Hoechst 33342, blue). (c) Representative flow cytometry analysis of primary fibroblasts treated for 48 hours with DMSO or rapamycin and stained for mito-chondrial membrane potential (TMRE, red). (d) Schematic of m.3243A>G mutant mitochondrial DNA restriction analysis assay. (e) Restriction analysis-based assessment of m.3243A>G heteroplasmy levels in cells treated for 48 hours with DMSO or rapamycin at 2 concentrations: 10 and 100 ng/ml. (f) Quantification of restriction analysis. Forty-eight hours of rapamycin treatment has no impact on heteroplasmy levels, as anticipated. (g) Western blot of lysates from primary fibroblast derived from pediatric mitochondrial disease patients and control fibroblast obtained from the Coriell Institute Cell Repository and quantification. Pediatric lines were as follows: Leigh Syndrome (LS), ataxia and cerebellar hypoplasia, complex I (ACH), and Kearns-Sayre Syndrome (KSS). *P < 0.05, ***P < 0.001. Bar: ~10 μm. MD, mitochondrial disease; mtDNA, mitochondrial DNA. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
MELAS/MIDD and controls cells then were treated for 48 hours with dimethylsulfoxide (DMSO), rapamycin, or cyclosporin A. Cyclosporin A was included as a control for the immunosuppressive therapy that patients were receiving before the start of this study. Cyclosporin A had no significant impact on pS6/S6, whereas 48 hours of rapamycin treatment reduced pS6 levels in all cell lines, as anticipated.
Rescue of mitochondrial morphology and membrane potential by short-term rapamycin treatment with no impact on heteroplasmy levels.
To examine the impact of mTOR inhibition on mitochondrial morphology and membrane potential, a general measure of mitochondrial function, we treated dermal fibroblasts with rapamycin or DMSO. Cells were stained with tetramethylrhodamine ethyl ester (TMRE), a marker of mitochondrial membrane potential, and 10-N-nonyl acridine orange, a marker of the inner mitochondrial membrane that acts as a membrane potential insensitive marker of mitochondrial mass (Figure 1b and c). MELAS/MIDD lines show reduced membrane potential, determined by TMRE staining intensity, and abnormal morphology, characterized by swelling, fragmentation, and the presence of depolarized (low TMRE staining) mitochondria (Supplementary Figure S3). Treatment with rapamycin attenuated these phenotypes in all MELAS/MIDD fibroblasts while having no overt impact on the control cells (Figure 1b and c). Electron microscopy also indicated that rapamycin treatment is associated with a partial rescue of mitochondria morphology and cytoskeletal fiber organization in fibroblasts from MELAS/MIDD patients (Supplementary Figure S4).
mTOR inhibition is associated with induction of autophagy and mitophagy. Previously, we showed that rescue of mitochondrial disease in a mouse model was independent of any direct changes to primary mitochondrial defects,8 arguing against the model that selective removal of defective mitochondria underlies the benefits of mTOR inhibition. Consistent with this model, the rescue of membrane potential was not associated with a change in levels of m.3243A>G heteroplasmy (Figure 1 d–f).
Hyperactive mTOR signaling in primary fibroblasts from pediatric mitochondrial disease patients.
To determine whether hyperactive mTOR is a shared feature in mitochondrial disease fibroblast lines, we obtained 3 control primary fibroblast lines and 5 pediatric mitochondrial disease primary fibroblast lines from the Coriell Institute Cell Repository (Camden, NJ). This set includes 3 Leigh syndrome lines, 1 ataxia and cerebellar hypoplasia Complex I line, and a Kearn-Sayre syndrome line (Table 2). Interestingly, we found that pediatric mitochondrial disease cells also showed a significant increase in pS6 compared with control fibroblasts. These results indicate that hyperactivated mTOR signaling is a shared feature of genetically distinct mitochondrial disorders (Figure 1g).
Table 2|.
Fibroblast cell lines obtained from the Coriell Institute Cell Repository
| Cell line ID |
Description | Mutation(s) | Sex | Age at sampling, yr |
Race | Passage frozen |
Publication | dbSNP ID |
|---|---|---|---|---|---|---|---|---|
| GM06225 | Kearns-Sayre Syndrome | - | Male | 10 | Caucasian | 5 | 15280047 | 14761 |
| GM01503 | Leigh Syndrome | MTATP6 | Female | 3 | Caucasian | 5 | 2376596; | 21779 |
| 7200213 | ||||||||
| GM03672 | Leigh Syndrome | MTATP6 | Female | 1 | Caucasian | 2 | 2376596 | 21068 |
| GM13411 | Leigh Syndrome | MTATP6; 8993T>G mtDNA; L156R | Male | 0.8 | Asian | 3 | 23665194; | 11776 |
| 8042671 | ||||||||
| GM24529 | Ataxia and cerebellar hypoplasia mitochondrial complex I deficiency; NUBPL | NUBPL; T311C; NADH:ubiquinone oxidoreductase Fe/S protein; assembly of mitochondrial complex I | Female | 14 | Caucasian | 2 |
dbSNP, Single Nucleotide Polymorphism Database; NADH, reduced nicotinamide adenine dinucleotide; NUBPL, nucleotide-binding protein-like.
Attenuation of early replicative senescence in MELAS/MIDD fibroblasts.
To determine whether long-term rapamycin treatment could rescue additional cellular phenotypes in MELAS/MIDD cells we performed replicative senescence assays to measure cellular replicative capacity. Primary MELAS/ MIDD fibroblast lines lose the ability to grow at early population doublings compared with control fibroblasts, indicating a dramatic reduction in proliferative capacity and premature senescence compared with control lines (Figure 2a). Rapamycin increased the proliferative capacity in both control and MELAS/MIDD lines, however, importantly, the relative increase in MELAS/MIDD cells was substantially greater than in controls (Figure 2a and b). Interestingly, cyclosporin A slightly increased the proliferative capacity in all cell lines, but to a similar level in control and MELAS/ MIDD cells.
Figure 2|. Mechanistic target of rapamycin (mTOR) inhibition attenuates replicative capacity defects in primary mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/maternally inherited diabetes and deafness (MELAS/MIDD) fibroblast lines.
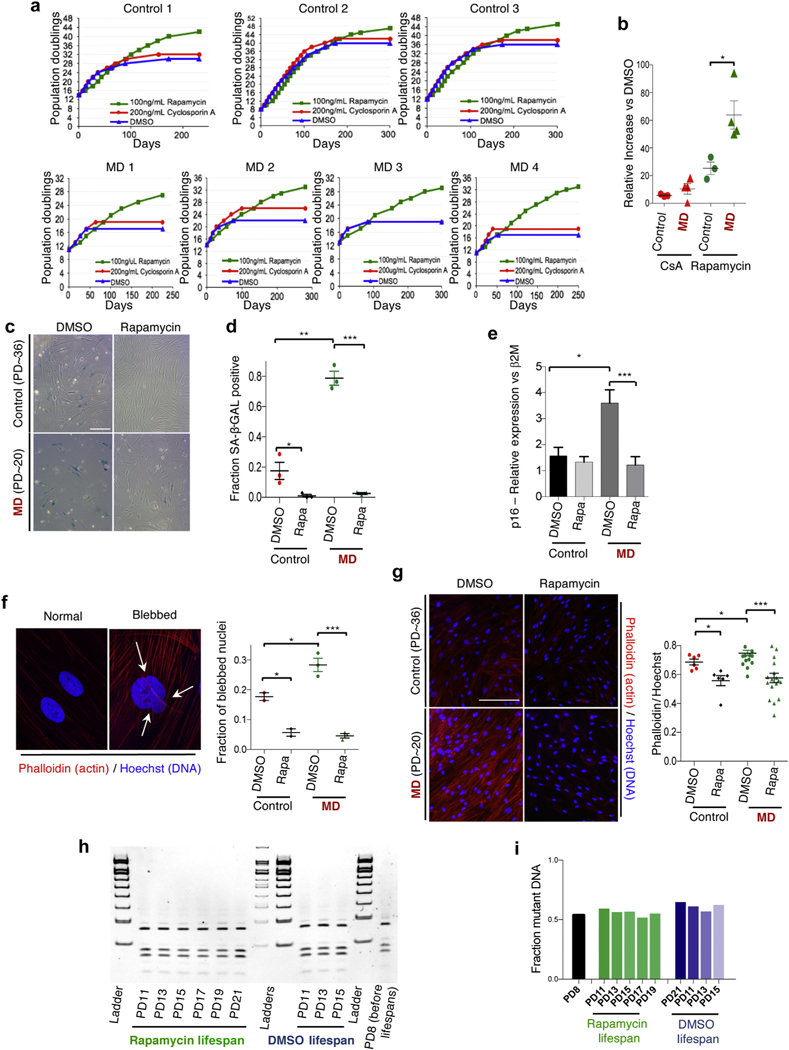
(a) Replicative capacity data from 3 controls and 4 MELAS/MIDD fibroblasts treated with dimethylsulfoxide (DMSO), rapamycin, or cyclosporin A (CsA). (b) Relative change in the replicative capacity of CsA and rapamycin-treated control and MELAS/MIDD primary fibroblast lines versus DMSO treatment. (c) Representative senescence-associated β-galactosidase (SA-β-Gal) staining of low population doubling (PD) DMSO and rapamycin-treated MELAS/MIDD and control fibroblasts. (d) Quantification of SA-β-Gal staining (data points are representative of individual cell lines, ≥6 fields were assessed for each line). (e) Senescence-associated factor p16INK4a (p16) shows increased expression in MD fibroblasts, which was attenuated by 48 hours of rapamycin treatment. (f) Representative images of normal nuclei and nuclei showing nuclear blebbing (arrows indicate nuclear invaginations or blebs). Staining shows actin cytoskeleton (Phalloidin, red) and nuclei (Hoechst 33342, blue). Quantification of nuclear blebbing (data points are representative of individual cell lines, ≥6 fields were assessed for each line). (g) Staining for actin cytoskeletal organization (Phalloidin, red) and nuclei (Hoechst, blue). Quantification of nuclear cytoskeletal signal normalized to DNA content (data points are representative of individual cell lines, ≥6 fields were assessed for each line). (h) Mitochondrial m.3243A>G heteroplasmy analysis across the lifespan of MD line 2. Samples were collected every 2 population doublings and assessed together at the completion of the lifespan study. (i) Quantification of (h). Mitochondrial m.3243A>G heteroplasmy did not change in either DMSO- or rapamycin-treated cells throughout the duration of the lifespan studies. *P < 0.05, **P < 0.01, ***P < 0.001. Bar ~ 50 uM. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
A recent study investigating senescence in the setting of nonphysiological models that induce mitochondrial dysfunction in cultured cells found that the presence or absence of pyruvate governs a cellular decision to proliferate or senesce.13 By using media supplemented with pyruvate we found that neither the shortened lifespan of MELAS/MIDD fibroblasts nor the lifespan increase resulting from rapamycin treatment were impacted, suggesting key differences between senescence in genuine mitochondrial disease cells versus models of mitochondrial stress (Supplementary Figure S5).
In addition to measuring the replicative capacity, we examined the effects of rapamycin on markers of senescence: senescence associated β-galactosidase (SA-β-Gal) staining, accumulation of actin-cytoskeleton stress fibers (detected by phalloidin staining), nuclear morphology defects (nuclear blebbing), expression of p16INK4a and expression of the senescence-associated factor p21/Cip1. In each case the MELAS/MIDD lines showed features consistent with premature cellular senescence that were attenuated by rapamycin treatment (Figure 2c–g and Supplementary Figure S6).
Attenuation of early replicative senescence in MELAS/MIDD fibroblasts.
To determine whether the observed benefits to replicative capacity result from changes in m.3243A>G heteroplasmy levels we collected DNA samples at each passage (every other population doubling [PD]) across the duration of the replicative lifespan study. At the completion of the study we assessed the mutant mtDNA load as a function of PD in MD1, the line with the highest levels of mutant DNA at the start of the study (Figure 2h). Surprisingly, m.3243A>G levels did not change throughout the duration of the study in either DMSO- or rapamycin-treated cells (Figure 2i), showing that our culture conditions did select against mutant DNA and indicating that the beneficial effects of rapamycin treatment are not a result of altered heteroplasmy levels.
Clinical impact of mTOR inhibition in transplant recipients with MELAS/MIDD.
Based on the in vitro studies we decided to stop calcineurin inhibitors in all patients and to introduce either rapamycin or everolimus. Only a few weeks after we introduced mTOR inhibitors we observed a dramatic improvement in the general condition of the 4 patients. Patients 1 and 2 started to gain weight quickly, without edema, and recover their appetites. Anorexia progressively disappeared, as well as the general asthenia. Clinical tests showed a marked improvement in the serum levels of prealbumin and albumin. The Karnofsky and ECOG scores of patients 1 and 2 increased from 30% to 60% and from 3 to 1, respectively, within 20 months (Figure 3, Table 1).
Figure 3|. Clinical and biological impact of mechanistic target of rapamycin (mTOR) inhibition in transplant recipients with mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/maternally inherited diabetes and deafness (MELAS/ MIDD).
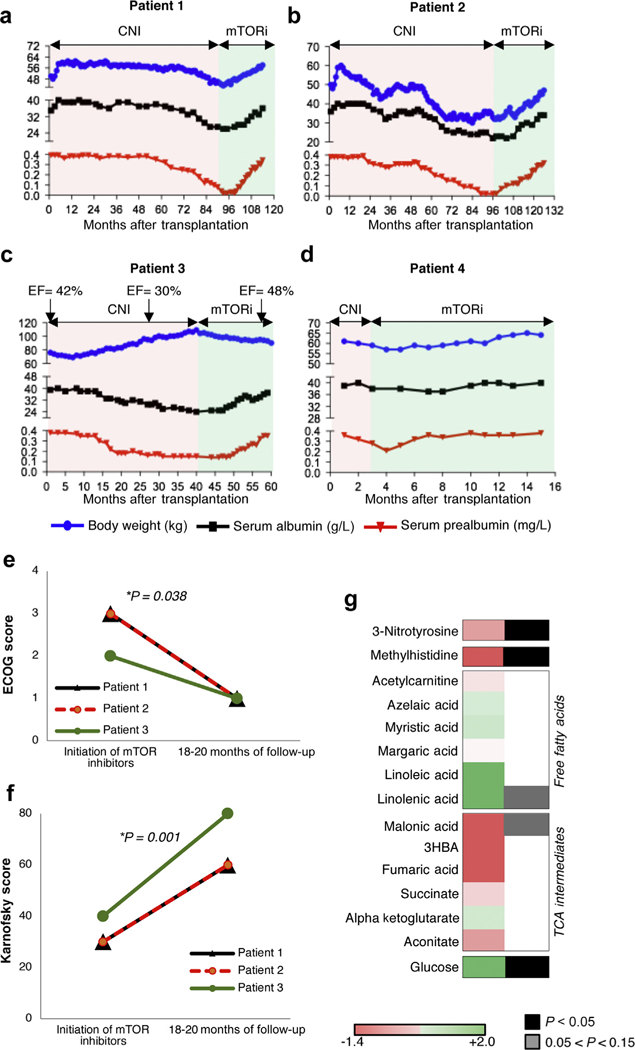
(a) Patient 1, (b) patient 2, (c) patient 3, and (d) patient 4. Pink parts of the graph show the period of exposure to calcineurin inhibitors (CNI), and green shows the period of exposure to mTOR inhibitors (mTORi). The blue dots show the body weight of the 4 patients over time, the black dots represent the serum albumin levels, and the red dots represent the serum prealbumin levels. In patient 3, the heart ejection fraction (EF, %) before and after the switch to mTOR inhibitors is added. (e) Eastern Cooperative Oncology Group (ECOG) and (f) Karnofsky scores at initiation of mTOR inhibitor treatment and at 18 to 20 months of follow-up evaluation. P values were calculated by paired t test. (g) Log10 average difference of amino acid metabolites levels between samples on sirolimus/everolimus and on calcineurin inhibitors.
Improvement also was visible in patient 3 with an enhancement of his general condition (Figure 3, Table 1). The patient lost weight owing to the reduction of the edema related to a progressive amelioration of the heart systolic ejection fraction from 30% to 48%. No additional treatments for congestive heart failure were added during the period, supporting a beneficial effect of mTOR inhibitors in this context. The Karnofsky and ECOG scores of patient 3 increased from 40% to 80% and from 2 to 1, respectively, within the 18 months. Patient 4 was switched only a few months after transplantation compared with the other patients, but we observed an improvement of his global status compared with the dialysis period. However, in this specific case it was difficult to separate the amelioration linked to the transplantation or the introduction of mTOR inhibitors, or both (Figure 3, Table 1). Importantly, in all patients the estimated glomerular filtration rate remained stable after the switch, meaning that the clinical improvement was not related to a gain of kidney function (Table 1).
Metabolomic impact of mTOR inhibition in transplant recipients with MELAS.
Metabolic profiling of patients’ sera also provided additional evidence of improved clinical outcome upon switching to sirolimus/everolimus (Figure 3g and Supplementary Figure S7). Serum 3-nitro-tyrosine, a marker of oxidative stress, was reduced significantly by mTOR inhibition (Figure 3g and Supplementary Figure S7), consistent with our in vitro results on mitochondrial membrane potential and morphology. Several markers of amino acid metabolism also were altered significantly by rapamycin treatment (Figure 3g and Supplementary Figure S7). In particular, levels of 1- and 3-methyl-histidine were reduced significantly upon switching to rapamycin, consistent with decreased muscle protein breakdown (Figure 3g and Supplementary Figure S7). Similar to our previous findings in Ndufs4−/− mice,8,9 free fatty acid levels generally were increased by rapamycin treatment, although not significantly, whereas tricarboxylic acid intermediates were reduced (Figure 3g and Supplementary Figure S7). Finally, rapamycin treatment increased the levels of circulating glucose in all 4 patients (Figure 3g and Supplementary Figure S7), also consistent with our prior findings in the Ndufs4−/− model.8,9 Notably, a robust reduction in glucose consumption also was observed in the fibroblasts treated with rapamycin, indicating that the altered glucose handling resulting from mTOR inhibition is at least partly cell autonomous (Supplementary Figure S8).
DISCUSSION
This study provides direct evidence supporting mTOR inhibition as a promising therapeutic strategy in human mitochondrial disease. Although this work will need to be followed up by studies involving larger sample sizes, and MELAS/MIDD patients who have not undergone organ transplantation, our results indicate that mTOR inhibition may be a safe and effective means to improve health and quality of life in transplant patients with MELAS/MIDD.
In addition, our cell-based studies provide new mechanistic insight into the cellular impact of MELAS/MIDD and the mechanism of benefit of rapamycin. We found S6 to be hyperphosphorylated in MELAS/MIDD fibroblasts and report premature cellular senescence to be one cellular pathologic feature of MELAS/MIDD disease. We also observed MELAS/ MIDD cells to have overtly defective mitochondrial membrane potential and morphology, which was rescued by short-term mTOR inhibition. We were able to show that the improvement with rapamycin was not associated with a reduction in relative levels of mutant mtDNA, arguing against selective mitophagy of dysfunctional mitochondria as a mechanistic explanation.
Defining the precise mechanism that mediates the benefits of mTOR inhibition in mitochondrial disease is an active area of research that may lead to more targeted intervention strategies. We focused here on fibroblasts, but we cannot formally exclude a positive impact of mTOR inhibitors on the immune system in this population.
The unique unbiased metabolomics profile of the patients before and after the switch to mTOR inhibitors provide additional evidence of mitochondrial improvement. We observed a decrease in the levels of 1 and 3-methyl-histidine, a marker of muscle protein breakdown, suggesting that mTOR inhibition in this context prevents muscle wasting in MELAS/MIDD patients. We also found a decrease in oxidative stress markers, arguing for mitochondrial respiratory chain improvement. Importantly, the results were consistent with those observed in the Ndufs4−/− mice before and after rapamycin treatment.8
Finally, our study suggests that prescreening of mTOR hyperactivation in patient samples and response to mTOR inhibitors may be a viable approach to personalized medicine in mitochondrial disease patients. More basic research is needed to determine which forms of mitochondrial disease will be refractory to mTOR inhibition, but it seems likely that those associated with hyperactive mTOR are the most likely to benefit.
METHODS
Patients and data collection
Our institutional review board approved the study and written informed consent was obtained from each patient. Between January 2006 and December 2015, there were 1753 adult kidney transplants performed at Necker Hospital. Among these, we identified 4 transplant recipients with MELAS/MIDD syndrome (baseline characteristics are summarized in Table 1). After transplantation, patients were monitored monthly during the first year, and then every 3 months. Each patient received an immunosuppressive regimen based on calcineurin inhibitors, steroids, and purine inhibitors. In all patients we stopped the calcineurin inhibitors for 1 week (wash-out period) before switching to mTOR inhibitors. After the switch, patients were followed up weekly for a month, biweekly for a month, monthly for the next 12 months, and then every 3 months. At all time points, patients had physical examinations, performance status scoring using the Karnofsky14,15 (on a scale from 0 to 100, with lower numbers indicating greater disability) and the ECOG14,15 indexes (a scale of 0 to 5, with 0 indicating no symptoms and higher scores indicating increasing symptoms), and biological samplings. Sera from the 4 patients were collected every year after transplantation before the switch to the mTOR inhibitors, and then for all visits after the switch.
Generation of primary dermal fibroblast cultures.
Punch skin biopsy specimens were collected from patients and healthy control individuals using standard methods. To generate dermal fibroblast cultures, biopsy specimens were minced and incubated at room temperature in 0.05% trypsin-ethylenediamine tetraacetic acid (25300054; Thermo Fisher Scientific, Waltham, MA) solution for 30 minutes with gentle shaking. Cells were collected by centrifuging at 700g for 10 minutes, resuspended in cell culture media containing 25% fetal bovine serum, and plated onto 24-well plates to establish lines. Fibroblast cultures were grown and maintained in 1x Eagle minimal essential medium (10–010-CV; Corning, Corning, NY) supplemented with 25% fetal bovine serum and penicillin/streptomycin (30–001-CI; Corning) to a final concentration of 100 IU penicillin and 500 μg/ml streptomycin.
Pediatric cell lines.
Pediatric mitochondrial disease cell lines were obtained from the Coriell Institute Cell Repository. Pediatric lines were as follows: Leigh Syndrome; ataxia and cerebellar hypoplasia, complex I; and Kearns-Sayre Syndrome. The Coriell cell line IDs were as follows: GM01503, GM03672, GM13411, GM24529, and GM06225 (see Table 2). All cell line characterizations, including patient data and relevant cell line publications, can be found on the Coriell Institute Cell Repository website by searching the cell line ID and opening the catalog data for the fibroblast cell line matching the identification (https://www.coriell.org/0/Sections/Search/).
Analysis of mitochondrial membrane potential and morphology.
Cells at similar PD (~10 ± 2) were plated 1:4 from confluent cultures onto coverglass chamber slides and allowed to grow until approximately 80% confluent. Media was replaced and supplemented with 100 ng/ml rapamycin (BP29631; Fisher, Hampton, NJ) in DMSO (BP321-1; Fisher), or equal volume DMSO, for 48 hours. Cells were stained for 15 minutes in media with 100 nmol/l TMRE (BDB564696; Fisher) and 5 mg/ml Hoechst 33342 (89139-126; Biotium, Fremont, CA) and imaged on a Leica SP5 confocal microscope (Leica, Wetzlar, Germany). Samples were treated and imaged in 1 session using identical imaging parameters. Flow cytometry analysis was performed by staining cells with only TMRE or 10-N-nonyl acridine orange, dissociating with trypsin-ethylenediamine tetraacetic acid-containing dye for 10 minutes at room temperature, collecting cells by centrifugation, resuspension in cold phosphate-buffered saline (PBS), and analysis on a BD Canto II flow cytometer (San Jose, CA) using 488-nm excitation with 585/42 band-pass and 530/30 band-pass filters for TMRE and 10-N-nonyl acridine orange, respectively. A single gate was set to cells using forward and side scatter and all settings were unchanged throughout data collection (this gate was applied to all samples). A total of 10,000 or more events were collected for every sample.
Analysis of S6 phosphorylation by Western blot.
Cells at similar PD (~10 ± 2) were plated 1:4 from confluent cultures and allowed to grow until approximately 80% confluent. Media was replaced with media containing 100 ng/ml rapamycin in DMSO, 200 ng/ml cyclosporin A (1101/100; R&D Systems, Minneapolis, MN) in DMSO, or equal volume DMSO for 48 hours. Cultures were rinsed with 1x PBS, treated for 10 minutes with 0.05% trypsin, collected by centrifugation at 4° C, and pellets were flash frozen on dry ice. Protein lysates were collected by directly adding 1x RIPA buffer (89900; Pierce, Waltham, MA) containing protease and phosphatase inhibitors (PI78441; Pierce) to cell pellets, sonicating in 10 one-second bursts, on ice, with an XL-2000 QSonica (Cole-Palmer, Vernon Hills, IL) at maximum output, and centrifuging to remove cell debris. Protein concentration was determined by BCA assay (PI23228; Pierce), equal protein was run on 4% to 12% Bis-Tris 26-well NuPage midigels (WG1403; Fisher), and transferred to nitro-cellulose blots (IB23001; Fisher). Blots were blocked in LI-COR Odyssey blocking buffer (427-40100; LI-COR, Lincoln, NE); probed with primary antibodies anti-pS6, anti-S6, and anti-glyceralde-hyde-3-phosphate dehydrogenase (4858P, 2217S, and 2118S; Cell Signaling, respectively); followed by secondary antibody IRDye800 donkey anti-rabbit (925-32213; LI-COR) and imaged using the LI-COR Odyssey Clx scanning imager as previously described.16 Data were quantified using National Institutes of Health ImageJ software (Bethesda, MD).17
Replicative capacity studies.
Excluding Supplementary Figure S3, all cell studies presented were performed in 1x Eagle minimal essential medium (10-010-CV; Corning) supplemented with 25% fetal bovine serum and penicillin/streptomycin (30-001-CI; Corning) at a final concentration of 100 IU penicillin and 500 μg/ml streptomycin. Fibroblast replicative capacity studies were performed using standard methods.18 Briefly, to start the replicative capacity studies, low PD cells at approximately 80% confluence were treated with 100 ng/ml rapamycin in DMSO, 200 ng/ml cyclosporin A in DMSO, or equal volume DMSO for 48 hours. Cells were split 1:4 and capacity was measured by splitting cells 1:4 (adding 2 PD) each time they reached confluence. Media was changed 2 to 3 times per week with fresh drugs. All drugs were stored in frozen aliquots to prevent degradation associated with freeze-thaw cycles.
The lifespans in Supplementary Figure S3 were performed as described earlier, with the exception of the cell media that was used, which was Eagle minimal essential medium with 1.5 g/l sodium bicarbonate, nonessential amino acids, L-glutamine, and pyruvate (10-009-CV; Corning) supplemented with 25% fetal bovine serum and penicillin/streptomycin.
SA-β-Gal, actin cytoskeleton staining, and nuclear blebbing.
SA-β-Gal staining was performed using Abcam kit ab65351 (Cambridge, UK) according to the manufacturer’s specifications. Images were collected using a Zeiss Axiovert 40CFL brightfield inverted microscope (Oberkochen, Germany). The fraction of SA-β-Gal-positive cells was determined by counting the total and SA-β-Gal-stained cells in 3 to 6 fields (20 x) per treatment per cell line. Average values were grouped to compare control and MELAS/MIDD fibroblasts. Cytoskeletal staining was performed by growing cells on glass coverslips, fixing cells for 10 minutes in 3.7% paraformaldehyde in PBS, permeabilizing in 1% Triton-X 100 (AC327371000; Fisher) for 5 minutes with gentle shaking, blocked for 30 minutes in 1% bovine serum albumin (BP9700100; Fisher), and stained in 1x PBS containing 1% bovine serum albumin, 5 U/ml phalloidin-CF594 (89427-138; Biotium), and Hoechst 33342 (5 mg/ml) for 30 minutes at room temperature. Cells were washed 3 times with 1x PBS, mounted onto slides using Everbrite mounting media (Biotium, Fremont, CA) containing 1 μg/ml Hoechst 33342, and imaged on a Zeiss Axiovert 40CFL brightfield inverted microscope with a 20x objective and 4’,6-diamidino-2-phenylindole and Texas Red filter cubes. All images were collected in 1 session using the same exposure settings. Actin (red) and DNA (blue) channel signal intensities were measured using ImageJ software. The ratio of red to blue channel intensity was calculated in 3 to 6 fields (20x) per treatment per cell line. Average values were grouped to compare control and MELAS/MIDD fibroblasts.
Nuclear blebbing was assessed using images collected from the phalloidin/Hoechst-stained slides. The fraction of blebbed nuclei was calculated in 3 to 6 fields (20x) per treatment per cell line. Averages for each sample were combined to compare treated and untreated control and MELAS/MIDD fibroblast lines.
A3243G genotyping by Sanger sequencing and restriction digestion.
Total DNA was extracted using the phenol/chloroform/isoamyl alcohol method following the manufacturer’s protocol (25666; Sigma, St. Louis, MO). A 183-base pair amplicon containing the A3243 locus was amplified using forward primer 5’-GCGCCTTCCCCCGTAAATGATATCATCTCAACTTAG-3’ and reverse primer 5’-AATGGGTA-CAATGAGGAGTAGGAGGTTGGCCATGG-3’ using Phusion High-Fidelity Polymerase (F-530S; Thermo Fisher Scientific) in a 2-cycle polymerase chain reaction (PCR) reaction. PCR products were submitted for Sanger sequencing by Macrogen (Seoul, South Korea, www.macrogenusa.com) and analyzed by restriction digestion using HaeIII according to the manufacturer’s specifications (R0108S; NEB, Ipswich, MA). Cleaved and uncleaved products were run on a 4% to 20% tris/borate/EDTA polyacrylamide gel (EC62255; Fisher), stained with GelGreen DNA dye (41007; Biotium), and imaged using blue-fluorescent excitation wavelength (Supplementary Figure S9).
Real-time quantitative PCR.
RNA was extracted from flash-frozen cell pellets using the TRIzol method (Thermo Fisher Scientific) cDNA was generated using the Superscript III First-strand synthesis kit (18080051; Thermo Fisher Scientific) according to the manufacturer’s specifications. Expression of p16 (CDKN2A, p16INK4a) was assessed using TaqMan probes (Thermo Fisher Scientific) hs00923894_m1 (p16) and Hs99999907_m1 (β-2-microglobulin, a control gene), and the TaqMan Gene Expression Master Mix (4369016; Thermo Fisher Scientific), as directed, on an Applied Biosystems StepOne real-time PCR machine (Foster City, CA) using the standard reverse-transcription PCR protocol. Relative expressionwas determined using the ΔΔ threshold cycle method.
Serum metabolomics.
Serum samples were obtained at each visit (see earlier) and 50 ul serum was analyzed via triple quadruple liquid chromatography-mass spectrometry in multiple reaction monitoring mode at the Northwest Metabolomics Research Center of the University of Washington. A total of 128 metabolites were identified successfully in all samples.
Glucose consumption.
Glucose consumption in Supplementary Figure S6 was measured using a Bayer Contour glucose monitoring system with test strips (23125600; Thermo Fisher Scientific). Briefly, media was replaced 24 hours before splitting at confluence and glucose concentration was measured immediately before splitting cells. Consumption was calculated as the final concentration minus the initial glucose mass in media.
Statistical analysis
All data were presented as means ± SEM. Comparisons between groups were performed using the 2-tailed Student t-test. P < 0.05 was considered significant. Statistical comparisons of capacity curves were performed using the log-rank test as indicated.
For the metabolomics analysis, the log10 of the area under the curve for each metabolite was centered on the mean log10 and scaled on the SD of each individual sample. Centered and scaled values were treated as repeated measures for each patient on either calcineurin inhibitors or mTOR inhibitors.
Supplementary Material
Figure S1. Dose-dependent effects of rapamycin in fibroblasts. Western blot of phosphorylated and total S6 in control (WI38 line) fibroblasts treated with varying concentrations of rapamycin for 48 hours. The 50% inhibitory concentration (IC50) of rapamycin for S6 phosphorylation was calculated to be approximately 0.3 nmol/l (A). Relative glucose use in WI38 fibroblasts treated with varying doses of rapamycin for 48 hours. The IC50 for rapamycin on glucose consumption is approximately 2.6 nmol/l (B). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure S2. Additional Western blot data. Western blot of phosphorylated and total PRAS40, a component of mechanistic Target of Rapamycin Complex 1 (mTORC1) using samples shown in Figure 1. MELAS fibroblasts show a significant reduction in phosphorylated PRAS40 that is attenuated by 48 hours of rapamycin treatment (A). Western blot of phosphorylated and total protein kinase C (PKC). There was a trend toward increased PKC phosphorylation (driven by 2 of the 4 lines), but this did not reach significance (B). Western blot of IGF1R processing and phospho-ERK (p42/44). IGF1R is produced as a pro-protein and processed by cleavage (C). MELAS fibroblasts have significantly lower levels of processed, compared with unprocessed, IGF1R, but this was not altered substantially by 48 hours of rapamycin treatment. Conversely, levels of phosphorylated ERK (p42/44 MAPK) were increased significantly by 48 hours of rapamycin treatment, but were not different between the control and MELAS lines. CsA, cyclosporin A; DMSO, dimethylsulfoxide; ERK, extracellular signal-regulated kinase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; MAPK, mitogen-activated protein kinase; MELAS, mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes.
Figure S3. Abnormal morphology and membrane potential of mitochondria in fibroblasts from patients with mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/ maternally inherited diabetes and deafness (MELAS/MIDD). Representative images of abnormal mitochondria morphology and membrane potential in the fibroblasts from a MELAS/MIDD patient, characterized by swelling, fragmentation, and the presence of depolarization as indicated by low tetramethylrhodamine ethyl ester (TMRE, red) staining relative to the less membrane potential-sensitive dye 10-N-nonyl acridine orange (10-NA0). Nuclear DNA was stained with the cell permeant dye Hoechst 33342 (blue). Low magnification is shown in the left column and higher magnification is shown in the right column. Arrows highlight overtly swollen mitochondria, as indicated by a balloon or ball-like appearance versus normal fibrillar organization. Bar: ~10 μm.
Figure S4. Mitochondrial and cytoskeletal defects in mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/maternally inherited diabetes and deafness (MELAS/MIDD) fibroblasts. Mitochondria in MELAS/MIDD fibroblasts have collapsed and abnormal cristae and cells are characterized by high levels of cytoskeletal fibers (A). Mitochondria are visually more intact and organized in rapamycin-treated MELAS/MIDD fibroblasts whereas cytoskeletal fibers are less distinct (B). Red arrows point to mitochondria with collapsed and abnormal cristae. Yellow arrows indicate mitochondria with healthy cristae morphology. Blue arrows point to regions of cytoskeletal fibers. DMSO, dimethylsulfoxide; MD, mitochondrial disease.
Figure S5. Replicative lifespan in the presence of pyruvate. Addition of pyruvate attenuated the early growth retardation associated with rapamycin treatment but had no impact on the short lifespan of mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/maternally inherited diabetes and deafness (MELAS/MIDD) cells or the increase in lifespan resulting from rapamycin treatment. Replicative lifespans of 2 control lines treated with dimethylsulfoxide (DMSO) or rapamycin in media supplemented with pyruvate (A). ***P < 0.0001, paired t test DMSO versus rapamycin. Replicative lifespans of 3 MELAS/MIDD fibroblast lines treated with DMSO or rapamycin in media supplemented with pyruvate (B). *P = 0.02, paired t test DMSO versus rapamycin.
Figure S6. p21 expression in fibroblasts from mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/maternally inherited diabetes and deafness (MELAS/MIDD). Western blot analysis of the senescence-associated marker p21/Cip1 and quantification. Western blot performed at the start of the lifespan study, before onset of senescence. MELAS/MIDD fibroblast lines show substantially increased levels of p21/Cip1 compared with control fibroblast lines. *P < 0.05. CsA, cyclosporin A; DMSO, dimethylsulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure S7. Metabolomic impact of mechanistic target of rapamycin (mTOR) inhibition in transplant recipients with mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/maternally inherited diabetes and deafness (MELAS/MIDD). Heatmap of the log10 change of 128 serum metabolites before and after the switch to mTOR inhibitors. Individual sample values per each metabolite were averaged across treatment per each patient. Data are represented as the difference between the average value on sirolimus/everolimus and the average value on calcineurin inhibitors period. Green, increase; red, decrease; white, unchanged. Black, P < 0.05; gray, 0.15 < P < 0.05.
Figure S8. Glucose use in primary fibroblast culture. Glucose use in primary fibroblasts was decreased robustly by treatment with rapamycin but was not effected by treatment with cyclosporin A. Glucose consumption decreased with replicative age in control and mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/maternally inherited diabetes and deafness (MELAS/MIDD) cells treated with dimethylsulfoxide (DMSO) or cyclosporin; rapamycin treatment decreased consumption but also slowed the replicative age-associated decline. Linear regression trendlines were added when more than 3 data points are available. Glucose use is reported in (ug glucose/h/confluent 15-cm plate) (see Methods section). MD, mitochondrial disease.
Figure S9. Schematic of heteroplasmy analysis assay. Oligonucleotide primer sequences, amplicon sequence, and restriction site location (A). Schematic of restriction analysis band pattern (B). Restriction analysis of early passage patient (C) and control fibroblast lines (D). Sanger sequencing chromatogram of the mutant site in the amplicon from patient 1 (E). PCR, polymerase chain reaction. Supplementary material is linked to the online version of the paper at www.kidney-international.org.
SIGNIFICANCE STATEMENT.
Mitochondrial diseases represent a challenge in human medicine. Recent studies have indicated that the mechanistic target of rapamycin (mTOR) pathway is up-regulated in a number of mitochondrial diseases, and its inhibition is an effective therapeutic strategy in animal models. Mitochondrial diseases are clinically and genetically heterogeneous and can lead to organ failure and transplantation. mTOR inhibitors are used in human beings as anti-transplant rejection drugs. We show that fibroblasts derived from patients with mitochondrial diseases show hyperactive mTOR and cellular and mitochondrial defects, attenuated by mTOR inhibition. We further found that patients switched to mTOR inhibitors for transplant management showed clinical improvement. This study provides direct evidence that mTOR inhibition is beneficial in human mitochondrial disease patients.
ACKNOWLEDGMENTS
This project received funding from the European Research Council under the European Union’s Horizon 2020 research and innovation program grant 679254 (GC); and this work was supported by the Emmanuel Boussard Foundation (London, UK) and the Day Solvay Foundation (Paris, France), INSERM, Assistance Publique-Hopitaux de Paris, and University Paris Descartes. Supported by National Institutes of Health grants AG017242, GM104459, and CA180126 (YS); American Federation for Aging Research Postdoctoral Fellowship, National Institutes of Health grant F32 AG050444–02, and National Institutes of Health grant K99 GM126147–01 (SCJ); by National Institutes of Health predoctoral training grant 6T32AG023475–13 (BG); and by National Institutes of Health training grant T32AG000057 (AB).
Footnotes
DISCLOSURE
All the authors declared no competing interests.
REFERENCES
- 1.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. [DOI] [PubMed] [Google Scholar]
- 2.Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012;13:878–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macmillan C, Lach B, Shoubridge EA. Variable distribution of mutant mitochondrial DNAs (tRNA(Leu[3243])) in tissues of symptomatic relatives with MELAS: the role of mitotic segregation. Neurology. 1993;43: 1586–1590. [DOI] [PubMed] [Google Scholar]
- 4.Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. N Engl J Med. 2012;366:1132–1141. [DOI] [PubMed] [Google Scholar]
- 5.Laloi-Michelin M, Meas T, Ambonville C, et al. The clinical variability of maternally inherited diabetes and deafness is associated with the degree of heteroplasmy in blood leukocytes. J Clin Endocrinol Metab. 2009;94: 3025–3030. [DOI] [PubMed] [Google Scholar]
- 6.Dimauro S, Mancuso M, Naini A. Mitochondrial encephalomyopathies: therapeutic approach. Ann N Y Acad Sci. 2004;1011:232–245. [DOI] [PubMed] [Google Scholar]
- 7.Schleit J, Johnson SC, Bennett CF, et al. Molecular mechanisms underlying genotype-dependent responses to dietary restriction. Aging Cell. 2013;12:1050–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson SC, Yanos ME, Kayser EB, et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science. 2013;342:1524–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson SC, Yanos ME, Bitto A, et al. Dose-dependent effects of mTOR inhibition on weight and mitochondrial disease in mice. Front Genet. 2015;6:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng X, Boyer L, Jin M, et al. Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria-related neurodegeneration. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kreis H, Oberbauer R, Campistol JM, et al. Long-term benefits with sirolimus-based therapy after early cyclosporine withdrawal. J Am Soc Nephrol. 2004;15:809–817. [DOI] [PubMed] [Google Scholar]
- 12.Canaud G, Bienaime F, Viau A, et al. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat Med. 2013;19:1288–1296. [DOI] [PubMed] [Google Scholar]
- 13.Wiley CD, Velarde MC, Lecot P, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23: 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5: 649–655. [PubMed] [Google Scholar]
- 15.Velarde-Jurado E, Avila-Figueroa C. [Methodological considerations for evaluating quality of life]. Salud Publica Mex. 2002;44:448–463. [PubMed] [Google Scholar]
- 16.Canaud G, Bienaime F, Tabarin F, et al. Inhibition of the mTORC pathway in the antiphospholipid syndrome. N Engl J Med. 2014;371: 303–312. [DOI] [PubMed] [Google Scholar]
- 17.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park SH, Kang HJ, Kim HS, et al. Higher DNA repair activity is related with longer replicative life span in mammalian embryonic fibroblast cells. Biogerontology. 2011;12:565–579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Dose-dependent effects of rapamycin in fibroblasts. Western blot of phosphorylated and total S6 in control (WI38 line) fibroblasts treated with varying concentrations of rapamycin for 48 hours. The 50% inhibitory concentration (IC50) of rapamycin for S6 phosphorylation was calculated to be approximately 0.3 nmol/l (A). Relative glucose use in WI38 fibroblasts treated with varying doses of rapamycin for 48 hours. The IC50 for rapamycin on glucose consumption is approximately 2.6 nmol/l (B). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure S2. Additional Western blot data. Western blot of phosphorylated and total PRAS40, a component of mechanistic Target of Rapamycin Complex 1 (mTORC1) using samples shown in Figure 1. MELAS fibroblasts show a significant reduction in phosphorylated PRAS40 that is attenuated by 48 hours of rapamycin treatment (A). Western blot of phosphorylated and total protein kinase C (PKC). There was a trend toward increased PKC phosphorylation (driven by 2 of the 4 lines), but this did not reach significance (B). Western blot of IGF1R processing and phospho-ERK (p42/44). IGF1R is produced as a pro-protein and processed by cleavage (C). MELAS fibroblasts have significantly lower levels of processed, compared with unprocessed, IGF1R, but this was not altered substantially by 48 hours of rapamycin treatment. Conversely, levels of phosphorylated ERK (p42/44 MAPK) were increased significantly by 48 hours of rapamycin treatment, but were not different between the control and MELAS lines. CsA, cyclosporin A; DMSO, dimethylsulfoxide; ERK, extracellular signal-regulated kinase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; MAPK, mitogen-activated protein kinase; MELAS, mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes.
Figure S3. Abnormal morphology and membrane potential of mitochondria in fibroblasts from patients with mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/ maternally inherited diabetes and deafness (MELAS/MIDD). Representative images of abnormal mitochondria morphology and membrane potential in the fibroblasts from a MELAS/MIDD patient, characterized by swelling, fragmentation, and the presence of depolarization as indicated by low tetramethylrhodamine ethyl ester (TMRE, red) staining relative to the less membrane potential-sensitive dye 10-N-nonyl acridine orange (10-NA0). Nuclear DNA was stained with the cell permeant dye Hoechst 33342 (blue). Low magnification is shown in the left column and higher magnification is shown in the right column. Arrows highlight overtly swollen mitochondria, as indicated by a balloon or ball-like appearance versus normal fibrillar organization. Bar: ~10 μm.
Figure S4. Mitochondrial and cytoskeletal defects in mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/maternally inherited diabetes and deafness (MELAS/MIDD) fibroblasts. Mitochondria in MELAS/MIDD fibroblasts have collapsed and abnormal cristae and cells are characterized by high levels of cytoskeletal fibers (A). Mitochondria are visually more intact and organized in rapamycin-treated MELAS/MIDD fibroblasts whereas cytoskeletal fibers are less distinct (B). Red arrows point to mitochondria with collapsed and abnormal cristae. Yellow arrows indicate mitochondria with healthy cristae morphology. Blue arrows point to regions of cytoskeletal fibers. DMSO, dimethylsulfoxide; MD, mitochondrial disease.
Figure S5. Replicative lifespan in the presence of pyruvate. Addition of pyruvate attenuated the early growth retardation associated with rapamycin treatment but had no impact on the short lifespan of mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/maternally inherited diabetes and deafness (MELAS/MIDD) cells or the increase in lifespan resulting from rapamycin treatment. Replicative lifespans of 2 control lines treated with dimethylsulfoxide (DMSO) or rapamycin in media supplemented with pyruvate (A). ***P < 0.0001, paired t test DMSO versus rapamycin. Replicative lifespans of 3 MELAS/MIDD fibroblast lines treated with DMSO or rapamycin in media supplemented with pyruvate (B). *P = 0.02, paired t test DMSO versus rapamycin.
Figure S6. p21 expression in fibroblasts from mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/maternally inherited diabetes and deafness (MELAS/MIDD). Western blot analysis of the senescence-associated marker p21/Cip1 and quantification. Western blot performed at the start of the lifespan study, before onset of senescence. MELAS/MIDD fibroblast lines show substantially increased levels of p21/Cip1 compared with control fibroblast lines. *P < 0.05. CsA, cyclosporin A; DMSO, dimethylsulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure S7. Metabolomic impact of mechanistic target of rapamycin (mTOR) inhibition in transplant recipients with mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/maternally inherited diabetes and deafness (MELAS/MIDD). Heatmap of the log10 change of 128 serum metabolites before and after the switch to mTOR inhibitors. Individual sample values per each metabolite were averaged across treatment per each patient. Data are represented as the difference between the average value on sirolimus/everolimus and the average value on calcineurin inhibitors period. Green, increase; red, decrease; white, unchanged. Black, P < 0.05; gray, 0.15 < P < 0.05.
Figure S8. Glucose use in primary fibroblast culture. Glucose use in primary fibroblasts was decreased robustly by treatment with rapamycin but was not effected by treatment with cyclosporin A. Glucose consumption decreased with replicative age in control and mitochondrial encephalopathy with lactic acidosis and stroke-like episodes/maternally inherited diabetes and deafness (MELAS/MIDD) cells treated with dimethylsulfoxide (DMSO) or cyclosporin; rapamycin treatment decreased consumption but also slowed the replicative age-associated decline. Linear regression trendlines were added when more than 3 data points are available. Glucose use is reported in (ug glucose/h/confluent 15-cm plate) (see Methods section). MD, mitochondrial disease.
Figure S9. Schematic of heteroplasmy analysis assay. Oligonucleotide primer sequences, amplicon sequence, and restriction site location (A). Schematic of restriction analysis band pattern (B). Restriction analysis of early passage patient (C) and control fibroblast lines (D). Sanger sequencing chromatogram of the mutant site in the amplicon from patient 1 (E). PCR, polymerase chain reaction. Supplementary material is linked to the online version of the paper at www.kidney-international.org.