

Variable lung disease was documented in 2 infants with heterozygous TBX4 mutations; their clinical presentations, pathology, and outcomes were distinct. These findings demonstrate that TBX4 gene mutations are associated with neonatal respiratory failure and highlight the wide spectrum of clinicopathological outcomes that have implications for patient diagnosis and management.
The diffuse developmental growth disorders in human lung morphogenesis, acinar dysplasia and congenital alveolar dysplasia, are thought to represent a spectrum of growth arrest at different stages in lung development and are generally fatal in the newborn period or cause severe respiratory disease in infancy.1-4 Although genetic causes of developmental growth disorders are not well understood, TBX4 and FGFR2 mutations were recently identified in infants with acinar dysplasia and lethal respiratory failure within the first day of life5, 6. Heterozygous TBX4 mutations have also been reported in patients with childhood onset pulmonary arterial hypertension (PAH)7, 8 and small patellar syndrome (SPS, also known as ischiocoxopodopatellar syndrome: OMIM 147891), a rare autosomal dominant disorder characterized by patellar aplasia or hypoplasia and anomalities of the pelvis and feet with less frequent craniofacial dysmophisms9. Recurrent microdeletions at 17q23.1q23.2 including the TBX2 and TBX4 were described in patients with microcephaly, growth retardation, heart defects, and limb abnormalities10, and TBX4 duplication was associated with congenital clubfoot11, 12.
TBX4 is a member of a family of conserved, T-box containing transcription factors which play diverse roles in vertebrate organogenesis, including formation of the lung. In the mouse, TBX4 is first expressed in the pulmonary mesenchyme at E9.25, as endodermal cells in the embryonic lung tubules interact with splanchnic mesenchymal cells during branching morphogenesis13. Although homozygous deletion of Tbx4 in mice is embryonic lethal at approximately E10.5, caused at least in part by failure of chorioallantoic fusion14, conditional deletion of Tbx4 in the embryonic pulmonary mesenchyme impaired branching morphogenesis13. Deletion of Tbx4 inhibited FGF10 expression by embryonic lung fibroblasts and disrupted FGFR2 IIIB signaling in pulmonary epithelial cells that is required for proliferation and migration13. Thus, TBX4 is critical for regulating reciprocal signaling between pulmonary mesenchymal and epithelial cells during lung morphogenesis.
Two infants with heterozygous mutations in TBX4 presented with respiratory failure at birth with markedly differing clinical outcomes. Although one infant died in the early neonatal period, the other is without need for ventilatory support at 26 months of age. Thus, TBX4 mutation is associated with a wide spectrum of clinicopathological outcomes that have implications for patient diagnosis and management.
Case Presentations
Case 1: Lethal neonatal respiratory failure
Infant A:
A 38 6/7 weeks female infant was born by vaginal delivery to a 32-year-old G1P1 mother after induction for fetal growth restriction. She was cyanotic with increased work of breathing that did not improve with continuous positive airway pressure (CPAP) and 100% oxygen. Low lung volumes, a small right-sided pneumothorax and clear lung fields were observed on initial chest radiograph. Decompression of the right pneumothorax failed to improve her clinical status. After intubation and positive pressure ventilation, her oxygen saturations remained low. Growth measures were notable for weight of 2.925 kg (25th percentile), length of 46 cm (5th percentile), and head circumference of 28.8 cm (<<3rd percentile). She was transported to a quaternary neonatal intensive care center and placed on high frequency oscillatory ventilation, milrinone, and inhaled nitric oxide and epoprostenol. Admission chest radiograph demonstrated low lung volumes and bilateral pneumothoraxes. Bilateral chest tubes were placed. There was no evidence of limb abnormalities by physical examination and radiographic images of the chest and abdomen did not demonstrate rib, vertebral or proximal extremity abnormalities. Osseous structures of the head were normal by examination and computed tomography (CT). She was treated with epinephrine, vasopressin, and hydrocortisone for systemic hypotension. Echocardiogram revealed severe right ventricular hypertension despite high dose inhaled nitric oxide and inhaled epoprostenol. At 52 hours of life, with an oxygenation index persistently greater than 40, she was placed on extracorporeal membrane oxygenation (ECMO) and treated for 3 weeks without improvement. She developed seizures, intracranial hemorrhage and left middle cerebral artery infarction. Additional information on the clinical presentation and course of this infant is included in a recently published case report15. She was removed from ECMO after 19 days and died shortly thereafter. An autopsy and whole exome sequencing were performed.
Case 2: Neonatal respiratory failure and chronic pulmonary disease
Infant B:
A 39 4/7 weeks male infant was born to a 29-year-old G1P1 mother by Cesarean section due to breech presentation. The infant was in respiratory distress at birth and treated with CPAP and oxygen. He was small for gestational age; weight 2.61kg (5th percentile), length 45.9cm (<1st percentile) and head circumference 33cm (4th percentile). Respiratory distress and oxygen requirement persisted. He was transferred to a quaternary neonatal intensive care center for further evaluation. No craniofacial limb, hand or foot dysmorphisms were noted on physical examination with no radiographs available to assess for characteristic radiographic pelvis and lower limb features of SPS. Echocardiography demonstrated a large secundum atrial septal defect (ASD) with left to right shunting. Chest CT showed only dependent atelectasis and diffuse decreased attenuation. Sleep studies demonstrated non-apneic hypoxemia and tachypnea that improved on 0.25 lpm nasal cannula oxygen. Nasal ciliary biopsy showed normal ciliary motility and ultrastructure. Bronchoscopic evaluation demonstrated laryngomalacia, a deep laryngeal notch, grade 1 subglottic stenosis and abnormal branching of the left upper lobe bronchus. A heterozygous ABCA3 variant (c.863G>A p.Arg288Lys) of uncertain clinical significance was found by a diffuse lung disease genetic panel evaluation. The infant was discharged home at 25 days of age on 0.25 lpm nasal cannula oxygen.
At 3 months of age, sleep studies demonstrated persistent non-apneic hypoxemia, tachypnea, and mixed central and obstructive sleep apnea (AHI 17.4; OI 10.8). No genetic abnormalities were found in PHOX2B associated with congenital central hypoventilation syndrome. He was admitted for inpatient evaluation with failure to thrive. Physical examination was notable for tachypnea, mild accessory muscle use and retractions. He was also noted to have bilateral undescended testes and two small hemangiomas. Chest radiograph and CT findings were consistent with interstitial pneumonia and aberrant lobular architecture with abnormal alveolar growth. Cardiac catheterization demonstrated an ASD without shunting. Mild pulmonary hypertension was noted with pulmonary vascular resistance measurments of 3.5 iWU (Fick) or 2.7 iWU (TDCI). Mean pulmonary artery pressures were 25 and 27 mmHg on the right and left, respectively. Right and left pulmonary artery web pressures were 5 and 9 mmHg. Angiography demonstrated appropriately sized bilateral pulmonary arteries with normal aborization pattern. Echocardiography performed in a similar time frame showed mild tricuspid valve regurgitation and estimated right ventricular pressure of 52 mmHg plus central venous pressure. All prior and subsequent echocardiograms showed similar or lower estimates of right ventricular hypertension. Following multiple episodes of cardiopulmonary arrest thought to be secondary to increases in vagal tone, he was placed on ECMO for 4 days. At the time of ECMO cannulation he had a pulseless arrest and no return of spontaneous circulation despite multiple doses of epinephrine. Echocardiogram obtained immediately after cannulation showed mild pulmonary hypertension with estimated right ventricular pressure of 32 mmHg plus central venous pressure. While on ECMO, bronchoscopic airway evaluation demonstrated stable subglottic narrowing, diffuse bronchitis of the right lower lobe, mucous plugging, subsegmental bronchial stenosis and increased neutrophils in the bronchioalveolar lavage fluid. Following decannulation from ECMO, right upper and middle lobe lung wedge biopsies were obtained. Tracheostomy and gastrostomy tubes were placed for chronic ventilation and nutritional support. The infant was discharged home at 8 months of age on 24-hour ventilatory support and 1 lpm supplemental oxygen. By 15 months of age he was weaned off mechanical ventilation and required 0.5 lpm supplemental oxygen via tracheostomy. His growth measures improved to 17th percentile for weight, 5th percentile for length, and 42nd percentile for head circumference. The most recent chest radiograph showed bilateral heterogeneous lung opacities and no repeat chest CT has been obtained. The most recent echocardiogram showed no evidence of right ventricular hypertension. DNA microarray was obtained since the heterozygous ABCA3 variant of uncertain clinical significance was not considered to alone be causative of the lung abnormalities.
Methods
Histology and immunostaining
Formalin fixed, paraffin embedded tissue sections were used for all studies. Sections were stained with hematoxylin and eosin for histopathologic analysis. Immunohistochemistry was performed on a Roche BenchMark XT instrument using Roche pancytokeratin (CKAe1Ae3, catalog# 760-2595) and cytokeratin 7 (catalog# 790-4462) antibodies with protease 1 and mild EDTA pretreatments, respectively. Immunofluorescence staining was performed on tissue sections that were deparaffinized in xylene and rehydrated in a series of graded alcohols. Control lung tissue from a 4-month-old donor was provided by Gloria Pryhuber, M.D. at the University of Rochester in conjunction with the LungMap project (https://www.lungmap.net/). Endogenous peroxidase activity was blocked in a solution of methanol/3% hydrogen peroxide. Antigen retrieval was performed in 0.1 M citrate buffer (pH 6.0) by microwaving. Slides were blocked for 2 hours at room temperature using 4% normal donkey serum in 0.1 M PBS containing 0.2% Triton X-100 and then incubated with primary antibodies diluted in blocking buffer for approximately 16 hours at 4°C. Primary antibodies included NKX2.1 (1:1000; RB TTF-1 1231, Seven Hills Bioreagents), ABCA3 (1:100; WMAM-17G524, Seven Hill Bioreagents), HOPX (1:100, SC-30216, Santa Cruz Biotechnology) and ACTA2 (1:2000; A5228, Sigma-Aldrich). Appropriate secondary antibodies conjugated to Alexa Fluor 488, 568 or 633 were used at a dilution of 1:200 in blocking buffer for 1 hour at room temperature. Nuclei were counterstained with DAPI (1 μg/ml) (Thermo-Fisher). Sections were mounted using ProLong Gold (Thermo-Fisher) mounting medium and coverslipped.
Confocal microscopy
Tissue sections stained by immunofluorescence were imaged on an inverted Nikon A1R confocal microscope (×20 and ×60 magnification) NA 1.27 objectives using a 1.2 AU pinhole. Maximum intensity projections of multilabeled Z stack images obtained sequentially using channel series across the 5μm thick sections were generated using Nikon NIS-Elements software.
Genetic analysis
DNA was extracted from the peripheral blood of infant B and frozen lung tissue from infant A using standard methods. Tissue from infant A underwent whole exome sequencing (WES) with trio analysis at Cincinnati Children's Hospital Medical Center (CCHMC) using SureSelect CRE targeted sequence reagent (Agilent). Paired-end sequencing (2×125bp) was performed on the Illumina HiSeq2500 platform with more than 95% coverage at 20x. Data analysis was performed using an in-house bioinformatics pipeline based on GATK3.5 and Alamut Batch 1.4.4. Clinically significant variants were determined based on ACMG-AMP 2015 variant interpretation guidelines. An Illumina Infinium assay for single nucleotide polymorphism (SNP) microarray analysis was performed for Infant B using the Infinium® CytoSNP-850Kv1.1 BeadChip platform. B-allele frequency and Log2R ratio were analyzed with the Illumina GenomeStudio analysis software, and DNA copy number changes were prioritized using cnvPartition software. Infant B also underwent a gene panel through The Johns Hopkins DNA Diagnostic Laboratory in which sequencing of the coding regions of ABCA3, AP3B1, CSF2RA, CSF2RB, DKC1, FOXF1, HPS1, HPS4, NKX2.1, SFTPA2, SFTPB, SFTPC, SLC7A7, TERC, TERT, TINF2 was performed along with deletion/duplication analysis of all genes except SFTPA2 and SFTPC. Sanger sequencing was also performed for the PHOX2B gene.
Results
Autopsy and genetic findings: Infant A
The infant had no external congenital abnormalities, specifically no craniofacial, limb or foot abnormalities were noted. Radiographic images were not available for analyses of the patella, pelvis and lower limbs, thus radiographic abnormalities associated with SPS can not be excluded. The heart was normally formed. Severe neonatal anoxic/ischemic brain damage was present. Lungs had abnormal lobation, with three lobes on the right and three lobes on the left. Hilar bronchial arterial relationships were normal. The lungs had a combined weight of 79.9 grams (normal for 39 weeks gestation = 49.4+14.8 grams). The lungs had histologic features of congenital alveolar dysplasia thought to represent growth arrest in the late canalicular/early saccular phase of lung development2, 4. Branching bronchi and bronchioles with accompanying pulmonary arteries were normally developed with malformed distal pulmonary lobules comprised of simplified saccular airspaces separated by thickened septa (Figure 1). The number of saccules varied among lobules and were lined by cuboidal cells with few flattened cells. Normally developed alveoli were not present. Although bronchioles focally abutted the pleura and interlobular septa (Figure 1, A), parenchymal development was overall more prominent than seen in acinar dysplasia characterized by lung growth arrest in the pseudoglandular or early canalicular phase of development2, 4. Some of the small pulmonary arteries had mild medial hypertrophy. Pathologic findings diagnostic of alveolar capillary dysplasia with misalignment of pulmonary veins (ACDMPV), including malposition of pulmonary veins adjacent to pulmonary arteries, marked medial hypertrophy of pulmonary arteries and arterioles and markedly reduced capillary density, were not present. The congenital alveolar dysplasia was accompanied by diffuse acute and chronic pneumonia with neutrophils admixed with mucus filling conducting airways and distal airspaces. Confocal immunofluorescence microscopy demonstrated aberrant cellular differentiation of lung epithelial cells lining the primitive saccules. The atypical epithelial cells had variable expression of NKX2.1 and a discrepancy between NKX2.1 and ABCA3 expression, which are normally co-expressed in alveolar type II (AT2) cells, was noted (Figure 1 and Figure 3 [available at www.jpeds.com]). Nuclear HOPX expression was lacking with cytoplasmic staining in only scattered rare cells indicating a lack of AT1 cell differentiation.
Figure 1: Distinct morphologic lung abnormalities in infants with TBX4 mutations.
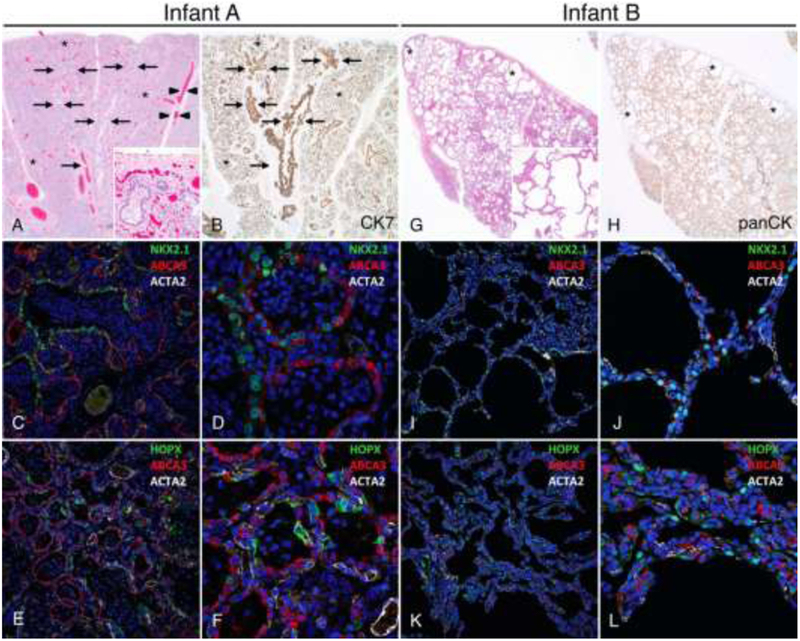
Lungs from Infant A (A-F) showing congenital alveolar dysplasia. H&E stained section (A) shows normally developed branching bronchi and bronchioles accompanied by arteries (arrows), veins appropriately located in the interlobular septa (arrowheads), and malformed lobules comprised of variable numbers of simplified saccular airspaces (*) with bronchioles focally abutting the pleura (inset). Epithelial cell immunostaining with cytokeratin 7 (B) highlights branching conducting airways (arrows) and variable numbers of simplified saccules comprising the malformed lobules (*). Confocal immunofluorescence low (C,E) and higher power images (D,F) show aberrant lack of co-expression of the alveolar type II cell (AT2) markers NKX2.1 and ABCA3 in the atypical epithelial cells lining distal saccular airspaces (C-D) and absence of nuclear staining for the alveolar type I cell marker (AT1), HOPX (E-F). Staining for smooth muscle actin (ACTA2) is also shown. Lungs from Infant B (G-L) showing alveolar growth abnormality. H&E stained section shows deficient alveolarization with variably enlarged and simplified alveoli (inset) and subpleural cystically dilated airspaces (*). Epithelial cell immunostaining with pancytokeratin (H) highlights the focally enlarged and simplified alveolar spaces that predominant in subpleural regions (*). In contrast to Infant A, confocal immunofluorescence low (I,K) and higher power images (J, L) of the lung biopsy from Infant B show co-expression of NKX2.1 and ABCA3 as seen in normal AT2 cell differentiation (I-J) and nuclear HOPX expression indicative of AT1 cell differentiation (K-L). Original magnification: 20x (A-B, G-H); 200x (A inset, G inset); 20X objective with pixel size of 0.63 μm (C, E, I, K); 60X objective with pixel size of 0.21 μm (D, F, J, L).
Figure 3 (online only): Distal epithelial cell differentiation in TBX4 mutant lungs.

Confocal immunofluorescence images of lungs from a 4 month old donor control lung (A-D), Infant A (E-H) and Infant B (I-L). Nuclear NKX2.1 and cytoplasmic ABCA3 are co-expressed in AT2 cells (A-B), and nuclear HOPX is expressed in AT1 cells (C-D) in the normal control lung. In contrast, the distal saccules comprising the lobules in Infant A are lined by atypical epithelial cells with a discrepancy in NKX2.1 and ABCA3 expression (E-F), and lack of nuclear HOPX expression (G-H). Enlarged and simplified alveoli in Infant B are lined by AT2 cells co-expressing NKX2.1 and ABCA3 (I-J) and AT1 cells expressing nuclear HOPX, Original magnification: 20X objective with pixel size of 0.63 μm (A, C, E, G, I, K), 60X objective with pixel size of 0.21 μm (B, D, F, H, J, L).
Whole exome sequencing identified a heterozygous pathogenic variant in TBX4: c.524_527del (p.Asn175Thrfs*52) which was not found in either parent (Figure 2). The four base pair deletion results in a frameshift starting at codon Asn175 causing a premature stop codon 52 base pairs downstream. This frameshift mutation occurs within the TBX4 DNA binding domain. Infant A was also found to have a variant within ABCA3: c.863G>A resulting in the substitution of arginine to lysine at position 288. The infant’s mother shared this ABCA3 variant and is healthy without pulmonary disease. Because both the inherited ABCA3 variant and the de novo TBX4 variant had similar allelic ratios of nearly 50%, and the DNA was extracted from lung tissue which contained a significant inflammatory cell infiltrate, the TBX4 alteration is most likely germline in origin.
Figure 2: TBX4 mutations.

A) Whole exome sequencing performed on Infant A identified a de novo four base pair deletion in exon 5 (c.524_527del) located within the T-box, the critical DNA binding domain of the resultant protein. B) Microarray analysis performed on Infant B identified a 2.2Mb deletion in the long arm of chromosome 17 (17q23.1-17q23.2; 58113570_60325222) encompassing the TBX2 and TBX4 genes.
Lung biopsy and genetic findings: Infant B
The lung biopsies from Infant B demonstrated an alveolar growth abnormality characterized by variably enlarged and simplified alveoli with focal cystically dilated airspaces predominanting in subpleural regions (Figure 1). Alveoli were lined by cuboidal AT2 and flattened AT1 cells and separated by thin septa with focal interstitial chronic inflammation. In contrast to infant A, confocal immunofluorescence studies demonstrated nuclear HOPX expression in AT1 cells and NKX2.1 was co-expressed with ABCA3, consistent with normal AT2 cell differentiation (Figure 1 and Figure 3). Accumulations of foamy macrophages with rare cholesterol clefts filling alveolar airspaces were focally observed. Ultrastructural analysis revealed normal lamellar bodies in AT2 epithelial cells as well as occasional abnormal lamellar bodies with features seen in ABCA3 related surfactant deficiency. Histopathologic patterns of surfactant homeostasis disorders including pulmonary alveolar proteinosis, chronic pneumonitis of infancy, desquamative interstitial pneumonia and nonspecific interstitial pneumonia were not present. Conducting airways were normally developed with focal peribronchiolar inflammation. Pulmonary veins were appropriately located within the interlobular septa and capillaries were correctly positioned subjacent to the alveolar epithelium.
Microarray analysis identified a 2.2Mb deletion on chromosome 17 (17q23.1-17q23.2) containing the entire TBX4 and TBX2 genes (Figure 2). Infant B also carried the same heterozygous ABCA3 variant (c.863G>A p. Arg288Lys) detected in Infant A.
Discussion
Genetic testing of 2 infants with neonatal respiratory failure identified pathogenic mutations in the TBX4 gene associated with congenital malformations of lung morphogenesis with differing clinical courses. Although treatment related lung injury related to positive pressure ventilation and oxygen supplementation may have contributed to the lung pathology, the presence of respiratory distress at birth with persistent oxygen requirement, the abnormal lung lobation in Infant A and the marked deficiency in acinar/alveolar development provide evidence that the lung phenotypes represent primary parenchymal disease. Exome sequencing of Infant A identified a heterozygous four base pair deletion in the TBX4 gene causing a frameshift predicted to impair the T-box DNA binding domain of the protein. This mutation was classified as likely pathogenic based upon the location in a functional domain of the protein and reports of pathologic variants in the TBX4 T-box domain causing autosomal dominant pulmonary hypertension found in the Human Gene Mutation Database (HGMD)7. A recent report describing a de novo heterozygous TBX4 missense mutation in a newborn with lethal diffuse lung disease6 together with the absence of this TBX4 variant in multiple databases, including HGMD, dbSNP, 1000 genomes, gnomAD, Exome Sequencing Project (ESP) and ClinVar, support the likelihood that this TBX4 variant represents a disease causing mutation. A heterozygous deletion in a region of 17q23 containing both TBX4 and TBX2 genes was identified in Infant B. In searching the above databases, 11 reported cases of microdeletions involving the TBX4 and TBX2 genes were identified7,10, 16. In one case, the infant presented in the newborn period with respiratory distress, growth restriction and microcephaly16. She developed apnea events that resulted in cardiovascular arrest requiring tracheostomy and gastrostomy placement for ongoing support. She also had mild laryngomalacia and mild pulmonary hypertension. Of the 11 reported cases with deletions involving TBX4 and TBX2 genes, seven had pulmonary hypertension, low birth weight and microcephaly, and six had postnatal growth failure. Clinical disease caused by deletions or mutations in the TBX2 gene alone was not found in our search, thus the potential contribution of the TBX2 deletion to lung disease in Infant B is uncertain. TBX4 mutations or deletions were previously reported in 30% of patients with childhood onset PAH with a lower frequency of 2% in adult onset PAH7. Recent genetic analysis of 130 childhood onset and 178 adult onset idiopathic PAH patients confirmed a significant enrichment of TBX4 mutations in pediatric onset PAH, with TBX4 mutations detected in 8% of patients8. Most of the predicted pathogenic variants localized to the conserved T-box domain as in the current case. The TBX4 mutations were inherited from unaffected parents in >50% of cases indicating incomplete penetrance. Interestingly, disease severity varied in TBX4 mutation carriers from mild/moderate PAH to progressive, fatal PAH or requiring transplantation similar to the marked difference in clinical pulmonary phenotypes in the current patients.
In the present studies, both infants shared the same R288K heterozygous ABCA3 variant. The mother of Infant A also has this same ABCA3 variant and is a healthy adult with no lung disease. According to the Grantham scale, the arginine to lysine change caused by this variant is considered conservative. This ABCA3 variant is known to dbSNP as rs117603931 and was found in gnomAD with a minor allele frequency (MAF) of 0.93% in non-Finnish Europeans and 0.83% in European Americans in the Exome Sequencing Project (ESP). Although reported in HGMD (CM064939) as a disease-causing mutation for fatal surfactant deficiency, ClinVar defines it as a benign variant without specified phenotype. There is a three to four-fold enrichment of this single ABCA3 variant among European-descent late-preterm and term infants with respiratory distress syndrome (RDS) with the variant predicted to lead to a 10.9% attributable risk for RDS17. In a cohort of pediatric patients with interstitial lung disease carrying heterozygous ABCA3 R288K variants, nine patients had respiratory symptoms that resolved after the neonatal period18. Functional characterization of the R288K ABCA3 variant after adenoviral-transduction into A549 cells demonstrated reduced ATPase activity that may impair phospholipid transport into lamellar bodies19. Based on all available information, the R288K variant is presently classified as a variant of uncertain clinical significance; however, there is mounting evidence that it may influence non-lethal, neonatal pulmonary disease17-21. Whether the ABCA3 R288K alleles contributed to the neonatal respiratory distress in the two infants in the present report is unclear, but it is possible that these ABCA3 variants exacerbate the lung disease in the setting of TBX4 mutations. Consistent with a causative role for TBX4 in developmental lung disease, deficient lung growth and maturation was present in both infants. Abnormal alveolarization was apparent in both infants with marked variation in severity of malformations despite shared TBX4 and ABCA3 mutations.
The present histopathologic findings demonstrate extensive, but distinct morphologic abnormalities in the lungs of infants with TBX4 associated congenital lung disease. In mice, TBX4 regulates FGF10 production by mesenchymal cells while activating FGFR2 in epithelial progenitor peripheral acinar buds which in turn produce SHH activating GLI2,3, FOXF1, and TBX2, 3, 4 transcription factors in subsets of embryonic lung fibroblasts. A gene network controlled by TBX transcription factors maintains NKX2.1 expression in epithelial progenitor cells of the peripheral acinar lung buds that is required for their growth and survival. The importance of this signaling network is exemplified by the severe lung malformations presently observed in two infants with TBX4 mutations, as well as those caused by mutations in NKX2.1 (Lung-Brain Thyroid Syndrome), FOXF1 (Alveolar Capillary Dysplasia), and defects associated with mutations in the SHH signaling pathway in Smith-Lemli-Opitiz syndrome (Whitsett et al., Physiological Reviews, in press, 2018). Present findings that TBX4 mutations disrupt acinar and alveolar development are consistent with the transcriptional roles of TBX family members previously identified in developing mouse lungs13.
The extent of lung malformations and clinical findings in Infants A and B were distinct. Aberrant lung morphogenesis in Infant A was diffuse but variable with regions of poor development of acinar structures to regions resembling the late canalicular/early saccular stage of development . In contrast, the histopathology in Infant B consisted of areas of near normal alveolarization adjacent to regions with deficient alveolar growth characterized by simplified, cystically dilated alveolar spaces. These distinct presentations and phenotypes may be related to the nature of the TBX4 mutations, to gene modifiers or to the additional loss of TBX2 in Infant B. It is unclear whether the missense mutation in TBX4 in Infant A causes haploinsufficiency or alters TBX4 functions in a manner distinct from the heterozygous deletion found in Infant B. Although both infants share the same potential disease associated ABCA3 allele that may influence lung disease pathogenesis, this ABCA3 mutation is unlikely to have caused the severe lung hypoplasia or disruption of alveolar development seen in both infants nor their differing phenotypes. Additionally, ABCA3 staining was present in lung epithelial cells in both infants and haploinsufficiency of ABCA3 is not known to cause lung malformation or significant pulmonary disease in term infants17-20.
Acknowledgments
Supported by NIH/NHLBI U01 HL122642 “Lung Map” Atlas Research Center and NIH/NHLBI U01 HL134745 Editing Alveolar Progenitor Cells for Correction of Monogenic Disease. The authors declare no conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Chow CW, Massie J, Ng J, Mills J, Baker M. Acinar dysplasia of the lungs: variation in the extent of involvement and clinical features. Pathology. 2013;45:38–43. [DOI] [PubMed] [Google Scholar]
- [2].Deutsch GH, Young LR, Deterding RR, Fan LL, Dell SD, Bean JA, et al. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med. 2007;176:1120–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kurland G, Deterding RR, Hagood JS, Young LR, Brody AS, Castile RG, et al. An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med. 2013;188:376–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Langston C, Dishop MK. Diffuse lung disease in infancy: a proposed classification applied to 259 diagnostic biopsies. Pediatr Dev Pathol. 2009;12:421–37. [DOI] [PubMed] [Google Scholar]
- [5].Barnett CP, Nataren NJ, Klingler-Hoffmann M, Schwarz Q, Chong CE, Lee YK, et al. Ectrodactyly and Lethal Pulmonary Acinar Dysplasia Associated with Homozygous FGFR2 Mutations Identified by Exome Sequencing. Human mutation. 2016;37:955–63. [DOI] [PubMed] [Google Scholar]
- [6].Szafranski P, Coban-Akdemir ZH, Rupps R, Grazioli S, Wensley D, Jhangiani SN, et al. Phenotypic expansion of TBX4 mutations to include acinar dysplasia of the lungs. American journal of medical genetics Part A. 2016;170:2440–4. [DOI] [PubMed] [Google Scholar]
- [7].Kerstjens-Frederikse WS, Bongers EM, Roofthooft MT, Leter EM, Douwes JM, Van Dijk A, et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. Journal of medical genetics. 2013;50:500–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhu N, Gonzaga-Jauregui C, Welch CL, Ma L, Qi H, King AK, et al. Exome Sequencing in Children With Pulmonary Arterial Hypertension Demonstrates Differences Compared With Adults. Circ Genom Precis Med. 2018;11:e001887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bongers EM, Duijf PH, van Beersum SE, Schoots J, Van Kampen A, Burckhardt A, et al. Mutations in the human TBX4 gene cause small patella syndrome. American journal of human genetics. 2004;74:1239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ballif BC, Theisen A, Rosenfeld JA, Traylor RN, Gastier-Foster J, Thrush DL, et al. Identification of a recurrent microdeletion at 17q23.1q23.2 flanked by segmental duplications associated with heart defects and limb abnormalities. American journal of human genetics. 2010;86:454–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Alvarado DM, Aferol H, McCall K, Huang JB, Techy M, Buchan J, et al. Familial isolated clubfoot is associated with recurrent chromosome 17q23.1q23.2 microduplications containing TBX4. American journal of human genetics. 2010;87:154–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Peterson JF, Ghaloul-Gonzalez L, Madan-Khetarpal S, Hartman J, Surti U, Rajkovic A, et al. Familial microduplication of 17q23.1-q23.2 involving TBX4 is associated with congenital clubfoot and reduced penetrance in females. American journal of medical genetics Part A. 2014;164A:364–9. [DOI] [PubMed] [Google Scholar]
- [13].Arora R, Metzger RJ, Papaioannou VE. Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PLoS genetics. 2012;8:e1002866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Naiche LA, Papaioannou VE. Loss of Tbx4 blocks hindlimb development and affects vascularization and fusion of the allantois. Development (Cambridge, England). 2003;130:2681–93. [DOI] [PubMed] [Google Scholar]
- [15].Reed BD, Arya S, Dufendach KR, Leino D. Refractory Respiratory Failure and Pneumothorax in a Full-Term Newborn. NeoReviews. 2018;19:e109–e11. [Google Scholar]
- [16].Nimmakayalu M, Major H, Sheffield V, Solomon DH, Smith RJ, Patil SR, et al. Microdeletion of 17q22q23.2 encompassing TBX2 and TBX4 in a patient with congenital microcephaly, thyroid duct cyst, sensorineural hearing loss, and pulmonary hypertension. American journal of medical genetics Part A. 2011;155a:418–23. [DOI] [PubMed] [Google Scholar]
- [17].Wambach JA, Wegner DJ, Depass K, Heins H, Druley TE, Mitra RD, et al. Single ABCA3 mutations increase risk for neonatal respiratory distress syndrome. Pediatrics. 2012;130:e1575–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wittmann T, Frixel S, Hoppner S, Schindlbeck U, Schams A, Kappler MV, et al. Increased risk of interstitial lung disease in children with a single R288K variant of ABCA3. Molecular medicine (Cambridge, Mass). 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wambach JA, Yang P, Wegner DJ, Heins HB, Kaliberova LN, Kaliberov SA, et al. Functional Characterization of ATP-Binding Cassette Transporter A3 Mutations from Infants with Respiratory Distress Syndrome. American journal of respiratory cell and molecular biology. 2016;55:716–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Naderi HM, Murray JC, Dagle JM. Single mutations in ABCA3 increase the risk for neonatal respiratory distress syndrome in late preterm infants (gestational age 34-36 weeks). American journal of medical genetics Part A. 2014;164a:2676–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Somaschini M, Presi S, Ferrari M, Vergani B, Carrera P. Surfactant proteins gene variants in premature newborn infants with severe respiratory distress syndrome. Journal of perinatology : official journal of the California Perinatal Association. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]