

Summary
In systemic lupus erythematosus (SLE), dsDNA antibodies associate with renal disease. Less is known about comorbidities in patients without dsDNA or other autoantibodies. Using an electronic health record (EHR) SLE cohort, we employed a phenome-wide association study (PheWAS) that scans across billing codes to compare comorbidities in SLE patients with and without autoantibodies. We used our validated algorithm to identify SLE subjects. Autoantibody status was defined as ever positive for dsDNA, RNP, Smith, SSA, and SSB. PheWAS was performed in antibody positive vs. negative SLE patients adjusting for age and race and using a false discovery rate of 0.05. We identified 1097 SLE subjects. In the PheWAS of dsDNA positive vs. negative subjects, dsDNA positive subjects were more likely to have nephritis (p = 2.33 × 10−9) and renal failure (p = 1.85 × 10−5). After adjusting for sex, race, age, and other autoantibodies, dsDNA was independently associated with nephritis and chronic kidney disease. Those patients negative for dsDNA, RNP, SSA, and SSB negative subjects were all more likely to have codes for sleep, pain, and mood disorders. PheWAS uncovered a hierarchy within SLE specific autoantibodies with dsDNA having the greatest impact on major organ involvement.
Keywords: systemic lupus erythematosus, electronic health records, phenome-wide association study, autoantibodies
Introduction
Autoantibodies play an important role in the pathogenesis of systemic lupus erythematosus (SLE) and are used for diagnosis and prognosis. Autoantibodies help clinicians cluster SLE patients to monitor for specific disease criteria. For example, epidemiologic studies have demonstrated the association between double-stranded DNA (dsDNA) antibodies and renal disease.1–4 These studies focused on clinical associations in patients with positive autoantibodies but have not examined comorbidities in patients that do not have these autoantibodies. Further, they have not evaluated the relative importance of SLE autoantibodies on ACR SLE criteria5 or comorbidities.
The electronic health record (EHR) serves as an efficient tool to conduct clinical research.6–8 EHRs provide longitudinal data on both ACR SLE disease criteria5 and comorbidities, complementing cohort and administrative database studies. Phenome-wide association studies (PheWAS) are a validated tool to conduct meaningful EHR-based research.9–13 Similar to a genome-wide association study scanning across a genome, PheWAS scans across billing codes in the EHR. PheWAS has uncovered novel genetic and phenotype associations in multiple autoimmune diseases including rheumatoid arthritis14–16 and SLE.17,18 We used PheWAS to test for differences in comorbidities in SLE patients with and without autoantibodies, specifically to assess for comorbidities that might be overrepresented in SLE patients without autoantibodies. We also determined the relative importance of SLE autoantibodies in their association with SLE manifestations to examine if a hierarchy of autoantibodies exists.
Materials and Methods
Study Population
Approval was obtained from the Institutional Review Board of Vanderbilt University Medical Center (VUMC) (#141222). We identified potential SLE subjects in the Synthetic Derivative, a de-identified, mirror image of the EHR, which contains over 2.8 million subjects with longitudinal data spanning several decades.19 The Synthetic Derivative contains all available information in the EHR including billing codes, demographics, inpatient and outpatient notes, laboratory values, radiology, pathology, and medication orders. The Synthetic Derivative does not contain outside records. The Synthetic Derivative reflects the patient population seen at VUMC, which is composed equally of males and females and is predominantly Caucasian (81%). We identified potential SLE patients within the Synthetic Derivative using our previously published, internally-validated algorithm of ≥ 4 counts of the SLE ICD-9 code (710.0) and a positive anti-nuclear antibody (ANA) with a titer of ≥ 1:160 while excluding ICD-9 codes for systemic sclerosis (710.1) and dermatomyositis (710.3).20 This algorithm has a positive predictive value of 89% and a sensitivity of 86%.
Autoantibodies
Chart review was conducted by rheumatologists to determine autoantibody status (AB, CC). Autoantibody status was defined as positive if ever positive, and negative if there was at least one assay and all were negative. All autoantibodies were measured via enzyme-linked immunosorbent assays with manufacturer values to determine positivity. Only autoantibody testing performed at VUMC was included, as outside labs could not be confirmed.
Phenome-wide association studies and statistics
In PheWAS, approximately 18,000 ICD-9 codes are condensed into 1,800 Phecodes that represent distinct clinical diagnoses. The Phecodes (version 1.2) and their corresponding ICD-9 codes are available at http://phewascatalog.org. To be a case, a subject has to have at least 2 counts of the Phecode on different days. A subject is a control if there are no counts of the ICD-9 code for that specific disease or related diseases. Subjects having 1 count of the code are excluded to reduce the possibility of coding errors or preliminary diagnoses that may be ultimately ruled out.21 For each Phecode, a logistic regression model is created with the code as the outcome and the option to add covariates with odds ratios (ORs) and 95% confidence intervals (95% CIs) reported. For a code to be used in the model, there must be at least 20 cases with that code.21 Analyses were performed using the PheWAS package21 in R version 3.2.5. We performed PheWAS comparing SLE subjects with and without dsDNA, RNP, Smith, SSA, and SSB autoantibodies, adjusting for current age and race/ethnicity. Race data was obtained from the EHR, which is a mixture of self-report and administrative entry. Prior studies have validated that these assignments represent self-report and genetic ancestry.22 Due to the very low number of Hispanics (n = 25) and Asians (n = 30) in our SLE EHR cohort, we combined these subjects into a third race group in addition to Caucasians and African Americans. We adjusted for multiple hypothesis testing using a false discovery rate (FDR) of 0.05 and an unadjusted p < 0.05 for additional analyses. There were 374 testable phenotypes for dsDNA positive vs. negative PheWAS, 268 for RNP, 278 for Smith, 290 for SSA, and 289 for SSB.
Autoantibody associations with SLE manifestations
We performed logistic regression to assess the impact of autoantibodies, age, sex, and race on SLE disease criteria.5 We assessed for differences in demographics in SLE patients with and without autoantibodies using the Mann-Whitney U test for continuous variables, as there were non-normal distributions in the data, and chi-square or Fisher’s exact test for categorical variables. Two-sided p values < 0.05 were considered to indicate statistical significance. Analyses were conducted using IBM SPSS software, version 24.0 (SPSS).
Chart review
To examine the most significant findings for the PheWAS, notably the increased fibromyalgia (FM)-related codes in the antibody negative vs. positive SLE subjects, we performed chart review to ensure the antibody negative subjects were not FM cases mislabeled as SLE. We randomly selected 50 SLE subjects without any antibodies and 50 SLE subjects with at least one antibody. Chart reviewed was conducted to ensure subjects were SLE cases, defined as a diagnosis by a specialist (rheumatologist, nephrologist, or dermatologist). We also noted if subjects had FM diagnosed by a rheumatologist and collected ACR SLE criteria5 if documented.
Results
Using our validated algorithm, we identified 1097 potential SLE subjects, who have been previously described.20 By definition, all patients had a positive ANA ≥ 1:160. SLE subjects were predominantly female (90%) and Caucasian (65%) with a current mean age of 50 ± 17 years and mean age at first SLE ICD-9 code of 40 ± 17 years. On average, SLE subjects had 9 years of EHR follow-up. Of the 1097 subjects, 14 (1%) had 5 SLE specific autoantibodies, 54 (5%) had 4, 65 (6%) had 3, 134 (12%) had 2, 439 (40%) with 1, 356 (32%) with none, and 35 (3%) with missing data.
PheWAS of dsDNA positive vs. negative SLE subjects
SLE subjects with positive vs. negative dsDNA autoantibodies are shown in Table 1. Of 1097 subjects, 521 had an ever positive dsDNA, 502 a negative dsDNA, and 74 with missing data. dsDNA positive vs. negative subjects were younger at time of analysis (47 ± 18 vs. 54 ± 16, p < 0.001) and at time of first SLE ICD-9 code (37 ± 17 vs. 43 ± 15, p < 0.001) and more likely to not be Caucasian (p < 0.001). There were no sex differences in positive vs. negative subjects (p = 0.79). In the PheWAS comparing dsDNA positive vs. negative subjects adjusting for age and race, dsDNA positive were more likely to be diagnosed with renal disease including nephritis (OR = 4.60, 95% CI 2.97 – 7.14, FDR p = 2.33 × 10−9), renal failure (OR = 2.30, 95% CI 1.68 – 3.15, FDR p = 1.85 × 10−5), and end stage renal disease (OR = 2.63, 95% CI 1.51 –4.58, FDR p = 1.25 × 10−2) (Figure 1, Table 2). dsDNA positive subjects were also more likely to have codes for ACR SLE disease criteria5 with pleurisy; pleural effusion (OR = 2.00, 95% CI 1.33 – 3.03, FDR p = 0.02) and thrombocytopenia (OR = 2.10, 95% CI 1.36 – 3.53, FDR p = 0.02
Table 1.
Demographics of antibody positive vs. negative SLE subjects.
| Demographics | dsDNA positivea (n = 521) | dsDNA negative (n = 503) | RNP positive (n = 183) | RNP negative (n = 490) | Smith positive (n = 119) | Smith negative (n = 569) | SSA positive (n = 235) | SSA negative (n = 524) | SSB positive (n = 137) | SSB negative (n = 609) |
|---|---|---|---|---|---|---|---|---|---|---|
| Current age Mean ± SD | 47 ± 18 | 54 ± 16** | 41 ±16 | 52 ± 16** | 42 ±15 | 50 ± 17** | 47 ±17 | 50 ± 17* | 47 ±19 | 49 ± 16 |
| Age at first SLE ICD-9 code Mean ± SD | 37 ± 17 | 43 ±15** | 33 ± 15 | 43 ±16** | 32 ± 14 | 42 ±17** | 38 ± 17 | 40 ±16* | 38 ± 18 | 40 ±16 |
| Female (%) | 90 | 90 | 87 | 91 | 87 | 91 | 92 | 90 | 88 | 91 |
| Race/ethnicity (%) | ||||||||||
| Caucasian | 45 | 55** | 17 | 83** | 11 | 89** | 27 | 73** | 16 | 84** |
| African American | 61 | 39 | 49 | 51 | 31 | 69 | 40 | 60 | 22 | 78 |
| Hispanic | 76 | 25 | 48 | 52 | 41 | 59 | 52 | 48 | 35 | 65 |
| Asian | 88 | 12 | 17 | 83 | 19 | 81 | 57 | 43 | 37 | 63 |
All antibody status defined as ever positive. The total number may not add up to 1097 SLE subjects due to the following missing data: 74 patients with missing dsDNA, 425 missing RNP, 410 missing Smith, 339 missing SSA, and 352 missing SSB.
p < 0.05, Mann Whitney U for continuous variables or chi-square tests for categorical variables
p < 0.001, Mann Whitney U for continuous variables or chi-square tests for categorical variables
Figure 1. Increased renal disease in dsDNA positive SLE subjects and increased pain and sleep-related codes in dsDNA negative SLE subjects.
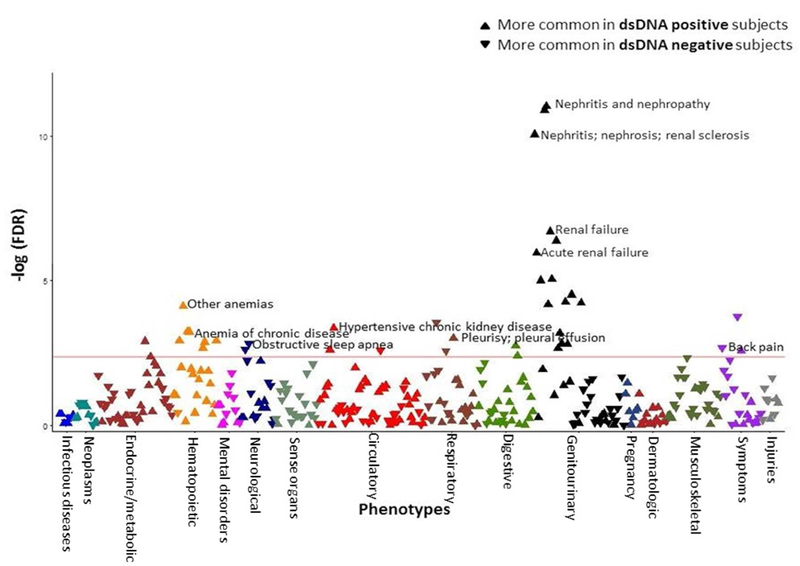
The x axis represents the PheWAS codes that are mapped to ICD-9 codes, organized and color-coded by organ system. The y axis represents the level of significance. Each triangle represents a PheWAS code. dsDNA negative subjects are the reference group. Triangles pointing down represent codes more common in dsDNA negative subjects. Triangles point up represent codes more common in dsDNA positive subjects. The PheWAS was adjusted for age and race/ethnicity, and the horizontal red line represents the false discovery rate (FDR) of 0.05. There were 42 codes that met the FDR of 0.05. dsDNA positive subjects had more codes related to renal disease and SLE disease criteria while dsDNA negative subjects had more codes related to sleep and pain disorders.
Table 2.
Selected codes from the PheWAS of dsDNA positive vs. negative SLE subjects.
| PheWAS codes | Phenotype present (≥ 2 or more instances of the PheWAS code)a | Phenotype absent (0 instances of the PheWAS code)a | Adjusted Odds Ratio for age and race (95% Confidence Interval) | False Discovery Rate pb |
|---|---|---|---|---|
| Codes favoring dsDNA positive subjects | ||||
| Nephritis and nephropathy in diseases classified elsewhere | 162 | 602 | dsDNA positive: 4.60 (2.97 – 7.14) dsDNA negative: 1.00 (ref) | 2.33 × 10−9 |
| Renal failure | 261 | 602 | 2.30 (1.68 – 3.15) | 1.85 × 10−5 |
| Other anemias | 275 | 585 | 1.87 (1.37 – 2.55) | 2.21 × 10−3 |
| End stage renal disease | 77 | 602 | 2.63 (1.51 – 4.58) | 0.01 |
| Pleurisy; pleural effusion | 130 | 739 | 2.00 (1.33 – 3.03) | 0.02 |
| Thrombocytopenia | 94 | 616 | 2.19 (1.36 – 3.53) | 0.02 |
| Codes favoring dsDNA negative subjects | ||||
| Obstructive sleep apnea | 41 | 820 | 0.30 (0.14 – 0.63) | 0.02 |
| Back pain | 196 | 699 | 0.59 (0.42 – 0.82) | 0.03 |
| Sleep disorders | 98 | 820 | 0.50 (0.32 – 0.78) | 0.03 |
| Enthesopathy | 54 | 742 | 0.41 (0.22 – 0.76) | 0.04 |
| Myalgia and myositis unspecified | 243 | 682 | 0.65 (0.48 – 0.88) | 0.05 |
Phenotype present indicates subjects who had the code listed on at least 2 instances vs. phenotype absent indicates subjects who did not have the code or related codes. Subjects with 1 instance of a code are excluded, so the total number of subjects for each PheWAS code does not add up to the 1097 SLE subjects. There are 74 subjects with a missing dsDNA.
Codes listed met the false discovery rate of 0.05.
In contrast, dsDNA negative subjects were more likely to have codes related to pain and sleep disorders including obstructive sleep apnea (OR = 0.30, 95% CI 0.14 – 0.63, FDR p = 0.02), back pain (OR = 0.59, 95% CI 0.42 – 0.82, FDR p = 0.03), and myalgia and myositis unspecified (OR = 0.65, 95% CI 0.48 – 0.88, FDR p = 0.05), which often represents patients diagnosed with fibromyalgia in our EHR16(Figure 1, Table 2).
PheWAS of RNP positive vs. negative subjects
SLE subjects with positive vs. negative RNP autoantibodies are shown in Table 1. Of 1097 subjects, 183 had a positive RNP, 489 negative, and 425 with missing data. RNP positive vs. negative subjects were younger at time of analysis (41 ± 16 vs. 52 ± 16, p < 0.001) and at time of first SLE ICD-9 code (33 ± 15 vs. 43 ± 16, p < 0.001) and more likely to not be Caucasian (p < 0.001). There were no sex differences in positive vs. negative subjects (p = 0.42). In the PheWAS comparing RNP positive vs. negative subjects adjusting for age and race, no codes met the FDR of 0.05. Fifteen codes met the unadjusted p < 0.05 (Supplemental Table 1). The most significant code that was more common in RNP positive vs. negative subjects was chronic kidney disease (OR = 2.51, 95% CI 1.25 – 5.01, p = 0.009). Since renal disease was an unexpected finding, we adjusted for dsDNA status to determine if dsDNA was driving this finding. When the PheWAS was adjusted for dsDNA, age, and race, the most significant code was for inflammatory arthritis (OR = 1.92, 95% CI 1.14 – 3.22, p = 0.01). All codes that were significant in the PheWAS adjusted for age and race remained significant when adjusted for dsDNA (Supplemental Table 2).
Similar to the dsDNA positive vs. negative PheWAS, RNP negative subjects were more likely to have codes for myalgia and myositis, unspecified (OR = 0.58, 95% CI 0.36 – 0.93, p = 0.02) and depression (OR = 0.52, 95% CI 0.32 – 0.99, p = 0.05).
PheWAS of Smith positive vs. negative subjects
SLE subjects with positive vs. negative smith antibodies are shown in Table 1. Of 1097 subjects, 119 had a positive Smith, 568 negative, and 410 with missing data. Smith positive vs. negative subjects were younger at time of analysis (42 ± 15 vs. 50 ± 17, p < 0.001) and at time of first SLE code (32 ± 14 vs. 42 ± 17, p < 0.001). All races/ethnicities were more likely to be Smith negative vs. Smith positive (p < 0.001). There were no sex differences in positive vs. negative subjects (p = 0.32). In the PheWAS comparing Smith positive vs. negative subjects adjusting for age and race, no codes met the FDR of 0.05. Seventeen codes met the unadjusted p < 0.05 (Supplemental Table 3). The most significant code, more common in Smith positive vs. negative subjects, was ascites non-malignant (OR = 4.18, 95% CI, 1.64 – 10.69, p = 0.003). Of these 21 subjects, all had ascites on chart review with 18/21 having nephritis and 8/21 having nephrotic range proteinuria. Other significant codes, more common in Smith positive subjects, were related to nephritis and cardiac codes. When we adjusted for dsDNA in addition to age and race, cardiac codes, specifically multiple codes for arrhythmias, became more significant while codes for nephritis were no longer significant (Supplemental Table 4).
PheWAS of SSA positive vs. negative subjects
SLE subjects with positive SSA vs. negative antibodies are compared in Table 1. Of 1097 subjects, 235 had a positive SSA, 523 negative, and 339 with missing data. SSA positive vs. negative subjects were significantly younger at time of analysis (47 ± 17 vs. 50 ± 17, p = 0.04) and at time of first SLE code (38 ±17 vs. 40 ± 16, p = 0.05). Asians and Hispanics were more likely to be SSA positive compared to African Americans and Caucasians (p < 0.001). There were no sex differences in positive vs. negative subjects (p = 0.66). In the PheWAS comparing SSA positive vs. negative subjects adjusting for age and race, no codes met the FDR of 0.05. Twenty-seven codes met the unadjusted p < 0.05 (Supplemental Table 5). The most significant code, which was more common in SSA negative subjects, was disorders of lipid metabolism (OR = 0.44, 95% CI 0.27 – 0.74, p = 0.002). Similar to the dsDNA and RNP PheWAS, SSA negative subjects were more likely to have pain, mood, and sleep disorder codes including cervicalgia, depression, and obstructive sleep apnea.
PheWAS of SSB positive vs. negative subjects
SLE subjects with positive SSB vs. negative antibodies are compared in Table 1. Of 1097 subjects, 136 had a positive SSB, 609 negative, and 352 with missing data. Of the subjects with a positive SSB, only 19 had a negative SSA. There were no significant differences in age at time of analysis (p = 0.29), age at first SLE code (p = 0.12), and sex (p = 0.09) when comparing SSB positive vs. negative subjects. All race/ethnicity groups were more likely to be SSB negative (p < 0.004). In the PheWAS comparing SSB positive vs. negative subjects adjusting for race, no codes met the FDR of 0.05. Eight codes met the unadjusted p < 0.05 (Supplemental Table 6). The most significant code, more common in SSB negative subjects, was vitamin D deficiency (OR = 0.38, 95% CI 0.18 – 0.78, p = 0.008). Serositis codes including pericarditis and pleurisy; pleural effusion were more common in SSB positive subjects. Similar to the other PheWAS, SSB negative subjects were more likely to have a sleep disorder code.
Chart review
With the findings of increased FM-related code in the antibody negative vs. positive SLE subjects, we performed chart review of antibody negative and positive SLE subjects to ensure the antibody negative subjects were not FM cases mislabeled as SLE. Of the random 50 SLE subjects who were antibody negative, 44 were SLE cases as defined by a rheumatologist with 1 subject having cutaneous lupus and 5 with an uncertain SLE diagnosis. The majority of SLE subjects (59%) had 4 or more SLE ACR criteria5 documented. Of the 44 SLE cases, 12 (27%) had a concomitant diagnosis of FM documented by a rheumatologist with an additional 3 having a possible diagnosis. None of the subjects were originally diagnosed as SLE and then called FM.
Of the random 50 SLE subjects with at least one positive antibody, 48 were SLE cases with 1 subject having mixed connective tissue disease and 1 subject with a questionable SLE diagnosis. Of the 48 SLE cases, 41 (85%) had 4 or more SLE criteria and 12 (25%) had a concomitant FM diagnosis with an additional 3 having a possible diagnosis. None of the 50 subjects were originally diagnosed as SLE and then called FM.
Autoantibody associations with SLE manifestations
In a logistic regression model for nephritis adjusting for sex, race, age, and autoantibodies (dsDNA, RNP, Smith), dsDNA was significantly associated with nephritis (OR = 3.27, 95% CI 2.01 – 5.32, p = 1.98 × 10−6) along with age (OR = 1.02, 95% CI 1.01 – 1.04, p = 0.002) and African American race (OR = 2.32, 95% CI 1.33 – 4.04, p = 0.003) (Figure 2A). In a model for chronic kidney disease (including acute and chronic renal failure, end stage disease, and dialysis) adjusting for sex, race, age, and autoantibodies, African American race (OR = 2.43, 95% CI 1.49 – 3.98, p = 3.8 × 10−4) and dsDNA (OR = 1.96, 95% CI 1.30 – 2.96, p = 0.001) were significantly associated with chronic kidney disease (Figure 2B).
Figure 2. Forest plots of logistic regression models for SLE criteria.
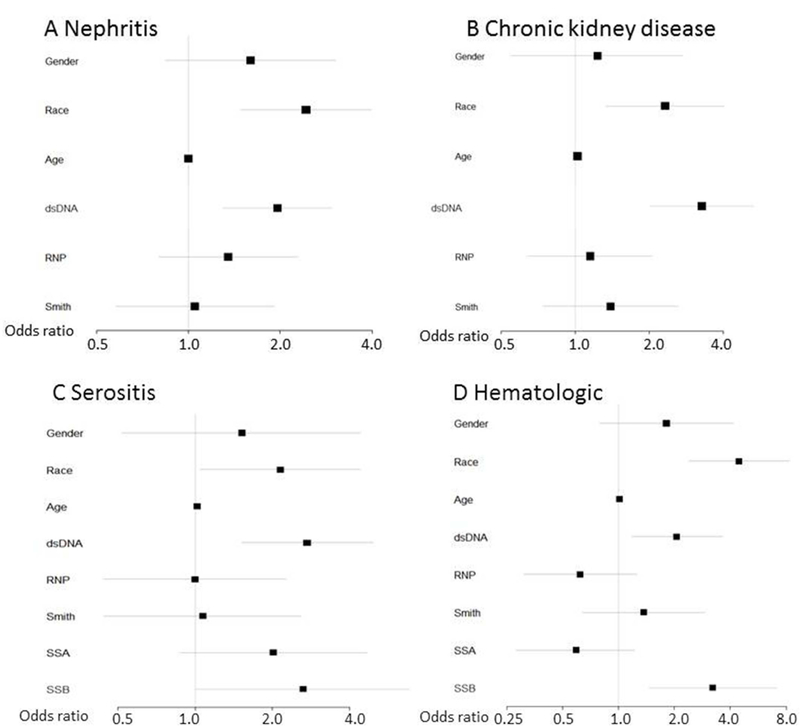
Logistic regression models for nephritis (A), chronic kidney disease (including acute and chronic renal failure, end stage renal disease, and dialysis) (B), serositis (including pericarditis and pleurisy/pleural effusion) (C), and hematologic criteria (thrombocytopenia, pancytopenia) (D) were created with covariates shown on the left. Odds ratios are across the bottom with horizontal lines depicting 95% confidence intervals. The reference group for gender was female and Caucasian for race.
For a model using a code for either pericarditis or pleurisy/pleural effusion, adjusting for sex, race, age, and autoantibodies (dsDNA, RNP, Smith, SSA, SSB), African American race (OR = 4.45, 95% CI 0.79 – 4.16, p = 2.4 × 10−6) , dsDNA (OR = 2.06, 95% CI 1.18 – 3.61, p = 0.01), and SSB (OR = 3.22, 95% CI 1.47 – 7.09, p = 0.004) were all associated with serositis (Figure 2C). To capture hematologic criteria, we combined codes for thrombocytopenia, aplastic anemia, and pancytopenia. Adjusting for sex, race, age and autoantibodies, African American race (OR = 2.15, 95% CI 0.52 – 4.41, p = 0.04), age (OR = 1.02, 95% CI 1.00 – 1.04, p = 0.02), dsDNA (OR = 2.73, 95% CI 1.52 – 4.93, p = 0.001) and SSB (OR = 2.63, 95% CI 1.01 – 6.82, p = 0.05) were associated with hematologic criteria (Figure 2D). For the above clinical associations, we examined if total number of autoantibodies or dsDNA were driving the findings. While controlling for total number of autoantibodies other than dsDNA, dsDNA, sex, race, and age, dsDNA remained associated with the above SLE manifestations (Figure 3). Lastly, we investigated if multiple codes that represent coronary artery disease (CAD) were associated with any of the autoantibodies. After adjusting for age, sex, and race, none of the autoantibodies were associated with CAD (data not shown).
Figure 3. Forest plots of logistic regression models for SLE criteria with total number of autoantibodies.
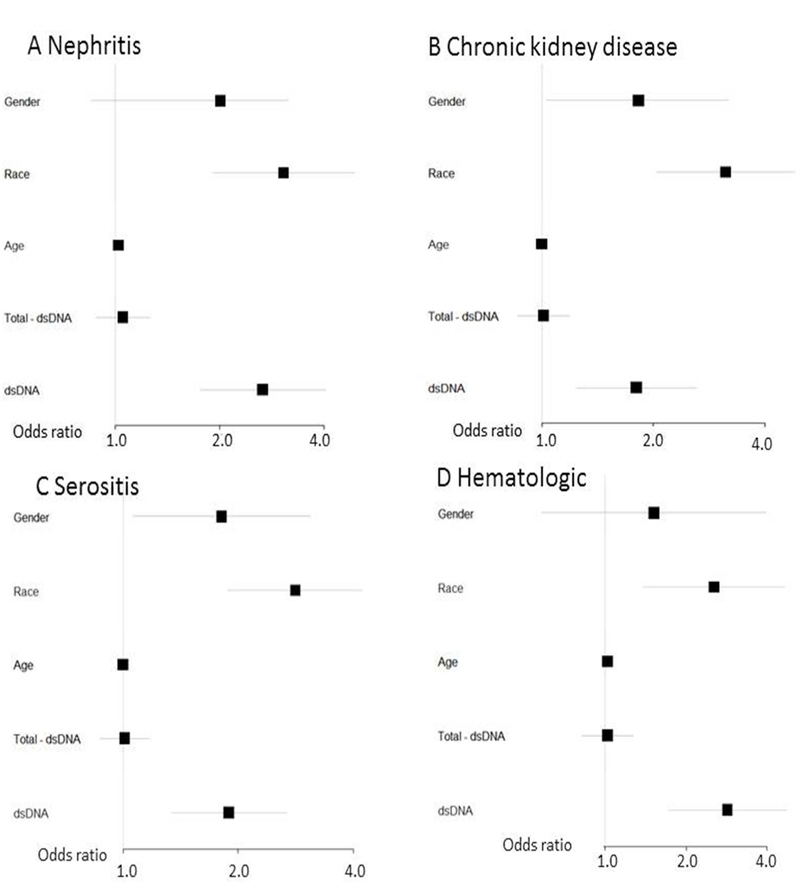
Logistic regression models for nephritis (A), chronic kidney disease (including acute and chronic renal failure, end stage renal disease, and dialysis) (B), serositis (including pericarditis and pleurisy/pleural effusion) (C), and hematologic criteria (thrombocytopenia, pancytopenia) (D) were created with covariates shown on the left. Total – dsDNA denotes total number of autoantibodies (RNP, Smith, SSA, SSB) not including dsDNA. Odds ratios are across the bottom with horizontal lines depicting 95% confidence intervals. The reference group for gender was female and Caucasian for race.
Discussion
This is the first study, to the best of our knowledge, that uses PheWAS to analyze differences in comorbidities based on autoantibody status in SLE. PheWAS comparing SLE patients with and without SLE specific autoantibodies demonstrated that autoantibody negative vs. positive SLE patients were more likely to have codes for pain, mood, and sleep disorders. While controlling for other autoantibodies, dsDNA was the most strongly associated with nephritis, chronic kidney disease, and multiple ACR SLE criteria.5
Ethnic minorities, particularly African Americans and Hispanics, are more likely to have SLE autoantibodies, specifically dsDNA and Smith, compared to Caucasians.23 Our study agrees with these finding as our African Americans and Hispanics with SLE were more likely than Caucasians to be both dsDNA and Smith positive. Studies have also focused on which ACR SLE criteria5 cluster with autoantibodies, notably dsDNA associating with renal disease.1–4 Our findings agree with these studies as dsDNA positive vs. negative subjects were more likely to have codes related to nephritis, thus validating PheWAS methodology in comparing SLE subjects in the EHR. We also uncovered that dsDNA positive subjects were more likely to have chronic kidney disease and end stage renal disease even after adjusting for race/ethnicity.
After evaluating antibody positive vs. negative subjects using PheWAS, we performed logistic regression to assess the impact of multiple autoantibodies on SLE manifestations. We uncovered a hierarchy within SLE autoantibodies where dsDNA, after controlling for the other autoantibodies and race, remained independently associated with nephritis and chronic kidney disease. After controlling for race and other autoantibodies, dsDNA, along with SSB, were significantly associated with serositis and hematologic manifestations. The association of SSB with hematologic manifestations has been shown in two cohort studies.24, 25
Further, dsDNA had a more significant impact than total number of autoantibodies on SLE manifestations. Studies have mainly examined the effect of one antibody on ACR SLE criteria5 in univariate analyses. Our analysis is unique in assessing the effect of multiple antibodies and demographics. Our findings suggest that dsDNA, compared to other autoantibodies, may be the most relevant in assessing a patient’s prognosis for major SLE manifestations.
In the PheWAS comparing antibody negative vs. positive subjects, antibody negative subjects were more likely to have codes for pain, mood, and sleep disorders, including a code that corresponds to fibromyalgia (FM). These findings agree with a PheWAS in rheumatoid arthritis where seronegative compared to seropositive subjects were more likely to have codes for FM.16 FM can coexist with SLE with FM prevalence rates from 22 to 33%.26–29 FM can be “mislabeled” as SLE, particularly in patients with a positive ANA. All the SLE subjects in this study were ANA positive. We hypothesized that autoantibody negative compared to positive SLE subjects may be more likely to have FM codes due to clinical uncertainty in the coding rheumatologist. Specifically, a rheumatologist may have doubt that the patient truly has a diagnosis of SLE without the more specific autoantibodies and then codes for alternative diagnoses such as FM. Alternatively, FM could be the diagnosis in some of the autoantibody negative subjects. We thus performed chart review on subjects who were antibody negative to ensure they had a SLE diagnosis. Of SLE cases who were antibody negative, the majority (59%) had 4 or more ACR SLE criteria5 documented. This rate was higher at 85% in the antibody positive SLE cases. This difference in number of ACR SLE criteria5 could suggest more doubt in the SLE diagnosis in the antibody negative vs. positive subjects which could affect coding patterns. Alternatively, physicians may not frequently review and document ACR SLE criteria at every visit unless this information is updated in a problem list. On chart review, problem lists rarely included ACR SLE criteria, demonstrating a limitation in collecting ACR SLE criteria from EHR data. Notably on chart review, none of the antibody-negative SLE cases were primary FM cases or cases where the rheumatologist initially diagnosed SLE and then changed the diagnosis to FM. These results demonstrate that FM was co-morbid with SLE and not an alternative diagnosis.
In addition to FM, antibody negative SLE subjects had more codes for mood and sleep disorders. Increased rates of depression and sleep disorders, 30 particularly obstructive sleep apnea, 30–32 have been described in SLE with studies reporting sleep disturbances between 56% and 80%.30, 33–36
For the antibody positive subjects, we found the expected renal associations with dsDNA and Smith. We did not find, however, the expected clinical associations of myositis and interstitial lung disease for RNP and sicca and photosensitivity for SSA and SSB. We hypothesize that this lack of association may be related to a lower power to detect these associations. For RNP, SSA, and SSB, we had fewer subjects (136–235) and more missing data (339–425) in contrast to 521 subjects with a positive dsDNA and 74 with missing data. This increased missingness is likely due to an older ANA reflex testing protocol. In addition, the lack of association could be due to limitations in the PheWAS methodology. PheWAS relies on billing codes, which are used differently by providers. For example, one rheumatologist may only code SLE and not code manifestations such as rash as separate codes. In contrast, another provider may use the code for SLE but also code additionally for manifestations. This inconsistency in coding may partly explain why specific manifestations were not found in the RNP, SSA, and SSB subjects. Further, some SLE manifestations such as photosensitivity are not accurately captured by a specific ICD-9 code. Lastly, the clinical associations for these antibodies rely on older, small cohort studies with 100 or fewer subjects.37–41 These studies may not have been adequately powered to identify differences in patients with and without autoantibodies.
While PheWAS confirmed both known associations with autoantibodies and captured novel associations with antibody negative SLE subjects, there are limitations to our study. We used a validated algorithm to identify SLE patients with a PPV of 89%. Although this algorithm has strong test characteristics, there is a possibility that some of our SLE patients may not have a SLE diagnosis. Another limitation of the EHR data is that disease activity and damage measures are not routinely collected in clinical practice in contrast to prospective cohort studies. Thus, we cannot adjust for disease activity or damage in PheWAS. Currently, there are no published EHR-based algorithms that assess for disease activity or damage in autoimmune diseases. Our future directions include developing these algorithms in SLE. Next, Phecodes only capture billing codes at VUMC. Patients can receive care in multiple systems, which may not be documented in the VUMC EHR. These potential missed diagnoses, however, would generally bias us to the null result. Lastly, our study was performed at a single institution’s EHR potentially limiting generalizability of our results to other groups of SLE patients. Using an EHR-based cohort to study SLE, however, captures a wider net of SLE patients in the health system. This methodology allows a unique data capture that may not be feasible in both cohort and administrative database studies.
Using PheWAS, we uncovered a hierarchy of autoantibodies where dsDNA, compared to other autoantibodies, was strongly associated with major organ involvement in SLE. SLE is a heterogeneous disease that poses significant diagnostic and treatment challenges. PheWAS serves as a novel EHR-based tool to better understand disease heterogeneity in SLE by identifying important comorbidities in subgroups of SLE patients.
Supplementary Material
Acknowledgments
Financial Support: Supported by grants NIH/NICHD 5K12HD043483-12 (Barnado), NIH/NIAMS 1 K08AR072757-01 (Barnado), NCRR/NIH UL1 RR024975, NCATS/NIH ULTR000445, NLM R01-LM010685 (Denny)
Footnotes
The Authors declare that there is no conflict of interest.
References
- 1.ter Borg EJ, Horst G, Hummel EJ, Limburg PC, Kallenberg CG. Measurement of increases in anti-double-stranded DNA antibody levels as a predictor of disease exacerbation in systemic lupus erythematosus. A long-term, prospective study. Arthritis Rheum 1990; 33: 634–43. [DOI] [PubMed] [Google Scholar]
- 2.To CH, Petri M. Is antibody clustering predictive of clinical subsets and damage in systemic lupus erythematosus? Arthritis Rheum 2005; 52: 4003–10. [DOI] [PubMed] [Google Scholar]
- 3.Artim-Esen B, Cene E, Sahinkaya Y, et al. Cluster analysis of autoantibodies in 852 patients with systemic lupus erythematosus from a single center. J Rheumatol 2014; 41: 1304–10. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman IE, Peene I, Meheus L, et al. Specific antinuclear antibodies are associated with clinical features in systemic lupus erythematosus. Ann Rheum Dis 2004; 63: 1155–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997; 40: 1725. [DOI] [PubMed] [Google Scholar]
- 6.Pathak J, Kho AN, Denny JC. Electronic health records-driven phenotyping: challenges, recent advances, and perspectives. J Am Med Inform Assoc 2013; 20: e206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritchie MD, Denny JC, Crawford DC, et al. Robust replication of genotype-phenotype associations across multiple diseases in an electronic medical record. Am J Hum Genet 2010; 86: 560–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowton E, Field JR, Wang S, et al. Biobanks and electronic medical records: enabling cost-effective research. Sci Transl Med 2014; 6: 234cm3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denny JC, Bastarache L, Ritchie MD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol 2013; 31: 1102–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denny JC, Crawford DC, Ritchie MD, et al. Variants near FOXE1 are associated with hypothyroidism and other thyroid conditions: using electronic medical records for genome- and phenome-wide studies. Am J Hum Genet 2011; 89: 529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hebbring SJ, Schrodi SJ, Ye Z, Zhou Z, Page D, Brilliant MH. A PheWAS approach in studying HLA-DRB1*1501. Genes Immun 2013; 14: 187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denny JC, Bastarache L, Roden DM. Phenome-Wide Association Studies as a Tool to Advance Precision Medicine. Annu Rev Genomics Hum Genet 2016; 17: 353–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bush WS, Oetjens MT, Crawford DC. Unravelling the human genome-phenome relationship using phenome-wide association studies. Nat Rev Genet 2016; 17: 129–45. [DOI] [PubMed] [Google Scholar]
- 14.Liao KP, Kurreeman F, Li G, et al. Associations of autoantibodies, autoimmune risk alleles, and clinical diagnoses from the electronic medical records in rheumatoid arthritis cases and non-rheumatoid arthritis controls. Arthritis Rheum 2013; 65: 571–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao KP, Sparks JA, Hejblum BP, et al. Phenome-wide association study of autoantibodies to citrullinated and non-citrullinated epitopes in rheumatoid arthritis. Arthritis Rheumatol 2017; 69: 742–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doss J, Mo H, Carroll RJ, Crofford LJ, Denny JC. Phenome-Wide Association Study of Rheumatoid Arthritis Subgroups Identifies Association between Seronegative Disease and Fibromyalgia. Arthritis Rheumatol 2017; 69: 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barnado A, Carroll RJ, Casey C, Wheless L, Denny JC, Crofford LJ. Phenome-wide association studies uncover a novel association of increased atrial fibrillation in males with systemic lupus erythematosus. Arthritis Care Res (Hoboken) 2018. [DOI] [PMC free article] [PubMed]
- 18.Barnado A, Carroll RJ, Casey C, Wheless L, Denny JC, Crofford LJ. Phenome-wide association study identifies marked increased in burden of comorbidities in African Americans with systemic lupus erythematosus. Arthritis Res Ther 2018; 20: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roden DM, Pulley JM, Basford MA, et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther 2008; 84: 362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnado A, Casey C, Carroll RJ, Wheless L, Denny JC, Crofford LJ. Developing Electronic Health Record Algorithms That Accurately Identify Patients With Systemic Lupus Erythematosus. Arthritis Care Res (Hoboken) 2017; 69: 687–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carroll RJ, Bastarache L, Denny JC. R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics 2014; 30: 2375–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dumitrescu L, Ritchie MD, Brown-Gentry K, et al. Assessing the accuracy of observer-reported ancestry in a biorepository linked to electronic medical records. Genet Med 2010; 12: 648–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alarcon GS, Friedman AW, Straaton KV, et al. Systemic lupus erythematosus in three ethnic groups: III. A comparison of characteristics early in the natural history of the LUMINA cohort. LUpus in MInority populations: NAture vs. Nurture. Lupus 1999; 8: 197–209. [DOI] [PubMed] [Google Scholar]
- 24.González-Naranjo LA, Betancur OM, Alarcón GS, et al. Features associated with hematologic abnormalities and their impact in patients with systemic lupus erythematosus: Data from a multiethnic Latin American cohort. Semin Arthritis Rheum 2016; 45: 675–83. [DOI] [PubMed] [Google Scholar]
- 25.Al Arfaj AS, Khalil N. Clinical and immunological manifestations in 624 SLE patients in Saudi Arabia. Lupus 2009; 18: 465–73. [DOI] [PubMed] [Google Scholar]
- 26.Iannuccelli C, Spinelli FR, Guzzo MP, et al. Fatigue and widespread pain in systemic lupus erythematosus and Sjogren’s syndrome: symptoms of the inflammatory disease or associated fibromyalgia? Clin Exp Rheumatol 2012; 30: 117–21. [PubMed] [Google Scholar]
- 27.Morand EF, Miller MH, Whittingham S, Littlejohn GO. Fibromyalgia syndrome and disease activity in systemic lupus erythematosus. Lupus 1994; 3: 187–91. [DOI] [PubMed] [Google Scholar]
- 28.Middleton GD, McFarlin JE, Lipsky PE. The prevalence and clinical impact of fibromyalgia in systemic lupus erythematosus. Arthritis Rheum 1994; 37: 1181–8. [DOI] [PubMed] [Google Scholar]
- 29.Di Franco M, Bazzichi L, Casale R, Sarzi-Puttini P, Atzeni F. Pain in systemic connective tissue diseases. Best Pract Res Clin Rheumatol 2015; 29: 53–62. [DOI] [PubMed] [Google Scholar]
- 30.Vakil M, Park S, Broder A. The complex associations between obstructive sleep apnea and auto-immune disorders: A review. Med Hypotheses 2018; 110: 138–43. [DOI] [PubMed] [Google Scholar]
- 31.Iaboni A, Ibanez D, Gladman DD, Urowitz MB, Moldofsky H. Fatigue in systemic lupus erythematosus: contributions of disordered sleep, sleepiness, and depression. J Rheumatol 2006; 33: 2453–7. [PubMed] [Google Scholar]
- 32.Chung WS, Lin CL, Kao CH. Association of systemic lupus erythematosus and sleep disorders: a nationwide population-based cohort study. Lupus 2016. 25: 382–8. [DOI] [PubMed] [Google Scholar]
- 33.Kasitanon N, Achsavalertsak U, Maneeton B, et al. Associated factors and psychotherapy on sleep disturbances in systemic lupus erythematosus. Lupus 2013; 22: 1353–60. [DOI] [PubMed] [Google Scholar]
- 34.Da Costa D, Bernatsky S, Dritsa M, et al. Determinants of sleep quality in women with systemic lupus erythematosus. Arthritis Rheum 2005; 53: 272–8. [DOI] [PubMed] [Google Scholar]
- 35.Greenwood KM, Lederman L, Lindner HD. Self-reported sleep in systemic lupus erythematosus. Clin Rheumatol 2008; 27: 1147–51. [DOI] [PubMed] [Google Scholar]
- 36.Abad VC, Sarinas PS, Guilleminault C. Sleep and rheumatologic disorders. Sleep Med Rev 2008; 12: 211–28. [DOI] [PubMed] [Google Scholar]
- 37.Naveau B, Dryll A, Peltier AP, Kahn MF, Ryckewaert A. [Evolutive aspects of Sharp’s mixed connective tissue disease. 23 cases (author’s transl)]. Nouv Presse Med 1981; 10: 2731–3. [PubMed] [Google Scholar]
- 38.Benhamou CL, Amor B, Menkes CJ, Delbarre F. [Evolution of connective tissue diseases with anti-ribonucleoprotein antibodies (in 28 patients) (author’s transl)]. Ann Med Interne (Paris) 1982; 133: 72–9. [PubMed] [Google Scholar]
- 39.von Muhlen CA, Tan EM. Autoantibodies in the diagnosis of systemic rheumatic diseases. Semin Arthritis Rheum 1995; 24: 323–58. [DOI] [PubMed] [Google Scholar]
- 40.Yasuma M, Takasaki Y, Matsumoto K, Kodama A, Hashimoto H, Hirose S. Clinical significance of IgG anti-Sm antibodies in patients with systemic lupus erythematosus. J Rheumatol 1990; 17: 469–75. [PubMed] [Google Scholar]
- 41.Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR. Mixed connective tissue disease--an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med 1972; 52: 148–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.