

Abstract
The human gut microbiota encodes β-glucuronidases (GUS) that play key roles in health and disease via the metabolism of glucuronate-containing carbohydrates and drugs. Hundreds of putative bacterial GUS enzymes have been identified by metagenomic analysis of the human gut microbiome, but less than 10% have characterized structures and functions. Here we describe a set of unique gut microbial GUS enzymes that bind flavin mononucleotide (FMN). First, we show using mass spectrometry, isothermal titration calorimetry, and x-ray crystallography that a purified GUS from the gut commensal microbe Faecalibacterium prausnitzii binds to FMN on a surface groove located 30 Å away from the active site. Second, utilizing structural and functional data from this FMN-binding GUS, we analyzed the 279 unique GUS sequences from the human microbiome project database and identified 14 putative FMN-binding GUSs. We characterized four of these hits and solved the structure of two, the GUSs from Ruminococcus gnavus and Roseburia hominis, which confirmed that these are FMN binders. Third, binding and kinetic analysis of the FMN-binding site mutants of these five GUSs show that they utilize a conserved site to bind FMN that is not essential for GUS activity, but can affect KM. Lastly, a comprehensive structural review of the PDB reveals that the FMN-binding site employed by these enzymes is unlike any structurally characterized FMN-binders to date. These findings reveal the first instance of an FMN-binding glycoside hydrolase and suggest a potential link between FMN and carbohydrate metabolism in the human gut microbiota.
Introduction
The human gut microbiome encodes about 5 million genes, outnumbering the human genome by 150-fold (1). Among the millions of genes in the gut microbiota are those that encode carbohydrate active enzymes (CAZymes), which serve key roles in the metabolism of dietary and endogenous polysaccharides in the human gut (2). Microbes utilize CAZymes to scavenge sugars from complex carbohydrates in the gut, and the metabolism of these sugars leads to the generation of short chain fatty acids that have been shown to play key roles in human physiology (3). Thus, an understanding of the structure and function of these microbial enzymes is essential for elucidating their roles in human health and disease.
One group of gut bacterial CAZymes are β-glucuronidases (GUSs). Microbial GUS enzymes are unique among CAZymes because they play roles in the metabolism of both polysaccharides and drug metabolites. For example, GUSs are capable of catalyzing the hydrolysis of GlcA-containing polysaccharides, such as heparin and hyaluronate, as well as small molecule drug glucuronides like SN-38-G, the active metabolite of the anticancer drug irinotecan, and NSAID glucuronides (4–7). Drug glucuronides are generated in the liver by uridine diphosphate glucuronosyl transferases (UGTs) and then secreted into the GI tract where they are processed by bacterial GUSs, which reverse the actions carried out by the host (Fig. SI) (8). Many other glucuronides, primarily glucuronate-containing polysaccharides, are present in the gut, including glycosaminoglycans, such as heparin and hyaluronate, as well dietary, bacterial, and plant polysaccharides like pectin, sphingans, and xylans (9). Given the importance of these diverse molecules in therapeutics, nutrition, and homeostasis of the gut microbiota, it is critical to understand the structure and function of the microbial enzymes that process them.
Utilizing metagenomic data and structural analyses, we recently catalogued 279 unique GUS enzymes from the Human Microbiome Project (HMP) stool sample database (4). Only a few bacterial GUSs from the human gut have a characterized structure and function, and most of these characterized GUSs do not have a clear function in the gut microbiota. Here we characterize the GUS from Faecalibacterium prausnitzii L2-6 and show that it binds flavin mononucleotide (FMN) at a distant surface site. While glycoside hydrolases are among the most well characterized enzymes in biochemistry, this is the first observation of a flavin-bound glycoside hydrolase. Utilizing this structural and functional data, we screened GUSs found in the HMP stool sample database for key FMN-binding residues and identified 14 additional FMN-binding GUSs. We characterized four of these biochemically and determined the crystal structures of two, which confirm a structurally conserved FMN-binding site. We further show by a comprehensive review of the PDB that the GUSs characterized here bind FMN unlike any previously characterized FMN-binding proteins. These data reveal the first association between FMN and a glycoside hydrolase, suggesting a link between FMN and carbohydrate metabolism in the human gut microbiota.
Results
Discovery of an FMN-binding GUS from the Human Gut Microbe F. prausnitzii L2-6
Utilizing structural, functional, and sequence data on characterized bacterial GUS enzymes (10–12), metagenomic analysis of the HMP stool sample database revealed 279 unique GUS enzymes in the human gut microbiome (4). Most of these proteins remain uncharacterized; thus, we initiated an effort to express and examine representative GUSs from the distinct classes discovered. Surprisingly, the gene synthesis, protein expression, and purification of a GUS from the human gut bacterium F. prausnitzii L2-6 (Fp2GUS) yielded a yellow protein product (Fig. 1A). Absorbance scan of purified Fp2GUS displayed a profile characteristic of a flavin-binding protein (Fig. 1A), and LC-MS analysis of purified Fp2GUS revealed the mass for flavin mononucleotide (FMN) (Fig. 1B). While FMN is bound upon expression and purification of Fp2GUS, absorbance scans revealed that the stoichiometry of binding was approximately 0.42:1 (FMN:Fp2GUS), suggesting that vacant FMN-binding sites may be present (Fig. S2). Thus, we utilized isothermal titration calorimetry (ITC) to measure the binding affinity of Fp2GUS for FMN using this partially occupied sample, which revealed a Kd of 1.05 μM (Fig. 1C). Together, these data establish that Fp2GUS is an FMN-binding glycoside hydrolase.
Fig. 1. Discovery of an FMN-binding GUS from the human gut microbe F. prausnitzii L2-6.
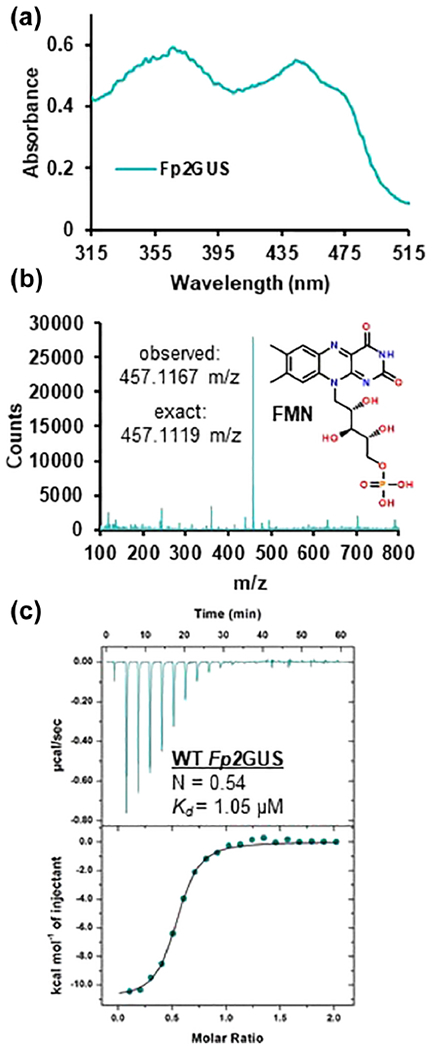
(A) Purified Fp2GUS is yellow and absorbance scan reveals a UV profile characteristic of a flavin-binding protein (B) Mass spectrum of purified Fp2GUS contains mass for FMN (observed mass: 457.1167 m/z, exact mass: 457.1119 m/z). (C) Titration of WT Fp2GUS with FMN monitored by isothermal titration calorimetry (ITC) reveals binding constant of 1.05 ± 0.06 μM and an FMN occupancy for WT Fp2GUS of 46% (N = 0.54).
Crystal Structure of Fp2GUS Reveals an FMN-Binding Site
To further understand the FMN-binding nature of Fp2GUS, we determined its crystal structure to 2.55 Å resolution (Table SI). The Fp2GUS crystal structure revealed an FMN-binding site approximately 30 Å away from the active site, formed by two alpha helices from the core TIM barrel fold (cyan) and two jellyroll-like β-sandwich domains (blue and green) (Fig. 2A). The FMN-binding site is primarily formed by Y154 and F179, which make π-π stacking interactions with the isoalloxazine ring of FMN (Fig. 2B, C). In addition to these two aromatic residues, K356 forms an ionic interaction with the phosphate of FMN, D151 forms a hydrogen bond with the amide nitrogen of the isoalloxazine ring, M161 participates in hydrophobic interactions with the isoalloxazine ring, and Y363 makes an edge-to-face π interaction with the isoalloxazine ring (Fig. 2B, C). Computational generation (DFT theory: wB97x-D 6-31G*) of the electrostatic potential map of FMN reveals that the most electron poor region of the isoalloxazine ring makes π-π stacking interactions with the negative faces of Y154 and F179 (Fig. 2C). In addition to the unprecedented FMN-binding site, Fp2GUS is also a trimer as identified from the crystal structure and size exclusion chromatography with multi-angle light scattering (Fig. S2D and S2E). Together, these structural data reveal that Fp2GUS binds to FMN via numerous π-π interactions at a site located 30 Å from the active site.
Fig. 2. Crystal structure of Fp2GUS reveals an FMN-binding site.
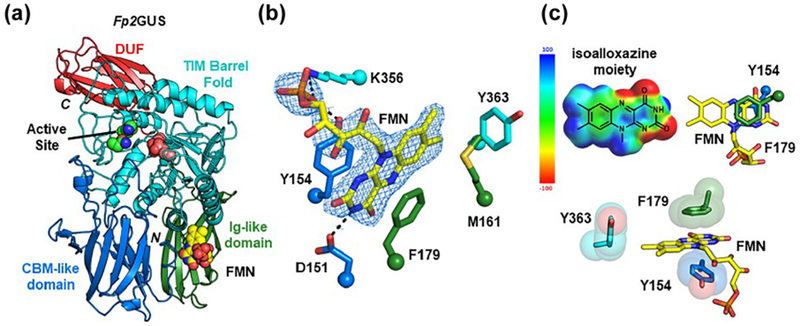
(A) Monomer of Fp2GUS with active site glutamates shown as deep salmon spheres, NxK motif shown as green spheres, and FMN shown as yellow spheres. (B) FMN-binding site of Fp2GUS with 2Fo-Fc density shown at 1.0 σ. (C) Electrostatic potential map (DFT theory: wB97x-D 6-31G*) of FMN highlights an array of π interactions between FMN and Fp2GUS, including π-π stacking between the electron poor region of the isoalloxazine moiety and the electronegative faces of Y154 and F179 (right and bottom), as well as an edge-to-face interaction with Y363.
FMN-Binding Site of Fp2GUS Integral to Protein Stability
To determine the role of the FMN-binding site in the structure and function of Fp2GUS, we performed site-directed mutagenesis of the following residues that contact FMN in the crystal structure: D151, Y154, F179, K356, and Y363 (Fig. 2B, C). Size-exclusion chromatography and SDS-PAGE analysis revealed that each Fp2GUS mutant yielded unstable protein products, with a smaller amount of full-length and soluble protein relative to wild-type (WT), suggesting that the FMN-binding site helps stabilize Fp2GUS (Fig. S3A and S3B). Absorbance scans of the intact peaks of the Fp2GUS mutants revealed either reduced or no flavin-binding profiles, suggesting reduced FMN binding (Fig. S3C). The K356A, D151A, and Y363A mutants of Fp2GUS still bind FMN with similar potency to the WT enzyme, suggesting that while these residues appear to form key contacts with FMN in the crystal structure, their overall contribution to binding FMN is negligible (Fig. S3D). Thus, it would appear that, for the K356A, D151A, and Y363A mutant forms of Fp2GUS, less of the protein was bound to FMN, but, for the fraction that did bind the cofactor, binding affinity remained similar to wild-type. In contrast, Y154A and F179A displayed significantly reduced binding to FMN, along with reduced stability (Fig. S3D). Finally, interestingly, while stability is lost upon mutation of the FMN-binding site, the catalytic activities of the Fp2GUS mutants are not significantly different from the WT protein (Fig. S3E). Together, these data suggest that the FMN-binding site of Fp2GUS plays a key role in the stability of the protein but does not affect catalysis.
Bioinformatic Analysis Identifies 14 Additional FMN-binding GUSs in the Human Gut
Using the structural and mutagenesis data from the Fp2GUS FMN-binding site as a guide, we analyzed GUS sequences from the HMP stool sample database to determine if other GUSs may be FMN-binders. Out of the 278 proteins examined, a total of 14 sequences, in addition to Fp2GUS, met these criteria (Fig. 3A and S4). Generation and analysis of a sequence similarity network (SSN) of GUS enzymes from the HMP stool sample database revealed that the one confirmed and 14 putative FMN-binding GUSs cluster into three clades, all of which contain GUSs exclusively from the previously defined No Loop (NL) structural category, which refers to the absence of an active site adjacent loop shown to play key roles in substrate specificity (Fig. 3A) (4). BLAST and SignalP 4.1 analysis of the putative FMN-binding GUS sequences reveal that they all come from bacteria in the class Clostridiales and do not contain signal peptide sequences, suggesting they are intracellular (13, 14). Further bioinformatic analysis utilizing the Enzyme Function Initiative Genome Neighborhood Tool (EFI-GNT) revealed two distinct genetic neighborhoods surrounding the FMN-binding GUS genes (Fig. 3B) (15). Four of the fifteen putative FMN-binders, including Butyrivibrio fibrisolvens, two strains of Roseburia inulinivorans, and Roseburia hominis are flanked by genes encoding a β-glucosidase (GH1), an α-L-rhamnosidase (GH78), and a xylose isomerase (ISO) (Fig. 3B). The remaining 11 FMN-binding GUS genes are flanked by AraC transcriptional regulators and MFS transporters, proteins that likely sense and transport glucuronate-containing molecules (Fig. 3B). Taken together, a family of FMN-binding GUS enzymes appears to be encoded by the human gut microbiome.
Fig. 3. Bioinformatic analysis predicts 14 additional FMN-binding GUS enzymes from the human gut microbiota.
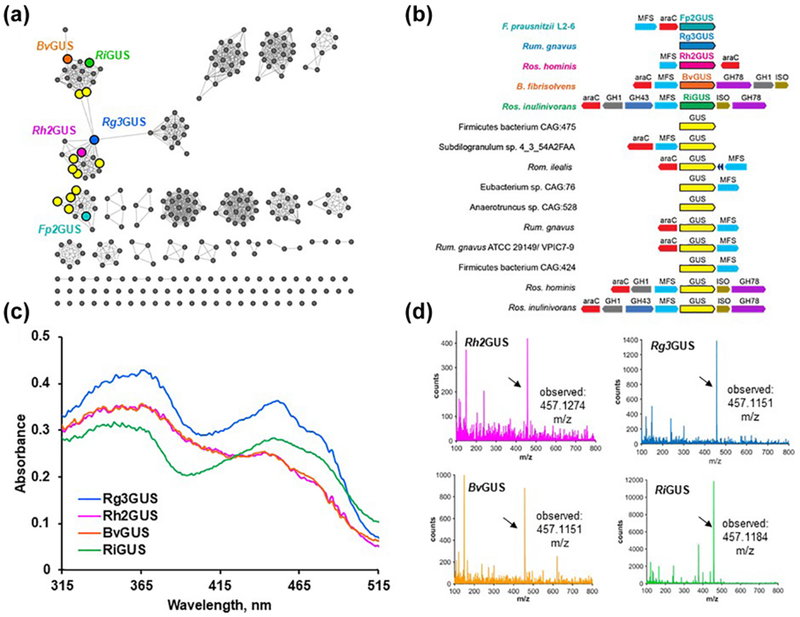
(A) SSN of the HMP GUSome with putative FMN-binders as larger, outlined circles and those that were synthesized and characterized further are labeled. BLAST/e-value/cut off value utilized to generate this SSN is 10−220 and sequences were acquired from the HMP stool sample database. (B) Genome neighborhood diagrams of putative FMN-binding GUSs (C) Absorbance scans of purified Rh2GUS, Rg3GUS, BvGUS, and RiGUS reveal flavin binding profile. (D) Mass spectra of purified Rh2GUS, Rg3GUS, BvGUS, and RiGUS reveal mass for FMN.
Biochemical Characterization Confirms FMN-Binding of Bioinformatic Hits
From the 14 additional putative FMN-binding GUS sequences identified, we selected the following four GUS genes to synthesize, express, and purify: Roseburia inulinivorans (RiGUS), Roseburia hominis (Rh2GUS), B. fibrisolvens (BvGUS), and Ruminococcus gnavus (Rg3GUS). These four enzymes share between 40-46% sequence identity with Fp2GUS (Fig. S5D). Upon expression and purification, all four of the selected sequences yielded yellow protein products with a flavin-binding absorbance profile (Fig. 3C), displayed the mass for FMN by LC-MS (Fig. 3D), and bound FMN with dissociation constants that range from 60 nM (Rg3GUS) to 1.27 μM (BvGUS) (Fig. 4A and Table S2). These results validate our structure-guided bioinformatic identification of FMN-binding GUSs from the gut microbiome.
Fig. 4. Binding studies and structural analysis demonstrate that GUS enzymes utilize a conserved motif to bind FMN.
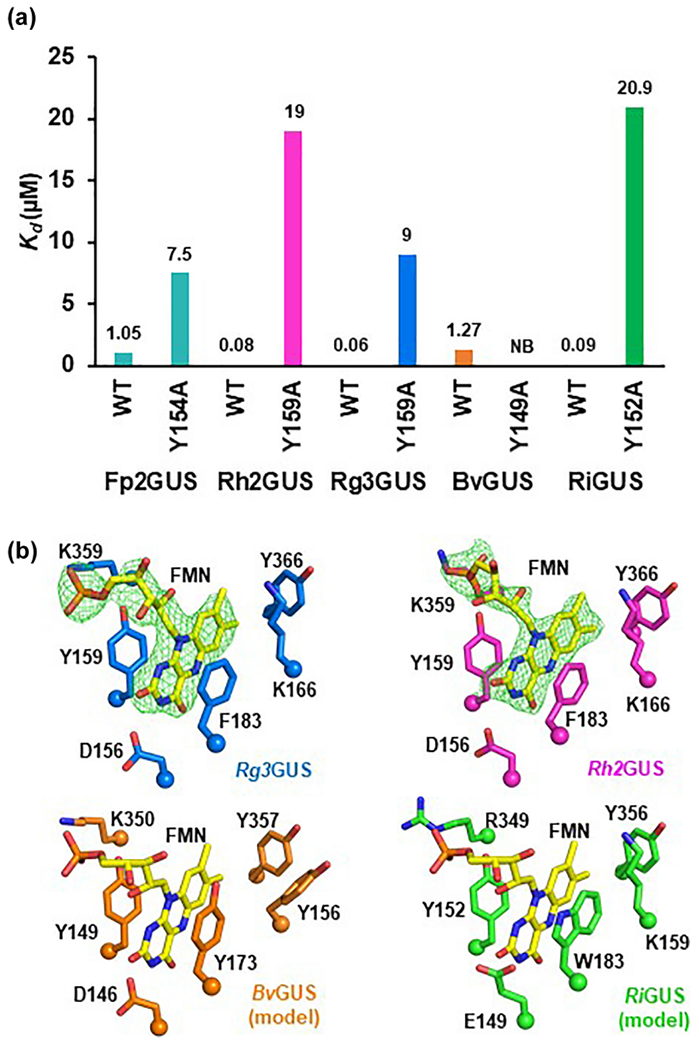
(A) Binding affinities (Kd) of GUS enzymes and FMN-binding site mutants for FMN as determined by isothermal titration calorimetry (B) Structures of FMN-binding sites as determined by crystallography for Rg3GUS (blue) and Rh2GUS (magenta) and by template-guided modeling for BvGUS (orange) and RiGUS (green).
To confirm that the FMN-binding sites of Rh2GUS, Rg3GUS, BvGUS, and RiGUS were similar in molecular nature to that identified in Fp2GUS, we mutated the residue corresponding to Y154 in Fp2GUS in each protein to alanine (Fig. 2B, 4B). Each of these mutants were no longer yellow or displayed significantly reduced yellow color, and no longer displayed a flavin-binding profile (Fig. S5C). Interestingly, unlike the FMN-binding site mutants of Fp2GUS, these mutants yielded stable protein products (Fig. S5A, B). The mutant GUSs Y152A RiGUS, Y159A Rg3GUS, and Y159A Rh2GUS were still capable of binding FMN, although they did so with much weaker affinities (Fig. 4A and Table S2). Together, these data demonstrate that FMN-binding GUSs utilize a conserved motif to bind FMN, and that mutation of FMN binding residues has differential effects on distinct GUS enzymes.
Structural Analysis of Rh2GUS and Rg3GUS Reveals a Conserved FMN-Binding Site
We next determined the crystal structures of Rh2GUS and Rg3GUS to 2.4 and 2.8 Å, respectively. Rh2GUS and Rg3GUS share high structural similarity with Fp2GUS, aligning with RMSD values of 1.6 Å and 1.8 Å over 624 Cα positions, respectively (Fig. S6A). Similar to Fp2GUS, Rh2GUS and Rg3GUS both contain an FMN-binding site that is approximately 30 ′ from the active site (Fig. 4B and S6B). The FMN-binding site of Rh2GUS and Rg3GUS is similar to Fp2GUS, except that a lysine replaces the methionine interacting with the isoalloxazine ring (Fig. 4B). While the quaternary structure of Rg3GUS is unclear based on the crystal structure (Fig. S6C), it appears that Rh2GUS may form either a unique dimer or tetramer based on its crystal structure (Fig. S6D). SEC-MALS analysis supports this conclusion, with predicted dimer-tetramer mixtures for both Rh2GUS and Rg3GUS (Fig. S7). Therefore, we conclude that FMN-binding GUSs in the gut microbiota contain similar FMN-binding sites, similar tertiary structures, but have distinct quaternary structures.
The FMN-Binding Site is Not Required for GUS Activity
To assess the role of the FMN-binding site in the catalytic function of these GUSs, we determined the catalytic properties of the WT and FMN-binding site mutants. Each GUS was able to hydrolyze the fluorescent reporter substrate 4-methylumbelliferyl glucuronide (4-MUG) (Table S3). The WT and FMN-binding mutants displayed nearly identical kcat values (Fig. 5A), but the FMN-binding mutants generally displayed higher KM values (Fig. 5B). For example, the FMN-binding mutants of Rh2GUS, Rg3GUS, and BvGUS all display significantly higher KM values than WT (Fig. 5B and Table S3). In contrast, the KM values of the FMN-binding mutants of Fp2GUS and RiGUS were not significantly different from the WT enzymes. Thus, these data confirm that the FMN-binding site is not necessary for catalytic function, but does increase the KM for some of these FMN-binding GUSs.
Fig. 5. Kinetic studies of FMN-binding GUSs show that FMN-binding site is not required for GUS activity.
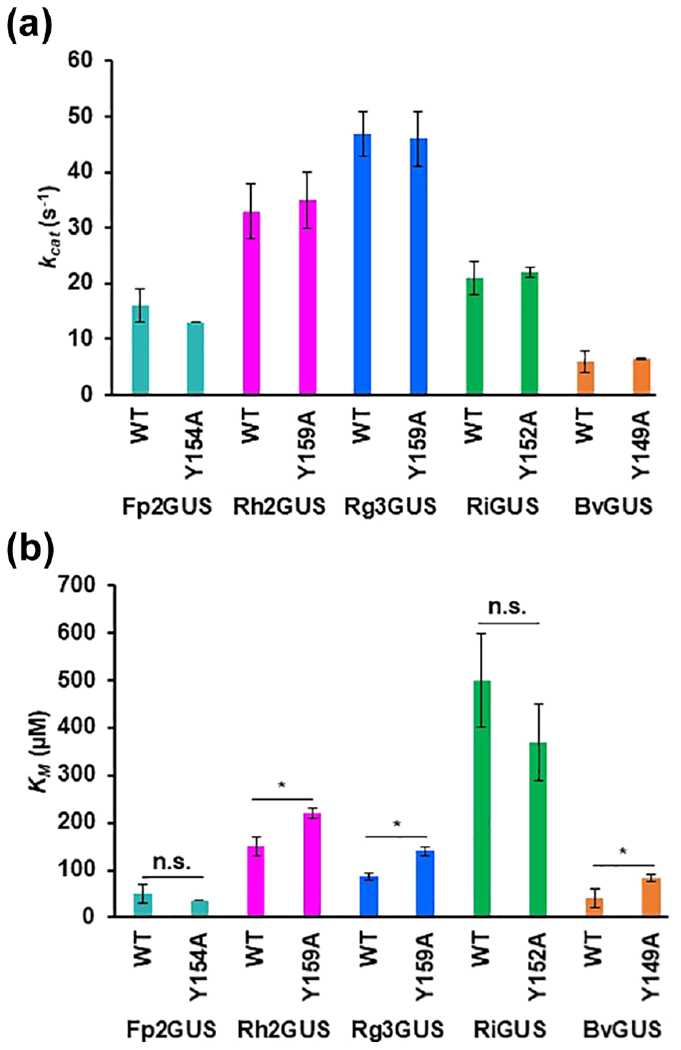
(A) Catalytic turnovers (kcat) of FMN-binding GUSs and their respective FMN-binding site mutants. (B) Michaelis constants (KM) of FMN-binding GUSs and their respective FMN-binding site mutants.
The GUS FMN-binding Site is Unique Among Characterized FMN-binding Proteins
Because the FMN-binding site does not affect catalysis, we performed a comprehensive search of the PDB in an attempt to deduce a function for this site by looking for similar structures. We examined the PDB for FMN-bound structures, which identified 1,056 deposited structures, with 438 non-redundant entries. We then visually inspected these 438 non-redundant structures in PyMOL to determine if any other FMN-binding proteins bind flavin in the same manner as the GUSs characterized in this work. In agreement with previous analyses, the most common fold we encountered was the TIM-barrel fold, and we use Old Yellow Enzyme (PDB: 1OYB) as an example of how FMN binds at the β-barrel core of the fold (Fig. 6). Interestingly, while the FMN-binding GUS enzymes characterized here are also TIM-barrel-containing proteins, they bind FMN on the exterior of this fold, not the interior, and two adjacent β-sandwich domains contribute to the FMN-binding site as well (Fig. 2A and 6). The second most common FMN-binding fold is the flavodoxin fold, named after the protein flavodoxin (PDB: 1FLD), which binds FMN at the edge of a β-sheet flanked on both sides by α-helices (Fig. 6). In addition to the TIM-barrel fold and flavodoxin fold, we identified 18 other structurally distinct FMN-binders out of the 438 non-redundant structures (Fig. S8). These other folds sample a wide variety of structural motifs to bind FMN, but none bind FMN outside the TIM barrel core as observed for the GUS enzymes characterized here (Fig. S8). Thus, the FMN-binding site in gut microbial GUS enzymes is unique.
Fig. 6. Structural comparison of FMN-binding proteins reveals that FMN-binding GUSs identified here bind FMN in a unique manner.
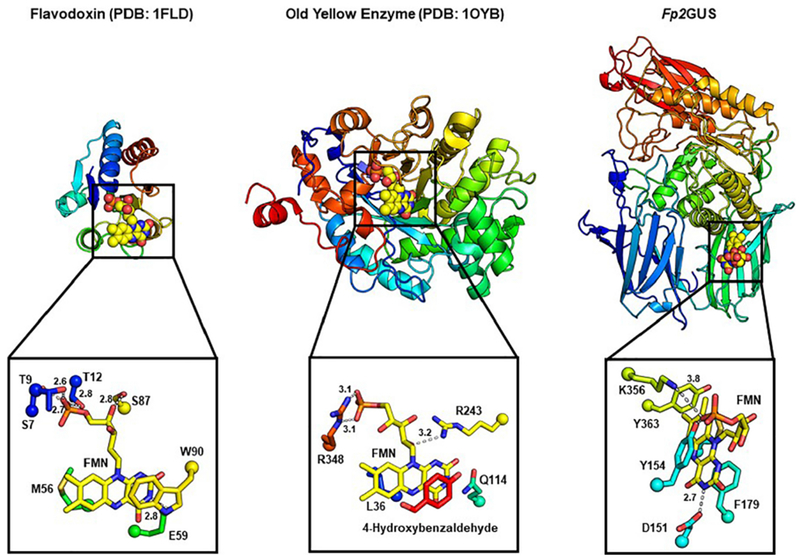
(A) Structure of flavodoxin (PDB: 1FLD) (B) Old Yellow Enzyme (PDB: 1OYB), and (C) Fp2GUS with zoom-in of their respective FMN-binding sites.
As an additional, distinct screen to confirm the novelty of this FMN-binding site, we performed a PDBeFold search to find similar structures to the GUS enzymes characterized here. The top hits were all previously characterized GUS enzymes, with the most similar being BfGUS (PDB: 3CMG) and BuGUS-3 (PDB: 6D1P). Inspection of these structures revealed the common GH2 GUS fold, but the absence of the key FMN-binding residues identified in the GUSs characterized here (Fig. S9B). Taken together, this structural analysis demonstrates that the GUSs characterized here are unique among FMN-binding proteins characterized to date.
Discussion
Here we structurally and functionally characterize a family of FMN-binding GUS enzymes. Characterization of the GUS from the human gut bacterium F. prausnitzii L2-6 revealed a novel FMN-binding glycoside hydrolase and its crystal structure guided the search for other FMN-binding GUS enzymes in the HMP stool sample database. We identified 14 more unique FMN-binders, four of which were characterized and confirmed to bind FMN. Our characterization of the WT and FMN-binding site mutants of these GUSs suggest that FMN plays a key role in the stability of Fp2GUS, but not for the other FMN-binders (Fig. S3A, S3B, S5A, and S5B). Furthermore, we show that mutating the FMN-binding site does not significantly affect enzyme activity, suggesting that the FMN-binding site is not essential for GUS function (Fig. 5). Together, these data reveal the first FMN-binding glycoside hydrolase, show that there is a family of FMN-binding glycoside hydrolases in the gut microbiota, the FMN-binding site is not required for the function of the enzyme, and reveal a novel FMN-binding site among FMN-binding proteins structurally characterized to date.
While previously characterized bacterial GUSs share similar tertiary structures to the FMN-binding GUSs discovered here, none contain the residues necessary to form the FMN-binding site (Fig. S9B). Fp2GUS shares RMSDs of 3.4 Å (across 528 Cα positions), 2.1 Å (624 Cα), and 2.7 Å (632 Cα) with E. coll GUS (EcGUS, PDB: 3LPF), B. fragilis GUS (BfGUS, PDB: 3CMG), and B. uniformis GUS 2 (BuGUS-2, PDB:5UJ6), respectively. The C-terminal domain of Fp2GUS, Rh2GUS, and Rg3GUS was disordered in the crystal structures elucidated here, with approximately 100 residues missing from each individual chain. This unresolved region may form a carbohydrate binding module (CBM), a structural feature seen at the C-terminus of two GUSs previously characterized from B. uniformis (4, 16). The active site of the FMN-binding GUSs characterized here is similar to previously characterized GUS enzymes, containing both the two conserved catalytic glutamates and the NxK motif (Fig. S9C) (4).
The FMN-binding GUSs characterized here add to the diverse quaternary structures discovered recently among gut bacterial GUSs. Five distinct oligomerization states have been previously observed, including three distinct tetramers and two unique dimers (Fig. S10). Here we uncover two additional tetrameric states, a trimer (Fp2GUS) and an apparent dimer-tetramer mix (Rh2GUS) (Fig. S2D and S6D). The oligomeric states of bacterial GUSs have previously been shown to play key roles in GUS function. For example, the tetrameric Loop 1 (L1) GUS enzymes (E. coli GUS, PDB: 3LPF, Fig. S9A) have small hydrophobic pockets around their active sites due to overlapping loops of adjacent protomers, limiting their substrate scope to lipophilic, small-molecule glucuronides (4). Most other bacterial GUSs have open, solvent-exposed active sites, allowing access to large, polar substrates like glucuronate-containing polysaccharides (4, 16). Both the trimer of Fp2GUS and the dimer-tetramer mix of Rh2GUS fit within this latter group of GUSs with solvent exposed active sites, suggesting that their preferred substrates are likely glucuronate-containing polysaccharides.
The cognate substrates of some of these FMN-binding GUSs may be extracellular polysaccharides (EPS). The EFI-GNT analysis of the GUS genes from B. fibirisolvens, two strains of R. inulinivorans, and R. hominis revealed nearby CAZymes, including a β-glucosidase (GH1), an α-l-rhamnosidase (GH78), and a xylose isomerase (ISO) (Fig. 3B). Together, these genes may coordinate the degradation of an EPS within the group known as sphingans. The repeating unit of sphingans contains α-linked rhamnose, β-linked glucose, and β-linked glucuronate moieties, all of which could be processed by the genes present in these genetic loci (Fig. S11) (17). Indeed, previous studies have associated orthologs of these enzymes with the catabolism of sphingans and related exopolysaccharides (18, 19). Detailed future studies with relevant polysaccharide substrates will be required to identify the cognate substrates of these FMN-binding GUSs.
The discovery of FMN-binding GUSs suggests a link between polysaccharide metabolism and FMN in the human gut. Interestingly, the pentose and glucuronate interconversion pathway contains a biosynthetic route that shunts glucuronate into the riboflavin metabolism pathway, responsible for the generation of FMN and FAD in bacteria (Fig. S12). A myriad of enzymes is needed to transform glucuronate to d-ribulose-5-phosphate, which can generate FMN in combination with GTP (Fig. S12). If glucuronate released by the FMN-binding GUSs is converted into FMN, it could serve as a positive feedback loop for GUS activity. However, this seems unlikely as we observed that FMN has only a small impact on the activity of these enzymes.
We note in our three structures of FMN-binding gut microbial GUS enzymes that a channel exists on the surface of each protein that provides access to N5 of the bound flavin, which is a key site for redox chemistry (Fig. S13). It is possible that a small molecule could access this site to utilize FMN for oxidation-reduction reactions. While this is speculative, two features of the FMN-bound GUS enzymes make this potentially accurate. First, the non-FMN binding GUS proteins have residues that block this channel, while the channel remains open in the FMN-bound GUS enzymes. Second, while many GUS enzymes have signal sequences and may be exported to the periplasm, all the FMN-binding GUS enzymes detected to date lack a signal sequence and thus are expected to remain intracellularly localized, a place where redox chemistry can be better controlled and utilized by the cell. Future work will be required to determine whether this channel provides functionally relevant access to the bound FMN of these particular GUS enzymes.
Structural and functional analysis reported here leaves the role of FMN in GUS function unclear. While FMN was a key factor for stability in Fp2GUS, it did not have a major effect on the activity or stability of the other four FMN-binding GUSs characterized. Based on structural comparison with previously characterized FMN-binding proteins, it also does not appear to be similar to any oxidoreductases or electron transport-like proteins characterized to date. Future studies will be necessary to unravel the role this cofactor plays in GUS function, whether it is present for stability like that observed for Fp2GUS, or impacts catalytic efficiency like that observed for some of the other FMN-binding GUSs characterized. Another possibility is that FMN may perform a completely different function from GUS activity. For example, these enzymes may have binding partners that can utilize FMN in ways that are not clear from studying these GUSs in an isolated system.
Conclusion
Here we characterize a unique set of FMN-binding GUS enzymes from the human gut microbiome. We determined the crystal structure of a GUS from F. prausnitzii and show that it binds FMN at a surface site approximately 30 Å from the active site. Using these structural data, we screened the HMP stool sample metagenomic database and identified 14 additional putative FMN-binders. We characterized four of these putative FMN-binding GUS enzymes in vitro and confirmed that they are bona fide FMN-binders, with binding affinities as low as 60 nanomolar. Site-directed mutagenesis of all five FMN-binders, and crystals structures of the FMN-binding GUS from R. hominis and R. gnavus, reveal a conserved FMN-binding site. Kinetic studies of the FMN-binding mutants suggest that the FMN-binding site is not necessary for GUS function, but mutations to this site can impact the KM. Lastly, a structural bioinformatic search demonstrates that no other characterized FMN-binders interact with FMN like that observed with these FMN-binding glycoside hydrolases.
Methods
Gene synthesis, expression, and purification of FMN-binding GUSs
Genes for Fp2GUS, Rh2GUS, Rg3GUS, BvGUS, and RiGUS were synthesized by BioBasic, incorporated into a pLIC-His vector via ligation independent cloning, and resultant plasmids were transformed into BL21-G E. coli cells. Glycerol stocks were made from overnights and snap frozen and stored at −80 °C. Verification of successful transformation and sample integrity were determined by DNA sequencing.
Cultures of 100 mL LB with ampicillin were inoculated with glycerol stock and incubated overnight at 37 °C with shaking at 225 rpm. For protein expression, 50 mL of the overnight, approximately 40 μL Antifoam 204, and 750 μL of 2000× ampicillin were added to 1.5 L LB in a 2.5 L Erlenmeyer flask and incubated at 37 °C at 225 rpm. At an OD was approximately 0.6, the temperature was reduced to 18 °C and induced with IPTG (100 mM) at an OD of approximately 0.8 and incubated overnight with shaking at 225 rpm. Cultures were spun down in a Sorvall Instruments RC-3B centrifuge at 4500 × g for 25 minutes in 1 L round, flat bottom plastic bottles. Cultures were resuspended in LB and transferred to a 50 mL falcon tube and spun down in a ThermoScientific Sorvall ST 40R centrifuge for 15 minutes at 5000 × g. Supernatant was discarded and proteins were stored at −80 °C until purification.
Cell pellets were lysed in 30 mL Nickel A buffer (20 mM KH2PO4, 500 mM NaCl, 50 mM imidazole, pH 7.4) with DNase, lysozyme, and a Roche EDTA-free protease inhibitor tablet. The resultant cell slurry was sonicated on a Fischer Scientific Sonic dismembrator model 500 twice with 1 s pulses for 1.5 minutes. The resultant lysate was subsequently spun down on a Beckman Coulter J2-HC centrifuge for 1 hour at 17000 rpm. The supernatant was subject to filtration with a 0.22 μm filter prior to purification.
Protein was first purified with an Aktaxpress FPLC (Amersham Bioscience) via a Ni NTA column. Protein was eluted in one step using Nickel B buffer (20 mM KH2PO4, 500 mM NaCl, 500 mM imidazole, pH 7.4). Fractions were collected and concentrated with a 50K centrifilter at 3000 × g for 15 minutes if necessary before size exclusion chromatography. The eluent was then subject to size exclusion chromatography on a HiLoadTM 16/60 Superdex 200 gel filtration column. Size exclusion buffer was utilized for elution (20 mM HEPES, 50 mM NaCl, pH 7.4). Fractions were collected and an SDS-PAGE gel was performed to assess purity and stability of the enzyme. Protein concentration was determined on a ND-1000 spectrophotometer and then snap frozen in liquid nitrogen and stored at −80 °C.
Side-directed mutagenesis of FMN-binding GUSs
All mutants were created via site-directed mutagenesis. Mutagenesis primers were synthesized by Integrated DNA technologies. Mutant plasmids were sequenced by Eton Bioscience to confirm successful mutagenesis.
Absorbance scans of WT and mutant GUSs and stoichiometry determination
Absorbance scans of WT and mutant GUSs were determined in a BMG labtech PHERAstar plate reader. All proteins were analyzed at 50 μM in 96-well Costar half area, clear, flat bottom UV-transparent plates. Resultant absorbance profiles were plotted in Microsoft Excel 2013.
Liquid chromatography-mass spectrometry
Protein samples were diluted in sizing buffer to approximately 10 μM and applied to a 0.22 um filter prior to analysis. Separation was carried out on a Viva C4 5μm 150×2.1 mm column. Solvent A was 0.1% formic acid in water and solvent B was 0.1% formic acid in acetonitrile. FMN-binding proteins were eluted using a linear gradient of 5% solvent B to 60% B over 20 minutes, held for one minute, and then an additional linear gradient from 60% to 95% B for 17 minutes. Samples were analyzed using an Agilent Technologies 6520 Accurate-Mass Q-TOF LC-MS instrument in positive-ion mode and the resultant data were analyzed in MassHunter Qualitative Analysis B0.06.00 software.
Isothermal titration calorimetry to determine FMN binding affinity
ITC experiments were performed on a MicroCal AutoITC-200. All experiments were performed at 25 °C with 120 s intervals, reference heat of 7 kcals/s, and 20 injections total. Protein and ligand were prepared in size exclusion buffer and all ITC data were corrected with a control experiment of ligand dilution into buffer. Protein and ligand concentrations were varied depending on the amount of FMN present as determined from absorbance scans.
Crystal formation, preparation, and data collection of FMN-binding GUSs
Fp2GUS was crystallized via the sitting drop method in Hampton Research 3-well Crystallization Plates (Swissci) at 11.1 mg/ml in 0.2 M magnesium chloride, 10% w/v PEG 3000, and 0.1 M sodium cacodylate. Incubation at 20 °C resulted in crystal formation after 11 days. Rg3GUS was crystallized by the hanging drop vapor diffusion method at 20.4 mg/mL in 0.13 M magnesium acetate and 10% PEG 8000. Incubation at room temperature yielded crystals of Rg3GUS after 2-3 days. Rh2GUS was crystallized by the hanging drop vapor diffusion method at 14.9 mg/ml in 0.2 M calcium acetate and 50% PEG 8000 at room temperature after approximately 30 days. All crystals were looped and cryoprotected in their crystallant plus 20% glycerol before storing in liquid nitrogen.
X-ray diffraction experiments were performed at GM/CA ID-D and ID-B beam sources. Standard collection methods were followed and resultant data were reduced in XDS and scaled in aimless. The structure of Fp2GUS was solved via molecular replacement in Phenix using the single component MR-Phaser program with B. fragilis GUS (3CMG) as the search model. Rg3GUS and Rh2GUS were solved by molecular replacement using the Fp2GUS structure as the search model. Final coordinates were deposited in the RCSB PDB with PDB codes 6MVF, 6MVG, and 6MVH for Fp2GUS, Rg3GUS, and Rh2GUS, respectively.
Bioinformatic analysis of HMP stool sample database for identification of FMN-binding GUSs
We utilized the previously generated database of GUS sequences from the HMP stool sample database to screen for putative FMN-binding GUSs (4). To identify other FMN-binding GUSs, we first performed pairwise sequence alignments of each GUS sequence against Fp2GUS and rejected those with less than 25% sequence identity. The remaining sequences were screened to with a length requirement of 700-800 residues and contained the FMN-binding site residues (or similar residues) identified in Fp2GUS: D, E, or other small residue at position 151, Y, F or W at position 154, F, Y, or W at position 179, K or R at position 356, and Y, F, or W at position 363 (Fig. S3).
In vitro kinetic assay for kcat and KM determination of FMN-binding GUSs
To assess the activities of WT and mutant GUSs, we measured their ability to hydrolyze the fluorescent reporter substrate 4-methylumbelliferyl glucuronide (4-MUG). Reactions were performed in black Costar 96-well plates with a flat, clear bottom and reaction volumes were as follows: 5 μL water, 5 μL buffer (25 mM HEPES, 25 mM NaCl pH 6.5 or 25 mM NaCH3CO2− and 25 mM NaCl pH 5.5), 5 μL FMN (25 μM for WT protein) or water (FMN-binding mutants), 5 μL GUS, and 30 μL 4-MUG (varying concentration). Reactions were initiated by addition of substrate and reactions were continuously monitored with excitation at 350 nm and emission at 450 nm in a BMG labtech PHERAstar plate reader. Initial velocities from the resultant data were fit by linear regression with a custom MATLAB program and kcat, KM, and kcat/KM were determined in SigmaPlot 13.0.
Supplementary Material
Supplementary Fig. 1. Overview of the potential roles of bacterial GUS in the gut microbiome.
Supplementary Fig. 2. (A) Absorbance scans of free FMN at varying concentrations. (B) Standard curve of FMN at absorbance of 365 nm. (C) Apparent stoichiometries of FMN:GUS based on protein concentrations and FMN concentrations as determined by standard curve analysis. (D) Trimer of Fp2GUS with active site and FMN shown as spheres, each chain rainbow colored from N-term (blue) to C-term (red). (E) Size-exclusion chromatography multi angle light scattering analysis of Fp2GUS confirms trimeric state in solution.
Supplementary Fig. 3. Characterization of WT and FMN-binding site mutants of Fp2GUS (A) FPLC traces of Fp2GUS FMN-binding mutants. (B) SDS-PAGE of WT and FMN-binding mutants of Fp2GUS (C) Absorbance scans of purified Fp2GUS FMN-binding mutants. (D) ITC binding parameters of WT and FMN-binding site mutants of Fp2GUS for FMN. (E) Kinetic parameters of WT and FMN-binding site mutants of Fp2GUS.
Supplementary Fig. 4. Bioinformatic search of the HMP stool sample database reveals 15 putative FMN-binding GUS enzymes.
Supplementary Fig. 5. Biochemical characterization of WT and FMN-binding site mutants of Rg3GUS, Rh2GUS, BvGUS, and RiGUS. (A) SEC traces for FMN-binding site mutants Y159A Rh2GUS, Y159A Rg3GUS, Y152A RiGUS, and Y149A BvGUS. (B) SDS-PAGE gel of WT and FMN-binding site mutants of Rh2GUS, Rg3GUS, RiGUS, and BvGUS. (C) Absorbance scans of WT and FMN-binding site mutants of Rh2GUS, Rg3GUS, RiGUS, and BvGUS. (D) Sequence identity matrix of the five characterized FMN-binding site mutants.
Supplementary Fig. 6. Structural comparison of FMN-binding GUSs and quaternary structure analysis. (A) Structural overlay of Fp2GUS, Rh2GUS, and Rg3GUS. (B) Tertiary structure analysis of Rh2GUS and Rg3GUS with catalytic residues and FMN shown as spheres. (C) Analysis of Rg3GUS asymmetric unit does not reveal obvious dimer or tetramer. (D) Putative dimer and tetrameric states of Rh2GUS.
Supplementary Fig. 7. SEC-MALS results of FMN-binding GUS enzymes.
Supplementary Fig. 8. Distinct FMN-binding proteins identified in the Protein Data Bank.
Supplementary Fig. 9. Structural comparison of FMN-binding GUSs with previously characterized GUS enzymes.
Supplementary Fig. 10. Structural comparison of quaternary structures of microbial GUS enzymes.
Supplementary Fig. 11. Chemical structure of a sphingan repeating unit, a potential substrate of the FMN-binding GUS enzymes.
Supplementary Fig. 12. Possible metabolic pathway from glucuronate to FMN.
Supplementary Fig. 13. Hypothetical substrate binding for redox chemistry at FMN-binding site.
Research highlights:
-
→
Discovery of an FMN-binding beta-glucuronidase from Faecalibacterium prausnitzii
-
→
Crystal structure of FMN-binding beta-glucuronidase reveals an FMN-binding site 30 Å away from the active site
-
→
Mutagenesis of the FMN-binding site suggests that it is not essential for catalytic function
-
→
The FMN-binding beta-glucuronidases bind FMN unlike any previously characterized FMN-binders
Acknowledgements
We would like to thank Marcey Waters for help generating the electrostatic potential map of FMN and the National Institutes of Health for funding this research (CA098468 and CA207416).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers
Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers 6MVF, 6MVG, and 6MVH for Fp2GUS, Rg3GUS, and Rh2GUS, respectively.
References
- 1.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Bork P, Ehrlich SD, Wang J, Antolin M, Artiguenave F, Blottiere H, Borruel N, Bruls T, Casellas F, Chervaux C, Cultrone A, Delorme C, Denariaz G, Dervyn R, Forte M, Friss C, Van De Guchte M, Guedon E, Haimet F, Jamet A, Juste C, Kaci G, Kleerebezem M, Knol J, Kristensen M, Layec S, Le Roux K, Leclerc M, Maguin E, Melo Minardi R, Oozeer R, Rescigno M, Sanchez N, Tims S, Torrejon T, Varela E, De Vos W, Winogradsky Y, and Zoetendal E (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 10.1038/nature08821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, and Henrissat B (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud D-J, and Bakker BM (2013) The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res 10.1194/jlr.R036012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pollet RM, D’Agostino EH, Walton WG, Xu Y, Little MS, Biernat KA, Pellock SJ, Patterson LM, Creekmore BC, Isenberg HN, Bahethi RR, Bhatt AP, Liu J, Gharaibeh RZ, and Redinbo MR (2017) An Atlas of β-Glucuronidases in the Human Intestinal Microbiome. Structure. 25, 967–977. e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pellock SJ, Walton WG, Biernat KA, Torres-Rivera D, Creekmore BC, Xu Y, Liu J, Tripathy A, Stewart LJ, and Redinbo MR (2018) Three structurally and functionally distinct β-glucuronidases from the human gut microbe Bacteroides uniformis. J. Biol. Chem. 10.1074/jbc.RA118.005414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saitta KS, Zhang C, Lee KK, Fujimoto K, Redinbo MR, and Boelsterli UA (2014) Bacterial β-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: Mode of action and pharmacokinetics. Xenobiotica. 10.3109/00498254.2013.811314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boelsterli UA, Redinbo MR, and Saitta KS (2013) Multiple NSAID-induced hits injure the small intestine: Underlying mechanisms and novel strategies. Toxicol. Sci. 131, 654–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dutton GJ (1966) Glucuronic Acid, free and combined, biochemistry, pharmacology, and medicine, Academic Press, New York [Google Scholar]
- 9.Porter NT, and Martens EC (2017) The Critical Roles of Polysaccharides in Gut Microbial Ecology and Physiology. Annu. Rev. Microbiol 10.1146/annurev-micro-102215-095316 [DOI] [PubMed] [Google Scholar]
- 10.Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh L-A, Mani S, and Redinbo MR (2010) Alleviating Cancer Drug Toxicity by Inhibiting a Bacterial Enzyme. Science (80-. ). 330, 831–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts AB, Wallace BD, Venkatesh MK, Mani S, and Redinbo MR (2013) Molecular insights into microbial β-glucuronidase inhibition to abrogate CPT-11 toxicity. Mol. Pharmacol. 84, 208–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wallace BD, Roberts AB, Pollet RM, Ingle JD, Biernat KA, Pellock SJ, Venkatesh MK, Guthrie L, O’Neal SK, Robinson SJ, Dollinger M, Figueroa E, McShane SR, Cohen RD, Jin J, Frye SV, Zamboni WC, Pepe-Ranney C, Mani S, Kelly L, and Redinbo MR (2015) Structure and Inhibition of Microbiome β-Glucuronidases Essential to the Alleviation of Cancer Drug Toxicity. Chem. Biol. 22, 1238–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990) Basic local alignment search tool. J. Mol. Biol 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 14.Petersen TN, Brunak S, von Heijne G, and Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 15.Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, and Whalen KL (2015) Enzyme function initiative-enzyme similarity tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta - Proteins Proteomics. 10.1016/j.bbapap.2015.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pellock SJ, Walton WG, Biernat KA, Torres-Rivera D, Creekmore BC, Xu Y, Liu J, Tripathy A, Stewart LJ, and Redinbo MR (2018) Three structurally and functionally distinct β-glucuronidases from the human gut microbe Bacteroides uniformis. J. Biol. Chem. 10.1074/jbc.RA118.005414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmid J, Sperl N, and Sieber V (2014) A comparison of genes involved in sphingan biosynthesis brought up to date. Appl. Microbiol. Biotechnol. 10.1007/s00253-014-5940-z [DOI] [PubMed] [Google Scholar]
- 18.Pham PL, Dupont I, Roy D, Lapointe G, and Cerning J (2000) Production of exopolysaccharide by Lactobacillus rhamnosus R and analysis of its enzymatic degradation during prolonged fermentation. Appl. Environ. Microbiol. 10.1128/AEM.66.6.2302-2310.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.HASHIMOTO W, and MURATA K (1998) α-<scp>L</scp> -Rhamnosidase of Sphingomonas sp. R1 Producing an Unusual Exopolysaccharide of Sphingan. Biosci. Biotechnol. Biochem. 10.1271/bbb.62.1068 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. Overview of the potential roles of bacterial GUS in the gut microbiome.
Supplementary Fig. 2. (A) Absorbance scans of free FMN at varying concentrations. (B) Standard curve of FMN at absorbance of 365 nm. (C) Apparent stoichiometries of FMN:GUS based on protein concentrations and FMN concentrations as determined by standard curve analysis. (D) Trimer of Fp2GUS with active site and FMN shown as spheres, each chain rainbow colored from N-term (blue) to C-term (red). (E) Size-exclusion chromatography multi angle light scattering analysis of Fp2GUS confirms trimeric state in solution.
Supplementary Fig. 3. Characterization of WT and FMN-binding site mutants of Fp2GUS (A) FPLC traces of Fp2GUS FMN-binding mutants. (B) SDS-PAGE of WT and FMN-binding mutants of Fp2GUS (C) Absorbance scans of purified Fp2GUS FMN-binding mutants. (D) ITC binding parameters of WT and FMN-binding site mutants of Fp2GUS for FMN. (E) Kinetic parameters of WT and FMN-binding site mutants of Fp2GUS.
Supplementary Fig. 4. Bioinformatic search of the HMP stool sample database reveals 15 putative FMN-binding GUS enzymes.
Supplementary Fig. 5. Biochemical characterization of WT and FMN-binding site mutants of Rg3GUS, Rh2GUS, BvGUS, and RiGUS. (A) SEC traces for FMN-binding site mutants Y159A Rh2GUS, Y159A Rg3GUS, Y152A RiGUS, and Y149A BvGUS. (B) SDS-PAGE gel of WT and FMN-binding site mutants of Rh2GUS, Rg3GUS, RiGUS, and BvGUS. (C) Absorbance scans of WT and FMN-binding site mutants of Rh2GUS, Rg3GUS, RiGUS, and BvGUS. (D) Sequence identity matrix of the five characterized FMN-binding site mutants.
Supplementary Fig. 6. Structural comparison of FMN-binding GUSs and quaternary structure analysis. (A) Structural overlay of Fp2GUS, Rh2GUS, and Rg3GUS. (B) Tertiary structure analysis of Rh2GUS and Rg3GUS with catalytic residues and FMN shown as spheres. (C) Analysis of Rg3GUS asymmetric unit does not reveal obvious dimer or tetramer. (D) Putative dimer and tetrameric states of Rh2GUS.
Supplementary Fig. 7. SEC-MALS results of FMN-binding GUS enzymes.
Supplementary Fig. 8. Distinct FMN-binding proteins identified in the Protein Data Bank.
Supplementary Fig. 9. Structural comparison of FMN-binding GUSs with previously characterized GUS enzymes.
Supplementary Fig. 10. Structural comparison of quaternary structures of microbial GUS enzymes.
Supplementary Fig. 11. Chemical structure of a sphingan repeating unit, a potential substrate of the FMN-binding GUS enzymes.
Supplementary Fig. 12. Possible metabolic pathway from glucuronate to FMN.
Supplementary Fig. 13. Hypothetical substrate binding for redox chemistry at FMN-binding site.