

SUMMARY
Irreversible inhibition of transcriptional cyclin-dependent kinases (CDKs) provides a therapeutic strategy for cancers that rely on aberrant transcription; however, lack of understanding of resistance mechanisms to these agents will likely impede their clinical evolution. Here, we demonstrate upregulation of multidrug transporters ABCB1 and ABCG2 as a major mode of resistance to THZ1, a covalent inhibitor of CDKs 7, 12, and 13 in neuroblastoma and lung cancer. To counter this obstacle, we developed a CDK inhibitor, E9, that is not a substrate for ABC transporters, and by selecting for resistance, determined that it exerts its cytotoxic effects through covalent modification of cysteine 1039 of CDK12. These results highlight the importance of considering this common mode of resistance in the development of clinical analogs of THZ1, identify a covalent CDK12 inhibitor that is not susceptible to ABC transportermediated drug efflux, and demonstrate that target deconvolution can be accomplished through selection for resistance.
In Brief
Gao et al. report ABC transporter upregulation as a major mechanism of acquired resistance to the THZ series of covalent CDK7/12/13 inhibitors and describe the generation of E9, which escapes drug efflux and whose target selectivity was confirmed by the acquisition of a CDK12-binding site mutation in E9-resistant cells.
Graphical Abstract
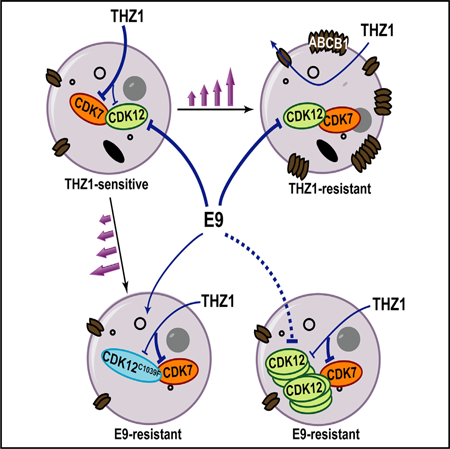
INTRODUCTION
Cancer cells that are reliant on aberrant transcription for their growth and survival present unique opportunities for therapeutic intervention (Sengupta and George, 2017). An especially vulnerable set of targets are the cyclin-dependent kinases (CDKs), which play critical roles in efficient gene transcription largely by regulating the activity of RNA polymerase II (RNAPII).Thus, targeting of specific CDKs such as CDK7, with THZ1, a novel covalent inhibitor of CDKs 7/12/13, has led to impressive responses in acute T cell leukemia (Kwiatkowski et al., 2014), MYCN-amplified neuroblastoma (NB) (Chipumuro et al., 2014), small-cell lung cancer (SCLC) (Christensen et al., 2014), and triple-negative breast cancer (Wang et al., 2015). THZ1 binds covalently to unique cysteines outside the ATP-binding domains of its target CDKs (Kwiatkowski et al., 2014), which provides an opportunity to achieve selectivity within the highly homologous CDK family and relative to other kinases. However, despite the potency shown byTHZ1 and other transcriptional CDK inhibitors in preclinical studies, resistance to these agents will inevitably arise as they enter clinical trials, underscoring the need to elucidate the basis of this impediment and devise means to overcome it.
RESULTS AND DISCUSSION
To identify stable molecular changes associated with the development of resistance to THZ1, we generated resistance models by continuously exposing MYCN-amplified, THZ1-sensitive (THZ1S) human NB cell lines (half maximal inhibitory concentration [IC50] values 2–16 nM) to escalating doses of the compound over 6–8 months, until they were proliferating in drug concentrations equal to 20–30 times the IC50 values for sensitive cells (Figure 1A). THZ1-resistant (THZ1R) cells resembled THZ1S cells morphologically and retained similar growth rates and cell-cycle profiles (Figures S1A and S2B). In contrast to the transcriptional downregulation observed in THZ1S NB cells (Chipumuro et al., 2014), neither RNAPII C-terminal domain (CTD) phosphorylation nor expression levels of the short-lived RNAs, MYCN and MCL1, were affected in THZ1R cells in the presence of THZ1 (Figure 1A). These results suggest that THZ1 resistance could arise from either acquired mutations of the binding targets of THZ1, inaccessibility of THZ1 to the targets, or from compensatory pathway activation, a less likely possibility due to the highly conserved nature of the CDKs targeted by THZ1.
Figure 1. ABC Transporter Upregulation Mediates THZ1 Resistance in NB and Lung Cancer Cells.

(A) Dose-responsecurvesforTHZ1Sand THZ1R NBcellstreated with THZ1 for72 hr(left). Forall dose-response curves, fractionsofviablecells relativeto DMSO-treated cells are shown. Western blot (WB) analysis of the indicated proteins in THZ1S and THZ1R cells treated with DMSO or THZ1 (right) (Kelly, 1 μM; NGP, LAN5, SK-N-BE2 [BE2], 300 nM) for 6 hr. (B) qRT-PCR analysis ofABC transporter expression in THZ1S and THZ1R cells (left). Expression was normalized to levels in THZ1S cells, with GAPDH used as an internal control. WB analysis of ABC proteins in THZ1S versus THZ1R cells (right). (C).Dose-response curves for THZ1S versus THZ1R lung cancer cells treated with THZ1 for 72 hr (left). WB analysis of the indicated proteins in THZ1S and THZ1R cells treated with DMSO or THZ1 (NCI-H82, H3122, 300 nM; PC9, 1 μM) for 6 hr (right). (D) qRT-PCR (left) and WB (right) analyses of ABCG2 in THZ1Sand THZ1R cells. (E).Viability ofTHZ1R Kelly NB cellstreated with THZ1 in combination with tariquidar at the indicated doses for72 hr (left). WB ofthe indicated proteins in THZ1R Kelly cells treated with THZ1 at the indicated concentrations in combination with tariquidar (125 nM) for 6 hr (right). (F).Dose-response curves forTHZ1SversusTHZ1R H82 SCLC cells treated with THZ1 in combination with KO-143 (200 nM) ortariquidar (125 nM) for72 hr(left). Viability of THZ1R PC9 cells treated with THZ1 in combination with the indicated doses of KO-143 for 72 hr (right). (G).Viability of THZ1R Kelly cells expressing either a control shRNA or two individual shRNAs against ABCB1 and treated with THZ1 (left). WB ofthe indicated proteins in THZ1R cells expressing either control (shCtrl) or ABCB1 shRNAs and treated with THZ1 alone (1 μM) or in combination with tariquidar (125 nM) for 6 hr (right). Error bars on all dose-response curves indicate mean values ± SD for three experiments. See also Figures S1-S3.
We first ruled out acquired mutations in CDK7 through direct sequencing of the whole gene, including the THZI-labeling Cys312 site (Figure S1C). Moreover, there were no significant changes in CDK7 transcript levels between sensitive and resistant cells (Figure S1D). Since THZ1 also covalently engages CDK12 at submicromolar concentrations, we ruled out kinase domain and THZ1-binding site mutations as well as altered expression of this kinase (Figures S1E and S1F). We next investigated mechanisms that might interfere with cellular accessibility of the compound to the target such as drug efflux pumps, specifically the ATP-binding cassette (ABC) family transporters, various members of which are overexpressed in NB (Yu et al., 2015). Moreover, CDK inhibitors are known to function as substrates for drug transporters (Cihalova et al., 2015; Robey et al., 2001), which may have accounted for their less than satisfactory performance in preclinical and early-phase clinical trials (Gorlick et al., 2012; Le Tourneau et al., 2010). Analysis of ABC transporter expression in THZ1S versus THZ1R cells indeed showed marked upregulation of the ABC sub-family B member 1 (ABCB1/MDR1/p-glycoprotein) in THZ1r cells (Figure 1B). Increased ABCB1 levels were retained in THZ1R cells grown in THZ1-free medium for up to 3 months, indicating stable resistance, which decreased gradually and was associated with a return of sensitivity to THZ1 (Figure S2A).
A pivotal question at this juncture was whether upregulation of ABC drug transporters serves as a resistance mechanism in transcription-factor-driven cancers other than NB. We therefore studied (1) NCI-H82 SCLC cells, which are sensitive to THZ1 through disruption of MYC-associated oncogenic signaling (Christensen et al., 2014), and (2) PC-9 and NCI-H3122 non small-cell lung cancer (NSCLC) cells, which express oncogenic MYC and also depend on mutant EGFR and translocated ALK, respectively, for survival (Lee and Wu, 2015; Riveiro et al., 2016). SCLC and NSCLC resistance models, generated in a similar manner to NB (Figure 1C, left), did not show downregulation of either RNAPII CTD phosphorylation or MYC levels compared with their sensitive counterparts (Figure 1C, right), and did not show mutations in CDKs 7/12 (not shown). Rather, instead of upregulation of ABCB1 levels as seen in THZ1R cells, ABCG2 (BCRP), another ABC family member with roles in chemotherapy resistance (Doyle and Ross, 2003), was upregulated in both SCLC and NSCLC cells (Figure 1D).
Exposure of THZ1R NB cells to a small-molecule inhibitor of ABCB1, tariquidar (Martin et al., 1999), rescued their sensitivity to THZ1 and led to growth inhibition (Figures 1E and S2B). Concomitant treatment with tariquidar also led to downregulation of RNAPII phosphorylation as well as MYCN and MCL1 expression (Figures 1E and S2C) and to induction of cell-cycle arrest, similar to that seen in THZ1S cells (Figure S2D). In addition, THZ1r NB cells were also cross-resistant to a known ABCB1 substrate, doxorubicin, an effect that could also be rescued with tariquidar (Figure S2E). This relationship was further supported by an efflux assay demonstrating that ABCB1 overexpression induced a decrease in the intracellular retention of doxorubicin in THZ1R versus THZ1S cells (Figure S2F). Treatment of THZ1R H82 SCLC cells with the ABCG2 inhibitor KO-143 (Allen et al., 2002), but not tariquidar, rescued their sensitivity to THZ1 (Figure 1F). This effect was also seen in THZ1R NSCLC cells (Figures 1F and S3A). To verify that these effects were truly specific to ABCB1 and ABCG2, we depleted each protein through shRNA-mediated knockdown in THZ1R cells, noting rescue of THZ1 sensitivity and on-target activity (Figures 1G, S2G, S3B, and S3C). Together, our results indicate that NB and lung cancer cells develop stable resistance to THZ1 through upregulation of ABC family members whose subtype appears to depend on cell lineage.
To identify compounds that might circumvent MDR-mediated resistance, we screened THZ1R cells against a panel of established and novel CDK inhibitors (Table S1). Given the activity of THZ1 against CDKs 7 and 12, we first tested THZ531, a THZ1 derivative with selectivity for CDK12 over CDK7 (IC50, 158 nM versus 8,500 nM) (Zhang et al., 2016). While THZ531 was quite potent in THZ1S cells, it was unable to overcome the upregulated ABCB1 expression in THZ1R cells, suggesting that it also serves as a substrate of ABC proteins. ABCB1 upregulation in THZ1R NB cells also imparted cross-resistance to the known ABCB1 substrate, dinaciclib. Interestingly, ABCG2-overexpressing THZ1R lung cancer cells were resistant only to THZ531, while remaining as sensitive as parental cells to other inhibitors, including dinaciclib and flavopiridol, suggesting that the mechanism of resistance in these cells arises from the specific substrate selectivity of the THZ class for this drug transporter protein (Table S1). Among all the inhibitors tested, a novel compound, E9, showed the most potent antiproliferative activity in THZ1R NB and lung cancer cells, with IC50 values ranging from 8 to 40 nM (Table S1 and Figure 2A). Moreover, E9 was more potent than other inhibitors, including ribociclib, palbociclib, and AZD5438 (Table S1). In vitro efflux assays established that E9 does not serve as a substrate for either ABCB1 or ABCG2 (Figure 2B), thus rendering it an attractive candidate to overcome THZ1 resistance.
Figure 2. The Covalent Inhibitor E9 Overcomes ABC-Mediated Resistance.
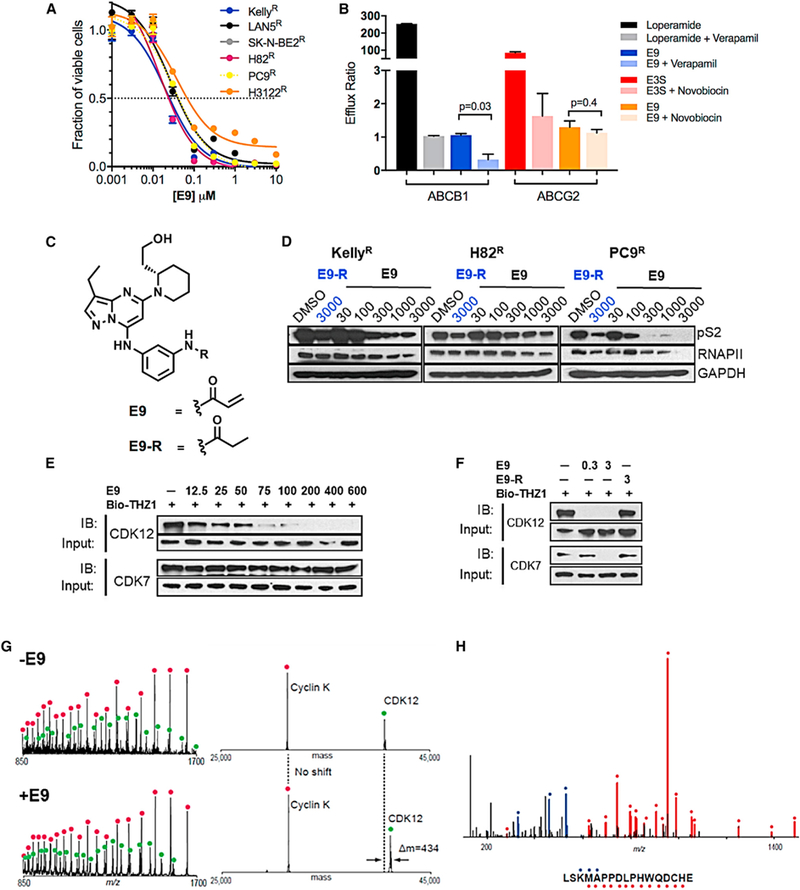
(A) Dose-response curves for THZ1R NB and lung cancer cells treated with E9 for 72 hr. Error bars indicate means ± SD, n = 3. (B) In vitro permeability analysis of E9 as a substrate of ABCB1 and ABCG2. MDR1-MDCK cells stably expressing ABCB1 and Caco-2 cells stably expressing ABCG2 were exposed to E9 (10 μM) aloneorin combination with inhibitorsofABCB1,verapamil (100 μM) orABCG2, novobiocin (50 μM), respectively, for2hrand the amount of E9 acrossacell monolayerquantified by liquid chromatographytandem massspectrometry.An efflux ratio>2 indicatesthatthetest compound isa potential substrate for either ABC transporter. Loperamide and E3S (10 μM) were used as positive controls for ABCB1 and ABCG2, respectively. Error bars represent means ± SD of three experiments. (C) Structures of E9 and E9-R. (D) WB of RNAPII CTD phosphorylation at Ser2 in NB and lung cancer cells treated with E9-R (3 μM) or E9 at the indicated doses for 6 hr. (E) WB of unengaged CDK12 and CDK7 in THZ1R Kelly NB cells treated with E9 at the Indicated doses for 6 hr. Cell lysates were subjected to a target engagement assay in which biotinylated THZ1 (bio-THZ1; 1 μM) was used to label unengaged CDKs. (F) WB of unbound CDK12 and CDK7 in THZ1R Kelly cells treated with E9 (0.3 and 3 μM) or E9-R (3 μM) for 6 hr followed by bio-THZ1 (1 mM). See also Table S1 and Figures S4 and S5. (G) Mass spectra (left) and zero-charge mass spectra (right) derived from CE-MS analysis of the CDK12/CCNK complex after treatment with DMSO (upper panels) or E9 (lower panels) for 1 hr at room temperature. The mass of CDK12 shifts after treatment with inhibitor, indicating covalent labeling. (H) MS/MS spectrum of the CDK12 peptide (residues 1025–1041) acquired during CE-MS analysis of E9-labeled CDK12/CCNK complex that was digested with GluC. Ions of type b and y are illustrated with blue and red glyphs, respectively (C*, E9 modified cysteine residue). No other E9 modified peptides were detected in this analysis.
E9 is a trisubstituted pyrazollopyridine with structural similarity to dinaciclib (which inhibits CDKs 1/2/5/9/12), except that E9 contains an acrylamide moiety that targets cysteine residues in CDKs 7, 12, and 13 (Figure 2C). We also generated a matched, reversible counterpart, E9-R, by replacing the acrylamide with a propyl amide that is incapable of forming covalent adducts with cysteines (Figure 2C). E9 led to a dose-dependent decrease in phosphorylated and total RNAPII in THZ1r NB and lung cancer models (Figures 2D and S4A), accompanied by decreased MYC and MCL1 expression, indicating its effect on transcription. E9-R, on the other hand, had a lesser effect and at a relatively higher dose. E9 also resulted in increased PARP cleavage (Figure S4A), and an increase in the subGI population in THZ1r lung cancer cells, while in NB cells, more of a G2/M arrest was seen after a 24-hr exposure to E9 (Figure S4B). To evaluate the ability of E9 to bind to CDK7 and CDK12, we performed a cell-based competitive pull-down assay using a biotinylated version of THZ1 (bio-THZI) (Figure 2E). Although E9 competed strongly with bio-THZI for binding to CDK12 at low nanomolar ranges in THZ1r cells, it failed to engage CDK7, even at 10-fold higher concentrations. The weak CDK7 binding was confirmed using a fixed time point biochemical assay where E9 showed an IC50 of >1 μM for the CDK7/CyclinH complex (Figure S4C). Since dinaciclib is also a reversible inhibitor of CDK2 and CDK9, we measured the ability of E9 to inhibit these two kinases through KINOMEscan profiling (Miduturu et al., 2011) (Figure S5A), with the expectation that E9 would also bind in a reversible fashion due to the absence of appropriately positioned cysteines in these kinases. While there was marked loss of activity against CDK2 compared with dinaciclib, E9 still retained fairly strong biochemical activity against CDK9 (Figure S4C; Table S1). Surprisingly, E9-R exhibited improved biochemical inhibition of CDK2, CDK7, and CDK9 compared with E9 (Figure S5B). However, this reversible inhibitory effect of E9-R did not translate to comparable antiproliferative activity in THZ1r cells (Figure S5C). Moreover, a competitive target binding assay demonstrated a lack of engagement of E9-R with CDK12 even at concentrations as high as 3 μM (Figure 2F). Finally, capillary electrophoresis mass spectrometry (CE-MS) showed that E9 modifies the CDK12 protein on Cys1039 (Figures 2G and 2H), confirming that it inhibits CDK12 through covalent binding.
We next sought to identify the functional target(s) of E9, whose inhibition resulted in antiproliferative activity in THZ1 s and THZ1 r cells. Given that E9-R shares activity with the parent compound against CDK9 (Figures S4C and S5B), but is inactive against CDK12 (Figure 2F) and less cytotoxic (Figure S5C), we hypothesized that targeting of CDK12 likely played a crucial role in the inhibitory activity of E9. Moreover, since the E9 scaffold is not a substrate of drug efflux pumps, E9-sensitive (E9S) cells would most likely develop resistance through on-target mechanisms. To test this prediction, we generated E9-resistant (E9R) Kelly and SK-N-BE2 NB cells from THZ1s cells by serial passage at increasing doses of the inhibitor (Figure 3A). The E9R NB cells did not acquire ABCB1 transporter overexpression (Figure S6A), yet the compound failed to inhibit RNAPII CTD phosphorylation in these cells (Figure 3B). Upregulation of ABCG2 was observed in SK-N-BE2 E9R cells (Figure S6A) but did not mediate resistance, as indicated by the lack of response to E9 when this agent was combined with KO-143 (Figure S6B). Hence, these data suggest that in E9R cells, the ability of the compound to inhibit 12 had become impaired.
Figure 3. E9 Induces Cytotoxicity by Covalently Targeting Cys1039 of CDK12.
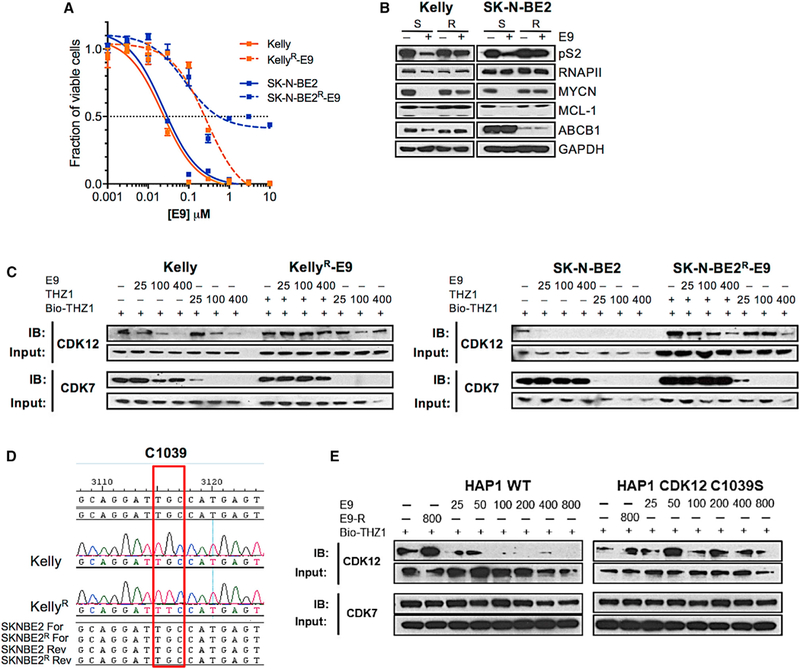
(A) Viability curves for E9S and E9R NB cells treated with E9 for 72 hr. Error bars represent means ± SD. (B) WB of the indicated proteins in E9S versus E9R cells treated with DMSO or E9 at concentrations to which the cells had become adapted (Kelly, 500 nM; SK-N-BE2, 300 nM) for 6 hr. (C) WB of unlabeled CDK12 and CDK7 in E9S and E9R cells treated with E9 followed by THZ1 for 6 hr at the indicated doses. Cell lysates were incubated with bio-THZ1 (1 μM) to label unengaged CDKs. (D) CDK12 sequence surrounding the THZ1-interacting Cys1039 in THZ1S and THZ1R cells showing the single point mutation that results in the substitution of Cys1039 for Phe in Kelly E9R cells (red box). (E) WB of unlabeled CDK12 and CDK7 in WT and C1039S HAP1 cells treated with E9-R (800 nM) or increasing concentrations of E9 as indicated for 6 hr. Cell lysates were incubated with bio-THZ1 (1 μM) to label unengaged CDKs. See also Figure S6.
To pinpoint the exact target of E9 in NB cells, we performed target engagement assays using E9 orTHZI in E9Sand E9R cells (Figure 3C). In E9S cells, E9 treatment led to a significant decrease in CDK12 binding at doses less than 25 nM in both NB cell types. In E9R cells, however, there was no appreciable decrease in CDK12 binding with E9 in Kelly cells, while a decrease could be seen in SK-N-BE2 cells, albeit at high doses (400 nM) (Figure 3C). By contrast, THZ1 treatment led to complete or near-complete loss of CDK7 binding in both lines, irrespective of their differential sensitivity toward E9. Since mutations in the target binding site are a common mode of resistance to kinase inhibitors, we analyzed the sequence of the region surrounding the Cys1039 of CDK12 in E9S versus E9R cells. Indeed, E9R Kelly NB cells had acquired a single point mutation, C1039F, at this critical E9 binding site (Figure 3D), thus explaining the loss of binding by E9, and the mechanism of resistance. Moreover, HAP1 chronic myeloid leukemia cells engineered to express a C1039S CDK12 mutation through CRISPR-cas9 knockin exhibited resistance to E9, further confirming the binding target of this compound (Figure 3E). The E9R SK-N-BE2 cells developed genomic amplification of CDK12 (Figure S6D), accounting for the significantly increased protein levels in these cells compared with their E9S counterparts and dose-dependent covalent inhibition of CDK12 activity (Figure 3C). Sequence analysis of the CDK13 gene in E9R cells showed no mutations, thus eliminating CDK13 as the functional target of E9 (data not shown). Thus, we determined through selection for resistance that E9 induces NB cell growth inhibition through covalent binding to CDK12. This finding was further confirmed by cross-resistance of E9R cells to the highly selective covalent CDK12 inhibitor, THZ531 (Zhang et al., 2016) and the absence of cross-resistance to either THZ1 or NVP2, more selective inhibitors of CDK7 (Kwiatkowski et al., 2014) and 9 (Barsanti et al., 2011) respectively, or the pan-CDK inhibitors dinaciclib and favopiridol (Figure S6C). Importantly, the specificity with which CDK12 functionality is maintained indicates an indispensable role for this kinase in the survival of NB cells and identifies another potential therapeutic target in this disease.
In summary, we report upregulation of the multidrug transporters ABCB1 and ABCG2 as a mechanism of acquired resistance to the THZ series of CDK inhibitors, and subsequent generation of E9, which is not a substrate of ABC transporters and which primarily engages CDK12. We observed that in THZ1r cells, ABCB1 overexpression also mediates ubiquitous cross-resistance to chemotherapy agents as well as most CDK inhibitors, while ABCG2 overexpression affects a limited group of inhibitors, including the THZ series of covalent agents. Our results would enable the selection of patients most likely to respond to this class of inhibitors given that the majority who relapse following standard chemotherapy have activation of the ABC family members in their tumors (Bugdeet al., 2017). In addition, since clinical applications of ABC transporter inhibitors are yetto be validated (Fletcher et al., 2010), inherent lack of susceptibility to ABC transporter-mediated drug efflux becomes a desirable feature for incorporation into the design and optimization of THZ analogs for clinical testing. To that end, E9, which functions as a potent non-covalent inhibitor of CDK9 and a covalent inhibitor of CDK12, while avoiding ABC transporter-mediated efflux, affords a convenient starting lead for further optimization.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rani E. George (rani_george@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
NBcell lines Kelly (from a female source), LAN5 (male), SK-N-BE2 (male) and NGP(male) were obtained from the Children’s Oncology Group (COG) cell line repository and genotyped at the DFCI Core Facility by standard methods. NSCLC cell lines PC-9 (male) and NCI-H82 (male) were obtained from the ATCC. The SCLC cell line, NCI-H3122 was obtained from the NCI Cell Line Repository and the gender of its source is not available. The cell lines were authenticated through STR analyses. Both types of cell lines were maintained in RPMI-1640 medium supplemented with 10% FBS. Mycoplasma testing was performed on a 6-monthly basis and all lines were negative.
METHOD DETAILS
Development of Resistant Cell Lines
Cells were originally grown in medium containing THZ1 at its corresponding IC50 value and split at a 1:5 ratio when they reached confluence. Doses of THZ1 were raised by 10 nM once the cells resumed normal growth rate after every one or two passages.
The cells were continuously cultured in THZI-containing medium. Most NB cell lines acquired substantial resistance to THZ1 within 3–6 months. Subsequently, the entire resistant population was subjected to characterization without single-cell cloning. Blinding was not performed at any stage of the study.
Cell Viability Assays
For viability assays with single agents, cells were plated in 96-well plates at 4 × 103 cells/well for 24 hours. Subsequently, the cells were treated with a test compound at doses ranging from 10 nM to 10 μM for 72 hours. Cell growth over 24 hours was determined using the CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer’s instructions. Results were reported as the relative fold-change in ATP with each group internally normalized to the respective vehicle control. Drug concentrations that inhibited 50% of cell growth were determined using GraphPad Prism (GraphPad Software Inc.). For the rescue experiments, the same procedure was followed, except that tariquidar or KO-143 (at a range of doses based on their IC50s) was simultaneously added to THZ1 or E9. Data is presented as the mean of duplicate wells ± SD.
Cell Cycle and Apoptosis Analyses
Cells(5×105) were seeded in 6-well plates for 24 h, followed by treatment with the test compounds for 24 h. For cell cycle analysis, the cells were trypsinized and fixed overnight with ice-cold 70% ethanol, treated with RNase A (0.5 mg/ml) and stained with propidium iodide (PI) (50 μg/ml). For apoptosis analysis, cells were collected by trypsinization and stained with PI and FITC-Annexin V (BD Biosciences) for 15 min. In both assays, flow cytometry (FACScalibur) was used to quantify the cell populations. The resultant data were analyzed using FlowJo software.
shRNA Knockdown
pLKO.1 plasmids containing shRNAs targeting ABCB1 and ABCG2 were obtained from the RNAi Consortium of the Broad Institute of MIT and Harvard and knockdown performed as described previously (Chipumuro et al., 2014), except that puromycin selection was omitted because it is a known substrate of ABCB1.
Western Blotting
The cells were collected by trypsinization and washed with PBS buffer. Cell lysates were prepared by using NP40 lysis buffer (Invitrogen) supplemented with cOmplete protease inhibitor cocktail, PhosSTOP phosphatase inhibitor cocktail and PMSF (1 mM). The lysates were cleared by centrifugation and resolved using Nupage gels and western blotted to detect proteins of interest. Antibodies to ABCB1, ABCG2, CDK12, CDK2, phospho-CDK2, cdc2/CDK1, phospho-CDK1, MYCN, cleaved PARP, GAPDH (Cell Signaling Technologies), MCL1, CDK7, RNAPII (Santa Cruz Biotechnologies), phospho-RNAPII S2, S5 (Bethyl Labratories) and phospho-RNAPII (S7) (Milipore) were used according to the manufacturers’ instructions.
Target Engagement Assay
To detect target labeling of CDK7 and CDK12, cells were treated with test compounds for 6 h at the indicated doses. Total cell lysates were prepared as for western blotting. To pull down CDK7 or CDK12,250 μg of total protein was combined with biotin-THZ1 at 1 μM and rotated at 4°C overnight. Streptavidin agarose resin (30 μl 50% slurry) was added and samples rotated for another 2 h at 4 °C. The resin was subsequently washed 3x with cell lysis buffer, and CDK7 and CDK12 released from the resin by boiling for 10 min in 2xgel loading buffer and resolved by western blotting. As a loading control, 25 μg of total protein was used.
qRT-PCR and Sequencing
qRT-PCR was performed using previously described methods (Chipumuro et al., 2014). For qRT-PCR using genomic DNA as the template, genomic DNA from 106 cells was prepared with the QiaAmp DNA mini kit. Genomic DNA (50 ng) was used as the template to amplify the gene of interest using the HotStar HiFidelity Polymerase kit (Qiagen). β-actin or GAPDH were used as internal control. PCR products were gel-purified using Qiaquick Gel Extraction Kit (Qiagen) and subjected to sequencing (primers available on request).
Doxorubicin Efflux Assay
Cells (5 ×105) seeded in 6-well plates and incubated for 24 h were treated with DMSO, tariquidar (125 nM) for 3 h and then cotreated with doxorubicin (20 μM) for 2 h, trypsinized and washed twice with ice-cold PBS buffer. Fluorescence intensity was quantified by flow cytometry (FACScalibur) and the data analyzed using FlowJo software.
In Vitro Permeability Assay for E9
Drug efflux assay in ABCB1-or ABCG2-expressing cells was performed by Cyprotex (Watertown, MA). Briefly, Madin-Darby canine kidney (MDCK) epithelial cells stably transfected with the ABCB1 gene were seeded on a MultiscreenTM plate (Millipore) to form a confluent monolayer over 4 days prior to the experiment. For apical to basolateral (A/B) permeability, E9 (10 μM) in the absence or presence of 100 μM verapamil (an ABCB1 inhibitor) was added to the apical (A) side and the amount of permeation on the basolateral (B) side was determined. For basolateral to apical (B/A) permeability, the test article in the absence and presence of 100 μM verapamil was added to the B side and the amount of permeation on the A side determined. The A-side buffer contained 100 μM Lucifer yellow dye in transport buffer (1.98 g/L glucose in 10 mM HEPES, 1x Hank’s Balanced Salt Solution), pH 7.4, while the B-side buffer had transport buffer at pH 7.4. MDCK-MDR1 cells were incubated with these buffers for 2 h, and the receiver-side buffer was removed for analysis by LC-MS/MS (using propranolol as an analytical internal standard). The rate of passage of E9 through this cell monolayer barrier in this bi-directional transport assay was used to determine the apparent permeability coefficient (Papp).
where dQ/dt is the rate of permeation of the drug across the cells, C0 the donor compartment concentration at time zero and A, the area of the cell monolayer. An efflux ratio is calculated from the mean apical to basolateral (A-B) Papp data and basolateral to apical (B-A) Papp data. Efflux Ratio = Re > 2 (in the absence of chemical inhibitor) indicated a potential substrate for the MDR-1 efflux transporter. Furthermore, reduction of Re ≤ 2 (in the presence of chemical inhibitor) confirmed the test article as a substrate for the ABCB1 efflux transporter. The final result was reported as the ± SD of 3 experiments. To assay the permeability of ABCG2, Caco-2 (human colorectal carcinoma) cells grown in tissue culture flasks, were trypsinized, suspended in medium, and the suspensions applied to membrane plate wells (96-well format). The cells were allowed to grow and differentiate for 3 weeks, with feeding at 2-day intervals. Monolayers were then prepared by rinsing both basolateral and apical surfaces twice with buffer at 37 °C (pH 7.4). Efflux assays were performed as for MDCK-MDR1 cells. Novobiocin (50 μM) was applied as the ABCG2 inhibitor. Loperamide (10 μM) and E3S (10 μM) were used as positive controls for ABCB1 and ABCG2, respectively.
KINOMEscan Profiling
KINOMEscan profling was performed by Ambit Biosciences (San Diego, CA).
Mass Spectrometry Analysis
CDK12/CCNK complex (10 μg) was treated with DMSO ora 5-fold molar excess of E9 for 1 hour at room temperature. Treated proteins were directly analyzed by CE-MS using a ZipChip CE system and autosampler (908 Devices, Boston, MA) interfaced to a QExactiv HF mass spectrometer (ThermoFisher Scientific, San Jose, CA). Protein solutions were loaded for 5 seconds and separation performed at 500V/cm on an HR chip (22 cm separation channel) for 6 minutes with a background electrolyte consisting of 1% formic acid in 50% acetonitrile. Pressure assist was utilized and started at 1 minute. The mass spectrometer recorded profile spectra (15k resolution, 1E6 target, lock mass enabled) from m/z 300–2000. Mass spectra were deconvoluted using MagTran version 1.03 b2 (Zhang and Marshall, 1998). To identify the labeled residue, treated protein was denatured with Rapigest (0.1% final concentration; Waters, Milford, MA), reduced (10 mM dithiothreitol), alkylated (22.5 mM iodoacetamide), and digested with GluC (Promega, Madison, WI) overnight at 37 °C. After cleaving Rapigest according to the manufacturer’s instructions, peptides were desalted using C18, dried by vacuum centrifugation, and reconstituted in 1% formic acid/50% acetonitrile with 100 mM ammonium acetate. Peptides were then analyzed by CE-MS using the system described above. Peptide solution was loaded for 30 seconds and separation performed at 500 V/cm on an HR chip for 10 minutes with a background electrolyte consisting of 1% formic acid in 50% acetonitrile. Pressure assist was utilized and started at 1 minute. The mass spectrometer was operated in data dependent mode and subjected the 5 most abundant ions in each MS scan (60k resolution, 1E6 target, lock mass enabled) to MS/MS (15k resolution, 2E5 target, 100 ms max inject time). Dynamic exclusion was enabled with a repeat count of 1 and an exclusion time of 6 seconds. MS/MS data was extracted to .mgf using multiplierz scripts (Askenazi et al., 2009; Parikh et al., 2009) and searched against a forward-reverse human NCBI refseq database using Mascot version 2.2. Search parameters specified fixed carbamidomethylation of cysteine, and variable oxidation (methionine) and E9 modification (cysteine). Precursor mass tolerance was set to 10 ppm and product ion tolerance was 25 mmu.
Synthesis of E9 and E9-R
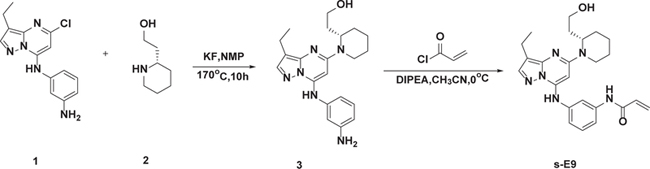
(S)-2-(1-(7-(3-Aminophenylamino)-3-Ethylpyrazolo[1,5-a]pyrimidin-5-yl)piperidin-2-yl)ethanol (3)
The mixture of 1 (250 mg, 0.87 mmol), 2 (168 mg, 1.3 mmol), KF (227 mg, 3.9 mmol) and NMP (2 mL) was stirred at 170 °C for 10 h. After completion, the residue was extracted with chloroform and iso-propanol (4:1) and the organic phase was washed twice with brine (50 mL), dried with Na2SO4, filtered and concentrated to remove the solvent under reduced pressure. The residue was purified by silica gel (MeOH/DCM = 0–20%) to obtain 3 (250 mg, yield 75.6%). LCMS (m/z): 381 [M + H]+;
(S)-N-(3-(3-Ethyl-5-(2-(2-Hydroxyethyl)piperidin-1-yl)pyrazolo[1,5-a]pyrimidin-7-ylamino)phenyl)acrylamide (E9)
To a solution of 3 (50 mg, 0.13 mmol) and DIPEA (0.2 mL), in CH3CN (2 mL) were added acrylyl chloride (15 mg, 0.17 mmol) in DCM (0.5 mL) dropwise. The mixture was stirred at 0°C for 1 h and after completion, concentrated to remove the solvent under reduced pressure, the residue was purified by prep-HPLC (C18 column, MeOH/H2O, containing 0.05%TFA) to obtain E9 (off-white solid, 33.7 mg, yield 59%).
HPLC: 96% (254 nm); LCMS (m/z): 435 [M + H]+ ;.1 H NMR (500 MHz, DMSO) 5 10.29 (s, 1H), 9.80 (s, 1H), 7.90 (d, J =18.7 Hz, 2H), 7.44 – 7.34 (m, 2H), 7.23 – 7.15 (m, 1H), 6.46 (dd, J = 17.0,10.1 Hz, 1H), 6.28 (dd, J = 17.0,1.9 Hz, 1H), 5.94 (s, 1H), 5.79 (dd, J =10.1, 1.9 Hz, 1H), 4.52 (s, 1H), 4.09 (s, 1H), 3.35 – 3.30 (m, 2H), 3.00 (t, J =12.1 Hz, 1H), 2.61 – 2.54 (m, 2H), 1.91 (td, J = 13.6, 5.4 Hz, 1H), 1.75–1.60 (m, 5H), 1.58 (s, 1H), 1.44 (d, J = 12.2 Hz, 1H), 1.22 (t, J =7.5 Hz, 4H).

(S)-N-(3-((3-Ethyl-5-(2-(2-Hydroxyethyl)piperidin-1-yl)pyrazolo[1,5-a]pyrimidin-7-yl)amino)phenyl)propionamide (E9-R)
To a solution of 3 (50 mg, 0.13 mmol) and DIPEA (0.2 mL) in CH3CN (2 mL) was added propionyl chloride (16 mg, 0.17 mmol) in DCM (0.5 mL) dropwise. The mixture was stirred at 0°C for 1 h and after completion, concentrated to remove the solvent under reduced pressure. The residue was purified by prep-HPLC (C18 column, MeOH/H2O, containing 0.05%TFA) to obtain E9-R (off-white solid, 25.7 mg, yield 45%).
HPLC: 98% (254 nm); LCMS (m/z): 437 [M + H]+ ;.1H NMR (500 MHz, DMSO) 5 9.99 (s, 1H), 9.79 (s, 1H), 7.87 (d, J =12.0 Hz, 2H), 7.37 – 7.28 (m, 2H), 7.13 (d, J = 7.8 Hz, 1H), 5.92 (s, 1H), 4.51 (s, 1H), 4.07 (s, 1H), 3.45 – 3.42 (m, 1H), 3.34 −3.31 (m, 1H), 3.17 (s, 1H), 3.00 (t, J = 12.9 Hz, 1H), 2.58 (q, J =7.5 Hz, 2H), 2.34 (q, J =7.5Hz,2H), 1.91 (td, J = 13.6, 5.3 Hz, 1H), 1.74–1.61 (m, 5H), 1.57 (s, 1H), 1.43 (d, J =12.0 Hz, 1H), 1.22 (t, J = 7.5 Hz, 3H), 1.09 (t, J = 7.5 Hz, 3H).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as the means ± SD of a minimum of 3 experiments except where indicated. Student’s t-test (two-tailed, unpaired) was used to compare the means for two groups, while one-way ANOVA was used in comparisons with multiple groups. Analyses were performed with GraphPad Prism 7.02 (GraphPad Software). P < 0.05 was considered significant. No data were excluded.
DATA AND SOFTWARE AVAILABILITY
The accession number for the sequence of the region surrounding CDK12 Cys1039 in E9 resistant Kelly cells that was reported in this paper is [GenBank]: [MG595150].
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ABCB1 | Cell signaling | Cat#12683S; RRID:AB_2715689 |
| ABCG2 | Cell signaling | Cat#4477S; RRID: AB_10544928 |
| CDK12 | Cell signaling | Cat#11973S; RRID:AB_2715688 |
| MYCN | Cell signaling | Cat#9405S; RRID:AB_10692664 |
| c-MYC | Santa Cruz | Cat#sc-40; RRID:AB_627268 |
| Cleaved PARP | Cell signaling | Cat#9541S; RRID:AB_331426 |
| GAPDH | Cell signaling | Cat#2118; RRID:AB_561053 |
| MCL1 | Santa Cruz | Cat#sc-819; RRID:AB_2144105 |
| CDK7 | Santa Cruz | Cat#sc-723; RRID:AB_2077155 |
| RNAPII | Santa Cruz | Cat#sc-899; RRID:AB_632359 |
| Phospho-RNAPII S2 | Bethyl | Cat#300–654A; RRID:AB_519341 |
| Phospho-RNAPII S5 | Bethyl | Cat#A304–408A; RRID:AB_2620602 |
| Phospho-RNAPII S7 | Millipore | Cat#04–1570; RRID:AB_10618152 |
| Bacterial and Virus Strains | ||
| TRCN0000059683: | Sigma-Aldrich | Clone ID:NM_000927.3–3500s1c1 |
| TRCN0000059684 | Sigma-Aldrich | Clone ID:NM_000927.3–1193s1c1 |
| TRCN0000059800 | Sigma-Aldrich | Clone ID:NM_004827.1–1718s1c1 |
| TRCN0000059802 | Sigma-Aldrich | Clone ID:NM_004827.1–544s1c1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tariquidar | Selleck | Cat#S8028; CAS:206873–63-4 |
| KO-143 | Sigma-Aldrich | Cat#K2144; CAS:461054–93-3 |
| THZ1 | Gray Lab (Kwiatkowski et al., 2014) | CAS:1604810–83-4 |
| THZ531 | Gray Lab (Zhang et al., 2016) | CAS:1702809–17-3 |
| AT7519 | MedChem Express | Cat#HY-50943; CAS:844442–38-2 |
| SNS-032 | Selleck | Cat#S1145; CAS:345627–80-7 |
| AZD5438 | ChemExpress | Cat#HY-10008; CAS:602306–29-6 |
| CR8 | Sigma-Aldrich | Cat#C3249; CAS:294646–77-8 |
| Dinaciclib | ChemExpress | Cat#HY-10492; CAS:779353–01-4 |
| E9 | This study | N/A |
| E9-R | This study | N/A |
| Flavopiridol | ChemExpress | Cat#HY-10005; CAS:146426–40-6 |
| LDC000067 | ChemExpress | Cat#HY-15878; CAS:1073485–20-7 |
| Ly2857785 | ChemExpress | Cat#HY-12293; CAS:1619903–54-6 |
| NVP2 | Gray Lab | N/A |
| P276–00 | Selleck | Cat#S8058; CAS:920113–03-7 |
| PD-0332991 | ChemExpress | Cat#HY-50767; CAS:827022–33-3 |
| PHA-848125 | Selleck | Cat#S2751; CAS:802539–81 −7 |
| R547 | ChemExpress | Cat#HY-10014; CAS:741713–40-6 |
| Ribociclib | ChemExpress | Cat#HY-15777B; CAS:1211441–98-3 |
| RO-3306 | ChemExpress | Cat#HY-12529; CAS:872573–93-8 |
| Critical Commercial Assays | ||
| DC protein Assay | Bio-Rad | Cat#5000111 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#G7573 |
| SuperScript III Reverse Transcriptase | Thermo Fisher | Cat#18080044 |
| RNeasy Mini Kit | Qiagen | Cat#74106 |
| QIAamp DNA mini Kit | Qiagen | Cat#51304 |
| Experimental Models: Cell Lines | ||
| Human: Kelly | Children’s Oncology Group | N/A |
| Human: LAN5 | Children’s Oncology Group | N/A |
| Human: SK-N-BE2 | Children’s Oncology Group | N/A |
| Human: NGP | Children’s Oncology Group | N/A |
| Human: PC9 | ATCC | N/A |
| Human: H3122 | NCI Tumor Cell Line Repository | N/A |
| Human: NCI-H82 | ATCC | ATCC® HTB-175™ |
| Human: HAP1 WT & transformed | Gray Lab (Zhang et al., 2016) | N/A |
| Oligonucleotides | ||
| RT-qPCR primer: ABCB1 Forward: TGACATTTATTCAAAGTTAAAAGCA |
This paper | N/A |
| RT-qPCR primer: ABCB1 Reverse: TGACATTTATTCAAAGTTAAAAGCA |
This paper | N/A |
| RT-qPCR primer: ABCG2 Forward: CTTCTTCCTGACGACCAACC |
This paper | N/A |
| RT-qPCR primer: ABCG2 Reverse: TCTGTAGTATCCGCTGATGTATTC |
This paper | N/A |
| RT-qPCR primer: ABCC1 Forward: AATACCAGCAACCCCGACTTCAC |
This paper | N/A |
| RT-qPCR primer: ABCC1 Reverse: GGTGTCATCTGAATGTAGCC |
This paper | N/A |
| RT-qPCR primers for ABCC3, ABCC4, CDK7, GAPDH, genomic qPCR primers for ABCB1, ABCG2 and CDK12, and sequencing primers for CDK7, CDK12 and CDK13, see Table S2. |
This paper | N/A |
| Software and Algorithms | ||
| FlowJo 10 | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo |
| GraphPad Prism 7.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
SIGNIFICANCE
The relatively recent generation of selective, potent small molecules that irreversibly inhibit transcriptional CDKs 7,12, and 13, represented by the THZ series of compounds, have spurred efforts to develop analogs for clinical testing. Our study brings timely insights into the basis for resistance to these agents that may be relevant to their clinical development and application. We show that neuroblastoma and lung cancer cells develop resistance to THZ1 and THZ531 through upregulation of the ABCB1 or ABCG2 drug transporters, implying that these compounds are substrates for these proteins. To identify compounds that would overcome such resistance, we generated a covalent transcriptional CDK inhibitor, E9, that is not a substrate for ABC proteins and hence escapes drug efflux, leading to growth inhibitory effects through covalent targeting of CDK12. The target specificity of E9 for CDK12 was confirmed by the acquisition of a cysteine binding site mutation (C1039F) in cells rendered resistant to E9. Thus, ABC transporter upregulation should be taken into account in the development of clinical analogs of THZ compounds. In addition, we suggest that cellular drug transporter status be investigated as a biomarker of response to THZ analogs in early-phase trials, which almost always enroll patients with relapsed/refractory disease often characterized by drug transporter upregulation following standard chemotherapy.
Highlights
ABC transporters mediate resistance to THZ series of transcriptional CDK inhibitors
E9 lacks substrate specificity for ABC transporters and thus overcomes THZ resistance
E9 induces cytotoxicity through covalent modification of C1039 of CDK12
ABC transporter status should be considered in the development of clinical THZ analogs
ACKNOWLEDGMENTS
The authors thank members of the George and Gray labs for helpful discussions. Y.G. is supported by a Friends for Life Neuroblastoma Fellowship; T.Z., N.S.G., and R.E.G. are supported by the NIH (R01 CA197336); Y.G., N.S.G., and R.E.G. are supported by a Department of Defense Translational Team Science Award (W81XWH-15-PRCRP-TTSA). N.S.G. is a scientific founder, SAB member, and equity holder in Syros Pharmaceuticals, which is developing Cdk7 inhibitors and has licensed IP from the Dana-Farber Cancer Institute (DFCI). T.Z. is an inventor of THZ1, which is licensed from DFCI to Syros. N.K. is an inventor of THZ1, which is licensed from DFCI to Syros.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and two tables and can be found with this article online at https://doi.org/10.1016/j.chembiol.2017.11.007.
REFERENCES
- Allen JD, van Loevezijn A, Lakhai JM, van der Valk M, van Tellingen O, Reid G, Schellens JH, Koomen GJ, and Schinkel AH (2002). Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 7,417–425. [PubMed] [Google Scholar]
- Askenazi M, Parikh JR, and Marto JA (2009). mzAPI: a new strategy for efficiently sharing mass spectrometry data. Nat. Methods 6, 240–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsanti P, Hu C, Jeff J, Keyes R, Kucejko R, Xiaodong L, Yue P, Pfister Kb, Sendzik M, Sutton J, and Lifeng W. (2011). Pyridine and pyrazine derivatives as protein kinase modulators. Patent WO2011012661,
- Bugde P, Biswas R, Merien F, Lu J, Liu DX, Chen M, Zhou S, and Li Y (2017). The therapeutic potential of targeting ABC transporters to combat multi-drug resistance. Expert Opin. Ther. Targets 21, 511–530. [DOI] [PubMed] [Google Scholar]
- Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, et al. (2014). CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 159, 1126–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS, Akbay EA, Altabef A, et al. (2014). Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 26, 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cihalova D, Staud F, and Ceckova M (2015). Interactions of cyclin-dependent kinase inhibitors AT-7519, flavopiridol and SNS-032 with ABCB1, ABCG2 and ABCC1 transporters and their potential to overcome multidrug resistance in vitro. Cancer Chemother. Pharmacol. 76, 105–116. [DOI] [PubMed] [Google Scholar]
- Doyle L, and Ross DD (2003). Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 22, 7340–7358. [DOI] [PubMed] [Google Scholar]
- Fletcher JI, Haber M, Henderson MJ, and Norris MD (2010). ABC transporters in cancer: more than just drug efflux pumps. Nat. Rev. Cancer 10, 147–156. [DOI] [PubMed] [Google Scholar]
- Gorlick R, Kolb EA, Houghton PJ, Morton CL, Neale G, Keir ST, Carol H, Lock R, Phelps D, Kang MH, et al. (2012). Initial testing (stage 1) of the cyclin dependent kinase inhibitor SCH 727965 (dinaciclib) by the pediatric pre-clinical testing program. Pediatr. Blood Cancer 59, 1266–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. (2014). Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Tourneau C, Faivre S, Laurence V, Delbaldo C, Vera K, Girre V, Chiao J, Armour S, Frame S, Green SR, et al. (2010). Phase I evaluation of seliciclib (R-roscovitine), a novel oral cyclin-dependent kinase inhibitor, in patients with advanced malignancies. Eur. J. Cancer 46, 3243–3250. [DOI] [PubMed] [Google Scholar]
- Lee JG, and Wu R (2015). Erlotinib-cisplatin combination inhibits growth and angiogenesis through c-MYC and HIF-1alpha in EGFR-mutated lung cancer in vitro and in vivo. Neoplasia 17, 190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, Berridge G, Mistry P, Higgins C, Charlton P, and Callaghan R (1999). The molecular interaction of the high affinity reversal agent XR9576with P-glycoprotein. Br. J. Pharmacol. 128, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miduturu CV, Deng X, Kwiatkowski N, Yang W, Brault L, Filippakopoulos P, Chung E, Yang Q, Schwaller J, Knapp S, et al. (2011). High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors. Chem. Biol. 18, 868–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh JR, Askenazi M, Ficarro SB, Cashorali T, Webber JT, Blank NC, Zhang Y, and Marto JA (2009). multiplierz: an extensible API based desktop environment for proteomics data analysis. BMC Bioinformatics 10, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveiro ME, Astorgues-Xerri L, Vazquez R, Frapolli R, Kwee I, Rinaldi A, Odore E, Rezai K, Bekradda M, Inghirami G, et al. (2016). 0TX015 (MK-8628), a novel BET inhibitor, exhibits antitumor activity in non-small cell and small cell lung cancer models harboring different oncogenic mutations. Oncotarget 7, 84675–84687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey RW, Medina-Perez WY, Nishiyama K, Lahusen T, Miyake K, Litman T, Senderowicz AM, Ross DD, and Bates SE (2001). Overexpression of the ATP-binding cassette half-transporter, ABCG2 (Mxr/BCrp/ABCP1), in flavopiridol-resistant human breast cancer cells. Clin. Cancer Res. 7, 145–152. [PubMed] [Google Scholar]
- Sengupta S, and George RE (2017). Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer 3, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, Yuzugullu H, Von T, Li H, Lin Z, et al. (2015). CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 163, 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DM, Huynh T, Truong AM, Haber M, and Norris MD (2015). ABC transporters and neuroblastoma. Adv. Cancer Res. 125, 139–170. [DOI] [PubMed] [Google Scholar]
- Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK, Ficarro SB, Elkins JM, Liang Y, Hannett NM, et al. (2016). Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol. 12, 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, and Marshall AG (1998). A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J. Am. Soc. Mass Spectrom. 9, 225–233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sequence of the region surrounding CDK12 Cys1039 in E9 resistant Kelly cells that was reported in this paper is [GenBank]: [MG595150].