

Summary
Axon loss determines persistent disability in multiple sclerosis patients. Here, we use in vivo calcium imaging in a multiple sclerosis model to show that cytoplasmic calcium levels determine the choice between axon loss and survival. We rule out the endoplasmic reticulum, glutamate excitotoxicity, and the reversal of the sodium-calcium exchanger as sources of intra-axonal calcium accumulation and instead identify nanoscale ruptures of the axonal plasma membrane as the critical path of calcium entry.
Keywords: axon degeneration, experimental autoimmune encephalomyelitis, multiple sclerosis, in vivo microscopy, calcium imaging, plasma membrane
Graphical Abstract
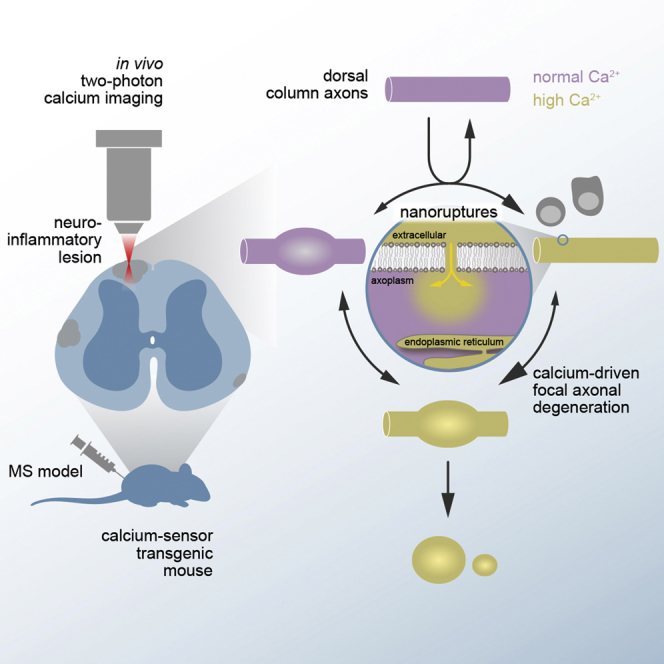
Highlights
-
•
Cytoplasmic calcium accumulations predict axon degeneration in neuroinflammation
-
•
Release from the axoplasmic reticulum does not explain calcium accumulations
-
•
Calcium influx from the extracellular space drives axon degeneration
-
•
Nanoscale ruptures allow entry of calcium across the axonal plasma membrane
Witte et al. identify cytoplasmic calcium accumulations as a key driver of axon degeneration in a model of multiple sclerosis. Calcium accumulates in the axoplasm because nanoscale ruptures of the axonal plasma membrane provide an entry path for extracellular calcium.
Introduction
Axon loss is a key determinant of persistent functional deficits in the common neuroinflammatory condition multiple sclerosis (Friese et al., 2014). We have previously shown that axons in multiple sclerosis and its animal model, experimental autoimmune encephalomyelitis (EAE), undergo focal axonal degeneration (Nikić et al., 2011). Focal axonal degeneration is a multi-step process that is initially characterized by axonal swellings and subcellular changes, such as mitochondrial damage, cytoskeletal disturbances, and impaired axonal transport (Nikić et al., 2011, Sorbara et al., 2014). These structural and functional changes, however, only progress to fragmentation in some axons, while they are fully reversible in others. Which molecular checkpoints decide whether focal axonal degeneration proceeds to irreversible axon degeneration or reverses toward recovery remain elusive.
Several lines of evidence suggest that the intra-axonal calcium concentration could be such a checkpoint. First, increased cytoplasmic calcium levels have been associated with axon degeneration processes following transection or contusion injuries (Williams et al., 2014, Vargas et al., 2015). Second, increased axonal calcium levels have been reported in neuroinflammatory lesions (Siffrin et al., 2010, Mossakowski et al., 2015). Finally, it has been shown that increased cytoplasmic calcium can trigger intra-axonal destruction pathways, such as the activation of calpain proteases (Yang et al., 2013).
Here, we now systematically explore the contribution and source of intra-axonal calcium in focal axonal degeneration using a spinal in vivo multiphoton imaging approach in combination with pre-existing and newly generated ratiometric calcium reporter mouse lines. By recording cytoplasmic calcium levels in individual axons that pass through neuroinflammatory lesions, we show that calcium accumulates in the axoplasm early during the focal axonal degeneration process. Following these axons over several hours then revealed that such calcium accumulations predict subsequent axonal fate: axons that clear calcium can recover, while those that maintain elevated calcium levels will likely degenerate. Monitoring and manipulating the calcium levels in distinct subcellular compartments then allowed us to identify the extracellular space—and not intracellular calcium stores—as the major source of the cytoplasmic calcium accumulation. Influx from the extracellular space occurs via nanoscale disruptions of the plasma membrane and therefore independent of classic calcium channels or pumps. Here, we thus propose calcium permeable membrane nanoruptures as a novel mechanism of inflammatory axon injury.
Results
Cytoplasmic Calcium Alterations Occur Early during Focal Axonal Degeneration and Predict Axonal Fate in Neuroinflammatory Lesions
To explore whether axoplasmic Ca2+ concentrations ([Ca2+]cyt) are altered in neuroinflammatory lesions, we induced EAE in Thy1-CerTN-L15 transgenic mice, in which neurons contain the genetically encoded calcium sensor CerTN in their cytoplasm (Heim et al., 2007). This fluorescence resonance energy transfer (FRET)-based sensor enables the ratiometric determination of calcium levels and in combination with spinal in vivo multiphoton imaging thus allowed us to record [Ca2+]cyt in individual dorsal column axons (Figure S1; see STAR Methods for details). While cytoplasmic calcium levels in healthy axons were tightly regulated, a substantial fraction of axons in acute EAE lesions showed elevated calcium levels. Notably, increased calcium levels were not only seen in fragmented (stage 2) and swollen (stage 1) axons, but also in about 10% of morphologically unaltered (stage 0) axons (Figures 1A–1C). This indicates that a dysregulation of [Ca2+]cyt occurs early during focal axonal degeneration.
Figure 1.

Cytoplasmic Calcium Levels Predict Axonal Fate in Neuroinflammatory Lesions
(A) In vivo multiphoton maximum intensity projection of spinal cord axons of healthy (left) and EAE (peak of disease, 2 days after onset; right) Thy1-CerTN-L15 mice. Top: grayscale images of YFP channel; bottom: ratiometric (yellow fluorescent protein/cyan fluorescent protein; YFP/CFP) images color coded for axonal cytoplasmic calcium ([Ca2+]cyt).
(B) Representative images of control axons in healthy spinal cord and normal-appearing (stage 0), swollen (stage 1), and fragmented (stage 2) axons in acute EAE (2–3 days after onset), color coded for [Ca2+]cyt.
(C) [Ca2+]cyt of single axons in healthy and acute EAE mice, plotted as YFP/CFP channel ratios and normalized to the mean of control axons. Top: percentage of axons with [Ca2+]cyt ≥3 SD above control mean, shown as mean ± SEM (tested per animal in n = 6 control and n = 11 EAE mice, Mann-Whitney U test).
(D) Multiphoton ratiometric time-lapse images of EAE lesions showing a normal-appearing axon rising in [Ca2+]cyt (top), a high [Ca2+]cyt axon undergoing focal axonal degeneration (middle), and the morphological recovery of a low [Ca2+]cyt axon (bottom).
(E) Transition probabilities of axons between morphological stages per observed axon hour in high [Ca2+]cyt (≥control mean + 3 SD, yellow) and low [Ca2+]cyt (magenta) states (n = 307, 8 mice, 1,201 axon hours).
Scale bars in (A), 25 μm, and in (B) and (D), 10 μm. ∗p < 0.05; ∗∗∗p < 0.001. See also Figure S1.
To assess how such dysregulated calcium levels influence axonal fate, we followed axon morphology and [Ca2+]cyt in a cohort of more than 300 axons in acute EAE lesions over time (n = 307 axons, 8 mice, imaged for a total of 1,201 “axon hours”). These experiments revealed that within an hour of observation about 4% of normal-appearing axons in neuroinflammatory lesions showed an intra-axonal calcium rise, and a substantial proportion of these axons (16% per hour) later progress to axonal swelling. In contrast, we rarely observed swelling of axons without a preceding calcium rise (Figures 1D and 1E). These results thus indicate that a rise of [Ca2+]cyt is the initiating step of focal axonal degeneration. Furthermore, cytoplasmic calcium levels also predict the subsequent progression along the focal axonal degeneration pathway: swollen [Ca2+]cyt-high axons had a higher probability of fragmentation and a lower probability of recovery compared to swollen axons that re-established cytoplasmic calcium homeostasis (Figures 1D and 1E). These results support the notion that elevations of cytoplasmic calcium trigger and drive axonal degeneration in neuroinflammatory lesions.
The Extracellular Space and Not Internal Stores Is the Major Source of Cytoplasmic Calcium Accumulation
Next, we explored which calcium sources are responsible for axoplasmic calcium accumulation. First, we investigated whether calcium release from the axonal endoplasmic reticulum (ER) initiates axonal degeneration, as has been shown in several axon injury models (Ouardouz et al., 2009, Li et al., 2013, Stirling et al., 2014, Villegas et al., 2014). To directly monitor axonal ER calcium [Ca2+]ER in vivo, we generated Thy1-TwitchER transgenic mice, which express ER-targeted Twitch-2B 54S+ (Thestrup et al., 2014), a FRET-based genetically encoded calcium indicator with an equilibrium dissociation constant in the range of resting axonal [Ca2+]ER (de Juan-Sanz et al., 2017) in neurons (Figures S2A and S2B). In vivo multiphoton imaging of Thy1-TwitchER mice allowed us to track changes of [Ca2+]ER induced by blocking ER calcium uptake (Figures S2C and S2D) or by contusion of spinal axons (Figures S2E, S2F, and S2G). We then measured [Ca2+]ER in individual axons in the healthy spinal cord and in acute spinal EAE lesions. In contrast to the changes in cytoplasmic calcium that precede morphological axon alterations, depletion of ER calcium was detected only in a small fraction of fragmented axons (Figures 2A–2C). Furthermore, when we induced ryanodine-receptor-mediated Ca2+ release from the ER by caffeine application to the spinal cord of healthy mice, we could efficiently deplete axonal ER calcium stores in vivo (Figures 2D and 2E; Video S1); however, such ER depletion did not result in sustained changes of [Ca2+]cyt in healthy axons (Figures 2F and 2G) or in axons that pass through neuroinflammatory lesions (Figures 2H and 2I). Taken together, these data indicate that calcium release from the axoplasmic reticulum occurs only late during focal axonal degeneration, and that—even when it occurs—ER depletion alone is insufficient to induce sustained changes of cytoplasmic calcium like those seen in focal axonal degeneration.
Figure 2.

Ca2+ Release from Axoplasmic Reticulum Does Not Cause Sustained Changes of Cytoplasmic Ca2+ Levels in Healthy and EAE Axons
(A) In vivo multiphoton maximum intensity projection of spinal cord axons of healthy (left) and EAE (2 days after onset, right) Thy1-TwitchER mice. Top: grayscale images of YFP channel. Bottom: ratiometric (YFP/CFP) images color coded for ER calcium levels ([Ca2+]ER).
(B) Ratiometric projection images showing [Ca2+]ER in healthy axons and normal-appearing (stage 0), swollen (stage 1), and fragmented (stage 2) axons in acute EAE lesions.
(C) [Ca2+]ER of single axons (YFP/CFP channel ratio normalized to the mean of control axons) in healthy spinal cord and different focal axonal degeneration stages in EAE (before and after application of 50 mM caffeine, respectively). Percentages (below) show the proportion of axons with depleted [Ca2+]ER < control mean – 3 SD, shown as mean ± SEM (tested per animal in n = 7 control and n = 7 EAE mice, paired t test control versus caffeine, Mann-Whitney U test control versus EAE stages 0–2 and EAE + caffeine, respectively).
(D, F, and H) Time-lapse projection images of axons in control spinal cord (D and F) and in an EAE lesion (H) before and after application of 50 mM caffeine. [Ca2+]ER is color coded in (D) and [Ca2+]cyt is color coded in (F) and (H).
(E, G, and I) Time course of control axon-normalized [Ca2+]ER (E) and [Ca2+]cyt (G and I) before and after caffeine application. Purple circles in (I) indicate low and yellow circles high [Ca2+]cyt axons at time point −50 s.
Presented as mean ± SD (n = 26 axons in E, 46 axons in G, 23 low versus 17 high [Ca2+]cyt axons in I). Scale bars in (A), (D), (F), and (H), 25 μm, and in (B), 10 μm. ∗∗p < 0.01; ∗∗∗p < 0.001. See also Figure S2 and Video S1.
In vivo multiphoton time-lapse of the healthy spinal cord axons of a Thy1-TwitchER and a Thy1-CerTN-L15 transgenic mouse upon application of 50 mM caffeine. Top: ratiometric (YFP/CFP) images color coded for ER calcium levels ([Ca2+]ER). Bottom: ratiometric (YFP/CFP) images color coded for cytoplasmic calcium levels ([Ca2+]cyt). Scale bar, 25 μm.
As ER calcium stores seemed insufficient to explain the cytoplasmic calcium rise in neuroinflammatory lesions, we next asked whether extracellular calcium accumulates in EAE axons. For this purpose, we locally removed extracellular calcium by incubating the exposed healthy or inflamed spinal cord with the Ca2+-chelator EGTA. While we observed no changes of [Ca2+]cyt in healthy axons (Figure S3), time-lapse in vivo imaging of acute EAE lesions revealed that extracellular calcium chelation normalized [Ca2+]cyt in both normal-appearing and swollen axons within 4 h (Figures 3A and 3B). Notably, normalization of [Ca2+]cyt was sufficient to induce morphological recovery of swollen axons and completely blocked axonal fragmentation in EAE lesions (Figure 3C). Influx of calcium from the extracellular space is thus the main driver of axon degeneration in neuroinflammatory lesions.
Figure 3.

Removal of Extracellular Ca2+ Reverses Cytoplasmic Calcium Accumulation and Prevents Axon Degeneration
(A) In vivo multiphoton maximum intensity projection of an acute EAE lesion (as ratiometric YFP/CFP images color coded for axonal [Ca2+]cyt), shown before (top, Pre) and after (bottom, 4 h EGTA) removal of extracellular Ca2+.
(B) [Ca2+]cyt plotted as YFP/CFP ratios of single-stage 0 (left) and stage 1 (right) axons before (Pre) and after (4 h EGTA) removal of extracellular calcium. Top: percentage of axons with [Ca2+]cyt ≥3 SD above control mean, shown as mean ± SEM (tested per animal in n = 5 control and n = 5 EAE mice, paired t test).
(C) Axon fate of [Ca2+]cyt-high stage 0 (left, 31 axons, 8 mice) and [Ca2+]cyt-high stage 1 (right, 66 axons, 8 mice) axons over 4 h with (EGTA) and without (artificial cerebral spinal fluid [aCSF]) removal of extracellular Ca2+ (chi-square test).
Scale bar in (A), 25 μm. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. See also Figure S3.
Nanoscale Ruptures of the Axonal Plasma Membrane Allow Extracellular Calcium Influx in Neuroinflammatory Lesions
Previous work has implicated glutamate excitotoxicity, opening of plasma membrane calcium channels, and reversal of the sodium-calcium exchanger as mediators of extracellular calcium influx in various forms of axon degeneration (Friese et al., 2014, Macrez et al., 2016). However, these canonical pathways of detrimental calcium overload did not seem to operate in focal axonal degeneration: neither the inhibition of the sodium-calcium exchanger (Figures S4A and S4C) or of voltage-gated calcium channels (Figures S4B and S4D) reduced cytoplasmic calcium levels or improved axonal fate in acute neuroinflammatory lesions, nor did the application of exogenous glutamic acid or glutamate agonists measurably alter cytoplasmic calcium levels or axonal fate (Figures S4E–S4G).
An alternative entry path for calcium into axons are disruptions of the plasma membrane, which have been previously described to occur in response to traumatic CNS injuries (Povlishock et al., 1983, Williams et al., 2014). To investigate whether such axonal membrane disruptions are also induced in neuroinflammatory lesions (in the absence of an obvious mechanical trigger), we subdurally applied fluorescently labeled macromolecules of different sizes (0.8 kDa cadaverine or 3, 10, and 70 kDa dextran) to the spinal cord of control and EAE animals. Fixed tissue analysis showed that all dyes remained excluded from the axoplasm in healthy animals, whereas a substantial proportion of axons in EAE lesions took up dye (Figures 4A and 4B). The fraction of labeled axons decreased with increasing size of the injected dye, and no significant axonal dye uptake was observed with the largest dye (70 kDa dextran), indicating that these membrane disruptions are <10 nm in size (Choi et al., 2010). Notably, ∼20% of normal-appearing axons, as well as many swollen and fragmented axons in EAE lesions showed cadaverine uptake, suggesting that such membrane nanoruptures occur already at the early stages of axon damage. Further analysis revealed that the presence of such nanoruptures was spatially correlated with local inflammation as the proportion of dye-positive axons as well as the extent of axon degeneration was higher in the densely infiltrated lesion center than in the lesion border (Figures S5A–S5C). In the lesion area, dye uptake was independent of axon caliber (Figure S5D) but appeared to be more pronounced in demyelinated axons compared to those axons that retain their myelin sheaths (Figure S5E). Investigating axonal dye uptake throughout the course of EAE further revealed that axonal membrane nanoruptures start with initial lesion formation at the onset of EAE symptoms and last to the chronic stage (at 15 days after EAE onset; Figures S5F and S5G). Axonal membrane damage thus occurs in lesions at different stages of the disease course and in axons at different stages of the degeneration process and can thus provide a persisting path for calcium entry during inflammatory axon damage.
Figure 4.

Ca2+ Enters through Nanoruptures of the Axonal Plasma Membrane
(A) Confocal projection of Thy1-CerTN-L15 axons (top) in healthy (left, control) and EAE (right) spinal cord after subdural injection of Texas-red-labeled dextran 3 kDa (bottom; red arrowhead indicates a dye-positive axon, white arrowhead a dye-negative axon).
(B) Percentage of dye-positive axons after application of fluorescent vital dyes with increasing sizes in control and EAE spinal cord (n = 5–9 animals per group, 78–205 axons per animal, means ± SEM, one-way ANOVA and Dunnett’s multiple comparison test).
(C) In vivo multiphoton projection of spinal cord axons in EAE Thy1-CerTN-L15 mice after subdural injection of Alexa-Fluor594-labeled cadaverine. Red arrowhead indicates dye uptake (left) in a [Ca2+]cyt -high axon (right).
(D) Left: [Ca2+]cyt levels of single dye-negative (gray dots) and dye-positive axons (red dots) in healthy and acute EAE mice, plotted as YFP/CFP channel ratios and normalized to the mean of control axons. Top: percentage of axons with [Ca2+]cyt ≥3 SD above control mean, shown as mean ± SEM (tested per animal in n = 7 control and n = 12 EAE mice, Mann-Whitney U test), Right: percentage dye-positive axons for [Ca2+]cyt-high (yellow area) and [Ca2+]cyt-low (magenta area) axons in the same control and EAE animals (means ± SEM, Mann-Whitney U test).
Scale bars in (A) and (C), 10 μm. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. See also Figures S4 and S5.
To directly test this hypothesis, we subdurally applied fluorescently labeled cadaverine in Thy1-CerTN-L15 mice and performed simultaneous in vivo imaging of dye uptake and [Ca2+]cyt. In line with our fixed-tissue analysis, we observed virtually no axonal uptake of cadaverine in healthy animals in vivo, whereas in EAE animals imaged at the peak of disease a substantial fraction of axons took up cadaverine (Figures 4C and 4D). At the same time, dye uptake and cytoplasmic calcium elevations correlated strongly on a single-axon level in vivo. For example, more than half of the normal-appearing axons that were dye positive also showed elevated calcium levels, while in the same lesions such calcium elevations are observed in only about 5% of normal-appearing axons that do not show dye uptake (Figure 4D). Membrane disruptions appear to be a major path of axonal calcium influx as about 60% of normal-appearing and swollen axons with elevated cytoplasmic calcium take up dye (Figure 4D).
Discussion
Our study identifies the cytoplasmic accumulation of calcium as a key driver of axon degeneration in neuroinflammatory lesions. This is in line with a number of previous studies of damaged axons after CNS trauma that have identified calcium overload as an important predictor of both the initial axonal interruption after mechanical impact (Williams et al., 2014), local axonal degeneration after trauma (Kerschensteiner et al., 2005, Knöferle et al., 2010), as well as of Wallerian degeneration, which finally removes the disconnected distal axon end (Vargas et al., 2015). Increased axoplasmic calcium levels can interfere with axon survival, by activating calcium-dependent proteases, such as calpains that break down the cytoskeleton, by promoting the secondary influx of other cations that lead to oncotic axonal swelling, and by interfering with the function of crucial organelles, such as mitochondrial ATP production (Friese et al., 2014, Bano and Ankarcrona, 2018). Our in vivo time-lapse observations (cf. Figures 1D and 1E), however, also emphasize that such cytoplasmic calcium accumulation is not necessarily a “point of no return,” as even a proportion of morphologically altered (swollen) axons manage to clear elevated calcium levels. This often results in subsequent morphological recovery, suggesting that also under the sustained stress of a neuroinflammatory lesion a “window of opportunity” exists to rescue axons in a metastable prodromal state of degeneration, just as we described previously for acute trauma (Williams et al., 2014). Hence, the fate of axons in neuroinflammatory lesions is likely decided by the net balance of calcium sources and calcium sinks with both sides of this balance being potentially amendable to therapy.
Our results further indicate that the extracellular space, in which the free calcium concentration is about 10,000-fold higher than in the axonal cytoplasm, is a major source of the cytoplasmic calcium accumulation. This is supported by our findings that removal of extracellular calcium by superfusion of the spinal cord with the calcium chelator EGTA normalized intraaxonal calcium levels and effectively halts axonal degeneration. This experiment per se does not exclude a role of internal calcium stores, as, for example, “store-operated calcium entry” (SOCE) opens a path for calcium from the extracellular space in response to calcium efflux from the ER (Li et al., 2013, Amcheslavsky et al., 2015). However, our direct measurements of ER calcium levels in mature axons show that such a depletion of the axonal ER calcium stores is not occurring early during focal axonal degeneration but can only be observed during late stages. This sets axon degeneration in inflammatory lesions aside from secondary axon injury after spinal laser lesions, where prior reports documented early involvement of ER stores (Stirling et al., 2014). Furthermore, in our experiments, the pharmacological induction of ER calcium depletion in mature spinal cord axons did not result in marked changes of the intra-axonal calcium levels or any overt signs of axon degeneration—irrespective of whether this depletion was done in a normal or a neuroinflammatory environment. These findings indicate that by itself depletion of axonal ER stores does not cause measurable increases in axoplasmic calcium in myelinated axons in vivo, which is in accordance with recent in vitro measurements (de Juan-Sanz et al., 2017). Hence, our data suggest that depletion of ER stores alone is unlikely to initiate axonal degeneration. How can these results be squared with the well-established fact that the ER contains micromolar concentrations of calcium (∼150 μM; de Juan-Sanz et al., 2017) and the prior reports of a role of ER-derived calcium in ischemic and traumatic axon degeneration in the spinal cord (Stirling et al., 2014, Ouardouz et al., 2003)? First, the amount of calcium contained in the axonal ER might be lower than often assumed—while the concentration appears to exceed axoplasmic levels by two orders of magnitude, the volume fraction of axonal ER is small compared to the somato-dendritic compartment, often forming a spare network of thin (<50 nm) tubules (Terasaki, 2018, Wu et al., 2017). While our imaging approaches lack resolution to confirm this morphology in vivo, confocal imaging in our Thy1-TwitchER mice showed that neuronal cell bodies and proximal dendrites are labeled much brighter than the corresponding axons, suggesting a lower volume fraction (Figures S2A and S2B). Still, the fact that, under non-inflammatory stress conditions, ER-derived calcium plays important roles suggests that other processes—for example, impaired clearance across the plasma membrane or into other organelles, such as mitochondria—contribute in other disease settings, but not in the neuroinflammatory lesions studied here.
Together these findings highlight how directly measuring calcium levels in different axonal compartments can illuminate our understanding of the role of calcium homeostasis in diseased axons. Indeed, when we used our approach to probe the mechanisms of calcium influx across the axonal plasma membrane, we found that canonical pathways of neuronal calcium influx such as opening of glutamate receptor and voltage-gated calcium channels or reverse action of the sodium-calcium exchanger are unlikely to drive calcium accumulation during focal axonal degeneration. In the case of glutamate, we found that, while exposure of the dorsal spinal cord to glutamic acid is sufficient to induce cytoplasmic calcium accumulation in neuronal cell bodies that can be found dispersed in the white matter, no such changes of the cytoplasmic calcium are induced in axons that run next to these neurons. One possible explanation might be the different densities of ionotropic glutamate receptors that are relatively abundant at synaptic and extra-synaptic locations on the cell body and dendrites but found only in confined nanocomplexes along mature axons in rodents and humans (Ouardouz et al., 2009, Christensen et al., 2016). Our data here suggest that the glutamate receptors in such nanocomplexes are insufficient to induce marked changes of the intra-axonal calcium levels or morphological alterations at least in mature CNS axons as probed here.
We also did not observe a protective effect of bepridil, a drug that blocks the sodium-calcium exchanger (Kapoor et al., 2003), on axonal calcium levels or the progression of focal axonal degeneration despite the fact that a similar local application of bepridil was able to completely block cytoplasmic calcium accumulation and axon damage induced by application of an exogenous nitric oxide (NO) donor. The latter finding is consistent with previous reports that have demonstrated a protective effect of sodium channel blockers and inhibitors of the sodium-calcium exchanger on nitric-oxide-mediated neurodegeneration (Kapoor et al., 2003). Taken together, these findings indicate that, while NO and its downstream products can effectively induce axon degeneration (Kapoor et al., 2003, Nikić et al., 2011), it is only one of several toxic mediators in EAE lesions and targeting NO actions alone is insufficient to prevent inflammatory axon degeneration at least in acute highly inflammatory EAE lesions. Similarly, blocking L- and T-type calcium channels with lomerizine, an intervention that limits secondary axon degeneration after optic nerve injury (Fitzgerald et al., 2009), could not prevent calcium accumulation and axon degeneration in acute neuroinflammatory lesions.
Indeed, overall our experiments failed to identify a pharmacologically defined pathway of calcium entry into axons in EAE lesions. Instead, experiments using dye exclusion as previously used in settings of blunt CNS trauma by others and us (Povlishock et al., 1983, Williams et al., 2014) revealed that also in neuroinflammation axonal membrane integrity is impaired. Based on the molecular weight of the excluded dyes, it appears that nanoscale (<10 nm; Choi et al., 2010) ruptures form in the axonal membrane. Such nanoruptures could arise based on several mechanisms: first, just as in CNS trauma, mechanical forces could be in play, e.g., based on osmotic imbalances that result in axonal swelling; still, such “mechanoporation” due to toxic edema seems unlikely in neuroinflammation, as the first evidence of calcium dyshomeostasis and dye influx are evident in our experiments before axonal shape changes are apparent. Therefore, a second scenario seems more likely, where membrane disruptions are due to toxic mediators, such as reactive species, which are abundant in neuroinflammatory lesions (e.g., Nikić et al., 2011, Mossakowski et al., 2015) and can cause lipid peroxidation, increased membrane rigidity, and altered membrane permeability (Di Domenico et al., 2017). Indeed, there is evidence for oxidized phospholipids in both oligodendrocyte membranes, as well as the membranes of some axonal spheroids in multiple sclerosis (MS) lesions (Haider et al., 2011). Similarly, secreted phospholipases, which are expressed by immune cells in MS and EAE lesions, could directly attack the plasma membrane (Kalyvas et al., 2009). Finally, the immune system employs a well-defined armamentarium of pore-forming proteins, such as the membrane attack complex, which is formed as part of the terminal complement pathway—a pathway that has been shown to be activated in both MS and a related animal model (Watkins et al., 2016, Michailidou et al., 2018). Perforin is another pore-forming protein that is released from cytotoxic granules of CD8+ T and natural killer (NK) cells, which has been implicated as a mediator of autoimmune-mediated attacks on oligodendrocytes, as well as on axons (Deb et al., 2009).
Indeed, it is worth noting that focal axonal degeneration can affect myelinated axons that are somewhat shielded from macromolecular exposure, suggesting that areas devoid of myelin, such as nodes of Ranvier, could be a site of initial damage, as we originally observed (Nikić et al., 2011). In line with these findings, we observed that demyelination further increased the probability of axonal dye uptake, probably because loss of the myelin sheaths facilitates access to the axonal membrane along the internode. That myelin can offer partial protection against membrane ruptures would argue that chronically demyelinated axons at advanced stages of the disease might be particularly vulnerable to such an insult. Indeed, while both membrane damage and axon degeneration were most pronounced in the densely infiltrated center of acute EAE lesions, we observed persisting dye uptake also at later lesion stages.
Independently of the exact entry pathway of calcium, our data demonstrate that even axons that contain high levels of cytoplasmic calcium can still spontaneously recover—as we previously observed after blunt trauma, where damaged axons could recover provided the duration of calcium dyshomeostasis remained below 2 h (Williams et al., 2014). While this opens only a very narrow window of intervention after trauma, the sustained and desynchronized time course of inflammatory axon damage suggests that in neuroinflammatory lesions therapeutically targeting membrane ruptures is more realistic. Indeed, the fact that axons can survive even after their membranes are disrupted implies that not only neutralizing membrane-attacking immune mediators or stabilizing membranes against such attacks are a future target to explore, but also the machinery of cellular membrane repair, which exists in axons and which is increasingly understood at the molecular level in non-neuronal cells (Andrews et al., 2014, Tang and Marshall, 2017). Hence, our findings establish a novel mechanism of inflammatory axon injury and identify the axonal plasma membrane as a therapeutic target structure in MS.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-MBP | Dako | Cat# A0623; RRID: AB_2650566 |
| Rabbit polyclonal anti-GRP78 BiP | Abcam | Cat# ab21685, RRID: AB_2119834 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen/Thermo Fisher Scientific | Cat# A-21245, RRID: AB_2535813 |
| Bacterial and Virus Strains | ||
| M. tuberculosis H37 Ra, desiccated | BD Difco | Cat# 231141 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MOG recombinant protein (N1-N125) | Nikić et al., 2011 | N/A |
| Incomplete Freund’s adjuvant | Sigma-Aldrich | Cat# F5506 |
| Pertussis toxin from Bordetella pertussis | Sigma-Aldrich | Cat# P7208 |
| Paraformaldehyde | Sigma-Aldrich | Cat# P6148 |
| NeuroTrace 435/455 | Invitrogen/Thermo Fisher Scientific | Cat# N21479 |
| Vectashield | Vector Laboratories | Cat# H1000 |
| Ketamine10% | WDT | VetCode QN01AX03 |
| Xylazine 2% | Serumwerk Bernburg | VetCode QN05CM92 |
| Medetomidin Cepetor | CP-pharma | VetCode QM05CM91 |
| Midazolam | Ratiopharm | PZN# 4921530 |
| Fentanyl | B. Braun Melsungen | PZN# 6900650 |
| EGTA (Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N’-tetraacetic acid) | Sigma-Aldrich | Cat# E3889 |
| Caffeine | Sigma-Aldrich | Cat# C0750 |
| Thapsigargin - Calbiochem | Merck | Cat# 586005 |
| Bepridil hydrochloride | Sigma-Aldrich | Cat# B5016 |
| L-Glutamic acid | Sigma-Aldrich | Cat# G1251 |
| Glycine | Sigma-Aldrich | Cat# G7126 |
| Spermine NONOate | Enzo Life Sciences | Cat# ALX-430-013 |
| Lomerizine dihydrochloride | Sigma-Aldrich | Cat# L6295 |
| NMDA | Sigma-Aldrich | Cat# M3262 |
| Kainate | Sigma-Aldrich | Cat# K0250 |
| AMPA | Sigma-Aldrich | Cat# A6816 |
| Alexa Fluor 594 Cadaverine | Invitrogen/Thermo Fisher Scientific | Cat# A30678 |
| Invitrogen Molecular Probes Dextran Texas Red, 3000 MW, Lysine Fixable | Invitrogen/Thermo Fisher Scientific | Cat# D3328 |
| Invitrogen Molecular Probes Dextran Texas Red, 10,000 MW, Lysine Fixable | Invitrogen/Thermo Fisher Scientific | Cat# D1863 |
| Invitrogen Molecular Probes Dextran Texas Red, 70,000 MW, Lysine Fixable | Invitrogen/Thermo Fisher Scientific | Cat# D1864 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.Thy1-CerTN-L15 | Heim et al., 2007 | N/A |
| Mouse: B6.Thy1-CFP-23 | Feng et al., 2000 | N/A |
| Mouse: B6.Cg-Tg(Thy1-YFP)HJrs/J | The Jackson Laboratory | Cat# JAX:003782; RRID: IMSR_JAX:003782 |
| Mouse: B6.Thy1-TwitchER 481 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: Twitch-2B 54S+ | Thestrup et al., 2014 | N/A |
| Plasmid: G-CEPIA1er (Vh-chain ER-signal peptide) | Suzuki et al., 2016 | Addgene Plasmid #58215 |
| Plasmid: Thy1-MCS | Caroni, 1997 | N/A |
| Software and Algorithms | ||
| Adobe Creative Suite 6 (Photoshop, Illustrator) | Adobe Systems | https://www.adobe.com/de/products/cs6.html |
| Fiji | SciJava | https://fiji.sc |
| GraphPad Prism, Version 6.0 and 7.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| SPSS Statistics for Windows, Version 22.0 | IBM Corporation | https://www.ibm.com/analytics/de/de/technology/spss/ |
Contact for Reagent and Resource Sharing
Further information and reasonable requests for resources and reagents may be directed to and will be fulfilled by, the Lead Contact, Martin Kerschensteiner (Martin.Kerschensteiner@med.uni-muenchen.de).
Experimental Model and Subject Details
All animal experiments were performed in accordance with regulations of the relevant animal welfare acts and protocols approved by the Animal Study Committee of the Regierung von Oberbayern. Mouse strains used in this study included wild-type and mutant mice. Genotypes and sources of transgenic mice are listed in the Key Resources Table. Both female and male adult mice (6-12 weeks of age) were used for the experiments. No differences between males and females were observed in this study. Experimental mice were kept and bred in the animal facilities of the institution under standard conditions. Animals were held in Eurostandard Type II long cages (Tecniplast) stored in an IVC rack system with a maximum of five mice per cage and provided with bedding and nesting material. Autoclaved food and water were supplied ad libitum. Mice were held at a 12 h light/12 h dark cycle. Mice were randomly assigned to experimental groups. Group sample sized were chosen based on previous studies.
Method Details
Generation Thy1-TwitchER mice
For measuring of endoplasmic reticulum calcium dynamics, we expressed ER targeted Twitch-2B 54S+, a ratiometric calcium sensor containing mCerulean3 and cpVenusCD and exhibiting a calcium equilibrium dissociation constant of 174.0 μM (Thestrup et al., 2014), under control of the regulatory elements of the Thy1-gene (Feng et al., 2000). To achieve ER targeting, an in-frame fusion-gene of the mouse Vh-chain ER-signal peptide and Twitch-2B 54S+ was created. This construct was inserted into an expression vector under the control of the Thy1-promoter (Feng et al., 2000). TwitchER transgenic mice on a C57Bl6 background were generated by pronuclear injection using standard protocols. Eleven founder lines were screened and one founder line (TwitchER 481) with bright subset labeling in DRG neurons was selected for this study.
Experimental autoimmune encephalomyelitis
Adult mice (6-12 weeks of age) were induced with EAE as previously described (Nikić et al., 2011). In short, we immunized animals with 250 μL of an emulsion containing 200-450 μg of purified recombinant myelin oligodendrocyte glycoprotein (MOG, N1-125, expressed in E. coli) and incomplete Freund’s adjuvant (Sigma Aldrich) with 5 mg/ml mycobacterium tuberculosis H37 Ra (BD Difco). Pertussis toxin (250-400 ng, Sigma Aldrich) was administered intraperitoneally on day 0 and day 2 after immunization. We assessed animal weight daily and assigned a score according to the severity of neurological deficits using an established EAE scoring scale: 0, no detectable clinical signs; 0.5, partial tail weakness; 1, tail paralysis; 1.5, gait instability or impaired righting ability; 2, hind limb paresis; 2.5, hind limb paresis with partial dragging; 3, hind limb paralysis; 3.5, hind limb paralysis and forelimb paresis; 4, hind limb and forelimb paralysis; 5, death.
Induction of spinal cord contusions
Spinal cord contusions were performed using the Infinite Horizons Impactor (Precision Systems and Instrumentation) as previously described (Williams et al., 2014). Briefly, pre-lesion images were obtained between L2 and L4, before a contusion injury was induced with a set force at 40 kDyne (which resulted in an actual force range of 40 to 45 kDynes). Imaging sites with significant subdural bleeding were not used for further analysis. Starting < 10 min after contusion injury, the injured spinal cord was imaged for 2 h every 15 min.
Tissue preparation and immunofluorescence
Mice were transcardially perfused with 4% (wt/vol) paraformaldehyde in phosphate buffered saline and spinal cords were post-fixed for 12-24 h. Next, lumbar spinal cords were isolated, cryoprotected using 30% (wt/vol) sucrose in phosphate buffered Saline (PBS) and cut using a cryostat into 30 μm thick longitudinal sections for dye analysis and 30 μm thick cross-sections for MBP immunofluorescence. For myelin basic protein (MBP) immunofluorescence, sections were first blocked with 10% goat serum in 0.1% (vol/vol) Triton X-100 in PBS and then incubated overnight with rabbit polyclonal MBP antibody (1:200, DAKO, A0623) at 4°C. MBP staining was visualized by incubating sections for 1 h with Alexa-Fluor-conjugated goat anti-rabbit (1:500, Thermo Fisher Scientific/Invitrogen). Sections were counterstained with the Nissl-like nucleic acid stain NeuroTrace 435/455 (Invitrogen), and then mounted in Vectashield (Vector Laboratories). For evaluation of Thy1-TwitchER mice, lumbar spinal cord and attached dorsal root ganglia (DRG) sections were cut into 30 μm transversal cryosections for immunostaining of ER structures and 40 μm section for overview scans. Sections were first mounted on-slide, blocked with 10% goat serum in 0.1% (vol/vol) Triton X-100 in PBS and then incubated for two days with a rabbit polyclonal antibody to GRP78 BiP (1:100, ab21685, Abcam) at 4°C. GRP78 BiP staining was visualized by incubating sections for 3 h with Alexa-Fluor-647-conjugated goat anti-rabbit (1:500, Thermo Fisher Scientific/Invitrogen) at room temperature and then mounted in Vectashield (Vector Laboratories).
Confocal microscopy
Upright confocal laser-scanning microscopes, FV1000 (Olympus) or SP8 (Leica), equipped with standard filter sets and laser lines were used for imaging fixed tissue. To assess dye uptake in spinal axons, 30 μm thick longitudinal cryosections were imaged with a 20x/0.85 NA oil immersion UPLSAPO objective (Olympus), 2x digital zoom, Z-spacing of 500 nm and a pixel resolution of 207 nm. To minimize bleed-through, all signals (NeuroTrace 435/455, YFP for axons, vital dye TexasRed) were scanned in frame-sequential mode. To assess the myelination status of spinal axons in Thy1-CerTN-L15 mice, 30 μm thick spinal cord transversal cryosections were imaged with a 60x/1.42 NA oil immersion UPLSAPO objective (Olympus) and 2.0x digital zoom. For spinal cord overview images of Thy1-TwitchER mice a tile scan was performed with a 20x/0.75 NA oil immersion HC PL APO CS2 objective (Leica). For high resolution images of GRP78 BiP labeled DRG neurons, single DRG neurons in 30 μm sections were imaged with a 63x/1.40 oil immersion HC PL APO CS2 objective (Leica) and a 3.5x digital zoom.
In vivo microscopy
We exposed the dorsal lumbar spinal cord for in vivo imaging and in vivo manipulation as previously described (Nikić et al., 2011, Romanelli et al., 2013). Animals were anaesthetized with ketamine-xylazine (ketamine 87 mg/kg, xylazine 13 mg/kg) or a mixture of medetomidin (0.5 mg/kg), midazolam (5 mg/kg) and fentanyl (0.05 mg/kg) intraperitoneally. A laminectomy was performed to gain access to the lumbar spinal cord. When exposed, the spinal cord was continuously superfused with artificial mouse cerebral spinal fluid (aCSF, 148.2 NaCl, 3.0 KCl, 1.4 CaCl2, 0.8 MgCl2, 0.8 Na2HPO4 and 0.2 NaH2PO4 in mM). The vertebral column was fixed using a spinal adaptor for a stereotaxic frame (Narishige STS-A, Davalos et al., 2008) and the spinal opening was framed using 2%–4% agarose diluted in aCSF to form a fluid reservoir throughout the imaging session. During imaging sessions anesthesia was maintained by regular injections and vital signs were checked between image acquisitions. In vivo microscopy was performed using Olympus FV1000-MPE and FV1200-MPE two-photon microscopy systems. All microscopes were equipped with femto-second pulsed Ti:Sapphire lasers (Mai Tai HP-DS, Spectra-Physics) and laser power was attenuated by acousto-optical modulators. Emission was detected with non-descanned conventional and gallium arsenide phosphide (GaAsP) detector photomultiplier tubes (PMTs, Hamamatsu).
To image cytoplasmic and ER calcium concentration, the genetically encoded calcium indicator proteins CerTN-L15 and Twitch-2B 54S+ were imaged using a two-photon wavelength of 840 nm to simultaneously excite both Cerulean and Citrine (CerTN-L15) and mCerulean3 and cpVenus (Twitch-2B 54S+), respectively. Fluorescence was collected in a cyan channel (here referred to as “CFP”) and yellow channel (“YFP”) using emission barrier filter pairs with 460-510 and 515-560 nm bandwidth (for conventional PMT detectors on the Olympus FV1000 MPE) or 460-500 and 520-560 nm bandwidth (for GaAsP detectors on the Olympus FV1000 MPE) or 455-490 and 526-557 nm (for GaAsP detectors on the Olympus FV1200 MPE). For imaging FRET signals together with vital dyes, emission light was first passed through a 565-nm short pass dichroic mirror, the CFP/YFP cube was positioned in front of GaAsP detectors and a 660-740 barrier filter was positioned in front of a conventional PMT to detect red signals. Images were acquired in 12 bit with an 25x/1.05 dipping cone water-immersion objective, pixel size of 0.26 μm pixel−1 or smaller, dwell time of 2.0 μs pixel−1 and laser power of 30-50 mW measured in the back focal plane. Images were acquired in volume stacks penetrating up to 60 μm into the tissue from the spinal cord surface. Both in healthy animals and EAE animals no signs of photodamage could be observed over an imaging period of 4h using these imaging conditions (data not shown). Animals showing signs of traumatic damage after laminectomy were excluded from the analysis.
To measure cytoplasmic or ER calcium levels in EAE, axons were analyzed that were in an inflamed area of the dorsal spinal cord. Local inflammation was assessed based on the presence of ongoing axonal degeneration (Nikić et al., 2011) and/or cellular infiltrates, labeled with nuclear stains as previously described (Romanelli et al., 2013). Acute EAE lesions (until 3d after clinical onset) were studied in animals that showed a clinical EAE score of 2 or higher, at which stage near confluent lesions are present in the superficial dorsal spinal cord (Sorbara et al., 2014).
Application of dyes and pharmacological substances
For bath application of pharmacological substances, the dura was carefully removed at the laminectomy site using a custom-made dura hook with a fine 30G needle. The spinal cord was exposed to EGTA (50mM in Ca2+ free, Mg2+ free aCSF applied from 0 to 240 min; Sigma-Aldrich), caffeine (50 mM in aCSF, from 0 - 2.5 min; Sigma Aldrich), thapsigargin (100 μM, 1% DMSO in aCSF, from 0-80 min; Calbiochem), bepridil (0.1mM, 1% DMSO in aCSF from 0 to 240 min, Sigma Aldrich), lomerizine (0.1 mM, 1% DMSO in aCSF from 0 to 240 min, Sigma Aldrich), glutamic acid (glutamic acid 100mM, glycine 10mM in aCSF, from 0 to 240 min, Sigma Aldrich), glutamate receptor agonists (NMDA 1 mM, glycine 0.1 mM, kainate 2 mM, AMPA 1mM in aCSF, from 0 to 120 min, Sigma Aldrich) or a nitric oxide donor (spermine NONOate, 50mM, Enzo Life Sciences). Images were acquired for pre-treatment time points in aCSF-bath solution and during treatment in vehicle or treatment solution, respectively. After the in vivo imaging was completed, animals were euthanized.
For dye application, 2 μL of the different fluorescently labeled vital dyes (Alexa-Fluor-594-conjugated cadaverine, and Texas-red-conjugated dextran 3, 10, and 70 kDa; all from Invitrogen /Thermo Fisher Scientific) were injected subdurally using a hand-held pulled glass micropipette as described previously (Williams et al., 2014). Two hours after dye-injection, mice were either imaged in vivo or transcardially perfused for confocal analysis of fixed tissue sections. Both in vivo and confocal images were always acquired contralateral to the side of dye injection to exclude any trauma-induced dye-uptake.
Quantification and Statistical Analysis
Processing and analysis of in vivo images
Images were post-processed using the open-source image analysis software, ImageJ/Fiji (http://fiji.sc), and the image-processing software, Photoshop (Adobe). In Fiji, the CFP and YFP (FRET) channels were individually visualized in a greyscale look-up table to determine axon morphology and diameter. We assessed axonal fragmentation in contrast-adjusted 3D image stacks by following individual axons in several slices to clearly distinguish fragmented axons from swellings. For Ca2+ measurements, cytosolic or ER fluorescence intensities were measured in the CFP and YFP channels, respectively. Per measured axon, three circular or one longitudinal region of interest in the axon were averaged for fluorescence intensity measurements (nearby non-axonal areas were selected for background correction). As we detected crosstalk from the CFP to the YFP channel (not vice-versa) when imaging Thy1-CFP-23 and Thy1-YFP-16 mice, we corrected the YFP signal by subtracting the measured crosstalk-fraction of the CFP signal (this value was determined for each microscope system used). Background-corrected YFP/CFP ratios were interpreted as a proxy of Ca2+ concentration as previously established for FRET-based calcium sensors (Mank and Griesbeck, 2008). Axons were considered to be Ca2+-elevated if YFP/CFP ratios showed elevation over the mean plus 3 standard deviations of the ratios measured in axons in the healthy spinal cord. We chose this binary scoring approach because the sigmoidal response curve of genetically encoded calcium indicators and their relatively low calcium equilibrium dissociation constant compared with extracellular calcium concentrations precludes an informative measurement of the actual calcium levels inside injured axons in our setting. For analyses of ER calcium levels, Ca2+ depletion was defined by a YFP/CFP ratio of 3 SD below the mean of the ratios measured in axons in the healthy spinal cord. For baseline characterizations of lesions, all axons in an image stack crossing a virtual medio-lateral line were measured. YFP/CFP ratios were normalized to the mean of control animals measured on the same system and time period. For time course analyses, only axons were chosen that could be followed over the time period of 2-4 h.
To analyze axonal Ca2+ together with dye-uptake in vivo, we first calculated YFP/CFP ratios as described above and then performed a bleed through-correction for the vital dye signal. To assess dye-uptake in the same axon, we calculated the intensity of the axonal vital-dye signal over local background. Axons with dye intensity above the mean + 3 SD of the control axon population in healthy spinal cords were scored as dye-positive. As in some cases dye uptake by nearby inflammatory cells in the EAE lesion increased the local background signal, each outlined axon was additionally scored as dye-positive or dye-negative by a blinded observer to validate the quantitative assessment. If axons were considered dye-positive by the observer but dye-negative according to the calculated ratio, local background was measured on the opposite side of the axon and used to calculate a new signal over background.
Ratiometric images presented in this work were processed as follows: in Fiji, maximum intensity projections of 3D stack sections were created from CFP and YFP images individually and a binary thresholded mask of axonal structures was generated from the image channel with higher signal-to-noise. Projection images were each multiplied by the binary mask and the resulting images divided by each other (YFP/CFP). The resulting image was pseudocolored with a custom look-up table spanning from dark blue via magenta to yellow hues. Images were exported as RGB images to Photoshop, despeckled and overlayed on the original greyscale maximum intensity projection, using the “Overlay” layer function.
Analysis of fixed tissue sections
Lesion areas in EAE animals were identified based on the increased cell density detected by NeuroTrace 435/455 labeling. In lesion areas, we subsequently outlined single axons and staged them as previously described (Nikić et al., 2011). Average dye signal intensity was determined within the outlined axons and in a similar area right next to the outlined axons. An axon was considered dye-positive when the ratio between signal intensity inside the axon and signal intensity outside the axon was greater than the mean value + 3 SD of the signal intensity measured in control axons in the healthy spinal cord. Similar to our in vivo analysis of dye uptake, axons were additionally scored by a blinded observer as dye-positive or -negative and new signals over background were calculated, if necessary, as described above. For a subset of animals (n = 5), images were analyzed by two different raters, which yielded comparable results. Rater A: 26 ± 7 (stage 0), 28 ± 5 (stage 1) and 16 ± 6 (stage 2); Rater B: 22 ± 2 (stage 0), 31 ± 14 (stage 1) and 17 ± 5 (stage 2) dye positive axons (in mean % ± SEM). Two sets of experiments performed by different experimenters (n = 4 and n = 5 animals, respectively) also resulted in similar dye uptake. Experimenter A: 18 ± 4 (stage 0), 28 ± 2 (stage 1) and 18 ± 4 (stage 2); Experimenter B: 11 ± 3 (stage 0), 21 ± 4 (stage 1) and 15 ± 2 (stage 2) dye positive axons (in mean % ± SEM).
Statistical analyses
Statistical details of experiments, including statistical test used and level of statistical significance can be found in the figure legends with number of animals/axons per group, respectively. Sample sizes were chosen according to previous in vivo imaging studies of spinal axons (Nikić et al., 2011, Williams et al., 2014). Statistical significance was calculated with SPSS Statistics for Windows (Version 22.0. IBM Corp) and Prism (Versions 6.0 and 7.0, Graphpad) using ANOVA and (paired) t tests (where normal distribution could be confirmed) or Kruskal-Wallis, Mann-Whitney U or Wilcoxon signed rank tests (where normal distribution could not be confirmed). Chi-square tests were used to compare distribution of axon stages. Normality of distribution was assessed with Shapiro-Wilk test. Obtained p values were corrected for multiple comparisons using Bonferroni correction or Dunnett’s multiple comparison test. Data were judged to be statistically significant when p < 0.05 and, if significant, reported in the figures using significance levels indicated in the figure legends.
Acknowledgments
We would like to thank A. Schmalz, L. Schödel, B. Fiedler, and Y. Hufnagel for excellent technical assistance and D. Matzek, B. Stahr, and N. and M. Budak for animal husbandry. Work in M.K.’s laboratory is financed through grants from the DFG (TRR128, Project B10 and B13), the European Research Council (ERC, FP/2007-2013; ERC Grant Agreement no. 310932), the BMBF (KKNMS), the DMSG, and the “Verein Therapieforschung für MS-Kranke e.V.” Work in T.M.’s lab is supported by the DFG (CIPSM EXC114, CRC870, Mi 694/8-1) and the ERC (FP/2007-2013; ERC Grant Agreement no. 616791). M.K. and T.M. are further supported by the DFG (Ke 774/5-1/Mi 694/7-1) and the Munich Cluster for Systems Neurology (SyNergy; EXC 1010). A.S. and J.L. were supported by the Graduate School of the TU München; A.-M.S., C.F.M., and J.P.B. were supported by the “FöFoLe” program at LMU Munich. P.R.W. was supported by the “Wings for Life” foundation.
Author Contributions
M.K. and T.M. conceived the experiments. M.E.W., A.-M.S., C.F.M., J.P.B., and P.S. performed and analyzed in vivo imaging of cytoplasmic calcium changes in axons. J.L. and A.S. performed and analyzed in vivo imaging of ER calcium changes in axons. R.N., P.R.W., A.S., and O.G. generated and characterized calcium sensors and reporter mouse lines. T.M., M.E.W., A.-M.S., C.F.M., J.L., and M.K. wrote the paper.
Declaration of Interests
The authors declare no competing interests.
Published: January 24, 2019
Footnotes
Supplemental Information includes five figures and one video and can be found with this article online at https://doi.org/10.1016/j.neuron.2018.12.023.
Contributor Information
Thomas Misgeld, Email: thomas.misgeld@tum.de.
Martin Kerschensteiner, Email: martin.kerschensteiner@med.uni-muenchen.de.
Supplemental Information
References
- Amcheslavsky A., Wood M.L., Yeromin A.V., Parker I., Freites J.A., Tobias D.J., Cahalan M.D. Molecular biophysics of Orai store-operated Ca2+ channels. Biophys. J. 2015;108:237–246. doi: 10.1016/j.bpj.2014.11.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews N.W., Almeida P.E., Corrotte M. Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Biol. 2014;24:734–742. doi: 10.1016/j.tcb.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bano D., Ankarcrona M. Beyond the critical point: an overview of excitotoxicity, calcium overload and the downstream consequences. Neurosci. Lett. 2018;663:79–85. doi: 10.1016/j.neulet.2017.08.048. [DOI] [PubMed] [Google Scholar]
- Caroni P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J. Neurosci. Methods. 1997;71:3–9. doi: 10.1016/s0165-0270(96)00121-5. [DOI] [PubMed] [Google Scholar]
- Choi J.J., Wang S., Tung Y.-S., Morrison B., 3rd, Konofagou E.E. Molecules of various pharmacologically-relevant sizes can cross the ultrasound-induced blood-brain barrier opening in vivo. Ultrasound Med. Biol. 2010;36:58–67. doi: 10.1016/j.ultrasmedbio.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen P.C., Samadi-Bahrami Z., Pavlov V., Stys P.K., Moore G.R.W. Ionotropic glutamate receptor expression in human white matter. Neurosci. Lett. 2016;630:1–8. doi: 10.1016/j.neulet.2016.07.030. [DOI] [PubMed] [Google Scholar]
- Davalos D., Lee J.K., Smith W.B., Brinkman B., Ellisman M.H., Zheng B., Akassoglou K. Stable in vivo imaging of densely populated glia, axons and blood vessels in the mouse spinal cord using two-photon microscopy. J. Neurosci. Methods. 2008;169:1–7. doi: 10.1016/j.jneumeth.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Juan-Sanz J., Holt G.T., Schreiter E.R., de Juan F., Kim D.S., Ryan T.A. Axonal endoplasmic reticulum Ca2+ content controls release probability in CNS nerve terminals. Neuron. 2017;93:867–881. doi: 10.1016/j.neuron.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb C., Lafrance-Corey R.G., Zoecklein L., Papke L., Rodriguez M., Howe C.L. Demyelinated axons and motor function are protected by genetic deletion of perforin in a mouse model of multiple sclerosis. J. Neuropathol. Exp. Neurol. 2009;68:1037–1048. doi: 10.1097/NEN.0b013e3181b5417e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico F., Tramutola A., Butterfield D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017;111:253–261. doi: 10.1016/j.freeradbiomed.2016.10.490. [DOI] [PubMed] [Google Scholar]
- Feng G., Mellor R.H., Bernstein M., Keller-Peck C., Nguyen Q.T., Wallace M., Nerbonne J.M., Lichtman J.W., Sanes J.R. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fitzgerald M., Bartlett C.A., Evill L., Rodger J., Harvey A.R., Dunlop S.A. Secondary degeneration of the optic nerve following partial transection: the benefits of lomerizine. Exp. Neurol. 2009;216:219–230. doi: 10.1016/j.expneurol.2008.11.026. [DOI] [PubMed] [Google Scholar]
- Friese M.A., Schattling B., Fugger L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014;10:225–238. doi: 10.1038/nrneurol.2014.37. [DOI] [PubMed] [Google Scholar]
- Haider L., Fischer M.T., Frischer J.M., Bauer J., Höftberger R., Botond G., Esterbauer H., Binder C.J., Witztum J.L., Lassmann H. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134:1914–1924. doi: 10.1093/brain/awr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim N., Garaschuk O., Friedrich M.W., Mank M., Milos R.I., Kovalchuk Y., Konnerth A., Griesbeck O. Improved calcium imaging in transgenic mice expressing a troponin C-based biosensor. Nat. Methods. 2007;4:127–129. doi: 10.1038/nmeth1009. [DOI] [PubMed] [Google Scholar]
- Kalyvas A., Baskakis C., Magrioti V., Constantinou-Kokotou V., Stephens D., López-Vales R., Lu J.-Q., Yong V.W., Dennis E.A., Kokotos G., David S. Differing roles for members of the phospholipase A2 superfamily in experimental autoimmune encephalomyelitis. Brain. 2009;132:1221–1235. doi: 10.1093/brain/awp002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor R., Davies M., Blaker P.A., Hall S.M., Smith K.J. Blockers of sodium and calcium entry protect axons from nitric oxide-mediated degeneration. Ann. Neurol. 2003;53:174–180. doi: 10.1002/ana.10443. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M., Schwab M.E., Lichtman J.W., Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat. Med. 2005;11:572–577. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- Knöferle J., Koch J.C., Ostendorf T., Michel U., Planchamp V., Vutova P., Tönges L., Stadelmann C., Brück W., Bähr M., Lingor P. Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc. Natl. Acad. Sci. USA. 2010;107:6064–6069. doi: 10.1073/pnas.0909794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Yang L., Selzer M.E., Hu Y. Neuronal endoplasmic reticulum stress in axon injury and neurodegeneration. Ann. Neurol. 2013;74:768–777. doi: 10.1002/ana.24005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrez R., Stys P.K., Vivien D., Lipton S.A., Docagne F. Mechanisms of glutamate toxicity in multiple sclerosis: biomarker and therapeutic opportunities. Lancet Neurol. 2016;15:1089–1102. doi: 10.1016/S1474-4422(16)30165-X. [DOI] [PubMed] [Google Scholar]
- Mank M., Griesbeck O. Genetically encoded calcium indicators. Chem. Rev. 2008;108:1550–1564. doi: 10.1021/cr078213v. [DOI] [PubMed] [Google Scholar]
- Michailidou I., Jongejan A., Vreijling J.P., Georgakopoulou T., de Wissel M.B., Wolterman R.A., Ruizendaal P., Klar-Mohamad N., Grootemaat A.E., Picavet D.I. Systemic inhibition of the membrane attack complex impedes neuroinflammation in chronic relapsing experimental autoimmune encephalomyelitis. Acta Neuropathol. Commun. 2018;6:36. doi: 10.1186/s40478-018-0536-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossakowski A.A., Pohlan J., Bremer D., Lindquist R., Millward J.M., Bock M., Pollok K., Mothes R., Viohl L., Radbruch M. Tracking CNS and systemic sources of oxidative stress during the course of chronic neuroinflammation. Acta Neuropathol. 2015;130:799–814. doi: 10.1007/s00401-015-1497-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikić I., Merkler D., Sorbara C., Brinkoetter M., Kreutzfeldt M., Bareyre F.M., Brück W., Bishop D., Misgeld T., Kerschensteiner M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2011;17:495–499. doi: 10.1038/nm.2324. [DOI] [PubMed] [Google Scholar]
- Ouardouz M., Nikolaeva M.A., Coderre E., Zamponi G.W., McRory J.E., Trapp B.D., Yin X., Wang W., Woulfe J., Stys P.K. Depolarization-induced Ca2+ release in ischemic spinal cord white matter involves L-type Ca2+ channel activation of ryanodine receptors. Neuron. 2003;40:53–63. doi: 10.1016/j.neuron.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Ouardouz M., Coderre E., Basak A., Chen A., Zamponi G.W., Hameed S., Rehak R., Yin X., Trapp B.D., Stys P.K. Glutamate receptors on myelinated spinal cord axons: I. GluR6 kainate receptors. Ann. Neurol. 2009;65:151–159. doi: 10.1002/ana.21533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povlishock J.T., Becker D.P., Cheng C.L., Vaughan G.W. Axonal change in minor head injury. J. Neuropathol. Exp. Neurol. 1983;42:225–242. doi: 10.1097/00005072-198305000-00002. [DOI] [PubMed] [Google Scholar]
- Romanelli E., Sorbara C.D., Nikić I., Dagkalis A., Misgeld T., Kerschensteiner M. Cellular, subcellular and functional in vivo labeling of the spinal cord using vital dyes. Nat. Protoc. 2013;8:481–490. doi: 10.1038/nprot.2013.022. [DOI] [PubMed] [Google Scholar]
- Siffrin V., Radbruch H., Glumm R., Niesner R., Paterka M., Herz J., Leuenberger T., Lehmann S.M., Luenstedt S., Rinnenthal J.L. In vivo imaging of partially reversible th17 cell-induced neuronal dysfunction in the course of encephalomyelitis. Immunity. 2010;33:424–436. doi: 10.1016/j.immuni.2010.08.018. [DOI] [PubMed] [Google Scholar]
- Sorbara C.D., Wagner N.E., Ladwig A., Nikić I., Merkler D., Kleele T., Marinković P., Naumann R., Godinho L., Bareyre F.M. Pervasive axonal transport deficits in multiple sclerosis models. Neuron. 2014;84:1183–1190. doi: 10.1016/j.neuron.2014.11.006. [DOI] [PubMed] [Google Scholar]
- Stirling D.P., Cummins K., Wayne Chen S.R., Stys P. Axoplasmic reticulum Ca(2+) release causes secondary degeneration of spinal axons. Ann. Neurol. 2014;75:220–229. doi: 10.1002/ana.24099. [DOI] [PubMed] [Google Scholar]
- Suzuki J., Kanemaru K., Iino M. Genetically encoded fluorescent indicators for organellar calcium imaging. Biophys. J. 2016;111:1119–1131. doi: 10.1016/j.bpj.2016.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S.K.Y., Marshall W.F. Self-repairing cells: how single cells heal membrane ruptures and restore lost structures. Science. 2017;356:1022–1025. doi: 10.1126/science.aam6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki M. Axonal endoplasmic reticulum is very narrow. J. Cell Sci. 2018;131:jcs210450. doi: 10.1242/jcs.210450. [DOI] [PubMed] [Google Scholar]
- Thestrup T., Litzlbauer J., Bartholomäus I., Mues M., Russo L., Dana H., Kovalchuk Y., Liang Y., Kalamakis G., Laukat Y. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat. Methods. 2014;11:175–182. doi: 10.1038/nmeth.2773. [DOI] [PubMed] [Google Scholar]
- Vargas M.E., Yamagishi Y., Tessier-Lavigne M., Sagasti A. Live imaging of calcium dynamics during axon degeneration reveals two functionally distinct phases of calcium influx. J. Neurosci. 2015;35:15026–15038. doi: 10.1523/JNEUROSCI.2484-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas R., Martinez N.W., Lillo J., Pihan P., Hernandez D., Twiss J.L., Court F.A. Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J. Neurosci. 2014;34:7179–7189. doi: 10.1523/JNEUROSCI.4784-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins L.M., Neal J.W., Loveless S., Michailidou I., Ramaglia V., Rees M.I., Reynolds R., Robertson N.P., Morgan B.P., Howell O.W. Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J. Neuroinflammation. 2016;13:161. doi: 10.1186/s12974-016-0611-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams P.R., Marincu B.-N., Sorbara C.D., Mahler C.F., Schumacher A.-M., Griesbeck O., Kerschensteiner M., Misgeld T. A recoverable state of axon injury persists for hours after spinal cord contusion in vivo. Nat. Commun. 2014;5:5683. doi: 10.1038/ncomms6683. [DOI] [PubMed] [Google Scholar]
- Wu Y., Whiteus C., Xu C.S., Hayworth K.J., Weinberg R.J., Hess H.F., De Camilli P. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc. Natl. Acad. Sci. USA. 2017;114:E4859–E4867. doi: 10.1073/pnas.1701078114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Weimer R.M., Kallop D., Olsen O., Wu Z., Renier N., Uryu K., Tessier-Lavigne M. Regulation of axon degeneration after injury and in development by the endogenous calpain inhibitor calpastatin. Neuron. 2013;80:1175–1189. doi: 10.1016/j.neuron.2013.08.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In vivo multiphoton time-lapse of the healthy spinal cord axons of a Thy1-TwitchER and a Thy1-CerTN-L15 transgenic mouse upon application of 50 mM caffeine. Top: ratiometric (YFP/CFP) images color coded for ER calcium levels ([Ca2+]ER). Bottom: ratiometric (YFP/CFP) images color coded for cytoplasmic calcium levels ([Ca2+]cyt). Scale bar, 25 μm.