

Abstract
Mutations in the sodium‐activated potassium channel gene KCNT1 have been associated with nonlesional sleep‐related hypermotor epilepsy (SHE). We report the co‐occurrence of mild malformation of cortical development (mMCD) and KCNT1 mutations in four patients with SHE. Focal cortical dysplasia type I was neuropathologically diagnosed after epilepsy surgery in three unrelated MRI‐negative patients, periventricular nodular heterotopia was detected in one patient by MRI. Our findings suggest that KCNT1 epileptogenicity may result not only from dysregulated excitability by controlling Na+K+ transport, but also from mMCD. Therefore, pathogenic variants in KCNT1 may encompass both lesional and nonlesional epilepsies.
Introduction
Sleep‐related hypermotor epilepsy (SHE), formerly known as nocturnal frontal lobe epilepsy, is characterized by focal seizures with various motor manifestations occurring during sleep.1 Autosomal dominant forms of SHE (ADSHE) have been associated with mutations of genes encoding subunits of the neuronal acetylcholine receptor (CHRNA4, CHRNB2, CHRNA2) or proteins of the mTORC1/GATOR1 complex (DEPDC5, NPRL2/3).1 Heterozygous missense mutations in KCNT1 were reported in a severe form of ADSHE with normal MRI, drug‐resistant seizures, intellectual disability, and psychiatric features.2 The KCNT1 gene encodes a sodium‐activated potassium channel that regulates excitability in neurons by contributing to the slow hyperpolarization that follows repetitive firing.
Malformations of cortical development (MCDs) have previously been documented in SHE,3 but only few studies have elucidated their genetic etiologies.4 Here, we report the co‐occurrence, not described previously, of mild MCDs (mMCDs) and KCNT1 pathogenic variants in four subjects with SHE.
Patients and Methods
We performed genetic diagnostic testing in one ADSHE patient (patient A.III.1) previously submitted to epilepsy surgery in whom mMCD was detected by histopathological examination, and in his family relatives (Fig. 1A). A cohort of further 36 published and unpublished patients with KCNT1‐related SHE, collected through data sharing with Epilepsy and Genetic Centers in Europe and North America, was reviewed for MRI abnormalities and/or surgical treatment. We collected two additional patients with de novo KCNT1 variants, one with ADSHE (patient B.III.2, Fig. 1B) and one with sporadic SHE (patient C). Both patients underwent epilepsy surgery before genetic testing. A fourth patient (B.III.1) belonging to family B (Fig. 1B) showed MCD at the MRI. Electro‐clinical phenotyping was performed by detailed clinical history and video‐EEG monitoring including seizure recording. All patients had had 1.5 or 3 Tesla brain MRIs. Epilepsy surgery was performed after long‐term scalp and intracranial video‐EEG monitoring. Histopathological diagnosis on FFPE neuropathological surgical specimens was made according to the classification of the International League Against Epilepsy (ILAE) Diagnostic Methods Commission.5 Sequencing was performed on genomic blood DNA using targeted gene panels comprising known epilepsy genes including those known to cause MCD‐related epilepsy (i.e., DEPDC5, NPRL2/3, MTOR).6 Informed consent from all participants and approval from the local ethics committees were obtained.
Figure 1.
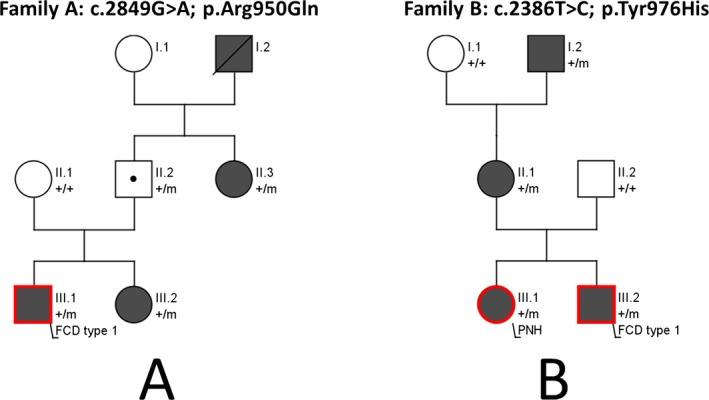
(A) Pedigree of family A (KCNT1 c.2849G>A; p.Arg950Gln). (B) Pedigree of family B (KCNT1 c.2386T>C; p.Tyr976His). The patient indicated with a dot is an unaffected carrier. Red contours indicate the presence of MCD, dark symbols indicate individuals with focal epilepsy. Individuals with KCNT1 mutations are indicated by m/+, and those negative by +/+. FCD, focal cortical dysplasia; PNH, periventricular nodular heterotopia.
Results
Genetic findings
In family A, genetic analysis revealed a c.2849G>A; p.Arg950Gln KCNT1 (NM_020822.2) missense mutation segregating in subjects A.III.1, A.III.2, inherited from their unaffected father, and found in A.II.3 (Fig. 1A), reflecting the incomplete penetrance seen in KCNT1 families.7 Family B (c.2386T>C; p.Tyr796His KCNT1) was previously reported.2 Patient C had a de novo c.2800G>A; p.Ala934Thr mutation. All KCNT1 variants were considered as pathogenic given their: a) recurrence in other published SHE cases; b) absence in control databases (gnomAD); c) familial segregation/de novo occurrence (Fig. 1A), and d) in vitro gain‐of‐function effect of the Tyr796His and Ala934Thr variants on the current magnitude.
Electro‐clinical phenotyping
Family A. Patient A.III.1 is a 23‐year‐old male with drug‐resistant sleep‐related frontal lobe seizures since 8 years of age (Table 1). Behavioral disturbances and neuropsychological deficits consistent with frontal lobe dysfunction appeared after epilepsy onset. 3T MRI was unremarkable. Epilepsy surgery was performed at age 14 to attempt to control seizures occurring several times per night, almost every night. After 2 years of postoperative seizure freedom, seizures reappeared with partially modified semiology (Table 1) reaching progressively the same frequency as before surgery (Engel class IV8). Histopathological examination (NeuN immunostaining) showed cortical dyslamination suggesting focal cortical dysplasia (FCD) type Ib (Fig. 2A). In patient A.III.2, seizures were drug‐resistant, whereas in patient A.II.3 they were well controlled by carbamazepine. In both patients, 1.5T MRI was normal. Familial history, clinical, and EEG features were consistent with ADSHE.
Table 1.
Electroclinical features, MRI, and pathological findings in patients with KCNT1‐related SHE and MCD
| Case | Mutation | Age/sex/ Ethnic backgr. | Age at seiz. onset | Seizure semiology | Epilepsy syndr. | Interictal scalp EEG findings | MRI Findings (Tesla) | Intracranial/ scalp video‐EEG findings | Surgery/ Age at surgery | Surgery outcome/follow‐up/ seiz. semiology | Histo pathol. | Current TRT | Comorbidities |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A.III.1 |
c.2849G>A; p.Arg950Gln |
23 y/M/ Danish |
8 y | Sleep‐related HMS | ADSHE | Bifrontal sharpwaves and bitemporal asynchronous sharpwaves |
Unremarkable (3 T) |
Sleep‐related HMS (asymmetric posturing, choking, retching, distressed breathing)IEEG: R mesial fronto‐central onset |
Resection of the R mesial fronto‐parietal regions/ 14 y |
Engel Class IV/ 8 y/sleep‐related HMS (tingling, choking, vocalizations) |
FCD type Ib |
CBZ, LTG, TPM, LCS, CNZ | Behavioral disturbances (impulsivity, attention deficit), poor executive functions |
| B.III.1 |
c.2386T>C; p.Tyr796His |
37y/F/ Italian |
3 y | Sleep‐related focal seizures with tonic posturing | ADSHE | L fronto‐temporal spikes, increased by sleep | L Periventric. nodular heterotopia with transmantlesigns (1.5 T) | Not performed | Not performed | – | – | CBZ, TPM |
Severe psychosis, learning disabilities, precocious puberty |
| B.III.2 |
c.2386T>C; p.Tyr796His |
35 y/M/ Italian |
3 y | Sleep‐related HMS, nocturnal wandering | ADSHE | R fronto‐temporal theta activity |
Unremarkable (1.5 T) |
Sleep‐related HMS (pelvic thrusting, tonic posturing, pedaling) IEEG: R ant. cingular onset with spread to R mesial superior frontal gyrus and orbital region |
Resection of the R anterior cingulate gyrus + lateral frontal cortex (F1,F2, post. F3)/21 y |
Engel Class II (at present/14 y/ rare diurnal seiz. with staring |
FCD type Ib |
CBZ, VPA, PB |
Severe psychosis, learning disabilities, precocious puberty |
| C |
c.2800G>A; p.Ala934Thr de novo |
27 y/F/ French |
1 y 8 m |
Sleep‐related R tonic motor seizures with consciousness impairment; infrequent daytime episodes with R lower limb atonia, sudden fall, consciousness impairment. |
SHE | Bilateral multifocal spikes |
Discrete temporal lobe asymmetry (R > L) (1,5 Tesla) Uninformative (sequelæ of the previous surgery) (3 Tesla) |
(before first surgery): Sleep‐related R sided tonic seiz., axial tonic seiz., “agitated” seiz. IEEG: L multifocal epileptogenic zone with centro‐parietal predominance (before 2nd surgery): Sleep‐related R side grimacing, R sided tonic seiz., R sided clonic jerks Scalp EEG: L multifocal (parieto‐ post. temporal occipital), R fronto‐temporal onset |
First surgery: L centro‐parietal resection/ 6 y 2nd surgery: L parieto‐occipital (+ temporo‐occipital junction) resection/ 25 y |
Engel Class IV/ 2 y (after 2nd surgery)/ Semiology unchanged |
FCD type Ia |
CBZ, LEV, PB, VNS |
Learning disabilities and delayed psychomotor development since 3 years of age. Worsening of the cognitive status and appearance of autistic features over the years. |
HMS, hypermotor seizures; IEEG, intracranial EEG; CBZ, carbamazepine; LTG, lamotrigine; TPM, topiramate; LCS, lacosamide; CNZ, clonazepam; LEV, levetiracetam; CLB, clobazam; PB, phenobarbital; VPA, valproic acid; VNS, vagus nerve stimulator; periventr., periventricular; ant., anterior; backgr, background; SE, status epilepticus; y, years; mo, months; seiz., seizure; R, right; L, left.
Figure 2.
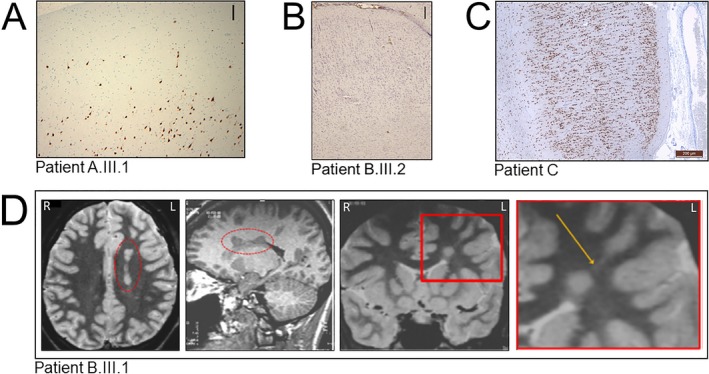
Neuropathological findings. (A) In individual A.III.1, NeuN staining highlights areas of underdeveloped or focal losses of cortical layer 2 and scattered malpositioned pyramidal type neurons in cortical layer 1, consistent with FCD type Ib (original magnification 100X). Scale bar: 250 μm. (B) In individual B.III.2, Nissl‐stained section shows a laminar disorganization of the cortex indicative of FCD type Ib. Scale bar: 250 μm. (C) In individual C, NeuN staining section shows an abnormal cortical radial lamination and neurons with microcolumnar disposition consistent with FCD type Ia. No dysmorphic neurons or balloon cells were observed. Scale bar 200 μm. (D) MRI of individual B.III.1 showing deep nodular heterotopia in the left frontal lobe. Axial T2 WI (first panel from the left) and sagittal 3D T1WI (second panel from the left) show a left periventricular nodular heterotopia (in the red dashed ellipse). Coronal IR T1WI (third panel from the left) and magnification of the area included in the red square (fourth panel from the left) show a single heterotopic nodule in the deep‐periventricular left frontal white matter with the same signal intensity of the grey matter. The nodule is connected by a radial band (arrow) with the normal‐appearing overlying cortex. R = right, L = left.
Family B. All four affected members of this family were diagnosed with ADSHE. Patient B.III.2 (previously described2) suffered from drug‐resistant sleep‐related seizures since the age of 3 years. Brain MRI (1.5T) was unremarkable. At 21 years of age, he underwent epilepsy surgery with resection of the right mesio‐lateral frontal region. Histopathological examination (Nissl staining) displayed cortical laminar disorganization indicative of FCD type Ib (not mentioned in the initial publication2) (Fig. 2B). Epilepsy improved and seizure semiology changed after surgery. At 14‐years follow up he has only rare diurnal seizures with staring (Engel Class II). Patient B.III.1 is the 37‐year‐old sister with drug‐resistant sleep‐related frontal seizures. Brain MRI (1,5T) showed left periventricular nodular heterotopia (PNH) connected by a radial band with the normal‐appearing overlying cortex (Fig. 2D). Both patients presented with learning disabilities, severe psychosis, and precocious puberty. Patients B.II.1 and B.I.2 had sleep‐related seizures consistent with ADSHE. Brain MRI (1.5T) was normal in patient B.II.1 and not available in patient B.I.2.
Patient C is a 27‐year‐old female with drug‐resistant sleep‐related frontal lobe seizures since the age of 20 months (seizures occurred almost every night). Brain MRI (1.5T) showed discrete temporal lobe asymmetry. Two epilepsy surgeries were performed at the age of 6 and 25 years, without improvement of her epilepsy (Engel class IV, follow‐up: 2 years). Histopathological analysis (NeuN immunostaining) after the second surgery showed abnormalities of cortical lamination and the presence of characteristic microcolumns in the occipital cortex consistent with FCD type Ia (Fig. 2C).
Discussion
In this study, we report the finding of mMCDs in patients with SHE due to pathogenic variants in KCNT1. Together with the GATOR1‐encoding genes,9 this study emphasizes the genetic continuum between apparently nonlesional and lesional focal epilepsies.
Our four patients present with electro‐clinical features compatible with SHE, and FCD type I or PNH. All patients suffered from severe refractory focal seizures, intellectual disability, and psychiatric disturbances consistent with the phenotype previously reported in KCNT1‐related SHE.2 In three unrelated patients, epilepsy surgery failed to achieve seizure freedom, although epilepsy severity markedly improved in one (Engel Class II). Neuropathological analysis detected dyslamination compatible with FCD type I, a type of FCD often associated with unsatisfactory seizure control after surgery, with less than 50% of patients achieving seizure freedom,10 probably due to the more diffuse nature of FCD type I as compared to FCD type II, which has a better postseizure outcome. The focal onset of seizures documented by intracranial EEG recordings in patients with FCD type I is likely to represent only a portion of a more diffuse epileptogenic network, which probably involves a larger amount of abnormal cortex.11 Nevertheless, the transient seizure‐free period followed by a change of seizure semiology observed in patients A.III.1 and B.III.2 indicates that the surgical intervention indeed impinged on the epileptogenic network, although the resection probably was not large enough to yield seizure freedom.
A role of KCNT1 in provoking widespread epileptogenesis that sustains seizure propensity after epilepsy surgery is also possible. Structural and molecular alterations (such as ion channel expression/function) in the perilesional region, may also contribute to the epileptogenicity of focal MCDs.12 The coexistence of two highly epileptogenic substrates (i.e., MCD and KCNT1 mutation) could underlie the epilepsy severity in these patients. Indeed, KCNT1 mutations and MCDs might reciprocally influence each other in developing the pathophysiological mechanisms that generate seizures, as suggested in patients with MCD carrying SCN1A mutations.13 An alternative hypothesis is the existence of a second‐hit somatic mutation that would support the focal cortical lesion, as previously demonstrated in DEPDC5‐related focal epilepsy,4, 14 a hypothesis that could not be verified here because of the lack of resected epileptogenic cortex. Given the poor epilepsy surgery outcome, our findings raise the issue whether surgery is an effective treatment in KCNT1‐related SHE, and, in a broader perspective, whether genetic testing should be included in the presurgical assessment of patients who are considered as possible candidates for epilepsy surgery.15
Interestingly, in the fourth nonoperated patient with ADSHE (patient B.III.1), MRI showed unilateral PNH, a type of MCD that has been shown to be associated with FCD type I in the cortex overlying the nodules.16 Negative MRI in three patients (A.III.2, A.II.3 and B.II.1) might depend on the limited sensitivity of current neuroimaging techniques to detect subtle abnormalities, especially FCD type I or, alternatively, it might suggest that KCNT1 variants can be associated with lesional and nonlesional epilepsies even within the same family.
The contribution of ion channels in the pathogenesis of MCDs has previously been reported. For instance, dysplastic neurons present in FCDs display strong Kv4.2 expression.17 Recently, neuropathological investigations in a child diagnosed with a severe KCNQ2‐related neonatal epileptic encephalopathy, revealed the presence of heterotopic neurons in the deep white matter.18 In a knockdown mouse model, KCNK potassium channels dysfunction, resulting in increased frequency of spontaneous calcium influx, impeded physiological neuronal migration in the developing cerebral cortex, possibly playing a role in the genesis of MCD via an activity‐dependent mechanism.19 Lastly, MCDs in patients with SCN1A and SCL35A2 mutations have been documented.13, 20
Our study supports an emerging view in which subtle structural brain changes (MRI‐undetectable) can occur in epilepsies caused by mutations in ion channel genes and further emphasizes the need for repeated and careful review of high‐resolution imaging for subtle abnormalities, that may not be immediately appreciated, in patients with KCNT1‐related focal epilepsies.
Author contributions
G.R. and R.S.M: concept and study design, data acquisition and analysis, drafting of the manuscript and figures; S.B.: concept and study design, drafting of the manuscript and figures; G.P., F.P., L.N., E.H., J.C., R.A.P., J.B, M.B., P.K., L.M.D., E.G.: data acquisition and analysis.
Conflict of Interest
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Acknowledgments
We thank Roberto Spreafico and Nadia Colombo for their helpful comments and suggestions on the manuscript. We are indebted to the patients and their families for their collaboration in performing this study. This work was supported by the European Research Council (ERC 682345 to SB).
Funding Information
This work was supported by the European Research Council (ERC 682345 to SB).
Funding Statement
This work was funded by European Research Council grant ERC 682345.
References
- 1. Tinuper P, Bisulli F, Cross JH, et al. Definition and diagnostic criteria of sleep‐related hypermotor epilepsy. Neurology 2016;86:1834–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Heron SE, Smith KR, Bahlo M, et al. Missense mutations in the sodium‐gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 2012;44:1188–1190. [DOI] [PubMed] [Google Scholar]
- 3. Nobili L, Francione S, Mai R, et al. Surgical treatment of drug‐resistant nocturnal frontal lobe epilepsy. Brain 2007;130:561–573. [DOI] [PubMed] [Google Scholar]
- 4. Baulac S, Ishida S, Marsan E, et al. Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann Neurol 2015;77:675–683. [DOI] [PubMed] [Google Scholar]
- 5. Blümcke I, Thom M, Aronica E, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE diagnostic methods commission. Epilepsia 2011;52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oates S, Tang S, Rosch R, et al. Incorporating epilepsy genetics into clinical practice: a 360° evaluation. NPJ Genom Med. 2018. May;10:13 10.1038/s41525-018-0052-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Møller RS, Heron SE, Larsen LH, et al. Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia 2015;56:e114–e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Engel J. Outcome with respect to epileptic seizures In: Engel J, Jr, ed. Surgical treatment of the epilepsies, 2nd ed New York: Raven Press, 1993:609–621. [Google Scholar]
- 9. Marsan E, Baulac S. Mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol Appl Neurobiol. 2018;44:6–17. [DOI] [PubMed] [Google Scholar]
- 10. Simpson SL, Prayson RA. Post‐surgical outcome for epilepsy associated with type I focal cortical dysplasia subtypes. Modern Pathol 2014;27:1455–1460. [DOI] [PubMed] [Google Scholar]
- 11. Frater JL, Prayson RA, Morris HH III, et al. Surgical pathologic findings of extratemporal‐based intractable epilepsy: a study of 133 consecutive resections. Arch Pathol Lab Med 2000;124:545–549. [DOI] [PubMed] [Google Scholar]
- 12. Wong M. Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development with abnormal glioneuronal proliferation. Epilepsia 2008;49:8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barba C, Parrini E, Coras R, et al. Co‐occurring malformations of cortical development and SCN1A gene mutations. Epilepsia 2014;55:1009–1019. [DOI] [PubMed] [Google Scholar]
- 14. Ribierre T, Deleuze C, Bacq A, et al. Second‐hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia‐associated epilepsy. J Clin Invest. 2018;128:2452–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stevelink R, Sanders MW, Tuinman MP, et al. Epilepsy surgery for patients with genetic refractory epilepsy: a systematic review. Epileptic Disord. 2018;20:99–115. [DOI] [PubMed] [Google Scholar]
- 16. Meroni A, Galli C, Bramerio M, et al. Nodular heterotopia: a neuropathological study of 24 patients undergoing surgery for drug‐resistant epilepsy. Epilepsia 2009;50:116–124. [DOI] [PubMed] [Google Scholar]
- 17. Aronica E, Boer K, Doorn KJ, et al. Expression and localization of voltage dependent potassium channel Kv4.2 in epilepsy associated focal lesions. Neurobiol Dis 2009;36:81–95. [DOI] [PubMed] [Google Scholar]
- 18. Dalen Meurs‐van Der Schoor C, van Weissenbruch M, Van Kempen M, et al. Severe neonatal epileptic encephalopathy and KCNQ2 mutation: neuropathological substrate? Front Pediatr 2014;2:136 10.3389/fped.2014.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bando Y, Hirano T, Tagawa Y. Dysfunction of KCNK potassium channels impairs neuronal migration in the developing mouse cerebral cortex. Cereb Cortex 2014;24:1017–1029. [DOI] [PubMed] [Google Scholar]
- 20. Winawer MR, Griffin NG, Samanamud J, et al. Somatic SLC35A2 variants in the brain are associated with intractable neocortical epilepsy. Ann Neurol 2018;83:1133–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]