

Abstract
Our objective was to examine the brain biopsies by histopathology and investigate the prognosis of patients with myelin oligodendrocyte glycoprotein antibody‐associated demyelinating pseudotumor. The clinical, MRI, and histological features of two patients with myelin oligodendrocyte glycoprotein antibody‐associated demyelinating pseudotumor were reviewed. Both patients were treated with steroid plus rituximab and followed up. The brain biopsies of both cases revealed T cells, macrophages, and complement‐mediated demyelination, which was in accord with multiple sclerosis‐like pathology. Moreover, both cases showed favorable response to steroid plus rituximab therapy. Our cases add a new variant to the myelin oligodendrocyte glycoprotein‐encephalomyelitis spectrum, which favorably responds to immunotherapy.
Introduction
Demyelinating pseudotumor, also called tumor‐like demyelinating lesions (TDLs), is a rare inflammatory demyelinating disease of the central nervous system.1 The pathogenesis of the disease is unclear. Due to the lack of a specific biomarker, TDLs are usually misdiagnosed as brain tumors. To raise the attention of neurologists to this emerging entity and to highlight the importance of testing for anti‐myelin oligodendrocyte glycoprotein (MOG) antibodies in the disease, here we present clinical and neuropathologic features of two TDLs cases associated with antibodies to MOG.
Case History
Case 1
A 45‐year‐old, nonsmoking and nondrinking man developed progressive apathy and cognitive impairment, followed by headache, dysarthria, left‐sided central facial paresis, and left limb weakness (Expanded Disability Status Score, EDSS, 7.5), without preceding infections, fever, or vaccinations. Brain T2 fluid‐attenuated inversion recovery‐magnetic resonance imaging (FLAIR‐MRI) showed a large lesion located in the white matter of the right frontal lobe and basal ganglia region adjacent to the lateral ventricle with patchy enhancement (Fig. 1A, B). Routine laboratory tests including serum inflammatory markers, infectious, and tumor agents were normal. Analysis of cerebrospinal fluid (CSF) showed normal levels of protein and negative oligoclonal bands (OCB). In addition, both anti‐aquaporin 4 (AQP4)‐IgG in serum and anti‐N‐methyl‐d‐aspartate receptor (NMDAR)‐IgG in CSF were negative. The brain biopsy was performed 2 weeks after the onset of symptoms for histological evaluation. The histological results were consistent with overlapping features of multiple sclerosis (MS) patterns I and II (Fig. 2, Case 1). After the brain surgery, the patient was treated with pulse methylprednisolone (1 g/day for 5 days and 0.5 g/day for 3 days) and rituximab (RTX, 375 mg/m2, 600 mg/month for 2 months). Five months after the treatment, the patient showed a favorable response. The EDSS decreased from 7.5 to 2.0, and cerebral lesions had shrunk remarkably on MRI (Fig. 1C).
Figure 1.
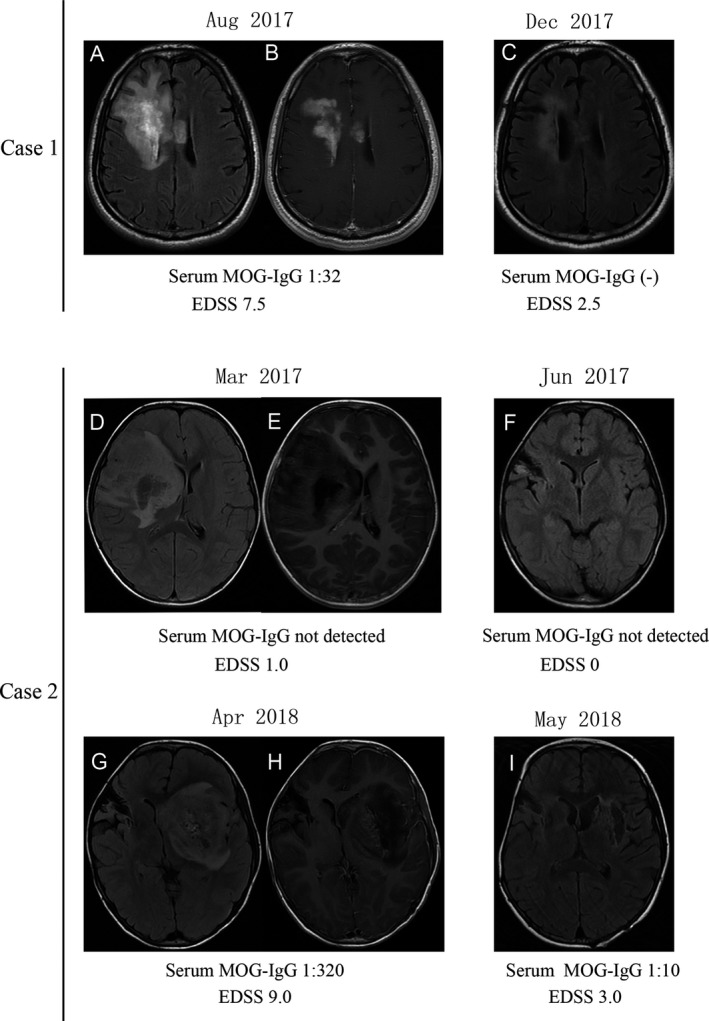
(A–C) Case 1 MRI findings showed large white matter lesions with patchy Gd‐enhancement located in the right frontal lobe and basal ganglia region. (A) axial‐fluid‐attenuated inversion recovery (FLAIR), (B), Gd‐enhanced axial T1, and the lesion obviously regressed during follow‐up (C, axial‐FLAIR). (D–I) Case 2 MRI showed a large edematous lesion in the white matter of the right frontal lobe and periventricular zone with linear Gd‐enhancement (D, axial‐FLAIR, E, Gd‐enhanced axial T1) that had regressed on follow‐up at 3 months (F, axial‐FLAIR). (G–I) One year later another large edematous lesion was seen in the left basal ganglia region with patchy enhancement (G, axial‐FLAIR, H, Gd‐enhanced axial T1) that regressed during follow‐up (I, axial‐FLAIR).
Figure 2.
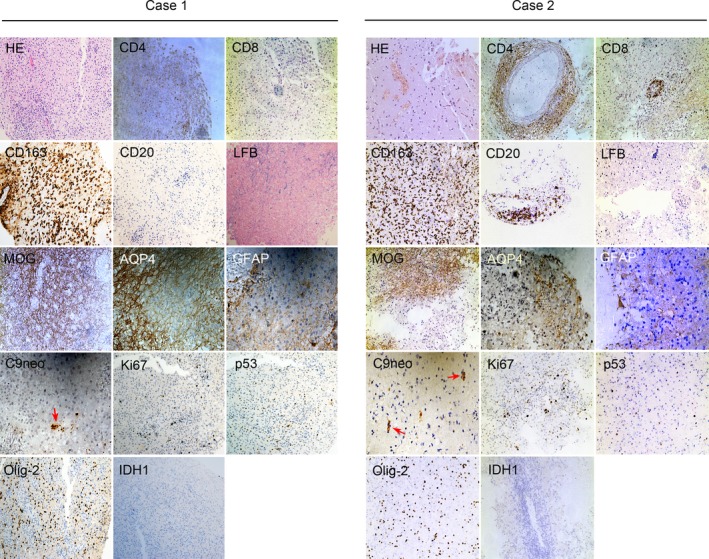
In Case 1 and Case 2, both of the right frontal lobe lesions revealed extensive inflammatory cells (HE). Perivascular and parenchymal CD4+ and CD8+ T cells dominated the inflammation, with many active macrophages (CD163+). However, there was a single CD20+ B cells in the lesions of case 1 and a few CD20+ B cells in the lesions of Case 2. A marked demyelinating lesion (LFB), loss of MOG immunoreactivity, a decrease of AQP4 expression and reactive GFAP + astrocytes, and mild complement deposition (C9neo, red arrows) were also seen in the lesion. There were scattered Ki67+ cells, few cells expressed p53 or Olig‐2 proteins, and none that expressed IDH1 protein were detected in the lesions of the two cases. HE, hematoxylin and eosin; LFB, luxol fast blue; MOG, myelin oligodendrocyte glycoprotein; AQP4, aquaporin‐4; GFAP, glial fibrillary acidic protein; IDH1, isocitrate dehydrogenase 1. Magnification: HE, CD4, CD8, CD163, CD20, LFB, MOG, Ki67+, p53, Olig‐2, IDH1 × 200; AQP4, GFAP, C9neo × 400.
Case 2
A 6‐year‐old girl first developed headache and vomiting without fever in March 2017, with EDSS 1.0. Brain T2 FLAIR‐MRI showed a large tumor‐like lesion located in the right frontal lobe and basal ganglia region with linear enhancement (Fig. 1D, E). A brain biopsy was performed 1 week after the onset of symptoms. Histological analysis correlated with MS pattern I/II features (Fig. 2, Case 2). The patient did not take any immunomodulating drugs after the brain biopsy. She recovered well, and her right brain lesions disappeared (Fig. 1F) in June 2017. However, in April 2018, she experienced facial asymmetry, dysphasia, drowsiness, and weakness in the limbs. Brain T2 FLAIR‐MRI showed a large tumor‐like lesion in the left basal ganglia region with patchy enhancement (Fig. 1G, H). Serum infectious, tumor agent, autoantibody and CSF analysis were negative. Then the patient was treated with methylprednisolone (80 mg/day for 6 days) and RTX (375 mg/m2, 200 mg/month for 2 months). Two months after the treatment, a favorable response was observed, including a decrease in EDSS (from 9.0 to 3.0) and lesion regression on MRI (Fig. 1I).
Antibodies to MOG
Two patients were tested for serum MOG‐IgG by an in‐house, cell‐based assay, as described previously.2 In brief, HEK293T cells were cotransfected with pIRES2‐EGFP plasmid which was subcloned with full‐length human MOG using Lipofectamine 2000 reagent according to the manufacturer's instructions (Thermo Scientific, Carlsbad, CA, USA). After 36 h, the MOG‐transfected cells, as well as untransfected negative control cells, were incubated at room temperature with centrifuged serum (1:50, diluted in Dulbecco's modified Eagle's medium (DMEM)) from patients and healthy controls for 30 min. If the serum anti‐MOG reactivity was weakly positive (less than 1:50), the serum was diluted to 1:10. The cells were then fixed with 4% paraformaldehyde for 20 min, blocked with 10% goat serum for 30 min, and immunolabeled with an AlexaFluor 546 secondary antibody against human IgG (1:1000; Thermo Scientific) for 1 h at room temperature. Before treatment, both case 1 and case 2 were positive for serum antibodies specific for MOG, with antibody titers of 1:32 and 1:320, respectively. After the treatment, anti‐MOG IgG converted to negative in case 1 and was reduced to a titer of 1:10 in case 2.
Neuropathological examination
Both of the patients’ brain biopsies were performed in the right frontal lobes. The brain tissues were fixed in 10% formalin and used for hematoxylin and eosin (HE), luxol fast blue (LFB), and immunohistochemical staining. In both patients, HE analyses showed extensive perivascular and parenchymal inflammatory cell accumulation, and LFB staining revealed profound demyelinating lesions. The immunohistochemical staining demonstrated that the infiltrated inflammatory cells consisted of a large number of CD4‐ and CD8‐positive T cells, and abundant CD163‐positive macrophages in both cases; however, only a single CD20‐positive B cell was seen in the lesions of case 1 and a few CD20‐positive B cells in the lesions of case 2 were seen. Furthermore, lesion regions were characterized by a loss of MOG immunoreactivity, a decrease in astrocytes expressing AQP4 and reactive gliosis staining by GFAP, and mild complement deposition (C9neo). In addition, in the lesion regions of the two cases, very few (5%) scattered Ki67+‐positive cells were observed, few expressed p53 protein or Olig‐2 protein, with no IDH1 protein in the perivascular space and adjacent parenchyma (Fig. 2).
Discussion
Here we report clinical, radiological, and pathological features of two patients with MOG antibody‐associated TDLs, who presented an MS pattern I/II‐like histopathology with T cells, macrophages, and complement‐mediated demyelination,3, 4 and who showed a favorable response to immunotherapy.
Neuropathologic reports of MOG antibody‐associated TDLs are scarce. MS pattern II pathology with complement deposition in the lesion has been described in a relapsing encephalomyelitis patient with a large white matter lesion and associated with MOG antibodies.5 Furthermore, the pathological features of severe inflammation, demyelination, and astrocyte and axon preservation were observed in two patients with MOG antibody‐associated demyelinating disease with large white matter lesions.6 In this study, MS‐like pathology (pattern I/II overlap), with a decrease in reactive astrocytes was observed. These results imply heterogeneous pathological features in the brain lesions of MOG‐associated demyelinating diseases. The underlying pathogenesis of MOG‐associated demyelinating diseases requires further study in future.
It is difficult to distinguish between TDLs and brain tumors on the basis of clinical or radiological evidence.1, 7 The two cases were initially diagnosed as brain glioma and thus received a brain biopsy. However, the disease was controlled by treatment with corticosteroid and RTX. Thus, a MOG antibody screen is of potential importance for TDLs in the central nervous system (CNS). Although the effect of RTX in MOG‐associated diseases is unclear, there are several reports that RTX can reduce relapses in this disease.8, 9, 10 In Case 2, the patient relapsed 1 year later, after she had not been treated with any immunomodulators. To date, our two patients have not relapsed after treatment with RTX. Therefore, RTX used as an acute treatment for our patients is also a preventative agent.
We acknowledge that there were several limitations in this study. Firstly, just as Waters et al. reported, a nonspecific IgG against human Fc might be associated with false positives.11 However, we used healthy donor serum and untransfected cells as controls in our MOG antibody test method.2 Furthermore, using the same method the patients’ titers dropped after treatment, suggesting that our result was not a false positive.
In conclusion, our cases add a new variant to the MOG‐encephalomyelitis spectrum. MOG antibody‐associated TDLs possess MS‐like histopathological features and show favorable responses to immunotherapy.
Conflict of Interest
The authors declare that no competing interests exist.
Acknowledgment
This study was supported by the National Natural Science Foundation of China (NO. 81771300 and 81701188).
Funding Information
This study was supported by the National Natural Science Foundation of China (NO.81771300 and 81701188).
Funding Statement
This work was funded by National Natural Science Foundation of China grants 81771300 and 81701188.
References
- 1. Kepes JJ. Large focal tumor‐like demyelinating lesions of the brain: intermediate entity between multiple sclerosis and acute disseminated encephalomyelitis? A study of 31 patients. Ann Neurol 1993;33:18–27. [DOI] [PubMed] [Google Scholar]
- 2. Chen L, Chen C, Zhong X, et al. Different features between pediatric‐onset and adult‐onset patients who are seropositive for MOG‐IgG: a multicenter study in South China. J Neuroimmunol 2018;321:83–91. [DOI] [PubMed] [Google Scholar]
- 3. Lucchinetti C, Bruck W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000;47:707–717. [DOI] [PubMed] [Google Scholar]
- 4. Metz I, Weigand SD, Popescu BF, et al. Pathologic heterogeneity persists in early active multiple sclerosis lesions. Ann Neurol 2014;75:728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spadaro M, Gerdes LA, Mayer MC, et al. Histopathology and clinical course of MOG‐antibody‐associated encephalomyelitis. Ann Clin Transl Neurol 2015;2:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou L, Huang Y, Li H, et al. MOG‐antibody associated demyelinating disease of the CNS: a clinical and pathological study in Chinese Han patients. J Neuroimmunol 2017;305:19–28. [DOI] [PubMed] [Google Scholar]
- 7. Kimura N, Kumamoto T, Hanaoka T, et al. Monofocal large inflammatory demyelinating lesion, mimicking brain glioma. Clin Neurol Neurosurg 2009;111:296–299. [DOI] [PubMed] [Google Scholar]
- 8. Whittam D, Cobo‐Calvo A, Lopez‐Chiriboga A, et al. Treatment of MOG‐IgG‐associated demyelination with rituximab: A multinational study of 98 patients. AAN Annual Meeting Abstract S13.003 2018.
- 9. Jarius S, Ruprecht K, Kleiter I, et al. MOG‐IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long‐term outcome. J Neuroinflammation 2016;13:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hacohen Y, Wong YY, Lechner C, et al. Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody‐associated disease. JAMA Neurol 2018;75:478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Waters P, Woodhall M, O'Connor KC, et al. MOG cell‐based assay detects non‐MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm 2015;2:e89. [DOI] [PMC free article] [PubMed] [Google Scholar]