

Abstract
Objective
Epilepsy treatment falls short in ~30% of cases. A better understanding of epilepsy pathophysiology can guide rational drug development in this difficult to treat condition. We tested a low‐cost, drug‐repositioning strategy to identify candidate epilepsy drugs that are already FDA‐approved and might be immediately tested in epilepsy patients who require new therapies.
Methods
Biopsies of spiking and nonspiking hippocampal brain tissue from six patients with unilateral mesial temporal lobe epilepsy were analyzed by RNA‐Seq. These profiles were correlated with transcriptomes from cell lines treated with FDA‐approved drugs, identifying compounds which were tested for therapeutic efficacy in a zebrafish seizure assay.
Results
In spiking versus nonspiking biopsies, RNA‐Seq identified 689 differentially expressed genes, 148 of which were previously cited in articles mentioning seizures or epilepsy. Differentially expressed genes were highly enriched for protein–protein interactions and formed three clusters with associated GO‐terms including myelination, protein ubiquitination, and neuronal migration. Among the 184 compounds, a zebrafish seizure model tested the therapeutic efficacy of doxycycline, metformin, nifedipine, and pyrantel tartrate, with metformin, nifedipine, and pyrantel tartrate all showing efficacy.
Interpretation
This proof‐of‐principle analysis suggests our powerful, rapid, cost‐effective approach can likely be applied to other hard‐to‐treat diseases.
Introduction
Epilepsy is a textbook example of a chronic disease – it affects many aspects of life, it is often difficult to detect and challenging to control, and it contributes to both morbidity and mortality. Epilepsy is life‐altering, in part, because it is unpredictable, and as such curtails daily activities like driving, attending school, and working. Currently, treatments include anti‐epileptic drugs, special diets, vagal nerve stimulation, and surgery; yet after decades using such treatment modalities and investing tremendous effort in research, ~30% of patients still have uncontrolled epilepsy and many take medications with intolerable side effects. Development of new antiseizure drugs has been significantly hampered by several issues, including the long lead time from discovery to market, challenges expected with insurance‐payer reimbursement, and regulatory drug‐safety hurdles. To circumvent these obstacles, drugs that are already FDA approved and known to target a pathway that is also disrupted in epilepsy are being considered as candidates for drug repositioning (DR).
Selecting compounds with therapeutic potential is not trivial and can be approached through techniques ranging from mechanism or hypothesis‐based prioritization to hypothesis‐free screens. An intermediate approach is DR.1, 2 DR itself also represents a continuum of approaches, ranging from experimental to fully computational.3 A largely experimental approach to DR relies on the list of gene expression pathways that differ in normal versus diseased tissue, as compared to the expression signature of drug‐repositioning candidate compounds. This correlation between candidate compounds’ expression signatures and differential gene expression can either be performed on the full transcriptional profile of differentially expressed (DE) genes,4 or targeted to individual modules of genes.5 Despite the inherent transcriptional differences between primary tissue and the in vitro cell lines available from the Connectivity Map,6 there have been numerous successful examples of DR using a range of primary tissues.4, 7 DR based on RNA‐Seq has not been reported for human epilepsy, so we took a full transcriptional profile DR‐based approach with human tissue from epilepsy‐affected brains and then tested candidate drug‐repositioning compounds in in vivo models. In temporal lobe surgical biopsy specimens, we compared differentially regulated expression pathways in electrically spiking tissue to nonspiking tissue and then identified 184 candidate antiseizure compounds (Fig. 1).
Figure 1.
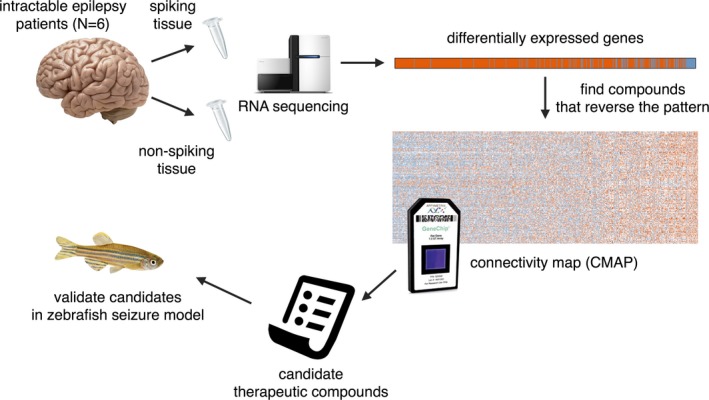
Overview of study design. Spiking and nonspiking tissue from N = 6 patients was obtained and assessed by RNA sequencing. Differentially expressed genes (spiking vs. nonspiking) were compared to gene expression profiles in the CMAP database, to identify compounds that reverse the differential expression signature, suggesting a potentially therapeutic activity. Four of these compounds were tested further for their antiseizure properties in a zebrafish model of seizures.
Next, we wanted to test the efficacy of some of these drugs in an animal model. The zebrafish model system is being increasingly used to study behavior. Not only do 70% of human genes have at least one counterpart in zebrafish, the zebrafish brain is remarkably similar to the mammalian brain. While popular mammalian model systems – such as mice – also have complex nervous systems and high homology to humans, it is not feasible to do high‐throughput chemical screens in these models. Conversely, the rapid development time and high number of offspring make high‐throughput screens with zebrafish much more practical. A little more than a decade ago, zebrafish were established as a simple vertebrate model for acute seizures.8 It was shown that the addition of pentylenetetrazole (PTZ) to the water of 7‐day postfertilization (dpf) zebrafish larvae was enough to induce significantly more movement in treated fish compared to controls (measured by the amount of distance swam). In addition, this increased swimming activity corresponded to epileptiform‐like activity in electrophysiology experiments performed on PTZ‐treated larvae. In follow‐up studies, it was also shown that this experimental method can be used to screen for drugs with antiseizure properties.9 Therefore, the zebrafish model system provides an opportunity to test the potential antiseizure drugs arising from our cell‐based chemical screen in a relatively short timeframe. We tested four of the most promising compounds in a zebrafish seizure assay and found three (metformin, nifedipine, and pyrantel tartrate) that had significant antiseizure properties in vivo. These drugs are FDA approved for human use, but none are specifically approved for seizures or epilepsy.
Methods
Patient recruitment and surgery
All patients underwent a routine, preoperative epilepsy workup. The results of the evaluations for individual cases were discussed along with recommended treatment plans at a multidisciplinary epilepsy conference. For patients included in this study, results were suggestive of unilateral mesial temporal lobe epilepsy, and therefore invasive phase II monitoring with intracranial electrodes was recommended. Patients underwent a two‐stage procedure: during the first stage they were implanted with electrodes to localize the seizure focus; during the second stage electrodes were removed and, if indicated, epileptogenic foci resected as per results from invasive recording. All implantation and resection surgeries were performed by two neurosurgeons experienced in epilepsy surgeries (MH, HK). Clinical information for all patients is summarized in Table 1.
Table 1.
Summary of patient clinical data
| Patient | Age of seizure onset | Age at surgery | Seizure type | # of AED trials | AEDs at time of biopsy | Final pathologic diagnosis | Outcome |
|---|---|---|---|---|---|---|---|
| 302 | 1 y.o. | 46 y.o. | Complex partial | 12 | 4 | Anterior Hippocampus: no diagnostic abnormality; Posterior Hippocampus: hippocampal sclerosis; Amygdala: rare abnormally clustered neurons and mild patchy gliosis | Engel IIB |
| 316 | 15 y.o. | 31 y.o. | Complex partial | 6 | 2 | Right temporal cortex: mesial temporal sclerosis, changes suggestive of focal cortical dysplasia; Anterior Hippocampus: mesial temporal sclerosis; Posterior Hippocampus: mesial temporal sclerosis; Amygdala: focal hemorrhage and granulation tissue consistent with prior electrode placement | Engel ID |
| 334 | 7 y.o. | 40 y.o. | Complex partial | 17 | 3 | Lateral Temporal Cortex: gliosis and changes related to recent dural grid monitoring; Temporal Pole: gliosis, predominantly involving the white matter; Posterior Lateral Temporal Cortex: gliosis and changes related to recent dural grid monitoring; Anterior Parahippocampal Gyrus: focal cortical dysplasia ILAE type Ib; Posterior Parahippocampal Gyrus: pigmented ganglioglioma WHO grade I, see comment, focal cortical dysplasia ILAE type IIa,; Amygdala: focal cortical dysplasia ILAE type Ib; Anterior Hippocampus: focally involved by ganglioglioma, patchy gliosis and changes related to recent dural grid monitoring; Posterior Hippocampus: focally involved by ganglioglioma, patchy gliosis and changes related to recent dural grid monitoring; comment – the pigmented ganglioglioma is “arising in the background of cortical dysplasia.” | Engel IA |
| 282 | 8 y.o. | 41 y.o. | Complex partial | 11 | 3 | Left Frontal Cortex: prominent gliosis, mostly involving white matter; Temporal Cortex: mild gliosis predominately involving the white matter; Lateral Temporal Cortex: mild gliosis involving white matter and focal areas of cortex; Amygdala: prominent gliosis; Hippocampus: gliosis predominately involving white matter | Engel IIC |
| 284 | 7 y.o. | 41 y.o. | Complex partial | 4 | 2 | Temporal Cortex: focal cortical dysplasia, gliosis; Amygdala: focal cortical dysplasia, gliosis; Hippocampus: focal cortical dysplasia, gliosis | Engel IA |
| 292 | 6 mo. | 50 y.o. | Complex partial | 8 | 2 | Amygdala: patchy marked superficial and deep cortical and white matter gliosis, with neuronal clustering; Frontal Cortex: rare superficial cortical and white matter neurons, with mild patchy gliosis of the superficial cortex and white matter; Hippocampus: rare white matter neurons and mild patchy gliosis of the cortex and white matter | Engel IB |
Table listing the relevant clinical information of patients from whom tissue samples were procured. Information includes age of onset of first seizure, age at which the biopsy was performed, classification of seizure type, the number of AED trials, the number of AEDs the patient was on at the time of surgery, a description of the pathology of biopsied areas, and the outcome following surgery as described using the Engel Surgical Outcome Score. (y.o. = years old) (mo. = months).
All patients agreed to the comprehensive research protocol (Research IRB ID #200112047) approved by the institutional review board at the University of Iowa, which included genetic investigation of resected brain tissues, as described in this study.
Intraoperative EEG was performed with multiple strip electrodes during the second surgery, typically covering the lateral and ventral temporal neocortex along the extent of the resection sites as well as the entire hippocampus to tailor resection extent. Brain samples from patients 282, 284, and 292 were evaluated by RNA‐Seq. After removal of the intracranial electrodes, patient 282 underwent resection of the posterior hippocampus, amygdala, and posterior lateral basal frontal cortex. Patient 284 underwent sided anterior temporal lobectomy with cortico‐amygdala‐hippocampectomy (resection of amygdala and hippocampus and resection/disconnection of the temporal pole). Patient 292 underwent resection of the anterior temporal lobectomy with cortico‐amygldala‐hippocampectomy and resection of dorsal medial frontal area.
Brain samples from patients 302, 316 and 334 were evaluated by RNA‐Seq from both spiking and nonspiking specimens. After removal of the intracranial electrodes, subjects 302 and 316 underwent tailored anterior temporal lobectomy (specifically, a cortico‐amygdala‐hippocampectomy) based on the invasive recording results and intraoperative EEG results. The surgery involved disconnection/resection of the temporal pole and temporal neocortex as well as resection of the amygdala and hippocampus. Subject 334 underwent extensive resection of the right temporal lobe epileptogenic zone, including a right cortico‐amygdala‐hippocampectomy and a mesial temporal cystic lesion, as well as resection of the ventral temporal cortex and middle and inferior temporal gyri, beyond the typical extent of the standard anterior temporal lobectomy.
Tissue procurement
Blood samples were obtained at the beginning of each case after induction of general anesthesia, intubation, and placement of appropriate arterial/venous lines and stored in an ice water bucket. All brain tissue samples, once resected, were separated into two pieces, one sent to the clinical pathology laboratory for tissue diagnosis and the other sent to the research laboratory. All separated tissue samples were immediately placed in pathology specimen cups, stored in a box with dry ice. At the destination research laboratory, specimens were stored in a ‐80 degree C freezer.
RNA sequencing
RNA from brain tissue was extracted using RNeasy Lipid Tissue Mini Kit (Qiagen, Cat# 74804). Blood RNA was extracted using Paxgene Blood RNA Kit (PreAnalytiX, Cat# 762164). Both RNA preparations followed the manufacturer's protocol. 500 ng total RNA of each sample was submitted to the genomics division of Iowa Institute of Human Genetics (IIHG). Twelve libraries were prepared using the Illumina TruSeq Stranded total RNA ribozero sample preparation kit (Illumina, Inc., San Diego, CA) and following Illumina's sample preparation guide, starting with 500 ng of input total RNA. The libraries were run on the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) and combined in equimolar amounts into one pool. The concentration of the pool was measured using the KAPA Illumina Library Quantification Kit (KAPA Biosystems, Wilmington, MA) and sequenced on the Illumina HiSeq 2500 sequencer with a 125 bp Paired‐End v4 SBS chemistry (Illumina).
Differential expression analysis
Transcript quantification
Samples yielded a mean of 88 million sequencing reads. Sequencing reads from FASTQ files were quantified with Kallisto,10 using the default settings and an index built from transcripts from GENCODE11 v24.
Differential expression analysis
A quasipoisson generalized linear mixed effects model (GLMM) was fit to each measure of transcript abundance (Kallisto counts), using the total sample counts as a covariate, and subject and sequencing batch as random effects. Variables of interest were tissue type and spiking status. Contrasts were tested using the glht() function in the multcomp R package12 to compare (1) spiking hippocampal to nonspiking cortical tissue and (2) spiking versus nonspiking hippocampal tissue. P‐values were adjusted for multiple testing using the Benjamini–Hochberg procedure.13 Transcripts were considered for further analysis if they showed FDR < 0.1 for both of the above contrasts and the direction of effect was consistent between contrasts. An annotated summary of DE transcripts is listed in Table S1.
Cluster analysis
Differentially expressed genes from Table S1 (spiking vs nonspiking tissue P‐value <0.05) were loaded into a STRING network using the STRINGapp14 within Cytoscape.15 A minimum edge interaction strength of 0.40 (medium‐confidence level in STRING) was required for an interaction to be drawn. The largest fully connected subcomponent of this graph was used for further analysis. The glay clustering algorithm within the clusterMaker application16 of Cytoscape revealed three clusters of DE genes. Functional enrichment analysis of each cluster was performed using the clusterProfiler library17 within R [https://stat.ethz.ch/pipermail/r-help/2008-May/161481.html]. From each cluster, genes were highlighted that were either well‐cited (>3 SD from the mean) in the epilepsy literature or that were the top two genes ranked by total number of edges with a citation count below the cluster's mean, a ratio of within cluster edges to total edges greater than the cluster's mean, and belonging to one of the top five annotated gene ontology terms for a cluster. Genes meeting this criteria were labeled in the network analysis and are shown in Table 2.
Table 2.
Differentially expressed genes highlighted in cluster analysis
| Symbol | P‐value | Coefficient | # Citations | Cluster | Edges | Name |
|---|---|---|---|---|---|---|
| High citation count degs | ||||||
| POLG | <1E‐16 | −18.3 | 56 | 1 | 29 | DNA polymerase gamma, catalytic subunit |
| KCNJ10 | <1E‐16 | 0.25 | 34 | 1 | 21 | Potassium voltage‐gated channel subfamily J member 10 |
| PSEN1 | 6E‐14 | 0.41 | 33 | 2 | 92 | Presenilin 1 |
| BRD2 | 4E‐4 | 0.55 | 12 | 3 | 82 | Bromodomain containing 2 |
| MEF2C | 1E‐3 | −0.26 | 12 | 3 | 46 | Myocyte enhancer factor 2C |
| CDK5 | <1E‐16 | −1 | 11 | 3 | 105 | Cyclin dependent kinase 5 |
| Highly connected, low citation count | ||||||
| CNTNAP4 | 1E‐5 | 0.50 | 1 | 1 | 19 | Contactin‐associated protein like 4 |
| SLC24A2 | 1E‐6 | 0.37 | 0 | 1 | 17 | Solute carrier family 24 member 2 |
| BTBD9 | 3E‐5 | −0.87 | 0 | 2 | 48 | BTB domain containing 9 |
| KLHL24 | 1E‐6 | 0.29 | 0 | 2 | 34 | Kelch like family member 24 |
| KIF19 | 5E‐9 | 0.68 | 0 | 3 | 41 | Kinesin family member 19 |
| L RRCC1 | 4E‐5 | 0.37 | 0 | 3 | 61 | Leucine rich repeat and coiled‐coil centrosomal protein 1 |
Table of genes from each cluster which pass citation threshold defined in methods. The P‐values and coefficients of expression are shown for each gene. Network properties such as cluster membership and total edge count are also displayed, along with the total number of citations in the epilepsy‐associated literature and the full gene name.
Cell‐type deconvolution
We used cell‐type specific expression levels18 as independent variables to predict the direction of differential expression (logistic regression) in our spiking tissues. We then used the model coefficients, their signs, and their significance to interpret the relative change in each cell type in the bulk tissue.
Drug repositioning
Gene‐level summaries of direction of effect were computed for the DE transcripts described above. First, for each contrast, the coefficient median was calculated, and the mean of these values was aggregated across all DE transcripts mapping to a single gene. This yielded a vector across all DE genes that indicated the direction of effect in healthy, nonspiking tissue. Using the CMap database,6 which provides gene expression values from cell lines cultured in the presence of each of a library of compounds, we performed a correlation test (cor.test() in R) on each CMap experiment (restricted to those genes that were DE in the current study). Thus, we obtained, for each drug tested in CMap, a P‐value and correlation coefficient that indicates the degree to which the drug perturbs gene expression in a way that is either correlated or anticorrelated with the gene expression signature observed in the current study. Compounds showing a correlation with P < 0.05 are provided in Table S2, along with PubMed IDs of papers mentioning both the drug and “epilepsy” or “seizure.”
Functional validation in zebrafish
Fish and embryo rearing
Zebrafish embryos and adults were reared as described previously,19 in the University of Iowa Zebrafish Facility. Embryos were staged by hours or days post fertilization at 28.5°C (hpf or dpf).20 Until the beginning of movement‐analysis experiments, larvae were raised in fish water (double distilled water supplemented with 0.30 g Crystal Sea (Marinemix) per liter).
Drug delivery
Concentrated stock solutions for pentylenetetrazole (PTZ, Sigma), metformin (Cayman Chemical), doxycycline (Cayman Chemical), nifedipine (Cayman Chemical), and pyrantel tartrate (TargetMol) were made by dissolving each in either ddH2O (PTZ, doxycycline, metformin) or DMSO (nifedipine, pyrantel tartrate). 7 dpf (or 5 dpf, in the case of the doxycycline treatment experiments) zebrafish larvae were placed individually into separate wells of a 96‐well plate in fish water. All water in each well was carefully drawn off, and 250 μL 1X Ringer's Solution (116 mmol/L NaCl, 2.9 mmol/L KCl, 1.8 mmol/L CaCl2, and 5 mmol/L HEPES) was pipetted into the well. Lastly, small volumes of concentrated stock solutions of drug (or vehicle) were delivered to their respective groups to reach their final concentrations: 5 mmol/L PTZ, 2 μmol/L metformin, 10 μmol/L doxycycline, 12 μmol/L nifedipine, and 75 μmol/L pyrantel tartrate. Solutions were mixed by gently tapping the plate. The larvae were divided such that all five groups had approximately equal representation on every plate (from 10 to 20 embryos per treatment group per plate).
Behavior analysis
Behavior was tracked using the ZebraLab system (Viewpoint). Larvae were pretreated with metformin, doxycycline, nifedipine, pyrantel tartrate, or vehicle during the first 30‐min behavior analysis (baseline), in which we measured total distance traveled and distance traveled at a velocity of greater than 20 mm/sec; movement greater than 20 mm/sec was classified as seizure behavior.21 After obtaining baseline behavior levels, PTZ, a proseizure drug, or vehicle was introduced into appropriate wells. Immediately after this addition, the second 30‐minute behavior analysis took place. The change in movement between the first and second half‐hour tests was analyzed. To measure the antiseizure effect of each drug in combination with PTZ, the average total distance traveled at a speed greater than 20 mm/sec was calculated for each treatment group. The average total movement at all speeds was used to determine the toxicity of each drug alone by comparing the net movement of vehicle‐treated controls and drug‐treated controls. Statistical analysis was performed using a two‐tailed student's t‐test on GraphPad Prism 7.
Study approval
Human
All patients agreed to the comprehensive research protocol (Research IRB ID #200112047) approved by the institutional review board at the University of Iowa, which included genetic investigation of resected brain tissues, as described in this study.
Animal studies
The University of Iowa Animal Care and Use Committee approved and oversaw all of the zebrafish studies.
Results
Differential expression
To identify transcripts DE in response to spiking, we carried out RNA‐Seq on brain tissue resected from six patients with intractable temporal lobe epilepsy. Six hundred and eighty‐nine transcripts from 628 genes were DE between spiking and nonspiking tissue (Table S1). Of these, 148 had been cited previously in publications that mention “epilepsy” or “seizure.” This represents a significant enrichment of literature‐supported genes (OR = 3.1, P < 2 × 10−16).
Some notable examples of genes previously linked to epilepsy that we detected as DE include POLG,22 PSEN1,23 KCNJ10,24 CACNA2D2,25 KCNH2,26 PLP1,27 MBP,28 MEF2C,29 SLC12A2,30 BRD2,31 NFKB1,32 QKI,33 CDK5,34, and NRXN1 35 (see Table S1 for a complete list). Most (51/90) GO terms enriched in our DE genes were also enriched among literature‐supported seizure genes (Table S2), meaning that, thematically, our results fit well with what is known about the molecular basis of epilepsy. Gene‐set enrichment analysis of DE genes not previously implicated in epilepsy ranked transcriptional regulators as the most over‐represented class of genes (P < 0.01), including SIN3A, SMARCC2, SMARCA5, CHD3, SETD1B, ARID2, BRD7, KAT2B, and BRCA1. Regulation via phosphorylation was also represented among DE genes, notably including PHLPP2, which dephosphorylates AKT at Ser473, the site important for mTORC2‐mediated AKT signaling to promote apoptosis. PHLPP2 expression is reduced in spiking tissue, suggesting a prosurvival response. MAL, an integral membrane protein involved in myelination, was among the DE genes without a previous association with epilepsy and further underscores the dysregulation of myelin‐centric processes in epilepsy. A full list of DE genes is found in Table S1.
Cluster analysis
We next performed a network and cluster analysis of DE genes using the STRING network. The fully connected subnetwork of DE genes using medium‐confidence STRING interactions (STRING score >0.4) was used as a starting point for clustering (440 genes). This network showed a significant enrichment of functional connectivity (P = 3.08 × 10−6) between DE genes. Cluster analysis was performed in Cytoscape15 using a greedy algorithm that maximizes modularity score in the clusterMaker application.16This analysis yielded three distinct clusters (Fig. 2A). Labeled genes in the network analysis are either highly cited in the epilepsy literature or represent highly connected potentially novel epilepsy‐associated genes (see specific criteria above).
Figure 2.
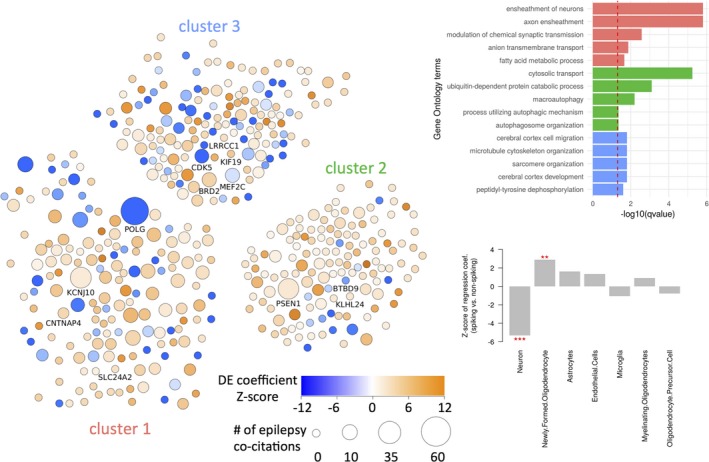
Network analysis and cell‐type changes in spiking bulk tissue. Differentially expressed genes were analyzed and clustered as a STRING network of proteins (A). Nodes are colored by the Z‐score of their differential expression coefficients, and sized relative to the number of times they are cited within the epilepsy literature. Labeled nodes are either highly cited or highly interconnected within the network and also contained within one of the top five gene sets annotated to the associated cluster. Each cluster was subject to functional enrichment analysis using the Gene Ontology (B), with only the top five terms reported. Using gene expression from purified cell samples, we inferred the relative abundance (w.r.t. nonspiking tissue) of cells types in spiking tissue. ***P < 0.001; **P < 0.01 (C).
Functional enrichment analysis of each cluster (Fig. 2B) yielded a number of multiple‐testing corrected significant gene sets from the Gene Ontology. Cluster 1 showed a strong enrichment of gene sets relating to myelination, a process previously reported in other RNA‐sequencing datasets in epilepsy.36 The genes POLG and KCNJ10 are two well‐known epilepsy‐associated genes in this cluster (Table 1). Two highly interconnected genes within this cluster and not previously reported within the epilepsy literature include CNTNAP4, a cell adhesion molecule in the neurexin family associated with psychiatric disease,37 and SLC24A2, a calcium transporter which has been observed to be mutated in ASD probands.38 Cluster 2 had a strong enrichment of gene sets relating to protein degradation, such as protein ubiquitination, and an overall enrichment of upregulated genes. Cluster 2 contained the well‐known Alzheimer's disease‐gene PSEN1, which has previously been linked with epilepsy.23 Two highly interconnected genes within cluster 2 included BTBD9, a gene associated with restless legs syndrome, and KLHL24, a ubiquitin ligase substrate receptor. Cluster 3 was enriched for annotations relating to brain development and cell migration, and harbored predominantly negatively regulated DE genes. Several epilepsy‐associated genes were present in this cluster, including MEF2C, BRD2, and CDK5, along with a highly interconnected centrosomal protein LRRCC1.
Myelination emerged as a theme from our literature‐supported epilepsy genes, and also in our gene expression results, pointing to a general upregulation of myelin‐associated genes in spiking tissue. This may suggest increased presence of oligodendrocytes in spiking tissue, potentially related to gliosis, an effect previously observed in temporal lobe epilepsy white matter pathology.39 Indeed, a follow‐up analysis, where we performed cell‐type deconvolution in spiking tissues, showed that neuron‐specific gene expression was generally attenuated (Z = −5.3, P = 1 × 10 − 7), while gene expression related to newly formed oligodendrocytes was increased (Z = 2.9, P = 0.004; see Fig. 2C). Expression related to astrocytes, oligodendrocyte precursors, myelinating oligodendrocytes, microglia, and endothelial cells did not differ significantly in spiking versus control tissues.
Drugs predicted to have anti‐ and proseizure properties
Based on their reported effects on gene expression in a panel of cultured cell lines,6 one hundred and eighty‐four compounds were predicted to have therapeutic properties in the context of seizures (Table S2), owing to their significant (P < 0.05) negative correlation with our observed epilepsy transcriptional signature (Fig. 3A). Meanwhile, 208 compounds were predicted as candidate proseizure compounds, based on their significant positive correlation with the epilepsy signature. While the magnitude of correlation values was low (−0.136 to 0.174), this is in line with observations from a previous drug repurposing study.40
Figure 3.
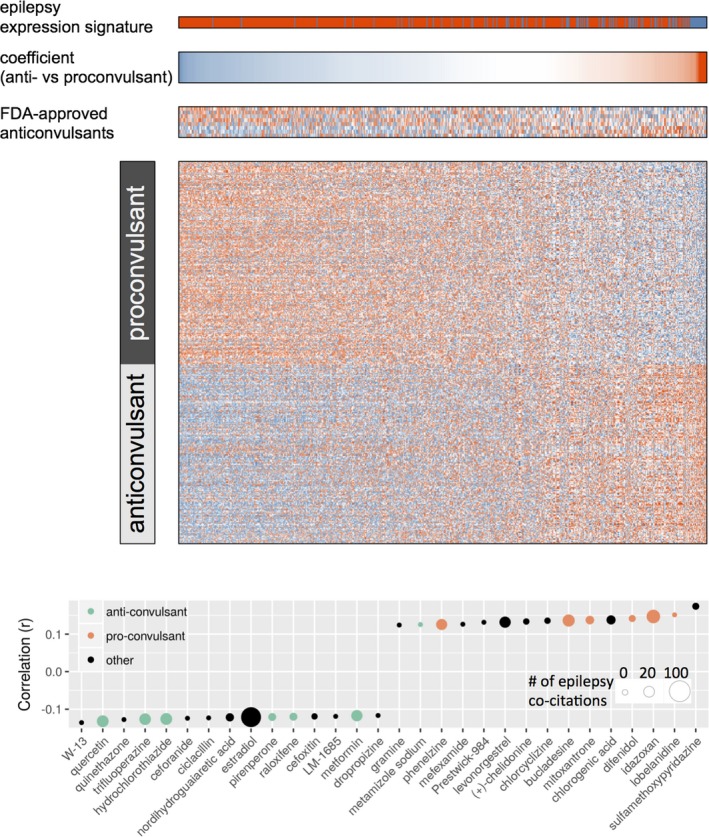
Finding compounds that mimic or reverse the epileptic gene expression signature. The expression matrix (A), a subset of the Connectivity Map, is divided into putative proseizure and antiseizure compounds. Predicted proseizure compounds are those that are significantly positively correlated with the epilepsy differential expression signature (top, spiking vs. non‐spiking tissue), while predicted antiseizure compounds are those significantly anticorrelated with the epilepsy signature. Genes (columns) are ordered by their coefficient value in predicting pro‐ from antiseizure compounds in a regularized logistic regression model. Positive coefficients indicate upregulation of the gene being associated with therapeutic properties, while negative coefficients indicate upregulation of the gene being associated with pathogenesis. The expression matrix of 8 FDA‐approved antiseizures are shown for reference. The top and bottom 15 most correlated/anticorrelated drugs are shown (B), with their color indicating literature‐reported anti‐ or proseizure activity.
We next tested whether known antiseizure drugs would have an overall negative correlation with our epilepsy expression signature. FDA‐approved anticonvulsants were identified in the Connectivity Map dataset, including carbamazepine, ethosuximide, gabapentin, phensuximide, primidone, topiramate, trimethadione, and vigabatrin. When correlating their expression profiles to our epilepsy expression signature, no clear direction of correlation was observed (Fig. 3A). Table S2 includes PubMed IDs of publications mentioning both the compound and “seizure” or “epilepsy.” Manual inspection of the literature associated with top pro‐ and anti‐epileptic candidate compounds supports that their predicted function is plausible (Fig. 3B). For instance, the literature supports the predicted properties of our top‐ranking anti‐epileptic candidates: quercetin,39 trifluoperazine,41 hydrochlorothiazide,42 pirenperone,43 raloxifene,44 and metformin.45 Similarly, the literature supports the compounds predicted to be proseizure: idazoxan,46 difenidol,47 mitoxantrone,48 bucladesine,49 and phenelzine.50
Structural properties of the compounds were investigated as predictors of their putative therapeutic (or pathogenic) properties. A random forest was trained using 881 NCBI PubChem structural characteristics, computed using the ChemmineR package, and the two class labels (proseizure or antiseizure). Default parameters were used in training. The out‐of‐bag classification errors of 49% (proseizure) and 56% (antiseizure) suggest that structural properties do not generally predict a compound's inferred therapeutic effect.
Validation of drug efficacy in zebrafish
Zebrafish are an effective model of epileptic seizures and, when used in combination with the proseizure compound PTZ, can be used to test potential antiseizure drugs.8, 51 Using zebrafish as our model system, we are able to test a large number of animals simultaneously. Furthermore, drugs can be added directly to the water, ensuring a consistent dose within treatment groups. From the list generated by our analysis, we tested whether seizures in larval zebrafish might be ameliorated by the predicted antiseizure properties of four compounds: doxycycline, metformin, nifedipine, and pyrantel tartrate. Many other compounds on the list are predicted to be more therapeutic, but their FDA approval has been discontinued or they are prohibitively expensive. For simplicity, fish treated with PTZ to induce seizures will henceforth be called “PTZ fish.” Fish not treated with PTZ will be called “control fish.”
Of the four drugs we tested, doxycycline had the least promising antiseizure potential. It had no effect on total distance moved (Fig. 4A) or seizure‐like behavior (velocity greater than 20 mm/sec) (Fig. 4A’), in either control fish or PTZ fish. By contrast, all other compounds showed some promise of being antiseizure.
Figure 4.
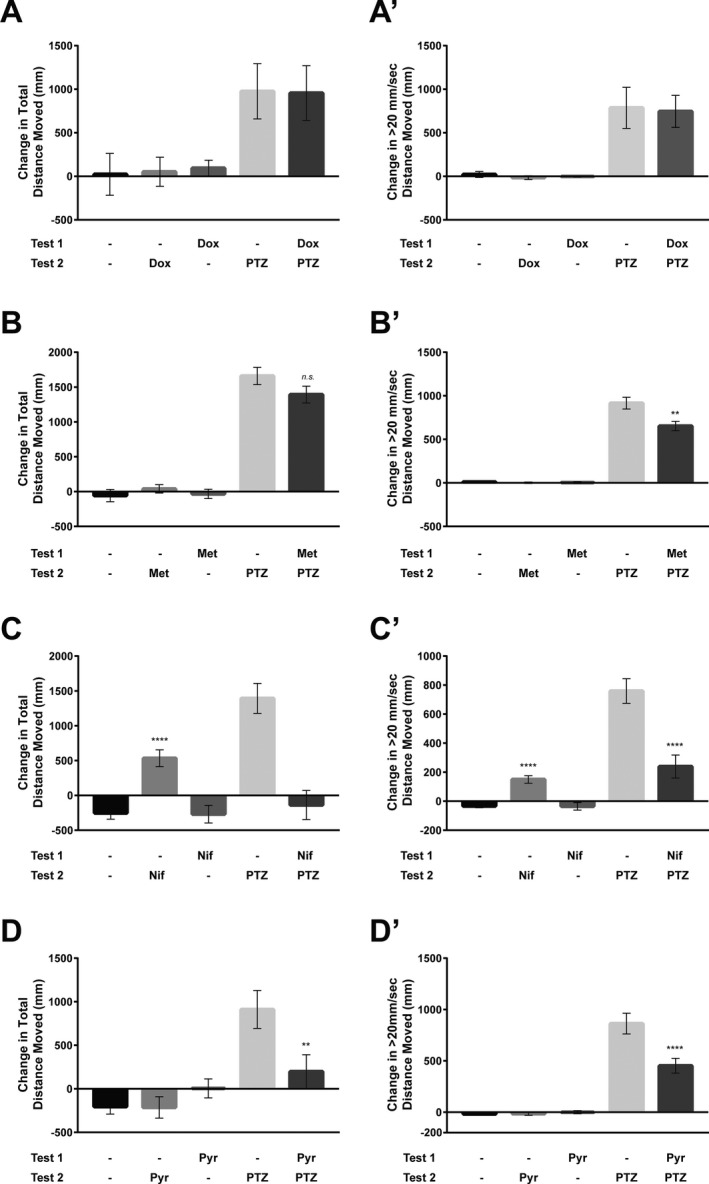
Larval zebrafish behavioral assays. (A) Doxycycline behavioral assay. The average total distance moved at all speeds (mm) for each treatment group (n = 7–8). In the first half‐hour behavioral assay (Test 1), larvae were treated with vehicle (−) or doxycycline (Dox). In the second half‐hour behavioral assay (Test 2), larvae were treated with vehicle, Dox, or PTZ. Movement greater than 20 mm/sec is shown in A’. (B) Metformin behavioral assay. The average total distance moved at all speeds (mm) for each treatment group (n = 30–66). In the first half‐hour behavioral assay, larvae were treated with vehicle or metformin (Met). In the second half‐hour behavioral assay, larvae were treated with vehicle, Met, or PTZ. Movement greater than 20 mm/sec is shown in B’. (C) Nifedipine behavioral assay. The average total distance moved at all speeds (mm) of each treatment group (n = 38). In the first half‐hour behavioral assay, larvae were treated with vehicle or nifedipine (Nif). In the second half‐hour behavioral assay, larvae were treated with vehicle, Nif, or PTZ. Movement greater than 20 mm/sec is shown in C’. (D) Pyrantel tartrate behavioral assay. The average total distance moved at all speeds (mm) of each treatment group (n = 38–40). In the first half‐hour behavioral assay, larvae were treated with vehicle or pyrantel tartrate (Pyr). In the second half‐hour behavioral assay, larvae were treated with vehicle, Pyr, or PTZ. Movement greater than 20 mm/sec is shown in D’. Bars represent mean ± SEM. ****P‐value < 0.0001. **P‐value < 0.03.
Metformin treatment of control fish had no effect on total movements or seizure‐like movement relative to vehicle treatment (Fig. 4B,B’). Interestingly, while metformin pretreatment of PTZ fish did not significantly affect their total movement (Fig. 4B) it did reduce their seizure‐type movement by roughly 28% (Fig. 4B’). This indicates metformin suppresses seizures without generally repressing motility or activity of the CNS.
Unexpectedly, nifedipine treatment of control fish induced higher total movement, and more seizure‐like movement (Fig. 4C,C’). This indicates that rather than being toxic to the fish, nifedipine can acutely induce movement. However, nifedipine pretreatment of PTZ fish reduced their total movement (by over 100%) (Fig. 4C) and also reduced their seizure‐type movement by roughly 68% (Fig. 4C’). Thus, nifedipine appears to inhibit PTZ‐induced seizure behavior while elevating overall motility.
Finally, pyrantel tartrate treatment of control fish had no effect of total movement or seizure‐type movement. However, pyrantel tartrate pretreatment of PTZ fish reduced total movement (by 78%) (Fig. 4D) and seizure‐type movement by about 48% (Fig. 4D’).
Discussion
This study applied transcriptomic analysis in epilepsy patient brain biopsy specimens with the aim of identifying potentially therapeutic compounds. In this study design, the control tissue comes from the same participants as the spiking tissue. This approach has the inherent drawback that the conversion to a nonspiking tissue transcriptional profile derived from cases may be less effective than a return to a true control profile. Weighing this drawback, in light of our patient population which has undergone extensive treatment for epilepsy, we chose to use the nonspiking tissues as controls to ensure identical environmental influences (e.g., drug exposures) and potentially reduce bias from comparing postmortem control tissue to that taken from living individuals. Weighing these two alternatives, we used the above described study design, through analyzing gene expression differences between seizing and nonseizing tissues we were able to identify several known epilepsy‐associated genes, such as POLG and PSEN1, along with several genes with low citation counts in the epilepsy literature but high functional enrichment in our network analysis, such as CNTNAP4 and BTBD9. Clustering of our DE gene network yielded three sets of genes that were distinct in both their functional annotations and in their characteristic direction of regulation. One emergent theme from our network analysis was an enrichment of differential expression of genes relating to myelination, and subsequent analysis suggested an increase in oligodendrocytes in spiking tissue. Overall loss or aberrant patterns of myelination exist in many epilepsy cases,52 linking the expression changes we see with a known clinical phenotype.
Additional observations from our network analysis were the upregulation of genes relating to the protein ubiquitination system and autophagy. Interestingly, the inhibition of protein degradation processes and autophagy in mouse models of status epilepticus are thought to play a protective role in mediating seizure‐induced brain damage.53 Several other studies have implicated the misregulation of protein ubiquitination processes as underlying some cases of epilepsy, therein identifying novel epilepsy‐associated disease genes.54, 55 While our study design does not allow for causality testing, the upregulation of genes relating to the protein ubiquitination system may be contributory to the phenotypes observed in our cases. Lastly, our network analysis also showed a downregulated grouping of genes related to cell migration and neuron development. Interestingly, the stimulation of neuronal migration has been directly observed in mouse models of epilepsy,56 and is thought to underlie the pathological phenotype of granule cell dispersion57 in cases of temporal lobe epilepsy. Taken together, our network analysis of genes differentially regulated in epilepsy recapitulates many known cellular processes underlying epilepsy and may inform future therapeutic targets.
Expanding on these analyses of epilepsy gene expression signatures, this study generated a list of 184 candidate anti‐epilepsy compounds. This list of possible seizure suppressing compounds includes 129 drugs that have been previously studied in some model of seizures and 55 that have never been studied in the context of seizures. 91 of these 184 compounds are already FDA approved for human use, but not for treating seizures or epilepsy. We selected four of these drugs (doxycycline, metformin, nifedipine, and pyrantel tartrate) to test for seizure suppression in vivo, in our zebrafish PTZ‐induced seizure model. These four drugs were selected from our list because they are all used clinically in humans, and can be taken orally. Doxycycline, a commonly used antibiotic, had no effect in our zebrafish seizure‐suppression model, while the other three drugs we tested suppressed seizure behavior significantly. An important caveat is that while it has been shown that swimming behavior can be used as a read‐out for seizure activity, only electrophysiological recordings can confirm true suppression of seizures.
Metformin is a commonly used diabetes medication that decreases liver glucose production.58 Although metformin has been reported to suppress seizures in some rodent models,45, 59, 60 it has not been studied in any clinical trial for human seizures or epilepsy, and is not currently FDA approved for use in epilepsy. The exact mechanism(s) of action of metformin is likely multifaceted and not fully elucidated, thus the mechanism whereby metformin suppresses seizures is not obvious. Nevertheless, metformin is widely used and typically well tolerated, making it an ideal candidate for testing as an antiseizure medication in humans.
Nifedipine is a widely prescribed antihypertensive medication, thought to act (predominantly) as an l‐type calcium channel blocker.61 Since nifedipine regulates ion movement, its role as a possible antiseizure and anti‐epileptic seems plausible. Nifedipine has been widely studied in rodent models of seizures (Table S2), and has been suggested as a possible therapy for human seizures, largely as an add‐on therapy for drug‐resistant epilepsy.62, 63, 64, 65, 66, 67, 68 A single randomized control trial was conducted in 1992, testing nifedinine as an add‐on agent in refractory epilepsy.69 This study enrolled 22 patients with refractory epilepsy but detected no effect with a single dose of nifedipine. Our results showing antiseizure activity in zebrafish, combined with results by others in rodents,70, 71, 72 and the small sample size and severe phenotype of epilepsy in these patients, suggest nifedipine should be reconsidered for clinical trials as a seizure suppressant in humans.
Pyrantel tartrate is an antiparasitic agent that acts by inhibiting fumarate reductase, and by directly acting on acetylcholine receptors at the neuromuscular junction of infecting helminths. Pyrantel tartrate is FDA approved for use in domestic animals and has been used to treat human parasitic infections.73 Unlike nifedipine and metformin (for which some rodent studies and human reports relate to seizures), a March 2018 PubMed search for “pyrantel and epilepsy” and “pyrantel and seizure” found no manuscripts that studied pyrantel in seizures. Thus, pyrantel tartrate represents a truly novel antiseizure drug candidate yielded by our screen.
Which human patient populations might be appropriate cohorts in which to test these new candidate antiseizure compounds? We selected the candidate drugs from experiments carried out on tissue from patients with refractory epilepsy. However, our in vivo screen used the proseizure compound pentylenetetrazol (PTZ), which inhibits the gamma‐aminobutyric acid (GABA)(A) receptor complex.74 Several groups of patients have seizures known to result from defects or imbalances in GABA‐ergic activity. These include patients with Dravet syndrome75, 76 (usually with SCN1A mutation) who can have impaired GABA‐mediated short‐interval intracortical inhibition, patients with Angelman syndrome77 (typically maternal deletion of 154q11.2‐q13 or UBE3A mutation) who can have GABA‐ergic expression anomalies, and in patients with Lennox–Gastaut syndrome (characterized by multiple seizure types, a specific electro‐encephalographic pattern, and mental regression) who often have mutations in the GABAR gene. These three patient populations are fairly easy to clinically phenotype, by genetic and EEG testing. We suggest they are an appropriate test population for initial clinical trials for the new potential antiseizure medications presented in Table S2, while acknowledging that the zebrafish PTZ model most closely mimics cases of acute seizures in humans, and may not translate to the aforementioned chronic epileptic diseases.
Our study demonstrates the power of combining patient samples, genome‐wide transcriptomic analyses, expression analysis from drug screens, and in vivo seizure medication screening. Using this combination, we quickly moved from the clinic and operating room to the elucidation of candidate antiseizure drugs that are already FDA approved This method has applicability not just for drug discovery, but for understanding seizure pathophysiology. These candidate antiseizure drugs could lead to in‐human clinical trials and rapidly and relatively inexpensively offer several new treatments for patients with previously untreatable seizures and epilepsy. The ability to consider already FDA‐approved drugs for treatment of patients with potentially devastating disease must be weighed against our lack of mechanistic understanding of the role of many of these molecules in seizures and epilepsy, or their safety for patients with these conditions.
Conflict of Interest
The authors all have a patent pending related to the use of the drugs described in this manuscript for epilepsy.
Author Contributions
Alex Bassuk oversaw all aspects of the manuscript. Alex Bassuk, Jacob Michaelson, and Rob Cornell conceived of the entire project outlined in Figure 1 together. Clinical analysis of patients and biopsy selection and collection were performed by Andrew Grossbach, Yasunori Nagahama, Mathew Howard, and Hiroto Kawasaki. Shu Wu prepared all of the human tissue for RNA‐Seq analysis. Jacob Michaelson designed all of the algorithms with assistance from Leo Brueggeman and input from Alex Bassuk. Leo Brueggeman performed cluster analysis and interpretation of RNA‐Seq data. Morgan Sturgeon and Russell Martin developed the novel zebrafish testing assay and performed all of the experiments under supervision by Rob Cornell and Alex Bassuk. All the authors contributed to the writing of the manuscript.
Supporting information
Table S1. Differentially expressed transcripts in spiking versus nonspiking tissue.
Table S2. List of drugs predicted to be pro‐ or antiseizure based on correlation between differentially expressed genes.
Acknowledgments
Funded by National Institutes of Health 5R01NS098590 (to AGB). AGB oversaw all aspects of the project and co‐wrote the manuscript with all the authors. JJM oversaw the computational experiments with LB. RAC oversaw all zebrafish experiments performed by MLS and RMM. All clinical analysis and resection were performed by AJG, YN, MAH, and HK. SW performed the RNA and DNA purification. The authors declare no competing interests. All data and materials will be available upon publication.
Funding Information
Funded by National Institutes of Health MH105527 (to AGB).
Funding Statement
This work was funded by National Institutes of Health grant MH105527.
Contributor Information
Jacob J. Michaelson, Email: jacob-michaelson@uiowa.edu
Alexander G. Bassuk, Email: alexander-bassuk@uiowa.edu
References
- 1. Iorio F, Rittman T, Ge H, et al. Transcriptional data: a new gateway to drug repositioning? Drug Discov Today 2013;18:350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mirza N, Sills GJ, Pirmohamed M, Marson AG. Identifying new antiepileptic drugs through genomics‐based drug repurposing. Hum Mol Genet 2017;26:527–537. [DOI] [PubMed] [Google Scholar]
- 3. Xue H, Li J, Xie H, Wang Y. Review of drug repositioning approaches and resources. Int J Biol Sci 2018;14:1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sirota M, Dudley JT, Kim J, et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med 2011;3:96ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Srivastava PK, van Eyll J, Godard P, et al. A systems‐level framework for drug discovery identifies Csf1R as an anti‐epileptic drug target. Nat Commun 2018;9:3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene‐expression signatures to connect small molecules, genes, and disease. Science 2006;313:1929–1935. [DOI] [PubMed] [Google Scholar]
- 7. Claerhout S, Lim JY, Choi W, et al. Gene expression signature analysis identifies vorinostat as a candidate therapy for gastric cancer. PLoS ONE 2011;6:e24662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baraban SC, Taylor MR, Castro PA, Baier H. Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c‐fos expression. Neuroscience 2005;131:759–768. [DOI] [PubMed] [Google Scholar]
- 9. Berghmans S, Hunt J, Roach A, Goldsmith P. Zebrafish offer the potential for a primary screen to identify a wide variety of potential anticonvulsants. Epilepsy Res 2007;75:18–28. [DOI] [PubMed] [Google Scholar]
- 10. Bray NL, Pimentel H, Melsted P, Pachter L. Near‐optimal probabilistic RNA‐seq quantification. Nat Biotechnol 2016;34:525–527. [DOI] [PubMed] [Google Scholar]
- 11. Harrow J, Frankish A, Gonzalez JM, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 2012;22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biom Z 2008;50:346–363. [DOI] [PubMed] [Google Scholar]
- 13. Benjamini Y, Hochberg Y. Controlling the false discovery rate ‐ a practical and powerful approach to multiple testing. J Roy Stat Soc B Met 1995;57:289–300. [Google Scholar]
- 14. Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality‐controlled protein‐protein association networks, made broadly accessible. Nucleic Acids Res 2017;45(D1):D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morris JH, Apeltsin L, Newman AM, et al. clusterMaker: a multi‐algorithm clustering plugin for Cytoscape. BMC Bioinformatics 2011;9:436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y, Chen K, Sloan SA, et al. An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 2014;34:11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Westerfield M. The Zebrafish book. A guide for the laboratory use of Zebrafish (Danio Rerio). 4th ed Eugene, OR: University of Oregon Press, 2000. [Google Scholar]
- 20. Kimmel CB, Ballard WW, Kimmel SR, et al. Stages of embryonic development of the zebrafish. Dev Dynam 1995;203:253–310. [DOI] [PubMed] [Google Scholar]
- 21. Winter MJ, Redfern WS, Hayfield AJ, et al. Validation of a larval zebrafish locomotor assay for assessing the seizure liability of early‐stage development drugs. J Pharmacol Toxicol Methods 2008;57:176–187. [DOI] [PubMed] [Google Scholar]
- 22. Anagnostou ME, Ng YS, Taylor RW, McFarland R. Epilepsy due to mutations in the mitochondrial polymerase gamma (POLG) gene: a clinical and molecular genetic review. Epilepsia 2016;57:1531–1545. [DOI] [PubMed] [Google Scholar]
- 23. Larner AJ. Presenilin‐1 mutation Alzheimer's disease: a genetic epilepsy syndrome? Epilepsy Behav 2011;21:20–22. [DOI] [PubMed] [Google Scholar]
- 24. Dai AI, Akcali A, Koska S, et al. Contribution of KCNJ10 gene polymorphisms in childhood epilepsy. J Child Neurol 2015;30:296–300. [DOI] [PubMed] [Google Scholar]
- 25. Pippucci T, Parmeggiani A, Palombo F, et al. A novel null homozygous mutation confirms CACNA2D2 as a gene mutated in epileptic encephalopathy. PLoS ONE 2013;8:e82154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sanguinetti MC. HERG1 channelopathies. Pflugers Archiv 2010;460:265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hobson GM, Garbern JY. Pelizaeus‐Merzbacher disease, Pelizaeus‐Merzbacher‐like disease 1, and related hypomyelinating disorders. Semin Neurol 2012;32:62–67. [DOI] [PubMed] [Google Scholar]
- 28. Hu X, Wang JY, Gu R, et al. The relationship between the occurrence of intractable epilepsy with glial cells and myelin sheath ‐ an experimental study. Eur Rev Med Pharmacol Sci 2016;20:4516–4524. [PubMed] [Google Scholar]
- 29. Vrecar I, Innes J, Jones EA, et al. Further clinical delineation of the MEF2C haploinsufficiency syndrome: report on new cases and literature review of severe neurodevelopmental disorders presenting with seizures, absent speech, and involuntary movements. J Pediatr Genet 2017;6:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loscher W, Puskarjov M, Kaila K. Cation‐chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology 2013;69:62–74. [DOI] [PubMed] [Google Scholar]
- 31. Pal DK, Greenberg DA. Major susceptibility genes for common idiopathic epilepsies: ELP4 in rolandic epilepsy and BRD2 in juvenile myoclonic epilepsy In: Noebels J. L., Avoli M., Rogawski M. A., Olsen R. W., Delgado‐Escueta A. V. eds. Jasper's basic mechanisms of the epilepsies. pp. 845–857. Bethesda, MD: Oxford University Press, 2012. [PubMed] [Google Scholar]
- 32. Teocchi MA, Ferreira AE, deda Luz Oliveira EP , et al. Hippocampal gene expression dysregulation of Klotho, nuclear factor kappa B and tumor necrosis factor in temporal lobe epilepsy patients. J Neuroinflamm 2013;10:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Taylor SM, Bennett GD, Abbott LC, Finnell RH. Seizure control following administration of anticonvulsant drugs in the quaking mouse. Eur J Pharmacol 1985;118:163–170. [DOI] [PubMed] [Google Scholar]
- 34. Dixit AB, Banerjee J, Tripathi M, et al. Synaptic roles of cyclin‐dependent kinase 5 & its implications in epilepsy. Ind J Med Res 2017;145:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen T, Giri M, Xia Z, et al. Genetic and epigenetic mechanisms of epilepsy: a review. Neuropsychiatr Dis Treat 2017;13:1841–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hansen KF, Sakamoto K, Pelz C, et al. Profiling status epilepticus‐induced changes in hippocampal RNA expression using high‐throughput RNA sequencing. Sci Rep 2014;6:6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karayannis T, Au E, Patel JC, et al. Cntnap4 differentially contributes to GABAergic and dopaminergic synaptic transmission. Nature 2014;511:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prasad A, Merico D, Thiruvahindrapuram B, et al. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3: Genes, Genomes, Genetics 2012;2:1665–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sefil F, Kahraman I, Dokuyucu R, et al. Ameliorating effect of quercetin on acute pentylenetetrazole induced seizures in rats. Int J Clin Exp Med 2014;7:2471–2477. [PMC free article] [PubMed] [Google Scholar]
- 40. Dudley JT, Sirota M, Shenoy M, et al. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Transl Med 2011;3:96ra76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hedges D, Jeppson K, Whitehead P. Antipsychotic medication and seizures: a review. Drugs Today 2003;39:551–557. [DOI] [PubMed] [Google Scholar]
- 42. Dashputra PG, Khapre MD. Potentiation of the antiepileptic action of diphenylhydantoin by chlorothiazide congeners. Ind J Med Res 1970;58:159–166. [PubMed] [Google Scholar]
- 43. Pawlowski L, Siwanowicz J, Bigajska K, Przegalinski E. Central antiserotonergic and antidopaminergic action of pirenperone, a putative 5‐HT2 receptor antagonist. Pol J Pharmacol Pharm 1985;37:179–196. [PubMed] [Google Scholar]
- 44. Pottoo FH, Bhowmik M, Vohora D. Raloxifene protects against seizures and neurodegeneration in a mouse model mimicking epilepsy in postmenopausal woman. Eur J Pharm Sci 2014;18:167–173. [DOI] [PubMed] [Google Scholar]
- 45. Zhao RR, Xu XC, Xu F, et al. Metformin protects against seizures, learning and memory impairments and oxidative damage induced by pentylenetetrazole‐induced kindling in mice. Biochem Biophys Res Comm 2014;448:414–417. [DOI] [PubMed] [Google Scholar]
- 46. Shouse MN, Langer J, Bier M, et al. The alpha 2‐adrenoreceptor agonist clonidine suppresses seizures, whereas the alpha 2‐adrenoreceptor antagonist idazoxan promotes seizures in amygdala‐kindled kittens: a comparison of amygdala and pontine microinfusion effects. Epilepsia 1996;37:709–717. [DOI] [PubMed] [Google Scholar]
- 47. Yang CC, Deng JF. Clinical experience in acute overdosage of diphenidol. J Toxicol Clin Toxicol 1998;36:33–39. [DOI] [PubMed] [Google Scholar]
- 48. Dinday MT, Baraban SC. Large‐scale phenotype‐based antiepileptic drug screening in a Zebrafish model of dravet syndrome(1,2,3). eNeuro. 2015;2:ENEURO‐0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hosseini‐Zare MS, Salehi F, Seyedi SY, et al. Effects of pentoxifylline and H‐89 on epileptogenic activity of bucladesine in pentylenetetrazol‐treated mice. Eur J Pharmacol 2011;670:464–470. [DOI] [PubMed] [Google Scholar]
- 50. Bhugra DK, Kaye N. Phenelzine induced grand mal seizure. Br J Clin Prac 1986;40:173–174. [PubMed] [Google Scholar]
- 51. Baraban SC, Dinday MT, Hortopan GA. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat Commun 2013;4:2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rodriguez‐Cruces R, Concha L. White matter in temporal lobe epilepsy: clinico‐pathological correlates of water diffusion abnormalities. Quant Imaging Med Surg 2015;5:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Engel T, Martinez‐Villarreal J, Henke C, et al. Spatiotemporal progression of ubiquitin‐proteasome system inhibition after status epilepticus suggests protective adaptation against hippocampal injury. Mol Neurodegener 2017;12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gross C. Degrading seizures (or, Playing Tag with Nedd4‐2). Epilepsy Curr 2017;17:241–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhu J, Lee KY, Jewett KA, et al. Epilepsy‐associated gene Nedd4‐2 mediates neuronal activity and seizure susceptibility through AMPA receptors. PLoS Genet 2017;13:e1006634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Orcinha C, Munzner G, Gerlach J, et al. Seizure‐induced motility of differentiated dentate granule cells is prevented by the central reelin fragment. Front Cell Neurosci 2016;10:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Houser CR. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res 1990;535:195–204. [DOI] [PubMed] [Google Scholar]
- 58. Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: an update. Ann Intern Med 2002;137:25–33. [DOI] [PubMed] [Google Scholar]
- 59. Sanchez‐Elexpuru G, Serratosa JM, Sanz P, Sanchez MP. 4‐Phenylbutyric acid and metformin decrease sensitivity to pentylenetetrazol‐induced seizures in a malin knockout model of Lafora disease. NeuroReport 2017;28:268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang Y, Zhu B, Zheng F, et al. Chronic metformin treatment facilitates seizure termination. Biochem Biophys Res Comm 2017;484:450–455. [DOI] [PubMed] [Google Scholar]
- 61. Curtis TM, Scholfield CN. Nifedipine blocks Ca2 + store refilling through a pathway not involving L‐type Ca2 + channels in rabbit arteriolar smooth muscle. J Physiol 2001;532(Pt 3):609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bacia T. Remission of seizures after therapy with calcium antagonist (nifedipine) in a patient with severe generalized epilepsy. Pol Tyg Lek 1994;49:122–123. [PubMed] [Google Scholar]
- 63. Chaisewikul R, Baillie N, Marson AG. Calcium antagonists as an add‐on therapy for drug‐resistant epilepsy. Cochrane Database Syst Rev 2001;4:C002750. [DOI] [PubMed] [Google Scholar]
- 64. Hasan M, Pulman J, Marson AG. Calcium antagonists as an add‐on therapy for drug‐resistant epilepsy. Cochrane Database Syst Rev 2013;28:CD002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kulak W, Sobaniec W, Wojtal K, Czuczwar SJ. Calcium modulation in epilepsy. Pol J Pharmacol 2004;56:29–41. [PubMed] [Google Scholar]
- 66. Sander JW, Trevisol‐Bittencourt PC. Nifedipine as an add‐on drug in the management of refractory epilepsy. Epilepsy Res 1990;6:82–84. [DOI] [PubMed] [Google Scholar]
- 67. Stefani A, Spadoni F, Bernardi G. Voltage‐activated calcium channels: targets of antiepileptic drug therapy? Epilepsia 1997;38:959–965. [DOI] [PubMed] [Google Scholar]
- 68. Straub H, Hohling JM, Kohling R, et al. Effects of nifedipine on rhythmic synchronous activity of human neocortical slices. Neuroscience 2000;100:445–452. [DOI] [PubMed] [Google Scholar]
- 69. Larkin JG, Besag FM, Cox A, et al. Nifedipine for epilepsy? A double‐blind, placebo‐controlled trial. Epilepsia 1992;33:346–352. [DOI] [PubMed] [Google Scholar]
- 70. Brahmane RI, Wanmali VV, Pathak SS, Salwe KJ. Role of cinnarizine and nifedipine on anticonvulsant effect of sodium valproate and carbamazepine in maximal electroshock and pentylenetetrazole model of seizures in mice. J Pharmacol Pharmacother 2010;1:78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. El‐Azab MF, Moustafa YM. Influence of calcium channel blockers on anticonvulsant and antinociceptive activities of valproic acid in pentylenetetrazole‐kindled mice. Pharmacol Rep 2012;64:305–314. [DOI] [PubMed] [Google Scholar]
- 72. Russo E, Constanti A, Ferreri G, et al. Nifedipine affects the anticonvulsant activity of topiramate in various animal models of epilepsy. Neuropharmacology 2004;46:865–878. [DOI] [PubMed] [Google Scholar]
- 73. Pereira VG, Mattosinho Franca LC. Successful treatment of Capillaria hepatica infection in an acutely ill adult. Am J Trop Med Hyg 1983;32:1272–1274. [DOI] [PubMed] [Google Scholar]
- 74. Hansen SL, Sperling BB, Sanchez C. Anticonvulsant and antiepileptogenic effects of GABAA receptor ligands in pentylenetetrazole‐kindled mice. Prog Neuropsychopharmacol Biol Psychiatry 2004;28:105–113. [DOI] [PubMed] [Google Scholar]
- 75. Braat S, Kooy RF. The GABAA receptor as a therapeutic target for neurodevelopmental disorders. Neuron 2015;86:1119–1130. [DOI] [PubMed] [Google Scholar]
- 76. Stern WM, Sander JW, Rothwell JC, Sisodiya SM. Impaired intracortical inhibition demonstrated in vivo in people with Dravet syndrome. Neurology 2017;88:1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Roden WH, Peugh LD, Jansen LA. Altered GABA(A) receptor subunit expression and pharmacology in human Angelman syndrome cortex. Neurosci Lett 2010;483:167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially expressed transcripts in spiking versus nonspiking tissue.
Table S2. List of drugs predicted to be pro‐ or antiseizure based on correlation between differentially expressed genes.