

Summary
The supply of quality juveniles via land‐based larviculture represents a major bottleneck to the growing finfish aquaculture industry. As the microbiome plays a key role in animal health, this study aimed to assess the microbial community associated with early larval development of commercially raised Yellowtail Kingfish (Seriola lalandi). We used qPCR and 16S rRNA gene amplicon sequencing to monitor changes in the microbiome associated with the development of S. lalandi from larvae to juveniles. We observed an increase in the bacterial load during larval development, which consisted of a small but abundant core microbiota including taxa belonging to the families Rhodobacteraceae, Lactobacillaceae and Vibrionaceae. The greatest change in the microbiome occurred as larvae moved from a diet of live feeds to formulated pellets, characterized by a transition from Proteobacteria to Firmicutes as the dominant phylum. A prediction of bacterial gene functions found lipid metabolism and secondary metabolite production were abundant in the early larval stages, with carbohydrate and thiamine metabolism functions increasing in abundance as the larvae age and are fed formulated diets. Together, these results suggest that diet is a major contributor to the early microbiome development of commercially raised S. lalandi.
Introduction
A global increase in consumer demand has resulted in aquaculture being one of the fastest growing sectors in the global economy, with recent estimates that 47% of world fish supplies are now sourced from aquaculture (FAO, 2018). Most fish farming is dependent on the land‐based production of juveniles (larviculture). However, high mortality rates (between 80% and 100% of hatched larvae) in marine fish larviculture systems often result in production bottlenecks (Planas and Cunha, 1999; Lee, 2003; Vadstein et al., 2013). While improvements in gamete quality, nutrition and physicochemical conditions in fish rearing systems have made larviculture for some species viable (Merchie et al., 1997; Sorgeloos et al., 2001), efficiencies remain relatively low (Vadstein et al., 2013), suggesting that other factors are important for successful larval rearing.
In recent years, there has been an increased awareness of the importance of the host microbiome for digestion, immune function and development of animals, including fish (Bates et al., 2006; Fraune and Bosch, 2010; Stephens et al., 2016; Egerton et al., 2018; Wang et al., 2018). It has been shown that early bacterial colonization of the fish gastrointestinal (GI) tract is caused by the uptake of waterborne (Han et al., 2010) and/or feed‐associated bacteria (Hansen and Olafsen, 1999). The fish species’ trophic level and diet preference (Sullam et al., 2012; Smith et al., 2015; Liu et al., 2016; Gajardo et al., 2017), as well as variations of environmental factors, such as temperature and salinity, can also influence the types of microorganisms that colonize the gut (Bledsoe et al., 2016; Dehler et al., 2017).
Yellowtail Kingfish (Seriola lalandi) is an economically significant aquaculture species, particularly in the Asia‐Pacific region. While extensive nutritional and physiological research has been carried out on S. lalandi (Chen et al., 2006; Miegel et al., 2010; Bowyer et al., 2014), recent attention has turned to the microbial communities associated with this species (Aguilera et al., 2013; Ramirez and Romero, 2017; Legrand et al., 2018). For example, a culture‐dependent study observed differences in microbial composition of the S. lalandi GI tract between juvenile and pre‐adult life history stages (Aguilera et al., 2013). More recently, Ramirez and Romero (2017) found that wild‐caught and farmed S. lalandi have distinct intestinal microbiomes and suggest this is primarily due to differences in diet availability. While these reports highlight the changing nature of the S. lalandi microbiome, nothing is known about the microbiome of this species during early ontogeny in a cultured environment.
The aim of this current study was to explore the bacterial community associated with the early developmental stages of S. lalandi larvae in a larviculture production facility using 16S rRNA gene amplicon sequencing and functional gene prediction (Fig. 1). As the intestinal microbiome of many species has been shown to be sensitive to host physiology, diet and the environmental conditions, we hypothesized that the S. lalandi microbiome varies throughout the early development of the fish. To determine how different sources of environmental bacteria influence the early microbiome development, the waterborne and feed‐associated bacterial communities were also investigated. The findings of this study contribute to a broader understanding of the microbial diversity associated with economically valuable fish species and provide a foundation on which to develop methods for beneficial microbiome manipulation within commercially raised S. lalandi larvae.
Figure 1.
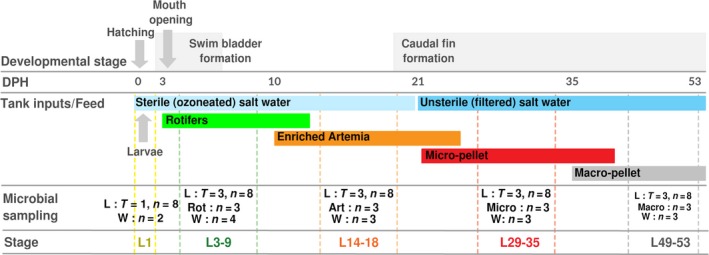
Experimental design and sampling methodology. Key developmental events (top), husbandry events (middle) and sampling procedure (bottom) during the course of the study, on a scale of days post‐hatching (DPH). The number of S. lalandi larvae or early juveniles (L), rotifers (Rot), Artemia (Art), micro‐pellet (Micro), macro‐pellet (Macro) and water (W) samples initially sampled at each time point is shown. Note to obtain sufficient biomass for L1 and L3‐9 stage larvae, one sample consisted of pooled individuals, for L14‐18, whole larvae were sampled, and for the juveniles, gut samples were dissected as indicated in the text and are shown in Fig. S1. T = number of time points (where there are more than one), n = number of samples per time point.
Results
Quantification of bacterial load associated with developing larvae
Quantitative PCR was used to estimate the bacterial load associated with specific development stages of S. lalandi larvae and juvenile stages. Bacterial load significantly differed across the larval life stages (one‐way ANOVA, P < 0.01, df = 4, Fig. 2), showing a general increasing trend with age. Pairwise comparisons between the larval stages showed a significant increase in bacterial load between the early pre‐feeding (L1 DPH) and rotifer feed (L3‐9 DPH) stages compared to all other later stages (i.e. L14‐18, L29‐35 and L49‐53 DPH).
Figure 2.
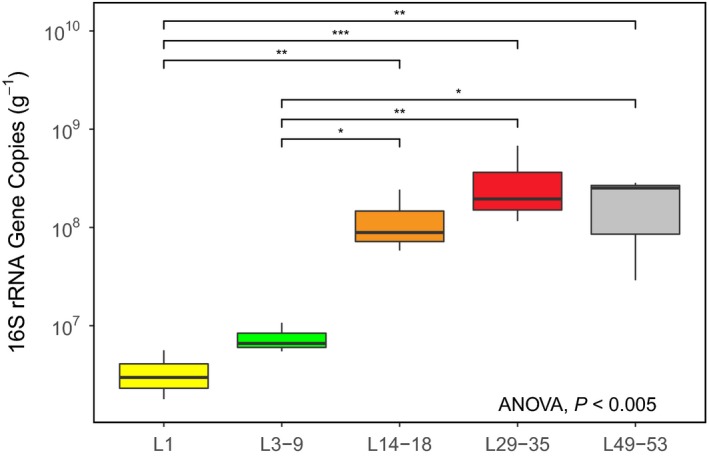
The 16S rRNA gene copy number: Biological triplicates of larvae samples at each life stage (as in Fig. 1) (normalized by the initial mass (g) of starting material. L = larvae/juvenile S. lalandi; numbers correspond to the days post‐hatching (DPH). Overall, there was a significant effect of life stage (ANOVA, < 0.005). Post hoc pairwise comparisons were performed using Tukey's HSD. The stages L1 and L3‐9, respectively, were significantly different from all later life stages (L14‐18, L29‐35 and L49‐53). Significantly different samples are indicated by asterisk. (*) *** P > 0.001; ** P < 0.001 > 0.01; * P < 0.01 > 0.05.
Bacterial richness, diversity and bacterial community composition
To characterize the early larval and intestinal microbial community associated with S. lalandi, we sampled larvae (L), feed (rotifers (Rot), Artemia (Art), micro‐pellet (Micro) and macro‐pellet [Macro]) and the rearing water (W) at specific stages that coincide with changes to the larval diet (Fig. 1). Sequencing of the V3 and V4 region of the 16S rRNA gene from 131 samples resulted in a total of 8 293 038 sequencing reads after quality filtering and chimera removal. This number was reduced to 5 911 072 reads after the removal of singletons, unclassified sequences, chloroplast and mitochondrial sequences. These sequences were assigned to 6259 OTUs at a 97% sequence similarity cut‐off. OTUs that appeared in less than three samples were removed (~1% of sequences), resulting in a total of 5 847 618 sequences, with a mean read depth of 43 351 sequences per sample, grouped into 2960 OTUs. After rarefaction, three samples (Ma_T1_n1, Ma_T2_n1, Ma_T3_n1) were not sufficiently sampled (< 10 000 sequences) and were thus removed from further analysis. The mean coverage estimate was 0.900288, indicating good sampling of the microbial communities (Fig. S5, Table S1).
Consistent with the qPCR results, pairwise comparisons between the larval stages showed a significant increase in bacterial richness (number of observed OTUs) between the early (L1, L3‐9, L14‐18 days after hatching (DPH)) and later (L29‐35, L49‐53 DPH) larval stages (one‐way ANOVA, P < 0.001, df = 4, Fig. 3A). Comparisons between the different feed samples (Fig. 3B) also revealed significant differences between the live feeds (rotifers and Artemia) and the micro‐pellet (P < 0.05) and between rotifers and the macro‐pellet (P < 0.001). There was a general increase in diversity with larval age that mirrored the diversity of the corresponding feed samples. Specifically, bacterial diversity (Shannon–Weaver diversity index) was significantly different in the L49‐53 juvenile stage from all other stages (one‐way ANOVA, P < 0.001, df = 4, Fig. 4A). Significant differences were also seen between the rotifer and macro‐pellet feed types (one‐way ANOVA, P < 0.05, df = 3, Fig. 4B). The bacterial richness (one‐way ANOVA, P < 0.01, df = 4, Fig. 3C) and diversity (one‐way ANOVA, P > 0.05, df = 4, Fig. 4C) of the rearing water were generally high across all stages.
Figure 3.
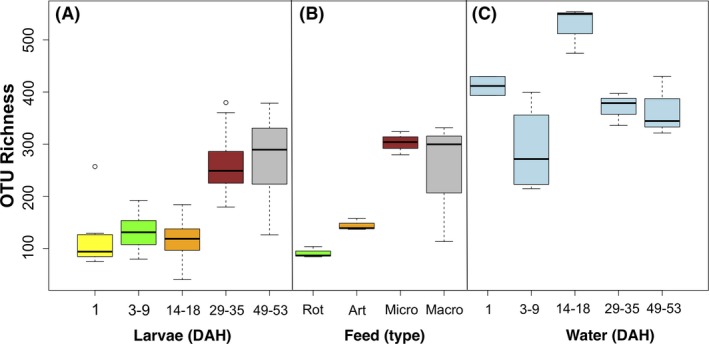
OTU richness: the average number of observed OTUs in (A) the larval microbial community throughout developmental stages, (B) the different feed types, (C) the rearing water during larval development. Colour of larvae (A) corresponds to colour of feed (B) at that stage. Significant differences (assessed by ANOVA with post hoc Turkey's HSD) between samples are indicated with an asterisk (*) *** P > 0.001; ** P < 0.001 > 0.01; * P < 0.01 > 0.05.
Figure 4.
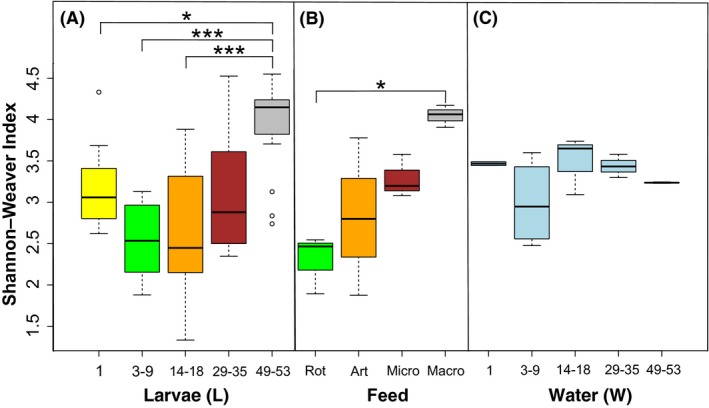
Shannon's diversity: the abundance and evenness of observed OTUs in (A) the larval microbial community throughout developmental stages, (B) the different feed types, (C) the rearing water during larval development. Colour of larvae (A) corresponds to colour of feed (B) at that stage. Significant differences (assessed by ANOVA with post hoc Turkey's HSD) between samples are indicated with an asterisk (*) *** P > 0.001; ** P < 0.001 > 0.01; * P < 0.01 > 0.05.
The bacterial community associated with the larvae, the feed and the rearing water was represented by over 600 genera assigned to 38 phyla. Of the 16 phyla with > 1% relative abundance, four (Proteobacteria, Firmicutes, Bacteroidetes and Parcubacteria) comprised 75% of the total OTU abundance (Fig. 5). Proteobacteria were more abundant in the early larvae (i.e. from sample L1 to L31); however, from 35 DPH (i.e. sample L35) their relative abundance decreased from > 50% to < 25% and was replaced by Firmicutes (up to 61%) (Fig. 5). Other abundant phyla present in the larvae include the Bacteroidetes (ranging from 4.5 to 8.0% across all larval samples), Actinobacteria, which had an average abundance across all samples of 6%, but showed sharp increases in the L7 and L9 stages (25.8–36.8%), and Fusobacteria (3.8% overall), which was enriched in the L29 and L31 stages (to 14.7–15.7%).
Figure 5.
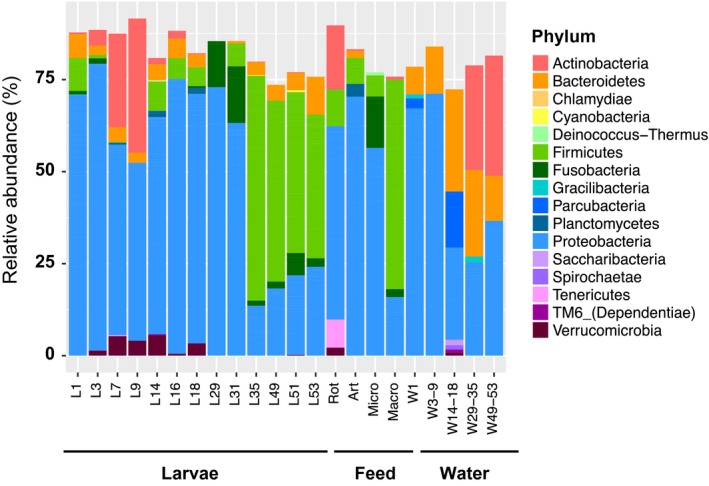
Phylum‐level bacterial taxonomic composition in the S. lalandi larviculture system. Relative abundances were calculated using a rarefied library (10 000 sequences per sample), and replicate samples were pooled into sample groups as presented on the x‐axis. Relative abundances are only shown for taxa representing > 1% of the rarefied data set sequences.
In the feeds, Proteobacteria dominated the bacterial community of the rotifer, Artemia and micro‐pellet feeds (57.4%, 65.3% and 78% respectively). In contrast, a high abundance of Firmicutes (71.5%) was detected for the macro‐pellet. In general, water samples had a high abundance of Proteobacteria and Bacteroidetes, with the abundance of Actinobacteria increasing during the later sampling times (i.e. 27.63% in W29‐35 to 33.83% in W49‐53).
Variation in bacterial community composition over larval development and identification of the core microbiota
Non‐metric multidimensional scaling (nMDS) based on Bray–Curtis dissimilarities of bacterial OTU profiles showed tight clustering of replicates from all larval stages, except for larvae from the L1 stage (PERMANOVA, Pseudo‐F = 7.3602, P = 0.001, Fig. 6A). Pairwise comparisons found significant differences between each larval stage (P < 0.05 for each pairwise comparison) with the exception of the comparison between L1 and the L3‐9 stages (P = 0.09). A trajectory of bacterial composition of the larval microbiome was seen between the larval stages, with the greatest divergence occurring as larvae move from the L14‐18 DPH stage to the L29‐35 DPH stage (P = 0.0002, Fig. 6B).
Figure 6.
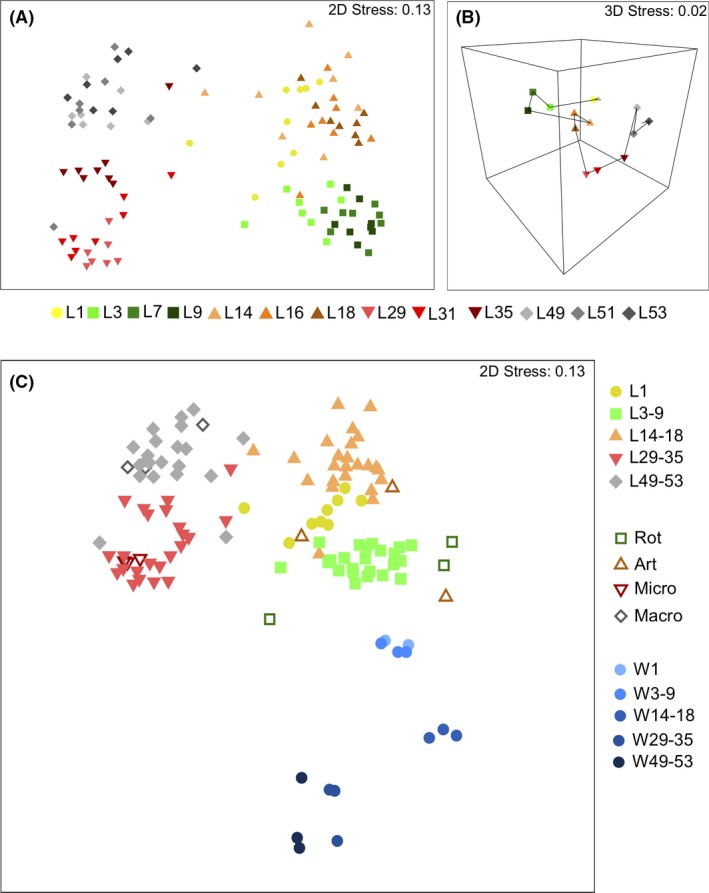
Non‐metric multidimensional scaling (nMDS) plots of: (A) Bray–Curtis distances in 2D of the larval intestinal microbiome across all developmental stages (L1 to L53). Each point represents individual larvae from its respective stage. (B) Bray–Curtis distances in 3D showing the trajectory (as indicated by arrows) of the larval intestinal microbiome over the duration of sampling. Each point represents eight replicate samples grouped into stage categories. Note the direction and length of the trajectory indicate difference. (C) Bray–Curtis distances of all samples in 2D, including larvae and both feed and rearing water. Feed samples are shown with hollow symbols (shape respective of related larval stage), and water samples with blue symbols (circles). Each point represents an individual sample.
Specific taxa that were differentially distributed between the larval stages were identified to the family (Table 1) and genus level (Table S5). Once the larvae began to feed, a significant increase in Rubritaleaceae, Rickettsiales and Micrococcaceae was observed, with the latter taxon decreasing again once the larvae transitioned from a rotifer to an Artemia diet. The greatest changes in bacterial community composition occurred between the L14‐18 and L29‐35 stage when the larvae moved from live feeds to formulated pellets. This included significant changes in 14 families within the phyla Firmicutes (including Lactobacillaceae, Clostridiaceae and Erysipelotrichaceae) and a significant increase in a number of Proteobacteria, including Acetobacteraceae, Colwelliaceae and Idiomarinaceae. In contrast, other Proteobacteria (e.g. Rhodobacteraceae, Hyphomicrobiaceae, Legionellaceae and Coxiellaceae), members of Actinobacteria and Rubritaleaceae significantly decreased in abundance between these two larval stages.
Table 1.
Summary of differential abundance (log 2) at family level between the larval stages.a
| Phylum | Family | L1(−) to L3–9 (+) | L3–9(−) to L14–L18(+) | L14–L18 (−) to L29–L35(+) | L29–35(−) to L49–53(+) |
|---|---|---|---|---|---|
| Actinobacteria | Micrococcaceae | 6.5469 | −6.4190 | ||
| Microbacteriaceae | 4.8697 | −4.1860 | |||
| Coriobacteriaceae | 7.8825 | ||||
| Corynebacteriaceae | −2.7468 | ||||
| Mycobacteriaceae | −6.5874 | ||||
| Propionibacteriaceae | −3.0615 | ||||
| Bacteroidetes | Bacteroidaceae | 7.1314 | |||
| Flavobacteriaceae | −2.5545 | ||||
| Marinilabiaceae | 7.3638 | ||||
| Firmicutes | Lachnospiraceae | −5.3064 | 6.3081 | ||
| Acidaminococcaceae | −5.5100 | ||||
| Veillonellaceae | −5.0716 | ||||
| Bacillaceae | −1.8452 | 1.2327 | |||
| Acidaminococcaceae | 9.0014 | ||||
| Clostridiales Family XIII | 6.6436 | ||||
| Paenibacillaceae | 6.8693 | ||||
| Lactobacillaceae | 5.0936 | ||||
| Planococcaceae | 5.2877 | ||||
| Peptostreptococcaceae | 4.8280 | ||||
| Clostridiaceae 2 | 6.6429 | ||||
| Erysipelotrichaceae | 4.6808 | ||||
| Streptococcaceae | 3.3091 | ||||
| Veillonellaceae | 4.0914 | ||||
| Clostridiaceae_1 | 3.8234 | ||||
| Staphylococcaceae | −1.9404 | ||||
| Thermoanaerobacterales Family III | 3.4843 | ||||
| Chlamydiae | Parachlamydiaceae | −8.1508 | |||
| Simkaniaceae | −6.9379 | ||||
| Fusobacteria | Fusobacteriaceae | 6.5622 | |||
| Verrucomicrobia | Rubritaleaceae | 7.1124 | −6.1291 | ||
| Proteobacteria | Alcaligenaceae | −6.0845 | 6.0186 | ||
| Rickettsiales | 8.1882 | ||||
| Enterobacteriaceae | −2.7626 | 2.0139 | −2.0885 | ||
| Phyllobacteriaceae | −4.4695 | ||||
| Bacteriovoracaceae | −6.9515 | ||||
| Colwelliaceae | −5.9603 | 6.9977 | |||
| Hydrogenophilaceae | 5.9285 | ||||
| Xanthomonadaceae | −2.1733 | ||||
| Thiotrichaceae | −4.1796 | ||||
| Aeromonadaceae | −3.3316 | ||||
| Moritellaceae | 5.7658 | ||||
| Campylobacteraceae | 5.2132 | ||||
| Acetobacteraceae | 7.0927 | ||||
| Rhodobacteraceae | −5.5359 | ||||
| Hyphomicrobiaceae | −8.6227 | ||||
| Vibrionaceae | 5.0881 | −3.1034 | |||
| Legionellaceae | −6.7005 | ||||
| Coxiellaceae | −6.6192 | ||||
| Idiomarinaceae | 7.7505 | ||||
| Tenericutes | Mycoplasmataceae | 8.4540 | −9.0815 | 8.0214 |
a. Analysis done using DESeq2 package, only differential abundances with P < 0.001 are displayed. Note: Negative values indicate a higher abundance in the earlier stage, compared to the later stage, and positive values indicate a higher abundance in the later stage compared to the earlier stage. A log2 fold difference is a 2X fold change.
While significant differences were identified in the larval microbiome between the developmental stages, 53 OTUs (representing 56% of the relative read abundance) were shared across all larval samples regardless of the developmental stage (Fig. 7A, Table S1). These OTUs were therefore identified as potentially core bacteria and included 27 Proteobacteria OTUs, 13 Firmicutes OTUs, seven Actinobacteria OTUs, four Bacteroidetes OTUs, one Cyanobacteria OTU and one Deinococcus–Thermus OTU. Included in the most abundant of these core bacteria are members of the Donghicola (OTU2), Lactobacillus (OTU28), Aeromonas (OTU4) and Photobacterium (OTU9) with the relative abundances of each fluctuating across the larval development (Table S1).
Figure 7.
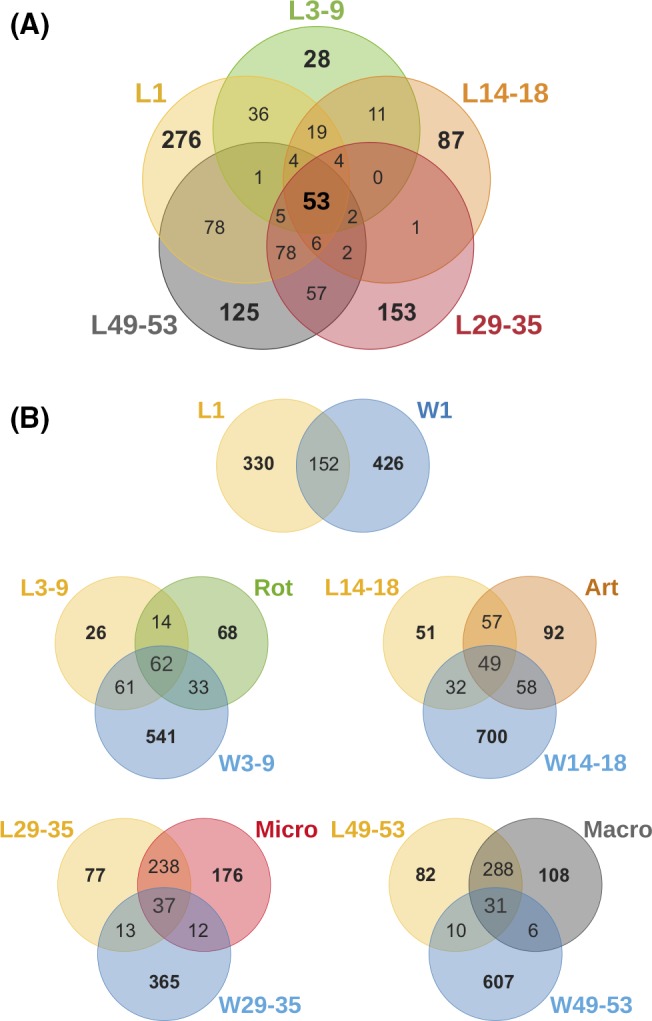
Venn diagrams were constructed exploring rarefied libraries of detected OTUs. Replicate samples were pooled to represent the total number of bacterial phylotypes identified with each sample category.
A. displays the extent of shared and exclusive OTUs between S. lalandi larvae at their five developmental stages.
B. displays the extent of shared and exclusive OTUs between S. lalandi larvae or early juveniles (L), the rearing water (W) and the feed (Rot, Art, Micro & Macro).
Influence of feed and water samples on larval microbiomes
We also observed significant differences in the microbial community associated with each of the sample types (larvae, feed and rearing water) (PERMANOVA, Pseudo‐F = 13.18, P = 0.001, Fig. 6C). With the exception of Artemia, each of the feed samples clustered together with their respective larval stage and these observations are supported by a high coefficient of correlation (Spearman's r s = 0.811, P = 0.0001). Similarly, the bacterial community associated with the early stage water samples (W1, W3‐9) showed the greatest resemblance to the community associated with the corresponding larval samples (Spearman's r s = 0.691, P = 0.004).
Early stage larvae tended to share a higher number of OTUs with water samples than with the feed (e.g. L3‐9 shared 8.5% OTUs with feed and 37% OTUs with water) (Fig. 7B). This pattern was reversed for the later stages (e.g. L49‐53 shared 70% OTUs with feed and only 2.5% with water). Spearman's rank correlations identified a total of 35 and 22 OTUs from the feed and water samples, respectively, that made a significant contribution to the OTUs present in the larval microbiome (Fig. S6). Of these feed‐associated OTUs, 10 were predominantly assigned to the Proteobacteria and were enriched in the early (L3‐9, L14‐18) larval stages, while 11 OTUs, predominantly belonging to the Firmicutes, were enriched in the later (L29‐35, L49‐53) larval stages (Table S2, Fig. S6A). The 22 OTUs (Table S3, Fig. S6B) originating from the rearing water showed a more variable pattern, with specific OTUs enriched at different larval stages.
Of the 53 OTUs identified as part of the core larval microbiome, 34 OTUs were found in the initial rearing water and 35 OTUs in the first rotifer feed. From the water, these include OTU11, assigned to Neptuniibacter sp., OTU1028 and OTU2, which both belong to the genus Donghicola, and are among the most abundant taxa associated with larvae especially in the early stages (Table S1). OTU3 (Glutamicibacter sp.), OTU81 (Vibrio sp.) and OTU6 (Nautella sp.) were among the most dominant of the core larval OTUs also associated with rotifers. Interestingly, the core larval microbiome included five OTUs (OTU205 (Lactobacillus sp.), OTU8677 (Chryseobacterium sp.), OTU327 (Psychrobacter sp.), OTU263 (Acinetobacter sp.) and OTU153 (Corynebacterium sp.)) that were neither found in the water nor in the first rotifer feed. This suggests that some microbial members appear to originate from the larvae either associated with eggs and/or from environmental contamination, and are maintained throughout development.
Functional prediction of larval metagenome throughout development
Functional profiles of the microbiome were predicted using Tax4Fun (Aßhauer et al., 2015), with approximately 44.4 ± 18.3% of all 16S rRNA sequences being incorporated into the analysis (see Table S4). Statistical analysis revealed that 203 KEGG pathways significantly differed in their abundance across the developmental stages, indicating a major change in the predicted microbiome functionality (Table 2). Differentially abundant pathway function included carbohydrate metabolism, amino acid metabolism, metabolism of cofactors and vitamins, lipid metabolism and metabolism of terpenoids and polyketides (Table 2). Specifically, pathways related to the metabolism of common sugars, including glucose (ko00010), galactose (ko00052) fructose and mannose (ko00051), starch and sucrose (ko00500) were predicted to be more abundant in later stage larvae (L29‐L53). Functions related to thiamine (vitamin B1) metabolism (ko00730) were also more abundant in the later larval stages. In contrast, functions related to lipid metabolism, including unsaturated fatty acids (ko01040), were significantly lower in the older larvae and more abundant in pre‐feeding (L1‐3) or larvae fed on live diets (L3‐L18).
Table 2.
Mean relative abundances (%) of selected KEGG pathways related to metabolism. Relative abundance of pathways significantly associated with a certain development stage is italicized. Relative abundances were predicted with Tax4Fun
| Pathway | Abundance (%) | ||
|---|---|---|---|
| L1 | L3‐L18 | L29‐L53 | |
| Amino acid metabolism | 12.359 ± 0.385 | 12.845 ± 0.707 | 10.639 ± 0.449 |
| Lysine biosynthesis (ko00300) | 0.879 ± 0.035 | 0.848 ± 0.026 | 0.928 ± 0.117 |
| Lysine degradation (ko00310) | 0.567 ± 0.034 | 0.517 ± 0.060 | 0.455 ± 0.045 |
| Biosynthesis of other secondary metabolites | 0.798 ± 0.036 | 0.813 ± 0.025 | 0.653 ± 0.088 |
| Penicillin and cephalosporin biosynthesis (ko00311) | 0.166 ± 0.018 | 0.160 ± 0.025 | 0.094 ± 0.026 |
| Streptomycin biosynthesis (ko00521) | 0.228 ± 0.009 | 0.237 ± 0.011 | 0.213 ± 0.017 |
| Carbohydrate metabolism | 12.677 ± 0.363 | 12.442 ± 0.805 | 13.746 ± 0.502 |
| Glycolysis/Gluconeogenesis (ko00010) | 0.883 ± 0.048 | 0.833 ± 0.045 | 1.100 ± 0.157 |
| Fructose and mannose metabolism (ko00051) | 1.204 ± 0.079 | 1.175 ± 0.204 | 1.428 ± 0.126 |
| Galactose metabolism (ko00052) | 0.487 ± 0.083 | 0.404 ± 0.092 | 0.856 ± 0.178 |
| Starch and sucrose metabolism (ko00500) | 1.407 ± 0.141 | 1.063 ± 0.165 | 1.815 ± 0.188 |
| Lipid metabolism | 3.570 ± 0.104 | 3.653 ± 0.312 | 3.408 ± 0.188 |
| Biosynthesis of unsaturated fatty acids (ko01040) | 0.257 ± 0.018 | 0.256 ± 0.030 | 0.181 ± 0.035 |
| Metabolism of cofactors and vitamins | 7.162 ± 0.131 | 7.254 ± 0.165 | 6.862 ± 0.344 |
| Thiamine metabolism (ko00730) | 0.574 ± 0.029 | 0.572 ± 0.035 | 0.660 ± 0.072 |
| Metabolism of terpenoids and polyketides | 2.683 ± 0.136 | 2.536 ± 0.239 | 2.249 ± 0.335 |
| Tetracycline biosynthesis (ko00253) | 0.050 ± 0.003 | 0.052 ± 0.006 | 0.041 ± 0.006 |
Discussion
The host–microbiome of fish plays a key role in growth, development and disease resistance (Kanther and Rawls, 2010; Nayak, 2010; Ran et al., 2016; Stephens et al., 2016; Egerton et al., 2018), yet there is limited information about the diversity and function of these microbes during the course of early development. Here, we examined the microbial community of S. lalandi during larval and juvenile development and assessed whether the bacteria from the feed or rearing water act as an inoculum for the S. lalandi gut microbiome.
We found the bacterial load of developing larvae significantly increased during the first three stages (i.e. up to L14‐L18), then remained stable. This pattern was reflected in the species richness, showing the increase in bacterial load is likely a result of the introduction of new microbial members and not just the growth of early colonizers. The relatively high levels of bacterial richness and diversity in S. lalandi larvae as early as 1 day post‐hatching are also consistent with the early microbiome colonization observed for other commercial fish species ((Califano et al., 2017; Li et al., 2017) and discussed below). Throughout larval development, only 53 OTUs were shared across all stages; however, these represented 56% of the read abundance, indicating a small, but abundant core community. Species of Streptococcus, Enterococcus and Lactobacillus, known to be components of the microbiome in healthy fish (Nayak, 2010; Sullam et al., 2012; Llewellyn et al., 2014) were identified as part of the core microbiome. These genera are typically capable of lactic acid fermentation and may contribute to the stability of the intestinal microbiome in Atlantic cod (Gadus morhua), Atlantic salmon (Salmo salar) and rainbow trout (Oncorhynchus mykiss) (as reviewed in Ringø and Gatesoupe (1998)). Members of the genera Aeromonas and Vibrio were also part of this core microbiome, but were more abundant during the pre‐feeding (L1) and L14‐18 stages. These bacterial genera are often found in aquatic environments and are known to contain species responsible for some of the most economically important diseases in marine aquaculture and fish production (Toranzo et al., 2005). While two sequences related to known pathogens (i.e. Vibrio tubiashii (OTU81) and Vibrio protolyticus (OTU945)) were detected, most sequences could only be classified to the genus level. Given there were no signs of disease in the larvae, this suggests that these taxa function either as opportunistic pathogens (Brown et al., 2012) or represent non‐pathogenic members of the genera (Austin et al., 1995).
Outside of the core microbiome, the composition of the bacterial community varied between each stage. Stage‐specific bacterial communities have also been observed for other fish and are thought to result from to a combination of stochastic and deterministic processes (Ingerslev et al., 2014; Burns et al., 2016; Yan et al., 2016; Lokesh et al., 2018). With respect to the S. lalandi investigated here, the most substantial gut microbiome changes occurred as the larvae transitioned from live feeds to the formulated pellet, suggesting that the temporal variation observed in this study was likely due to the changing diet. However, it is important to note that despite attempts to avoid contamination, these microbiome differences may also be due to a higher abundance of skin or seawater microbes in the younger larval samples, which due to their small size could not be dissected. Nevertheless, other studies have also shown a strong link between fish diet and microbiome membership (Lauzon et al., 2010; Gajardo et al., 2017; Li et al., 2017; Kashinskaya et al., 2018). For example, Li et al. (2017) observed that the microbiome of the gibel carp (Carassius auratus gibelio) was consistent between larvae fed the same diet, and the strongest microbiome shift occurred when the larvae were transitioned from Artemia to a formulated pelleted feed. In contrast, diet and rearing water appeared to have limited impact on the microbiome of cod larvae (Gadus morhua) (Bakke et al., 2015). Combined, these different observations suggest that both age and the environment can influence microbiome development and the relative influence of either factor may depend on the fish species, reinforcing the importance of system‐ and species‐specific studies.
We found 31% of the total OTUs observed in the early pre‐feeding stage larvae were shared with those in the early stage rearing water (W1; Fig. 7B). This included taxa belonging to Neptuniibacter, Donghicola and Marivita that are present in the top five most abundant OTUs in pre‐feeding (L1), but in low abundance in older larval and juvenile stages. This pattern has also been observed for channel catfish and other fish species, indicating that the bacterial community in the rearing water has a strong influence at the pre‐feeding stage, while the larvae are still reliant for their development on their endogenous yolk sac. However, in each of these systems the rearing water has less influence once the larvae begin feeding (Bledsoe et al., 2016; Califano et al., 2017). Interestingly, despite the strong influence of the water microbiota, a large proportion of the pre‐feeding stage OTUs (including five belonging to the core microbiota) appear to have originated from the larvae themselves. While environmental contamination cannot be ruled out, it is plausible that these bacteria represent early egg colonizers that have remained despite ozone sterilization prior to entering the hatchery facility. Indeed, several studies have shown early colonization of newly released eggs (Lauzon et al., 2010; Llewellyn et al., 2014; Lokesh et al., 2018). Future investigation of the microbial community associated with the fertilized eggs and the broodstock environment will be necessary to determine the true source of these early S. lalandi microbiome members.
The high community similarity between the diets and the corresponding larval samples indicates direct introduction of the feed‐associated bacteria into the intestine, rather than indirect modulation of the intestinal microbiome by the feed ingredients. The bacterial taxa that became enriched in the early larval stages of the live feed were largely members of the family Rhodobacteraceae, which are common marine symbionts involved in both carbon and sulfur biogeochemical cycling (Simon et al., 2017). The transition of larvae onto the diet of formulated pellet feeds however, was characterized by a reduction in Proteobacteria and an enrichment of Firmicutes. These included members of the Bacillaceae and Clostridiaceae, which are commonly associated with the gut microbiota of animals and known to play an important role in the fermentation of organic matter. Fusobacteriaceae were also identified as a main contributor to the gut microbiota from the formulated feed. While this bacterial group is commonly associated with the gastrointestinal tract of freshwater fish, it has also been found in some cultivated marine species (Hennersdorf et al., 2016; Tarnecki et al., 2017). Although the specific role of Fusobacteriaceae for S. lalandi is yet to be determined, these bacteria have previously been associated with carnivorous fish (Liu et al., 2016; Michl et al., 2017), indicating that they may assist in the breakdown of the animal protein components of their diet. Interestingly, the pattern of a shift from a Proteobacteria to a Firmicutes in the larval microbiota reflects recent observation of the gut microbiome in adult S. lalandi, where Firmicutes were higher in abundance in aquaculture‐raised kingfish and Proteobacteria more abundant in wild‐caught individuals (Ramirez and Romero, 2017). This suggests that the composition of the gut microbiome of aquaculture‐raised animals may be influenced by the introduction of formulated feed, regardless of their age. However, future work will be required to determine both the metabolic activity of these feed‐associated bacteria and their ability to directly colonize the fish intestinal mucosa.
We used Tax4Fun (Aßhauer et al., 2015) to predict the gene functions associated with the microbiome during larval development at different major diet changes (i.e. pre‐feeding, live feed and formulated pellets). While confirmation of the functional predictions is required, we observed pathways related to the metabolism of common sugars were more abundant in later stage larvae (L29‐L53). These pathways were similarly enriched in aquaculture‐raised adult kingfish (Ramirez and Romero, 2017) and are likely to be a reflection of the comparatively higher carbohydrate content present in formulated diets than in live feed or wild prey. Of particular note is the high representation of pathways related to the metabolism of starch (ko00500), which is a major carbohydrate constituent of the formulated pellets used here (see reference in Supplementary Material). In contrast, functions related to lipid metabolism were significantly lower in the older larvae and more abundant in pre‐feeding (L1‐3) or larvae fed on live diets (L3‐L18). Interestingly, this includes the gene functions involved in the biosynthesis of unsaturated fatty acids (ko01040), such as the essential omega‐3 fatty acids, that cannot be produced by marine fish larvae, yet are vital for their neural and retinal development (Kristin et al., 2013).
Predicted functions involved in amino acid and vitamin metabolism were more abundant in larvae fed on live diets (L3‐L18) possibly reflecting a higher demand for these functions compared to larvae fed on formulated pellets that are generally optimized in terms of micro‐/macro‐nutritional requirements (Kristin et al., 2013). However, in some cases deviations from this trend were observed. For example, functions related to thiamine (vitamin B1) metabolism (ko00730) are higher in older larvae (L29‐L53) fed on formulated pellets and may be a result of its role as an essential cofactor in carbohydrate metabolism. In addition, while lysine biosynthesis functions (ko00300) were significantly enriched in older larvae fed on formulated pellets, degradation genes (e.g. ko00310) were generally more abundant in younger larvae during pre‐feeding or live feed stages. Lysine can be a limiting amino acid in many formulated feeds (Mukhtar et al., 2017), and the enrichment of functions related to its biosynthesis suggests that GI microorganisms could act as a supplementary source for this important amino acid.
Gene functions related to secondary metabolite production were more prevalent in the early stage larvae. Specifically, the microbiomes of pre‐feeding stage larvae (L1) had a significantly higher proportion of functions related to the synthesis of beta‐lactam antibiotics (ko00311). Functions related to the production of other common antibiotics, including tetracycline (ko00253) and streptomycin (ko00521), were significantly more abundant during the live feeding stages (L3‐L18). These antibiotics are commonly used to treat infections with Gram‐negative bacteria, including several disease‐causing vibriosis (Shaw et al., 2014), suggesting that the microbes associated with early larval stages have the potential to produce antibiotics that can protect against pathogenic invaders. However, it is important to note that these are functional predictions only and will need to be confirmed in future studies with more direct measures.
In conclusion, our study contributes to a growing body of work on animal microbiome development. We found that within a commercial larviculture setting, the S. lalandi larvae are rapidly colonized by a diverse range of bacteria. Despite a core bacterial community being observed across all larval developmental stages, distinct shifts in the microbiome membership were seen between individual larval stages coinciding with dietary changes. We found shifting to a formulated pellet had the strongest influence on the gut microbiome. During this stage, a high proportion of taxa were shared between the gut and feed samples, suggesting the feed may serve as a continual inoculum for the fish gut microbiome. While the viability of these bacteria is yet to be determined, these results give support to the possibility of supplementing the feed with specific microbial taxa as a means of establishing a functionally beneficial microbiome and improving the performance of reared S. lalandi larvae (Merrifield et al., 2010; Nayak, 2010). Moreover, the findings here provide an important foundation on which to further investigate the link between the early gut microbiome and the heath of adult S. lalandi.
Experimental procedures
Rearing of Yellowtail Kingfish (S. lalandi), sampling and total bacterial community extraction
This study was conducted under the authorization of NSW Primary Industries (Fisheries) Animal Care and Ethics Committee (ACEC) (REF: 93/1 – Port Stephens). Yellowtail Kingfish (S. lalandi) larvae were reared at the NSW Port Stephens Fisheries Institute (PSFI) according to existing commercial production‐scale procedures (Fielder and Heasman, 2011) and further detailed in Supporting information (See Supporting data file: Rearing of Yellowtail Kingfish (S. lalandi)).
The sampling scheme for microbial community analysis of the S. lalandi intestine aimed to identify the bacteria present at each larval developmental stage that coincides with changing larval feed (Fig. 1). The analysis targeted the bacterial community in (i) the S. lalandi larvae, (ii) the feeds (separately: rotifers, Artemia, micro‐ and macro‐pellet) and (iii) the rearing water. Across the developmental timeline, eight independent replicate samples were collected. In addition, three replicate samples of each feed type and a total of 15 rearing‐water samples were collected during larval development (Fig. 1).
Seriola lalandi larvae (L) were randomly sampled across the commercial rearing tanks at one time point at the pre‐feeding stage (1 DPH, labelled L1), and samples were then taken at three time points across each of the established developmental stages: rotifer feeding (3–9 DPH, labelled L3‐9), Artemia feeding (14–18 DPH, labelled L14‐18), micro‐pellet feeding (29–35 DPH, labelled L29‐35), macro‐pellet feeding (49–53 DPH, labelled L49‐53) (Fig. 1 and Fig. S1). Samples from L1 and L3‐9 consisted of ~0.05 g (wet weight) larval pools (due to low biomass of individual larvae), corresponding to approximately 10–12 larvae, while samples from L14‐18 consisted of individual larvae. Larval pools and individual larvae were washed with sterile artificial seawater to remove externally attached microbial cells. Samples from L29‐35 and L49‐53 consisted of the individual gastrointestinal tract, as dissection was possible from the juvenile stages. Larval pools, individuals and intestines, were transferred to sterile 1.5 ml polypropylene tubes, snap‐frozen in liquid nitrogen and stored at −20°C. Rearing‐water samples (0.5 l) were sequentially filtered through a 0.45 and 0.22 μm pore‐size filter, with the aid of a 50 ml syringe. Pre‐filtering with 0.45 μm was performed to ensure the removal of any small feed or larval particles. The final 0.22 μm filters were transferred to sterile tubes snap‐frozen in liquid nitrogen and stored at −20°C. Triplicate samples (0.3 g) of each feed type (rotifers, Artemia, micro‐pellet and macro‐pellet) were collected prior to their addition to the tanks. Individual feed samples were snap‐frozen in liquid nitrogen and stored at −20°C until further processing.
Total community DNA was extracted from all samples using the DNeasy® Blood & Tissue Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. To promote cell lysis before extraction, all samples were initially incubated with 10 mg ml−1 lysozyme for 30 min (with vortexing every 15 min) at 37°C (Califano et al., 2017) followed by a treatment with 20 mg ml−1 proteinase K for 2.5 h at 56°C (with vortexing every 30 min). Extracted DNA was quantified using a NanoDrop (ND‐1000) spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
16S rRNA gene copy number determination and analysis
Quantitative PCR amplification of the 16S rRNA gene using the 338F’ (Lane, 1991) and 528R’ (Callens et al., 2018) primers was used to determine absolute gene copy number by comparison to a 10‐fold dilution series of the standard fragment (see Fig. S2). The PCR mixture contained 1X PerfeCTa SYBR FastMix (Quantabio, Beverly, MA, USA), 300 nM each primer and 1 μl DNA template with a total volume of 10 μl. The PCR cycling conditions were as follows: activation at 95°C for 5 min, annealing at 40 cycles of 95°C for 15 s and extension at 60°C for 30 s followed by melt curve analysis from 65 to 95°C at 0.5°C intervals. Amplification and analysis were performed using the Mic qPCR Cycler (BioMolecular Systems, Australia) and micpcr software (version 2.6.3). Default settings included using the LinRegPCR method to perform baseline correction and to determine the fluorescence threshold to calculate C q and efficiency per sample.
Samples were amplified in biological triplicates with each sample having technical duplicates. Technical duplicates were repeated if they had a standard error >0.5 C t. The final gene copy numbers per sample were normalized by the amount of biological material extracted. One‐way analysis of variance (ANOVA) tests were performed on log‐transformed copy numbers. Post hoc pairwise comparisons using the multcomp package (version 1.4.8) were performed using the Tukey's test to determine significant difference between the groups of interest.
16S rRNA gene sequencing and analysis
The V3 and V4 regions of the 16S rRNA gene were amplified from the total community DNA using the 341F’ and 785R’ primers (Klindworth et al., 2013). The PCR was performed as described previously (Nielsen et al., 2017). PCR products were quantified using gel electrophoresis before being sequenced on the Illumina MiSeq platform with a 2 × 300 bp chemistry at the Ramaciotti Centre for Genomics (UNSW, Sydney, NSW, Australia).
16S rRNA gene reads were processed according to Granzow et al. (2017). In brief, sequence data were initially quality‐filtered and trimmed using trimmomatic version 0.36 truncating reads if the quality dropped below 15 in a sliding window of 4 bp (Bolger et al., 2014). usearch version 9.2.32 (Edgar, 2010) was used for further processing as described by Wemheuer and Wemheuer (2017) to merge and quality‐filter sequencing reads, excluding reads with < 400 or > 500 nucleotides, in addition to reads with more than one ambiguous base or an expected error of more than 1. Processed reads for all samples were then concatenated and clustered into operational taxonomic units at 97% sequence similarity using the UPARSE algorithm implemented in Usearch. Chimeric sequences were removed de novo during clustering and subsequently in reference mode using Uchime with the SILVA database (https://www.arb-silva.de/browser/) (SILVA SSURef 128 NR) as a reference. Remaining OTU sequences were then taxonomically classified by BLASTN (Camacho et al., 2009) against the SILVA database with an E‐value cut‐off of 10−20. All non‐bacterial OTUs were removed along with non‐BLAST aligned and singleton OTUs. Finally, processed sequences were mapped on OTU sequences to calculate the distribution and abundance of each OTU in every sample. Only OTUs occurring in more than two samples were considered for further statistical analysis.
Artificial metagenomes were predicted from the OTU table using Tax4Fun with short read mode disabled (Aßhauer et al., 2015). Prior to calculation, OTU sequences were reclassified against the SILVA SSURef 123 NR database with an E‐value cut‐off of 10−20. Tax4Fun transforms the SILVA‐based OTUs into a taxonomic profile of KEGG organisms, which is normalized by the 16S rRNA copy number (obtained from NCBI genome annotations). Spearman's correlation analysis of functional profiles derived from relevant available metagenome sequences and profiles deduced from 16S rRNA gene sequences revealed a median of the correlation coefficient of 0.76 for the mammalian gut system. This suggests that Tax4Fun provides a good prediction of functional profiles that may be obtained using metagenomic shotgun sequencing approaches. However, it should be noted that predictions are used for hypothesis building and require future confirmation using direct methods.
Diversity measures and statistical analysis
Measures of community diversity, i.e., OTU richness and Shannon's diversity, were calculated in R (version 3.3.3) using the vegan package (Oksanen et al., 2014). For the diversity measures, each sample was randomly subsampled (rarefied) to a total of 10 000 counts to account for uneven sequencing depth among the samples. Subsampling was performed 100 times, and the average was taken to reduce randomization effects on our subsampled data as in Nielsen et al. (2017). One‐way ANOVA followed by pairwise comparisons using the Tukey's test was performed to determine significant difference between groups. Relative abundances were calculated on the rarefied library (10 000 counts/sample) at the phylum level in R using the phyloseq package, and then plotted using the package ggplot2. Only taxa that represented > 1% of the rarefied data set sequences were displayed. To identify differentially abundant taxa between the larval stages, the DESeq2 package was employed considering P < 0.001 (Love et al., 2014), with the package presenting the log 2 fold change (2X) in bacterial abundance between stages.
For beta‐diversity analysis, the rarefied OTU table was square‐root‐transformed, before measures of community similarity, calculated as Bray–Curtis distances, were created using PRIMER v6 (Anderson et al., 2008) and visualized using non‐metric multidimensional scaling (nMDS) (Clarke and Gorley, 2006). Community‐level similarity was compared among larval, feed and water samples using permutational multivariate analysis of variance (Anderson, 2001) with the PERMANOVA add‐on (Anderson et al., 2008), which used 9999 permutations of residuals under a reduced model. Samples were further compared using the RELATE function with the Spearman rank correlation method. In addition, BVSTEP analysis was employed to find the smallest subset of OTUs from the feed and water with a multivariate pattern that matched the pattern of the larval community. This involved a between‐samples similarity matrix (Bray–Curtis distances) that used the minimum number of OTUs from the water and feed with a Spearman rank correlation of P > 0.90 to the larval OTU similarity matrix. A stepwise procedure with 9999 permutations was used to randomly select subsets of OTUs, with five random starts each (Clarke and Warwick, 1998). The selected OTU subset was removed from the matrix, and the procedure was repeated eight times.
To identify predicted functions and pathways highly associated with development stage of the larvae, a multipattern analysis was applied. For this purpose, the multipatt function from the IndicSpecies package was employed (De Caceres and Legendre, 2009). The resulting biserial coefficients (R) of each function/pathway with a particular stage were corrected for unequal sample size using the function r.g (Lubomír and Milan, 2006). P‐values were subsequently adjusted for multiple testing using the Benjamini–Hochberg correction (Benjamini and Hochberg, 1995).
Sequence data deposition
Raw 16S rRNA gene sequencing data were deposited in the NCBI Short Read Archive (SRA) under the accession number 454775.
Conflict of interest
None declared.
Supporting information
Table S1. 53 Core larval OTUs by larval relative abundance.
Table S2. Summary of key OTUs identified from the feed that correlate with OTUs in the larvae.
Table S3. Summary of key OTUs identified from the water that correlate with OTUs in the larvae.
Table S4. Predicted abundances of KEGG pathways and results of the indicator analysis.
Acknowledgements
The authors wish to thank Dr Ezequiel Marzinelli for advice on experimental design and statistical analysis and Luke Vandenberg for assistance in hatchery management and sampling.
Microbial Biotechnology (2019) 12(2), 275–288
Funding Information
No funding information provided.
References
- Aguilera, E. , Yany, G. , and Romero, J. (2013) Cultivable intestinal microbiota of yellowtail juveniles (Seriola lalandi) in an aquaculture system. Lat Am J Aquat Res 41: 395–403. [Google Scholar]
- Anderson, M.J. (2001) A new method for non‐parametric multivariate analysis of variance. Austral Ecol 26: 32–46. [Google Scholar]
- Anderson, A. , Gorley, R.N. and Clarke, K.R. (2008) . In Permanova + Primer: Guide to Software and Statistical Methods. Limited, P.‐E. (ed.). PRIMER‐E Ltd; Plymouth, UK. [Google Scholar]
- Aßhauer, K.P. , Wemheuer, B. , Daniel, R. , and Meinicke, P. (2015) Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31: 2882–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin, B. , Stuckey, L.F. , Robertson, P.A.W. , Effendi, I. , and Griffith, D.R.W. (1995) A probiotic strain of Vibrio alginolyticus effective in reducing diseases caused by Aeromonas salmonicida, Vibrio anguillarum and Vibrio ordalii . J Fish Dis 18: 93–96. [Google Scholar]
- Bakke, I. , Coward, E. , Andersen, T. , and Vadstein, O. (2015) Selection in the host structures the microbiota associated with developing cod larvae (Gadus morhua). Environ Microbiol 17: 3914–3924. [DOI] [PubMed] [Google Scholar]
- Bates, J.M. , Mittge, E. , Kuhlman, J. , Baden, K.N. , Cheesman, S.E. , and Guillemin, K. (2006) Distinct signals from the microbiota promote different aspects of zebrafish gut differentiation. Dev Biol 297: 374–386. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. , and Hochberg, Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc Ser B (Methodological) 57: 289–300. [Google Scholar]
- Bledsoe, J.W. , Peterson, B.C. , Swanson, K.S. , and Small, B.C. (2016) Ontogenetic characterization of the intestinal microbiota of channel catfish through 16S rRNA gene sequencing reveals insights on temporal shifts and the influence of environmental microbes. PLoS One 11: e0166379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. , and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer, J.N. , Booth, M.A. , Qin, J.G. , D'Antignana, T. , Thomson, M.J.S. , and Stone, D.A.J. (2014) Temperature and dissolved oxygen influence growth and digestive enzyme activities of yellowtail kingfish Seriola lalandi (Valenciennes, 1833). Aquacult Res 45: 2010–2020. [Google Scholar]
- Brown, S.P. , Cornforth, D.M. , and Mideo, N. (2012) Evolution of virulence in opportunistic pathogens: generalism, plasticity, and control. Trends Microbiol 20: 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns, A.R. , Stephens, W.Z. , Stagaman, K. , Wong, S. , Rawls, J.F. , Guillemin, K. , and Bohannan, B.J. (2016) Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J 10: 655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califano, G. , Castanho, S. , Soares, F. , Ribeiro, L. , Cox, C.J. , Mata, L. , and Costa, R. (2017) Molecular taxonomic profiling of bacterial communities in a gilthead seabream (Sparus aurata) hatchery. Front Microbiol 8: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callens, M. , Watanabe, H. , Kato, Y. , Miura, J. and Decaestecker, E. (2018) Microbiota inoculum composition affects holobiont assembly and host growth in Daphnia . BMC Microbiome 6: 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. , and Madden, T.L. (2009) BLAST+: architecture and applications. BMC Bioinformatics 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, B.N. , Qin, J.G. , Kumar, M.S. , Hutchinson, W. , and Clarke, S. (2006) Ontogenetic development of the digestive system in yellowtail kingfish Seriola lalandi larvae. Aquaculture 256: 489–501. [Google Scholar]
- Clarke, K.R. and Gorley, R.N. (2006) PRIMER v6: User Manual/Tutorial (Plymouth Routines in Multivariate Ecological Research). PRIMER‐E. Plymouth Marine Laboratory.
- Clarke, K.R. , and Warwick, R.M. (1998) Quantifying structural redundancy in ecological communities. Oecologia 113: 278–289. [DOI] [PubMed] [Google Scholar]
- De Caceres, M. , and Legendre, P. (2009) Associations between species and groups of sites: indices and statistical inference. Ecology 90: 3566–3574. [DOI] [PubMed] [Google Scholar]
- Dehler, C.E. , Secombes, C.J. and Martin, S.A.M. (2017) Seawater transfer alters the intestinal microbiota profiles of Atlantic salmon (Salmo salar L.). Sci Rep 7, 13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461. [DOI] [PubMed] [Google Scholar]
- Egerton, S. , Culloty, S. , Whooley, J. , Stanton, C. , and Ross, R.P. (2018) The gut microbiota of marine fish. Front Microbiol 9: 873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAO (2018) The State of World Fisheries and Aquaculture (SOFIA) 2018 In Food and Agriculture Organization of The United Nations, p. 227.
- Fielder, D. and Heasman, M. (2011). In Hatchery Manual for the Production of Australian Bass, Mulloway and Yellowtail Kingfish. Industries, N.D.o.P. (ed.). Taylors Beach, NSW: Port Stephens Fisheries Institute. [Google Scholar]
- Fraune, S. , and Bosch, T.C.G. (2010) Why bacteria matter in animal development and evolution. BioEssays 32: 571–580. [DOI] [PubMed] [Google Scholar]
- Gajardo, K. , Jaramillo‐Torres, A. , Kortner, T.M. , Merrifield, D.L. , Tinsley, J. , Bakke, A.M. , and Krogdahl, A. (2017) Alternative protein sources in the diet modulate microbiota and functionality in the distal intestine of Atlantic Salmon (Salmo salar). Appl Environ Microbiol 83: e02615–e02616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzow, S. , Kaiser, K. , Wemheuer, B. , Pfeiffer, B. , Daniel, R. , Vidal, S. , and Wemheuer, F. (2017) The effects of cropping regimes on fungal and bacterial communities of wheat and faba bean in a greenhouse pot experiment differ between plant species and compartment. Front Microbiol 8: 902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, S. , Liu, Y. , Zhou, Z. , He, S. , Cao, Y. , Shi, P. , et al (2010) Analysis of bacterial diversity in the intestine of grass carp (Ctenopharyngodon idellus) based on 16S rDNA gene sequences. Aqucult Res 42: 47–56. [Google Scholar]
- Hansen, G.H. , and Olafsen, J.A. (1999) Bacterial interactions in early life stages of marine cold water fish. Microbial Ecol 38: 1–26. [DOI] [PubMed] [Google Scholar]
- Hennersdorf, P. , Kleinertz, S. , Theisen, S. , Abdul‐Aziz, M.A. , Mrotzek, G. , Palm, H.W. , and Saluz, H.P. (2016) Microbial diversity and parasitic load in tropical fish of different environmental conditions. PLoS One 11: e0151594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingerslev, H.C. , von Gersdorff Jørgensen, L. , Lenz Strube, M. , Larsen, N. , Dalsgaard, I. , Boye, M. , and Madsen, L. (2014) The development of the gut microbiota in rainbow trout (Oncorhynchus mykiss) is affected by first feeding and diet type. Aquaculture 424–425: 24–34. [Google Scholar]
- Kanther, M. , and Rawls, J.F. (2010) Host‐microbe interactions in the developing zebrafish. Curr Opin Immunol 22: 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashinskaya, E.N. , Simonov, E.P. , Kabilov, M.R. , Izvekova, G.I. , Andree, K.B. and Solovyev, M.M. (2018) Diet and other environmental factors shape the bacterial communities of fish gut in an eutrophic lake. J Appl Microbiol. doi.org/10.1111/jam.14064 [DOI] [PubMed] [Google Scholar]
- Klindworth, A. , Pruesse, E. , Schweer, T. , Peplies, J. , Quast, C. , Horn, M. , and Glockner, F.O. (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next‐generation sequencing‐based diversity studies. Nucleic Acids Res 41: e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristin, H. , Manuel, Y. , Ivar, R. , Clara, B. , C., C.L.E. and Marisol, I. (2013) Fish larval nutrition and feed formulation: knowledge gaps and bottlenecks for advances in larval rearing. Rev Aquacult 5, S26–S58. [Google Scholar]
- Lane, D. (1991) 16S/23S rRNA sequencing In Nucleic acid Techniques in Bacterial Systematics. Stackebrandt E., and Goodfellow M. (eds). New York, NY: John Wiley & Sons. [Google Scholar]
- Lauzon, H.L. , Gudmundsdottir, S. , Petursdottir, S.K. , Reynisson, E. , Steinarsson, A. , Oddgeirsson, M. , et al (2010) Microbiota of Atlantic cod (Gadus morhua L.) rearing systems at pre‐ and posthatch stages and the effect of different treatments. J Appl Microbiol 109: 1775–1789. [DOI] [PubMed] [Google Scholar]
- Lee, C.‐S. (2003) Biotechnological advances in finfish hatchery production: a review. Aquaculture 227: 439–458. [Google Scholar]
- Legrand, T. , Catalano, S.R. , Wos‐Oxley, M.L. , Stephens, F. , Landos, M. , Bansemer, M.S. , et al (2018) The inner workings of the outer surface: skin and gill microbiota as indicators of changing gut health in yellowtail kingfish. Front Microbiol 8: 2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Zhou, L. , Yu, Y. , Ni, J. , Xu, W. , and Yan, Q. (2017) Composition of gut microbiota in the Gibel Carp (Carassius auratus gibelio) varies with host development. Microb Ecol 74: 239–249. [DOI] [PubMed] [Google Scholar]
- Liu, H. , Guo, X. , Gooneratne, R. , Lai, R. , Zeng, C. , Zhan, F. , and Wang, W. (2016) The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci Rep 6: 24340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewellyn, M.S. , Boutin, S. , Hoseinifar, S.H. , and Derome, N. (2014) Teleost microbiomes: the state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front Microbiol 5: 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokesh, J. , Kiron, V. , Sipkema, D. , Fernandes, J.M.O. and Moum, T. (2018) Succession of embryonic and the intestinal bacterial communities of Atlantic salmon (Salmo salar) reveals stage‐specific microbial signatures. Microbiology Open 13: e00672. doi.org/10.1002/mbo3.672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. , and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubomír, T. , and Milan, C. (2006) Statistical determination of diagnostic species for site groups of unequal size. J Veg Sci 17: 809–818. [Google Scholar]
- Merchie, G. , Lavens, P. , and Sorgeloos, P. (1997) Optimization of dietary vitamin C in fish and crustacean larvae: a review. Aquaculture 155: 165–181. [Google Scholar]
- Merrifield, D.L. , Dimitroglou, A. , Foey, A. , Davies, S.J. , Baker, R.T.M. , Bøgwald, J. , et al (2010) The current status and future focus of probiotic and prebiotic applications for salmonids. Aquaculture 302: 1–18. [Google Scholar]
- Michl, S.C. , Ratten, J.‐M. , Beyer, M. , Hasler, M. , LaRoche, J. , and Schulz, C. (2017) The malleable gut microbiome of juvenile rainbow trout (Oncorhynchus mykiss): diet‐dependent shifts of bacterial community structures. PLoS One 12: e0177735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miegel, R.P. , Pain, S.J. , van Wettere, W.H.E.J. , Howarth, G.S. , and Stone, D.A.J. (2010) Effect of water temperature on gut transit time, digestive enzyme activity and nutrient digestibility in yellowtail kingfish (Seriola lalandi). Aquaculture 308: 145–151. [Google Scholar]
- Mukhtar, B. , Malik, M.F. , Shah, S.H. , Azzam, A. and Slahuddin, I.L. (2017) Lysine supplementation in fish feed. Int J Appl Biol Foren 1, 26–31. [Google Scholar]
- Nayak, S.K. (2010) Role of gastrointestinal microbiota in fish. Aquacult Res 41: 1553–1573. [Google Scholar]
- Nielsen, S. , Wilkes Walburn, J. , Verges, A. , Thomas, T. , and Egan, S. (2017) Microbiome patterns across the gastrointestinal tract of the rabbitfish Siganus fuscescens . PeerJ 5: e3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen, J. , Blanchet, F.G. , Kindt, R. , Legendre, P. , Minchin, P.R. , O'Hara, R.B. , et al (2014). Vegan: Community Ecology Package. R Package Version 2.2‐0. URL http://CRAN.Rproject.org/package=vegan
- Planas, M. , and Cunha, I. (1999) Larviculture of marine fish: problems and perspectives. Aquaculture 177: 171–190. [Google Scholar]
- Ramirez, C. , and Romero, J. (2017) The microbiome of Seriola lalandi of wild and aquaculture origin reveals differences in composition and potential function. Front Microbiol 8: 1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, C. , Hu, J. , Liu, W. , Liu, Z. , He, S. , Dan, B.C. , et al (2016) Thymol and Carvacrol affect hybrid Tilapia through the combination of direct stimulation and an intestinal microbiota‐mediated effect: insights from a germ‐free zebrafish model. J Nutr 146: 1132–1140. [DOI] [PubMed] [Google Scholar]
- Ringø, E. , and Gatesoupe, F.‐J. (1998) Lactic acid bacteria in fish: a review. Aquaculture 160: 177–203. [Google Scholar]
- Shaw, K.S. , Rosenberg Goldstein, R.E. , He, X. , Jacobs, J.M. , Crump, B.C. , and Sapkota, A.R. (2014) Antimicrobial susceptibility of Vibrio vulnificus and Vibrio parahaemolyticus recovered from recreational and commercial areas of Chesapeake Bay and Maryland Coastal Bays. PLoS One 9: e89616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, M. , Scheuner, C. , Meier‐Kolthoff, J.P. , Brinkhoff, T. , Wagner‐Dobler, I. , Ulbrich, M. , and Klenk, H.P. (2017) Phylogenomics of Rhodobacteraceae reveals evolutionary adaptation to marine and non‐marine habitats. ISME J 11: 1483–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, C.C.R. , Snowberg, L.K. , Gregory Caporaso, J. , Knight, R. , and Bolnick, D.I. (2015) Dietary input of microbes and host genetic variation shape among‐population differences in stickleback gut microbiota. ISME J 9: 2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorgeloos, P. , Dhert, P. , and Candreva, P. (2001) Use of the brine shrimp, Artemia spp., in marine fish larviculture. Aquaculture 200: 147–159. [Google Scholar]
- Stephens, W.Z. , Burns, A.R. , Stagaman, K. , Wong, S. , Rawls, J.F. , Guillemin, K. , and Bohannan, B.J.M. (2016) The composition of the zebrafish intestinal microbial community varies across development. ISME J 10: 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullam, K.E. , Essinger, S.D. , Lozupone, C.A. , O'Connor, M.P. , Rosen, G.L. , Knight, R. , et al (2012) Environmental and ecological factors that shape the gut bacterial communities of fish: a meta‐analysis. Mol Ecol 21: 3363–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnecki, A.M. , Burgos, F.A. , Ray, C.L. , and Arias, C.R. (2017) Fish intestinal microbiome: diversity and symbiosis unravelled by metagenomics. J Appl Microbiol 123: 2–17. [DOI] [PubMed] [Google Scholar]
- Toranzo, A.E. , Magariños, B. , and Romalde, J.L. (2005) A review of the main bacterial fish diseases in mariculture systems. Aquaculture 246: 37–61. [Google Scholar]
- Vadstein, O. , Bergh, Ø. , Gatesoupe, F.J. , Galindo‐Villegas, J. , Mulero, V. , Picchietti, S. , et al (2013) Microbiology and immunology of fish larvae. Rev Aquacult 5: S1–S25. [Google Scholar]
- Wang, A.R. , Ran, C. , Ringø, E. , and Zhou, Z.G. (2018) Progress in fish gastrointestinal microbiota research. Rev Aquacult 10: 626–640. [Google Scholar]
- Wemheuer, B. , and Wemheuer, F. (2017) Assessing bacterial and fungal diversity in the plant endosphere. Methods Mol Biol 1539: 75–84. [DOI] [PubMed] [Google Scholar]
- Yan, Q. , Li, J. , Yu, Y. , Wang, J. , He, Z. , Van Nostrand, J.D. , et al (2016) Environmental filtering decreases with fish development for the assembly of gut microbiota. Environ Microbiol 18: 4739–4754. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. 53 Core larval OTUs by larval relative abundance.
Table S2. Summary of key OTUs identified from the feed that correlate with OTUs in the larvae.
Table S3. Summary of key OTUs identified from the water that correlate with OTUs in the larvae.
Table S4. Predicted abundances of KEGG pathways and results of the indicator analysis.