

Abstract
Lysophosphatidic acid (LPA) is a blood-derived bioactive lipid with numerous biological activities exerted mainly through six defined G protein-coupled receptors (LPA1-LPA6). LPA was first identified as a vasoactive compound because it induced transient hypertension when injected intravenously in rodents. Here, we examined the molecular mechanism underlying the LPA-induced hypertensive response. The LPA-induced hypertensive response was significantly attenuated by pretreatment with a Rho kinase inhibitor, which blocks Gα12/13 signaling. Consistent with this, the response was weakened in KO mice of LPA4, a Gα12/13-coupling LPA receptor. KO mice of another Gα12/13-coupling LPA receptor, LPA6, also showed an attenuated LPA-induced hypertensive response. However, LPA6 KO mice also displayed attenuated pressor responses to an adrenergic agent and abnormal blood vessel formation. Using several LPA analogs with varied affinity for each LPA receptor, we found a good correlation between the hypertensive and LPA4 agonistic activities. Incubated mouse plasma, which contained abundant LPA, also induced a hypertensive response. Interestingly the response was completely abolished when the plasma was incubated in the presence of an ATX inhibitor. Together, these results indicate that circulating LPA produced by ATX contributes to the elevation of blood pressure through multiple LPA receptors, mainly LPA4.
Introduction
Lysophosphatidic acid (LPA: 1- or 2-acyl-sn-glycerol-3-phosphate) is a bioactive lipid that can induce a number of cellular responses, including cell proliferation, migration and cytoskeletal reorganization, most of which are mediated through six defined G-protein-coupled receptors (GPCRs) specific to LPA1,2. So far six LPA receptors have been identified, including three that belong to the endothelial differentiation gene (EDG) family (LPA1–3) and another three that belong to the P2Y family (LPA4–6). LPA is continuously produced in the blood, where both an LPA-producing enzyme, autotaxin (ATX), and its substrate, lysophosphatidylcholine (LPC), are present3. Importantly, when plasma is isolated and incubated in vitro, a large amount of LPA is produced by the action of ATX4.
LPA was originally identified as a vasoactive lipid in soybean extract5. Tokumura et al. reported that when LPA was intravenously injected in rodents, such as rats and guinea pigs, it induced transient hypertension6. In addition, an i.v. injection of the incubated plasma induced a hypertensive response as was observed for LPA. Human hypertensive patients showed significantly higher levels of LPA in plasma, suggesting that LPA is associated with hypertension7. The molecular mechanism underlying the increase of blood pressure by LPA, however, remains largely unknown. LPA-induced hypertension was still observed in LPA1 KO, LPA2 KO and LPA1/LPA2 DKO mice8, suggesting the involvement of other LPA receptor(s) (LPA3–6).
LPA receptor agonists also serve as useful tools to evaluate the role of LPA receptor subtypes. To date, a number of compounds with structures similar to LPA have been developed in several laboratories and were shown to have distinct profiles in activating each LPA receptor9,10. We previously designed and synthesized LPA analogs similar to 2-acyl-LPA (LPA with a fatty acid at the sn-2 position), so-called “T-series” compounds, in which a ring structure derived from carbohydrates is introduced as a scaffold instead of a glycerol backbone11. These analogs have restricted conformational flexibility due to the sugar ring structure and show unique activity for LPA1–3 in Ca2+ and migration assays. Some compounds, such as T13, were potent ligands for LPA312. The reactivity of the T-series compounds to LPA4, LPA5 and LPA6 have not been examined so far.
To clarify the molecular mechanism underlying the LPA-induced pressor response, we examined whether the LPA receptors cloned so far (LPA1–6) are involved in LPA-induced transient hypertension by utilizing a combination of LPA receptor KO mice and LPA analogs. Here we report that LPA4 is the major hypertensive LPA receptor. Furthermore, we demonstrate that LPA6 signaling is crucial for normal vasoactivity and vascular development.
Results
LPA administration induces transient hypertension in mice
As was demonstrated in other experimental animals, administration of LPA (18:1-LPA, 1.4 mg/kg, i.v.) in ICR mice produced a weak hypotension followed by a transient hypertension that lasted for a minute (Fig. 1A). This LPA-induced hypertension was observed regardless of mouse strain and anesthetic (data not shown). The LPA-induced hypertensive response was dose-dependent and was observed at concentrations as low as 0.014 mg/kg for oleoyl (18:1)-LPA (Fig. 1B). The hypertensive activity of LPA was dependent on the acyl chain of LPA. Among the five LPA species tested, 18:1- LPA was the most potent in elevating blood pressure and myristoyl (14:0)-LPA was the least potent.
Figure 1.

LPA induces transient hypertension in mice. (A) Original recording of mice blood pressure. LPA (1-oleoyl, 1.4 mg/kg) was intravenously injected into anesthetized mice to monitor change in blood pressure. Arrow indicates the time point of injection of LPA. (B) The increase of mean artery pressure (MAP) in mice injected with the indicated dose of five LPA species was analyzed. Unsaturated LPA (18:1- and 20:4-LPA) showed potent hypertensive activity. Data represents change in MAP as mean ± S.E. (n = 4).
LPA induces a transient hypertension via the Gα12/13-Rho/ROCK pathway
To reveal the intracellular signals underlying LPA-produced hypertension, we tested two GPCR signaling inhibitors (Y-27632 and PTX). We found that Y-27632, an inhibitor of a Rho kinase that is activated downstream of Gα12/13-Rho signaling, significantly and dose-dependently attenuated the hypertensive activity of LPA (Fig. 2A). In contrast, PTX, which inactivates Gαi/o proteins, had no effect. We observed that bradycardia evoked by acetylcholine i.v. administration was attenuated by PTX pretreatment (data not shown), confirming that Gαi/o proteins were inactivated. These results suggest that LPA induces a transient hypertension via putative LPA receptor(s) coupling with Gα12/13 protein.
Figure 2.

Putative hypertensive LPA receptor(s) couple with Gα12/13 protein. (A) Effects of GPCR signaling inhibitor to LPA-induced hypertension. Y-27632 significantly inhibited LPA-induced hypertension. Increase in MAP represent as mean + S.E. (n = 7 Control group, n = 3 Y27632 and PTX group **P < 0.01, ***P < 0.001). (B,C) AP-TGFα release responses of LPA receptors (LPA1-6). LPA1-6 expressing HEK293 cells were stimulated with 18:1-LPA (3 μM). (B) Cells were transfected with control siRNA, Gα12/13 siRNA or TACE siRNA. (C) Cells were pretreated with Y27632 (10 μM). Data represents as mean + S.D. (n = 3, *P < 0.05, **P < 0.01, ***P < 0.001, n.s.: not significant).
Unlike other GPCR assay methods, the TGFα-shedding assay, a novel assay system for detecting GPCR activation that we developed recently13, effectively detects Gα12/13 signaling as well as Gαq signaling. Importantly, activation of all six LPA receptors was detectable using this assay. To evaluate possible coupling of each LPA receptor to Gα12/13 protein, we knocked down both Gα12 and Gα13 by siRNA. Simultaneous knockdown of Gα12 and Gα13 dramatically decreased the activity of LPA4 and LPA6, as was observed for knockdown of TNFα-converting enzyme (TACE) (Fig. 2B), a crucial enzyme in the TGFα-shedding assay (Fig. 2B). In agreement with this observation, Y-27632 significantly reduced the activities of LPA3, LPA4 and LPA6 (Fig. 2C). In LPA3-expressing cells, Gα12/13 knockdown partially inhibited the TGFα-shedding responses. These data indicate that LPA4 and LPA6 are the LPA receptors that mainly coupled with Gα12/13 protein.
Attenuated LPA-induced hypertensive response in both LPA4- and LPA6-deficient mice
To identify the LPA receptors involved in the pressor effect, we tested five single LPA receptor KO mice (Lpar1, Lpar2, Lpar3, Lpar4 and Lpar6 null). Consistent with a previous report8, administration of LPA in Lpar1 and Lpar2 null mice induced a similar hypertensive response as was observed in wild-type mice (Fig. S1). Similar results were obtained with Lpar1/Lpar2 double KO mice (data not shown). The LPA-evoked pressor response was also not affected in LPA3-deficient mice, although the hypotensive response was attenuated (Supplementary Fig. 1). These data suggest that Edg-type LPA receptors (i.e. LPA1, LPA2 and LPA3) are not involved in the LPA-induced hypertensive response, at least in mice. We next tested LPA4 and LPA6 KO mice and found that they showed an impaired pressor response to LPA (Fig. 3A). The dose-response curve of LPA-induced hypertension is shown in Fig. 3B. LPA4-deficient mice showed a slightly but significantly lowered response to various LPA dosages. In LPA6-deficient mice a lower dosage of LPA (0.014 mg/kg) induced hypertensive responses similar to those in wild-type mice. However, the responses induced by higher LPA dosages were significantly lower in LPA6-deficient mice. To address the redundant role of LPA4 and LPA6 in the pressor effect of LPA, we tried to generate mice lacking both Lpar4 and Lpar6 (Lpar4/Lpar6-double null mice). However, Lpar4/Lpar6-double null pups produced by intercrossing Lpar4+/− Lpar6+/− female and Lpar4+/Y Lpar6+/− male mice were not born (Table 1). Although Lpar4+/− Lpar6−/− and Lpar4−/Y Lpar6+/− mice, which retained only one wild-type allele, were born fertile, the number of offspring was less than the value expected from the Mendelian ratios. Further mating experiment using Lpar4−/− Lpar6+/− female and Lpar4−/Y Lpar6+/− male also failed to generate Lpar4/Lpar6-double null mice, suggesting that complete loss of Lpar4/6 results in embryonic lethality or death after parturition, while a single remaining wild-type allele is sufficient for normal development and reproduction. We thus could not test Lpar4/Lpar6-double null mice for LPA-induced transient hypertension.
Figure 3.

LPA-induced hypertension is attenuated in LPA4- and LPA6-deficient mice. (A) Original recording of mice blood pressure. LPA (1.4 mg/kg) was intravenously injected into mice. Arrow indicates the time point of injection of LPA. (B) The increase of MAP in KO mice and their control littermates. Mice were injected with the indicate dose of LPA. Data represents change in MAP as mean ± S.E. (n = 4 Lpar4+/+, n = 3 Lpar4−/−, Lpar6+/− and Lpar6−/− mice, *P < 0.05, **P < 0.01, ***P < 0.001).
Table 1.
Number of offspring of each genotype.
| Offspring Genotype (Lpar4)/(Lpar6) | Total | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of offspring (percentage of total) | |||||||||||||
| Parental genotype (Lpar4)/(Lpar6) | Female | Male | |||||||||||
| Female × Male | (+/+)/(+/+) | (+/+)/(+/−) | (+/+)/(−/−) | (+/−)/(+/+) | (+/−)/(+/−) | (+/−)/(−/−) | (+/Y)/(+/+) | (+/Y)/(+/−) | (+/Y)/(−/−) | (−/Y)/(+/+) | (−/Y)/(+/−) | (−/Y)/(−/−) | |
| (+/−)/(+/−) × (+/Y)/(+/−) | 15 (11.7) | 20 (15.6) | 6 (4.7) | 14 (10.9) | 18 (14.1) | 3 (2.3) | 13 (10.2) | 21 (16.4) | 5 (3.9) | 6 (4.7) | 7 (5.5) | 0 (0) | 129 |
| Offspring Genotype ( Lpar4 )/( Lpar6 ) | Total | ||||||||||||
| Number of offspring (percentage of total) | |||||||||||||
| Parental genotype (Lpar4)/(Lpar6) | Female | Male | |||||||||||
| Female × Male | (+/−)/(+/+) | (+/−)/(+/−) | (+/−)/(−/−) | (−/−)/(+/+) | (−/−)/(+/−) | (−/−)/(−/−) | (+/Y)/(+/+) | (+/Y)/(+/−) | (+/Y)/(−/−) | (−/Y)/(+/+) | (−/Y)/(+/−) | (−/Y)/(−/−) | |
| (+/−)/(+/−) × (−/Y)/(+/−) | 13 (10.7) | 27 (22.3) | 4 (3.3) | 9 (7.4) | 6 (5.0) | 0 (0) | 11 (9.1) | 21 (17.4) | 11 (9.1) | 8 (6.6) | 11 (9.1) | 0 (0) | 120 |
| Offspring Genotype ( Lpar4 )/( Lpar6 ) | Total | ||||||||||||
| Number of offspring (percentage of total) | |||||||||||||
| Parental genotype (Lpar4)/(Lpar6) | Female | Male | |||||||||||
| Female × Male | (+/−)/(+/+) | (+/−)/(+/−) | (+/−)/(−/−) | (−/Y)/(+/+) | (−/Y)/(+/−) | (−/Y)/(−/−) | |||||||
| (−/−)/(+/−) × (−/Y)/(+/−) | 6 (20.7) | 0 (0) | 8 (27.6) | 6 (20.7) | 9 (27.6) | 0 (0) | 29 | ||||||
Abnormal vasoactivity and vascular development in LPA6-deficient mice
To examine whether the attenuated hypertensive response induced by LPA in LPA4 and LPA6-deficient mice results from their abnormal vasculature, we next examined the vasoactivity of these mice. We confirmed that both LPA4- and LPA6-deficient mice had normal blood pressure and heart rate under normal conditions (Supplementary Fig. 2). LPA4-deficient mice showed hypertensive responses to phenylephrine similar to those observed in wild-type mice (Fig. 4A). LPA4-deficient mice also showed normal pressor responses to norepinephrine (Fig. 4B), indicating that their vasoactive response is not affected in LPA4-deficient mice despite the presence of some abnormalities in the blood vascular system of neonates as previously reported14. By contrast, LPA6-deficient mice displayed significantly lowered responses to both phenylephrine and norepinephrine (Fig. 4C and D), raising the possibility that they have abnormal vasculature that results in impaired vasoactivities. We thus analyzed postnatal retinal blood vessel formation in LPA6-deficient mice, a widely used evaluation system for physiological angiogenesis. Isolectin B4 staining, which visualizes the vascular network in the retina, revealed a decreased vascular density and branching in LPA6-deficient mice (Fig. 5A). In addition, LPA6-deficient vessels extended few filopodia at the vascular front where most endothelial tip cells are located (Fig. 5B). Quantitative analyses of the retinal vessels showed 22% less EC coverage, 26% less branching points and 35% fewer tip cells number in LPA6-deficient mice (Fig. 5A,B). Accordingly, an LPA6 signal was found to be essential for normal vasculature. Thus, we could not conclude that LPA6 is involved in LPA-induced hypertension using LPA6-deficient mice, even though they had weakened LPA-induced pressor responses (Fig. 3B).
Figure 4.

Vasoactive response is impaired in LPA6-deficient mice. (A,C) The increase of MAP in LPA4-deficient (A) and LPA6-deficient mice (C) injected with the indicated dose of phenylephrine. Data represents change in MAP as mean ± S.E. (n = 5 Lpar4+/+, n = 3 Lpar4−/−, n = 4 Lpar6+/− and Lpar6−/− mice, **P < 0.01) (B,D) The increase of MAP in LPA4-deficient (B) and LPA6-deficient mice (D) injected with norepinephrine (0.03 mg/kg). Data represents change in MAP as mean ± S.E. (**P < 0.01, n.s.: not significant).
Figure 5.

LPA6-deficient mice show abnormal vascular structure in retina. (A) Isolectin-B4 angiography in wild-type and LPA6-deficient mice at P6. Lpar6−/− mice displayed a reduction in the density of the vascular network correlating with a decreased number of branches. Data are shown as mean ± S.E. (**P < 0.01) (B) Magnification view of angiogenic front in retina. Arrow indicates the tip cell. The number of tip cells were decreased in Lpar6−/− mice. Data are shown as mean ± S.E. (**P < 0.01) Scale bars: 500 μm (A); 50 μm (B).
Hypertensive activity of LPA analogs, T-series compounds
To determine the LPA receptors involved in LPA-induced hypertension, we next used a pharmacological approach using LPA analogs called T-series compounds (Fig. 6A). The T-series compounds were designed to identify specific active conformations of the glycerol backbone of LPA by using carbohydrates with a fixed ring structure as scaffolds11. We tested the hypertensive activity of six T-series compounds in mice and found that T8 was the most potent in increasing blood pressure (Fig. 6B), with an activity almost equal to that of LPA (18:1). T7, an LPA analog with an acyl-chain and phosphate in the opposite position to T8, also showed a pressor effect but it was less potent than T8 and LPA. In contrast, T10, T17 and T19, which are stereoisomers of T8, and T16, which is a stereoisomer of T7, were much less potent. These data indicate that putative LPA receptor(s) involved in the LPA-induced hypertensive response strictly recognize the structure of these LPA analogs.
Figure 6.

The structure-activity relationship of T-series compounds as a vasopressor and as an LPA receptor agonist. (A) Structures of T-series synthesized based on 2-Oleoyl LPA as the lead compound. (B) Hypertensive activity of the T-series compounds. The indicated dose of LPA and T-series compound were intravenously injected into ICR mice, and the potencies of these compounds to induce hypertension was analyzed. Data represented as mean ± S.E. (n = 3) (C) Agonist activity of T-series compounds against the six LPA receptors. HEK293 cells expressing LPA1–6 were stimulated with the indicated concentration of LPA and T-series. Data represented as mean ± S.E. (n = 3) (D) MAP increases in LPA4-deficient mice injected with the indicated dose of T7 and T8. Data represents change in MAP as mean ± S.E. (n = 3).
The best vasopressor, T8, is a potent ligand for LPA4
Next, we evaluated the ability of the T-series compounds to activate the six LPA receptors (Fig. 6C, Table 2). For LPA1, LPA2 and LPA6, LPA was found to be the best ligand, while some T-series compounds were the best in activating LPA3, LPA4 and LPA5. The weaker vasopressor compounds (T16, T17 and T19) were found to be potent in activating LPA1, LPA3 and LPA5 (EC50 < 100 nM). For example, T16 and T17 were potent agonists for both LPA1 and LPA3, and T19 was a potent agonist for LPA5, indicating that LPA1, LPA3 and LPA5 are not the LPA receptors involved in the LPA-induced hypertensive response. Interestingly, T8, the most potent vasopressor compound, was found to be a potent ligand for both LPA4 and LPA6. The second most potent pressor substance, T7, activated LPA4, but not LPA6.
Table 2.
Estimates of EC50, Emax and RIA in TGF-α shedding assay.
| LPA (18:1) | T7 | T8 | T10 | T16 | T17 | T19 | ||
|---|---|---|---|---|---|---|---|---|
| LPA1 | Emax (%) | 13.7 | 12.5 | 10.5 | 15.4 | 11.7 | 11.6 | 9.5 |
| EC50 (nM) | 10 | 35 | 130 | 390 | 12 | 21 | 170 | |
| RIA | 1 | 0.26 | 0.062 | 0.030 | 0.71 | 0.41 | 0.043 | |
| LPA2 | Emax (%) | 31.2 | 18.5 | 33.9 | 34.3 | 29.7 | 33.6 | 36.1 |
| EC50 (nM) | 11 | 1000 | 3800 | 2200 | 160 | 310 | 2500 | |
| RIA | 1 | 0.006 | 0.003 | 0.005 | 0.066 | 0.038 | 0.005 | |
| LPA3 | Emax (%) | 25.1 | 28.5 | 25.7 | 24.5 | 25.1 | 25 | 21.8 |
| EC50 (nM) | 97 | 13 | 73 | 120 | 7.6 | 11 | 93 | |
| RIA | 1 | 8.5 | 1.4 | 0.77 | 13 | 8.9 | 0.90 | |
| LPA4 | Emax (%) | 10.6 | 10.4 | 12.3 | 18.2 | 12.5 | 10.8 | 10.1 |
| EC50 (nM) | 11 | 7.5 | 8.2 | 730 | 250 | 270 | 84 | |
| RIA | 1 | 1.4 | 1.5 | 0.025 | 0.050 | 0.040 | 0.12 | |
| LPA5 | Emax (%) | 22.2 | 21.2 | 25.3 | 25.5 | 18.7 | 26.7 | 25.7 |
| EC50 (nM) | 72 | 390 | 270 | 85 | 240 | 87 | 12 | |
| RIA | 1 | 0.18 | 0.30 | 0.98 | 0.26 | 0.99 | 6.7 | |
| LPA6 | Emax (%) | 10 | ND | 16.8 | 22.6 | ND | ND | 7.8 |
| EC50 (nM) | 47 | >1000 | 520 | 4000 | >1000 | >1000 | 970 | |
| RIA | 1 | ND | 0.15 | 0.027 | ND | ND | 0.038 |
ND: not determined.
We then injected LPA4-preferred compounds (T7 and T8) into LPA4-deficient mice. Hypertensive activity of T8 dramatically disappeared in LPA4-deficient mice (Fig. 6D), showing the involvement of LPA4 in T8-induced hypertension. Because T8 was a potent ligand for LPA6 (Fig. 6C) and LPA6 is intact in LPA4-deficient mice, it is reasonable to assume that LPA6 is not the receptor involved in LPA-induced hypertension. By contrast, hypertensive activity of T7 was significantly attenuated in LPA4 KO mice, confirming again the involvement of LPA4 (Fig. 6D). However, the T7–induced hypertension still remained in LPA4 KO mice, raising the possibility that LPA target(s) other than LPA4 are involved. Such targets do not include LPA6 because T7 was found to be a poor agonist for LPA6 (Fig. 6C).
ATX-producing LPA in incubated mouse plasma induces transient hypertension
Incubated plasma is known to contain vasoactive compounds in rats and guinea pigs15. LPA has been assumed to be an active component of the pressor response because abundant LPA is accumulated in incubated plasma by the action of ATX. In fact, injecting mice with incubated mouse plasma induced transient hypertension (Fig. 7A), as was observed with LPA (Fig. 1A). After 3 hr incubation, the level of plasma LPA was markedly increased to about ~10 µM, which corresponds to the LPA dosage (0.014 mg/kg) in Fig. 1B. This LPA level was significantly lowered by the ATX inhibitor (ONO-8430506) treatment (Fig. 7B). Interestingly, the hypertensive response was not induced by incubated mouse plasma prepared in the presence of the ATX inhibitor (Fig. 7A). These results clearly showed that LPA produced in the incubated plasma by ATX is responsible for the hypertensive activity of the plasma.
Figure 7.
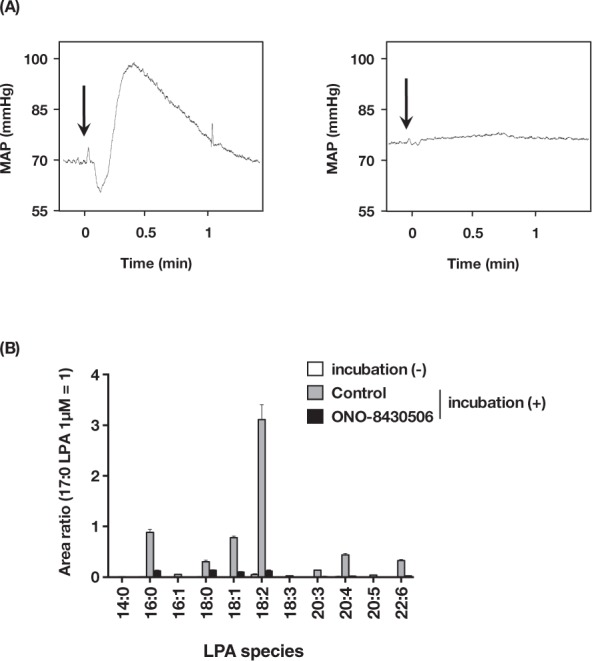
ATX-producing LPA in incubated mouse plasma induces transient hypertension. (A) Change in blood pressure after administration of incubated mouse plasma. Each incubated plasma (left: control, right: containing ONO-8430506) was intravenously injected into mice. Arrow indicates the time point of injection. (B) LPA levels in incubated mouse plasma. Plasma samples containing vehicle or ONO-8430506 (10 μM) were incubated at 37 °C for 3 hr. Data represents relative abundance, which is the ratio between analyte and internal standard (17:0 LPA, 1 μM) peak area, as mean + S.E. (n = 3).
Discussion
In this study we examined the mechanism underlying the pressor activity of LPA using several tools, including an inhibitor of an LPA-producing enzyme, five LPA receptor deficient mice and LPA analogs with different affinities for each LPA receptor. First, when mouse plasma was incubated with an ATX inhibitor, we were unable to detect the hypertensive activity of the plasma. Production of LPA in the plasma was completely suppressed by the inhibitor, showing clearly that LPA is the hypertensive substance in the incubated plasma.
Among the five LPA receptor deficient mice (LPA1–4, and LPA6), LPA4 and LPA6-deficient mice had a partially but significantly attenuated LPA-induced hypertensive response (Fig. 3). Comparison between the hypertensive and the LPA receptor agonistic activities of T-series compounds suggested that LPA4 is the most probable hypertensive LPA receptor (Fig. 6 and Table 2). Notably, the hypertensive activity of the most potent vasopressor compound T8 was almost completely suppressed in LPA4-deficient mice. Although we did not test LPA5-deficient mice, the involvement of LPA5 could be excluded since one of the T-series compounds, T19, which was found to be a potent agonist for LPA5, was a poor inducer of hypertension. The present study also suggests that LPA has other target(s) than LPA4 to induce transient hypertension, since LPA- and T7-induced hypertension was not suppressed completely in LPA4-deficient mice (Figs 3B and 6D). Although LPA activates ion channels such as TRPV116, this activity does not appear to be involved in hypertension because TRP channel blockers (A784168, capsazepine and AMG9810) had no effect on the hypertensive activity of LPA (Supplementary Fig. 3). Because Y-27632 dramatically suppressed LPA-induced hypertensive response (Fig. 2), GPCR-type LPA receptors other than LPA1–6 that couple with Gα12/13 may be involved.
Use of an ATX inhibitor clearly showed that LPA is the factor in the incubated plasma that induces transient hypertension (Fig. 1D). Unlike incubated plasma which contains a high concentration of LPA (~ several µM level), the LPA level in the fresh plasma is quite low (Fig. 7B). Thus, it remained unclear if endogenous LPA in the circulation controls blood pressure. The LC-MS/MS analysis revealed that the concentration of circulating LPA in healthy mice was several tens of nM (Fig. 7B). The minimum dose of LPA to induce obvious hypertension was 0.01 mg/kg (Fig. 1B), which corresponds with about ~ 500 nM circulating LPA. Therefore, endogenous circulating LPA seems to be slightly lower than the minimum concentration to induce hypertension. Mouse plasma contains mainly unsaturated LPA, such as 18:2-LPA, which was reported to be a potent inducer of hypertension in rat6. In the present study, unsaturated LPA, such as 18:1-LPA and 20:4-LPA, induced hypertension more efficiently than saturated LPAs (14:0, 16:0 and 18:0) in mice (Fig. 1B). In addition, the concentration of LPA and ATX are markedly elevated in various pathological conditions. For example, unsaturated plasma LPA, such as 18:2, 20:4 and 22:6, was selectively increased in patients with acute coronary syndrome17,18. Upon blood coagulation, 20:4-LPA is known to be produced in activated platelets18. The ATX level increases in normal pregnant women in the third trimester and to a higher extent in women facing preterm delivery19. Therefore, it would be quite interesting to examine whether the increased unsaturated LPA affects the blood pressure in such pathological conditions.
The precise molecular mechanisms by which LPA leads to elevate blood pressure via LPA4 remain to be elucidated. In agreement with previous studies using rats20, we confirmed that pretreatment by indomethacin (an inhibitor of prostanoid synthesis) and hexamethonium (a nicotinic acetylcholine receptor antagonist that acts in autonomic ganglia) had no effect on the LPA-induced hypertensive response in mice (data not shown), showing that prostanoids and the autonomic nervous system are not involved. In this study, we showed that pretreatment with Y27632, a Rho kinase (ROCK) inhibitor, significantly suppressed LPA-induced hypertensive response (Fig. 2A). LPA4 is known as a Gα12/13-coupled GPCR according to our present data (Fig. 2B). Recent studies have shown that the Rho-ROCK pathway is a novel therapeutic target in the treatment of various cardiovascular diseases, such as pulmonary hypertension and cerebral vasospasm21,22. Activation of Rho downstream of G12/13 is known to elicit an actomyosin-dependent contraction of aortic smooth muscle cells in an intracellular Ca2+-independent manner. Thus, it is possible that an ATX/LPA/LPA4 axis operates upstream of the Gα13-Rho-ROCK pathway, possibly in aortic smooth muscle cells, and is a promising drug target.
ATX-deficient mice die around embryonic day 9.5–10.5 with profound vascular defects in both yolk sac and embryo, indicating that LPA produced by ATX has a critical role in embryonic vascular development23. However, none of the single LPA receptor-deficient mice showed the same phenotype, although LPA4-deficient mice were partially lethal due to impaired blood and lymphatic vessel formation. Thus, it remains unclear how LPA regulates embryonic vascular development. Both LPA4 and LPA6 couple mainly with Gα12/13 protein (Fig. 2B) to activate Rho-ROCK signaling and act to coordinately regulate cell motile activity24. In this study, we showed that LPA6-deficient mice were viable but had obvious vascular abnormalities (Fig. 5), which is partially similar to the phenotype of LPA4-deficient mice. In contrast, we were unable to produce Lpar4/Lpar6-double null mice. It is also notable that the number of offspring carrying a single wild-type Lpar4 allele on an Lpar6-deficient background was less than expected (Table 1). These data raise the possibility that both LPA4 and LPA6 generate angiogenic signaling, which is essential for embryonic vascular development and is missing in ATX-deficient mice. Interestingly only LPA6-deficient mice exhibited impaired pressor response to various vasopressors (Fig. 4). Therefore, further studies are necessary to investigate the precise and differential roles of LPA4 and LPA6 signaling in embryonic vascular development.
In summary, we propose a novel mechanism of LPA-induced transient hypertension in which unsaturated LPA produced by ATX stimulates mainly LPA4 and induces the hypertensive response through Gα12/13-Rho-ROCK signaling.
Methods
Reagents
LPA (1-myristoyl (14:0), 1-palmitoyl (16:0), 1-stearoyl (18:0), 1-oleoyl (18:1), 1-arachidonyl (20:4)) was purchased from Avanti Polar Lipids. LPA and T-series compounds were dissolved in PBS containing 0.1% fatty acid free BSA (Sigma) and stocked at −20 °C. Biotinylated Griffonea Simplicifolia I isolectin B4 was purchased from Vector Laboratories. PTX and Y-27632 were from Calbiochem and Wako, respectively. The ATX inhibitor (ONO-8430506)4 was kindly donated by ONO Pharmaceutical Company.
Mouse breeding
Mice (C57BL6 and ICR, male, 8 weeks) were purchased from SLC Japan. LPA1, LPA2, LPA3 and LPA4 knockout (KO) mice were established as described previously14,25,26. LPA6 KO mice with a mixed 129/Sv and C57BL/6 were obtained from Deltagen (San Carlos, CA). Mice were housed under specific pathogen-free conditions in an air-conditioned room and fed standard laboratory chow ad libitum. All mice were treated in accordance with the protocol approved by the Animal Ethics Committee of the Graduate School of Pharmaceutical Sciences, Tohoku University, Japan.
Whole-mount staining and immunofluorescence staining
Immunostaining of flat-mount retinas was performed according to a previously described method27.
Measurement of blood pressure in mice
Male mice anesthetized with urethane (1.5 mg/kg, i.p.) were placed on a heating plate at 40 °C. Under a stereoscopic microscope, the trachea was exposed and cannulated. Subsequently, a polyethylene-tipped cannula (PE-60 tubing) was inserted into the left carotid artery to monitor arterial pressure. The arterial cannula was connected to a transducer and blood pressure signals were recorded using PowerLab4/25 (Bio Research Center, Nagoya, Japan). To analyze acute blood pressure response, a second catheter was placed in the right femoral vein to infuse agonists. Mice received a bolus injection (100 µl/time) at 5–10 min intervals. For pharmacological studies, PTX (30 µg/kg, i.v.) was dissolved in PBS and administered 24 hr and 48 hr before injection of LPA. Mice were treated with saline dilutions of Y-27632 (0.1–10 mg/kg, i.v.) 5 min before injection of LPA.
LC-MS/MS analysis
Lipids were extracted from plasma using methanol (including 17:0-LPA as internal standard; final concentration was 100 nM) as described previously28 and stored at −80 °C. LC-MS/MS analysis was performed according to a previously described method with minor modifications28. In this study, we used an LC-MS/MS system that included an Ultimate3000 HPLC and TSQ Quantiva triple quadropole mass spectrometer (Thermo Fisher Scientific). LPA analyses were performed in the multiple reactive monitoring (MRM) in negative mode28. LC was performed using a reverse phase column (CAPCELL PAK C18 (1.5 mm I.D. x 250 mm, particle size was 3 µm)) with a gradient elution of solvent A (5 mM ammonium formate in 95% (v/v) water, pH 4.0) and solvent B (5 mM ammonium formate in 95% (v/v) acetonitrile, pH 4.0) at 200 µL/min. Gradient conditions were as follows: hold 50% B for 0.2 min, followed by a linear gradient to 100% B over 11.8 min, hold 100% B for 5 min, return to the initial condition over 0.5 min, and maintain for 2.5 min until the end of run (total run time 20 min).
AP-TGFα shedding assay
This assay was conducted according to a previously described method with several modifications13. To improve signal detection, HEK293 cells were transfected with Gα chimeric proteins and treated with the LPA1–3 antagonist, Ki16425. The siRNAs were transfected into cells by using Lipofectamine RNAiMAX. We validated siRNA mediated knockdown of Gα12 and Gα13, previously13. Two days post-transfection, cells were co-transfected with mouse FLAG-LPA1–6, AP-TGFα and Gα chimera by using Lipofectamine2000. After 24 hr, cells were resuspended in HBSS buffer, seeded in 96 well assay plates and stimulated with ligands and 10 µM Ki16425 for 1 hr. 80 µl of conditioned media were transferred to new 96 well plates and mixed with an equal volume of p-NPP solution. AP activity was calculated by the measurement of absorbance at 405 nm with a microplate reader (Molecular Devices).
Calculations
Agonist activities in the reporter gene and shedding assays were estimated as described previously29. The EC50 value and Emax values were calculated by fitting a logistic equation to the data by nonlinear regression analysis. The RIA (relative intrinsic activity) values, which indicate the relative potency of an agonist to LPA, were determined.
Statistical analysis
Unpaired Student’s t-test, one-way ANOVA followed by Tukey’s post hoc test and multiple comparisons t-test were used for the statistical analysis. A value of P < 0.05 was considered statistically significant.
Supplementary information
Acknowledgements
We thank ONO Pharmaceutical Co., Ltd for the gift of the ATX inhibitor. This work was supported by Grant-in-Aid for Scientific Research on Innovative Areas JP15H05899 (J.A.), KAKENHI grant JP17K08264 (A.I.); the PRIME JP17gm5910013 (A.I.) and the LEAP JP17gm0010004 (K.K., A.I. and J.A.) from the Japan Agency for Medical Research and Development (AMED). J.C. was supported by NIH R01 NS084398.
Author Contributions
K.K. designed and performed most of the experiments; H.M. contributed to LC-MS analysis; A.I. performed AP-TGFα shedding assay; H.Y. performed Whole-mount staining and immunofluorescence staining; M.K. synthesized T-series compounds; J.C. provided the LPA3 KO mice, manuscript discussions and editing. S.I. and T.S. provided the LPA4 KO mice and supervised the research; K.K. and J.A. wrote the manuscript, with feedback from all of the authors. J.A. supervised all aspects of the study.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Kuniyuki Kano, Email: k-kano@m.tohoku.ac.jp.
Junken Aoki, Email: jaoki@m.tohoku.ac.jp.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-019-39041-4.
References
- 1.Aikawa S, Hashimoto T, Kano K, Aoki J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J. Biochem. 2015;157:81–89. doi: 10.1093/jb/mvu077. [DOI] [PubMed] [Google Scholar]
- 2.Kihara Y, Mizuno H, Chun J. Lysophospholipid receptors in drug discovery. Exp. Cell Res. 2015;333:171–177. doi: 10.1016/j.yexcr.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aoki J, Inoue A, Okudaira S. Two pathways for lysophosphatidic acid production. Biochimica Et Biophysica Acta-Molecular and Cell Biology of Lipids. 2007;1781:513–518. doi: 10.1016/j.bbalip.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Saga H, et al. A Novel Highly Potent Autotaxin/ENPP2 Inhibitor Produces Prolonged Decreases in Plasma Lysophosphatidic Acid Formation In Vivo and Regulates Urethral Tension. PLoS ONE. 2013;9:e93230–e93230. doi: 10.1371/journal.pone.0093230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tokumura, A. et al. Identification of vasopressor phospholipid in crude soybean lecithin. 13, 468–72 (1978). [DOI] [PubMed]
- 6.Tokumura A, Fukuzawa K, Tsukatani H. Effects of synthetic and natural lysophosphatidic acids on the arterial blood pressure of different animal species. Lipids. 1978;13:572–574. doi: 10.1007/BF02533598. [DOI] [PubMed] [Google Scholar]
- 7.Yao CS, et al. Significant association between lower pulse pressure and increasing levels of a novel type of phospholipid. Genet. Mol. Res. 2014;13:2922–2930. doi: 10.4238/2014.February.21.16. [DOI] [PubMed] [Google Scholar]
- 8.Panchatcharam M, et al. Lysophosphatidic Acid Receptors 1 and 2 Play Roles in Regulation of Vascular Injury Responses but Not Blood Pressure. Circulation Research. 2008;103:662–670. doi: 10.1161/CIRCRESAHA.108.180778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kano K, Arima N, Ohgami M, Aoki J. LPA and its analogs-attractive tools for elucidation of LPA biology and drug development. Curr. Med. Chem. 2008;15:2122–2131. doi: 10.2174/092986708785747562. [DOI] [PubMed] [Google Scholar]
- 10.Jiang G, Inoue A, Aoki J, Prestwich GD. Phosphorothioate analogs of sn-2 radyl lysophosphatidic acid (LPA): metabolically stabilized LPA receptor agonists. Bioorg Med Chem Lett. 2013;23:1865–1869. doi: 10.1016/j.bmcl.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Tamaruya Y, et al. Identifying Specific Conformations by Using a Carbohydrate Scaffold: Discovery of Subtype-Selective LPA-Receptor Agonists and an Antagonist. Angew. Chem. Int. Ed. 2004;43:2834–2837. doi: 10.1002/anie.200454065. [DOI] [PubMed] [Google Scholar]
- 12.Aikawa S, et al. Autotaxin-lysophosphatidic acid-LPA3 signaling at the embryo-epithelial boundary controls decidualization pathways. EMBO J. 2017;36:2146–2160. doi: 10.15252/embj.201696290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inoue AA, et al. TGFα shedding assay: an accurate and versatile method for detecting GPCR activation. Nat Methods. 2012;9:1021–1029. doi: 10.1038/nmeth.2172. [DOI] [PubMed] [Google Scholar]
- 14.Sumida H, et al. LPA4 regulates blood and lymphatic vessel formation during mouse embryogenesis. Blood. 2010;116:5060–5070. doi: 10.1182/blood-2010-03-272443. [DOI] [PubMed] [Google Scholar]
- 15.Wurm H, Kenner T. Some properties of a high molecular vasopressor substance generated in human serum by incubation. Basic Res Cardiol. 1978;73:1–9. doi: 10.1007/BF01914651. [DOI] [PubMed] [Google Scholar]
- 16.Nieto-Posadas, A. et al. Lysophosphatidic acid directly activates TRPV1 through a C-terminal binding site. Nat Chem Biol, 10.1038/nchembio.712 (2011). [DOI] [PubMed]
- 17.Dohi T, et al. Increased lysophosphatidic acid levels in culprit coronary arteries of patients with acute coronary syndrome. Atherosclerosis. 2013;227:323–328. doi: 10.1016/j.atherosclerosis.2013.01.032. [DOI] [PubMed] [Google Scholar]
- 18.Kurano M, et al. Possible involvement of minor lysophospholipids in the increase in plasma lysophosphatidic Acid in acute coronary syndrome. Arterioscler Thromb Vasc Biol. 2015;35:463–470. doi: 10.1161/ATVBAHA.114.304748. [DOI] [PubMed] [Google Scholar]
- 19.Tokumura A, et al. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002;277:39436–39442. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- 20.Tokumura, A., Kume, T., Fukuzawa, K. & Tsukatani, H. Cardiovascular effects of lysophosphatidic acid and its structural analogs in rats. 219, 219–24 (1981). [PubMed]
- 21.Shi J, Wei L. Rho Kinases in Cardiovascular Physiology and Pathophysiology: The Effect of Fasudil. J. Cardiovasc. Pharmacol. 2013;62:341–354. doi: 10.1097/FJC.0b013e3182a3718f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimokawa H, Sunamura S, Satoh K. RhoA/Rho-Kinase in the Cardiovascular System. Circulation Research. 2016;118:352–366. doi: 10.1161/CIRCRESAHA.115.306532. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka M, et al. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J. Biol. Chem. 2006;281:25822–25830. doi: 10.1074/jbc.M605142200. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi K, et al. Lysophosphatidic acid (LPA) signaling via LPA4 and LPA6 negatively regulates cell motile activities of colon cancer cells. Biochemical and Biophysical Research Communications. 2017;483:652–657. doi: 10.1016/j.bbrc.2016.12.088. [DOI] [PubMed] [Google Scholar]
- 25.Contos JJA, et al. Characterization oflpa2 (Edg4) and lpa1/lpa2 (Edg2/Edg4) Lysophosphatidic Acid Receptor Knockout Mice: Signaling Deficits without Obvious Phenotypic Abnormality Attributable to lpa2. Molecular and Cellular Biology. 2002;22:6921–6929. doi: 10.1128/MCB.22.19.6921-6929.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye X, et al. LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature. 2005;435:104–108. doi: 10.1038/nature03505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yukiura H, Kano K, Kise R, Inoue A, Aoki J. Autotaxin overexpression causes embryonic lethality and vascular defects. PLoS ONE. 2015;10:e0126734. doi: 10.1371/journal.pone.0126734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okudaira M, et al. Separation and quantification of 2-acyl-1-lysophospholipids and 1-acyl-2-lysophospholipids in biological samples by LC-MS/MS. J Lipid Res. 2014;55:2178–2192. doi: 10.1194/jlr.D048439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ehlert FJ, Griffin MT, Sawyer GW, Bailon R. A simple method for estimation of agonist activity at receptor subtypes: comparison of native and cloned M3 muscarinic receptors in guinea pig ileum and transfected cells. J. Pharmacol. Exp. Ther. 1999;289:981–992. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.