

Abstract
Wilms tumor (WT) is the most common type of renal malignancy in children. Survival rates are low and high-risk WT generally still carries a poor prognosis. To better elucidate the pathogenesis and tumorigenic pathways of high-risk WT, the present study presents an integrated analysis of RNA expression profiles of high-risk WT to identify predictive molecular biomarkers, for the improvement of therapeutic decision-making. mRNA sequence data from high-risk WT and adjacent normal samples were downloaded from The Cancer Genome Atlas to screen for differentially expressed genes (DEGs) using R software. From 132 Wilms tumor samples and six normal samples, 2,089 downregulated and 941 upregulated DEGs were identified. In order to identify hub DEGs that regulate target genes, weighted gene co-expression network analysis (WGCNA) was used to identify 11 free-scale gene co-expressed clusters. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were annotated using KEGG Orthology Based Annotation System annotation of different module genes. The Search Tool for the Retrieval of Interacting Genes was used to construct a protein-protein interaction network for the identified DEGs, and the hub genes of WGCNA modules were identified using the Cytohubb plugin with Cytoscape software. Survival analysis was subsequently performed to highlight hub genes with a clinical signature. The present results suggest that epidermal growth factor, cyclin dependent kinase 1, endothelin receptor type A, nerve growth factor receptor, opa-interacting protein 5, NDC80 kinetochore complex component and cell division cycle associated 8 are essential to high-risk WT pathogenesis, and they are closely associated with clinical prognosis.
Keywords: high-risk Wilms tumor, the cancer genome atlas, weighted gene co-expression network analysis, survival analysis
Introduction
Wilms tumor (WT), or nephroblastoma, is a type of pediatric renal malignancy that typically occurs in children, representing 6–14% of childhood tumors (1,2). With the advancement of multimodal therapies for WT, the 5-year overall survival rate has reached its peak (2,3). Unfortunately, high-risk WT, including anaplastic histology WT subtypes, rhabdoid tumor, metastatic renal sarcoma and carcinoma, still have a generally poor prognosis (4). Additionally, current treatment strategies for high-risk nephroblastoma include surgery, radiation therapy and chemotherapy; however, a targeted cure is still lacking (5). Therefore, novel therapeutic methods targeting specific mechanisms of high-risk nephroblastoma carcinogenesis are required to improve treatment efficiency and avoid the side effects of traditional therapy.
Advances in RNA sequencing technologies have revealed the complexity of the human genome. Investigation of the RNA transcriptome is one of the most important challenges facing biology today, as RNAs represent novel potential biomarkers and drug targets (6–9). Presently, accumulating evidence suggests that numerous key mRNAs identified in WT are closely associated with the pathogenesis of this tumor, including programmed cell death, reversion inducing cysteine rich protein with kazal motifs, TIMP metallopeptidase inhibitor 3, tropomyosin 1 and phosphatase and tensin homolog (10). However molecular biomarkers that may be predictive of a curative effect and prognosis in high-risk WT have not been reported.
The Cancer Genome Atlas (TCGA), a publicly available database, provides the gene expression profiles of >10,000 specimens from >25 different tumor types to be used for biological discovery (11). In the present study, weighted gene co-expression network analysis (WGCNA) and other analyses were performed using TCGA datasets to identify hub genes associated with clinical features of WT. These hub genes have the potential to be biomarkers of high-risk WT tumorigenesis.
Materials and methods
Study population
Datasets were manually retrieved from TCGA database (12). The primary tumor site for high-risk WT was the kidney. Regarding experimental strategy, RNA sequencing (seq) was used. Taken together, a total of 132 WT and 6 normal samples, including the clinical information and mRNA-seq data, were downloaded via the Data Transfer Tool (provided by GDC Apps; http://tcga-data.nci.nih.gov/). The sequenced data were all derived from IlluminaHiSeq RNA-Seq platforms. The present study followed the publication guidelines provided by TCGA (http://cancergenome.nih.gov/publications/publicationguidelines).
Differentially expressed gene (DEG) analysis
Prior to statistical analysis, the high-risk WT RNA-seq data derived from 138 WT samples were normalized across all matrixes using the ‘batch’ package (13). In addition, tumor sample and normal sample data were merged and expressed data, which closed to zero, were deleted. Differential expression (DE) for genes, including all six normal control samples, was tested with the ‘DESeq’ and ‘edgR’ packages in R software version 3.4.0 (14,15). The empirical probability distribution of the fold-changes associated with significant DEs (P<0.05) was used to define a |log2 fold-change| with a threshold >2.0 (16). To remove potential noise, all fold-change values associated with comparisons that were not considered significant by the ‘Limma’ package or potentially significant by threshold-filtering were converted to ‘zero’, which in the log2 scale corresponds to the complete absence of differential regulation among all samples, with the function f(C)=IC (X), where:
Finally, to maintain every one data corresponding to per samples, the retained fold-change values were calculated by subtracting the median normalized log2 expression values of the normal samples from the respective WT samples.
Construction of gene co-expression network
WGCNA is a systems biology method for describing the correlation patterns among genes across microarray samples. WGCNA may be used for identifying modules of highly correlated genes, summarizing such clusters using a module eigengene or an intra-modular hub gene, relating modules to one another and to external sample traits, and for calculating module membership measures. Correlation networks facilitate network-based gene screening methods that may be used to identify candidate biomarkers or therapeutic targets (17–19).
To identify the interactions between the DEGs, the WGCNA, which used the topological overlapping measurement, was performed to identify the co-expression modules with a threshold of power cutoff of 14 and a module size cutoff ≤30 (13). All DEGs were used, and Pearson's correlation was calculated for all pairs of selected genes. The correlation data were converted into the adjacency matrix with a power function; therefore, the correlation strength between two genes, xi and xj, was defined as aij=| 0.5 * (1+cor(xi, xj))|β, where xi and xj represent the expression values of the probes. The parameter β was determined by the criterion that the resulting adjacency matrix approximately met a scale-free topological feature, according to the proposed model-fitting index. The row index u (u=1,…, m) represents sample measurements. This was further transformed into a topological overlap matrix (TOM), which captures not only the direct interaction among two genes, but also the indirect interactions throughout all the other genes in the network. In the present study, it was possible to identify two functions of adjacency matrices. At first, the TOM is defined as follows:
Therefore, it was possible to calculate the node connectivity. Secondly, the other function was performed as a matrix into hierarchical clustering of the transcript profiles for identifying the modules, and it may be defined as follows:
Functional annotation and protein-protein interactions (PPIs)
To identify Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways associated with each DEG, the KEGG Orthology Based Annotation System (KOBAS) was used to annotate the function of different module genes, with a false discovery rate thresholds <0.05 (20). Cytoscape software was used to construct a co-expression network of hub DEG-mediated pathways (21). These DEG-pathway network data were subsequently exported as Microsoft Excel files (Microsoft Corporation, Redmond, WA, USA).
In order to obtain protein interactions between DEGs of different modules, we used the Search Tool for the Retrieval of Interacting Genes online tool to construct a PPI network for the DEGs (minimum required interaction score >0.4) (22). In addition, Cytoscape software was used to visualize the PPI networks. Finally, the Cytohubb plugin was used to identify the hub genes in the PPI network (23).
Survival analysis
In order to identify prognostic hub mRNA signatures, survival curves of all the clinical data from patients in TCGA were combined with those of hub DEGs using log-rank tests. The ‘survival’ package was adopted to produce the survival curves in R software (24). This analysis was based on Kaplan-Meier univariate survival analysis (24). P<0.05 was considered to indicate a statistically significant difference.
Results
Patient characteristics
The detailed clinical characteristics of the WT study population are summarized in Table I. In the present study, the median age of patients with WT was 3 years old and the median survival time was 77.5 months. In addition, there were more female patients (54.4%) compared with male patients (45.6%). The distribution of race among patients demonstrated that Caucasian represented largest proportion (68.4%). Regarding the tumor stage, it was identified that the patients with stage II and III represented the largest proportion (33%) compared with other stage groups. As demonstrated in Table I, the distribution of histological classification illustrated that the patients with favorable-histology WT were increased compared with those with diffusely anaplastic WT. Regarding adverse events, patients with WT suffered more relapses (18.8%) compared with patients with progression (2.1%).
Table I.
Clinical characteristics of 138 patients with Wilms tumor.
| Variable | Patients with Wilms tumor (n=138) |
|---|---|
| Age, years | |
| Median | 3 |
| IQR | 2-5 |
| Overall survival time, months | |
| Median | 77.5 |
| IQR | 44.75–109 |
| Sex | |
| Male | 63 (45.4%) |
| Female | 75 (54.6%) |
| Race | |
| Caucasian | 94 (68.4%) |
| African American | 24 (17.7%) |
| Other | 20 (13.9%) |
| Ethnicity | |
| Local | 86 (62.2%) |
| Hispanic or Latino | 11 (8%) |
| Other | 41 (29.8%) |
| Tumor stage | |
| Stage I | 28 (20%) |
| Stage II | 45 (33%) |
| Stage III | 46 (33%) |
| Stage IV | 10 (7.6%) |
| Stage V | 9 (6.4%) |
| Histology classification | |
| Favorable-histology Wilms tumor | 117 (85.7%) |
| Diffusely anaplastic Wilms tumor | 21 (14.3%) |
| Adverse event | |
| Progression | 3 (2.1%) |
| Relapse | 26 (18.8%) |
| None | 109 (79.1%) |
IQR, interquartile range.
Gene expression profiles in high-risk WT
Using high-risk WT expression profiles from the TCGA database, significant DEGs in 138 tumor samples compared with six normal samples were identified. There were 2,089 downregulated and 941 upregulated DEGs identified by the DESeq package, accounting for 68.94 and 31.06% of all DEGs, respectively. In total, 4,894 DEGs were identified using the edgR package, among which 2,044 DEGs were downregulated in high-risk WT tumor samples and 2,850 were upregulated. According to the Venn diagram, a total of 2,921 overlapping DEmRNAs were identified, among which 1,987 were downregulated DEGs and 934 were upregulated, accounting for 68.02 and 31.98% of all overlapping DEGs, respectively. These genes were DEGs, as computed by the DESeq and edgR algorithms (Fig. 1). The top 10 upregulated and downregulated mRNAs with a |log2foldchange|>2.0 are presented in Table II.
Figure 1.
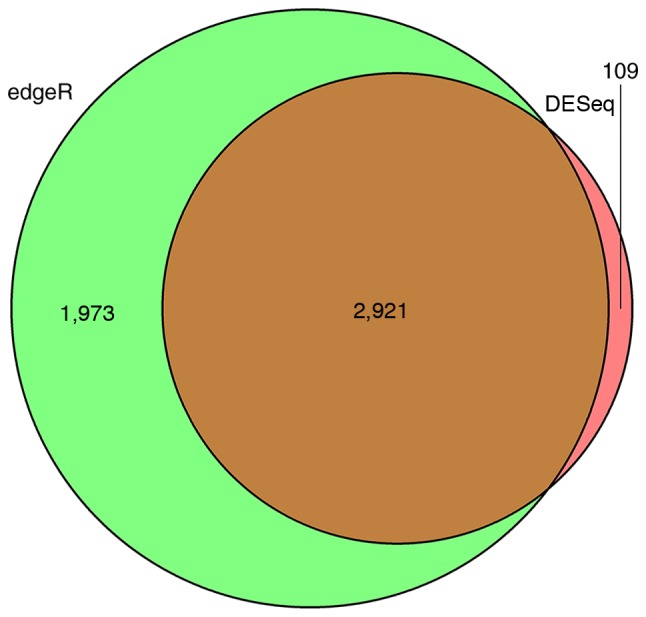
Venn Diagram for selecting identical DEGs obtained by the different algorithms. Number of dysregulated expression genes identified by edgR (pale green) and DESeq (light coral), and overlapping DEGs (brown). DEGs, differentially expressed genes.
Table II.
The top 10 upregulated and downregulated genes.
| Gene symbol | logFC | Adjusted P-value |
|---|---|---|
| LIN28B | 13.36619486 | 5.42×10−6 |
| SIX2 | 12.52402043 | 7.06×10−47 |
| DGKK | 12.24930467 | 4.66×10−16 |
| VSTM2B | 12.04214637 | 6.34×10−9 |
| GPAT3 | 11.61327772 | 1.85×10−9 |
| CHRNA1 | 11.52619613 | 2.15×10−13 |
| DLK1 | 11.46063048 | 1.56×10−3 |
| COL2A1 | 10.55873377 | 1.68×10−44 |
| PCDH15 | 10.33793967 | 2.40×10−25 |
| GATA4 | 10.29733454 | 1.25×10−7 |
| Gene symbol | logFC | Adjusted P-value |
| UMOD | −18.37509732 | 9.61×10−65 |
| AQP2 | −13.96005987 | 1.11×10−32 |
| KNG1 | −12.65948815 | 3.72×10−41 |
| FXYD4 | −12.04659281 | 4.80×10−61 |
| GP2 | −11.12440106 | 9.70×10−28 |
| SLC9A4 | −10.52222348 | 1.56×10−29 |
| CLCNKA | −10.26143663 | 1.07×10−23 |
| BSND | −10.11652616 | 8.55×10−22 |
| HRG | −10.11203117 | 3.38×10−19 |
| SEMG2 | −9.973583814 | 3.12×10−6 |
FC, fold-change.
Construction of weighted gene co-expression modules
To explain the association between the 2,921 overlapping DEGs, WGCNA, a systems biology method, was used to screen potential biomarkers and therapeutic targets via gene co-expression network construction. DEGs involved in similar pathways or with the same biological function tended to have the same expression cluster. As demonstrated in Fig. 2 and Table III, a total of 11 co-expressed modules were identified with a power cutoff of 14 and a module size cutoff ≤30. Therefore, the grey colored clusters represent the non-clustering genes in WGCNA. As six colored DEG modules, specifically black, blue, magenta, red, turquoise and yellow, were more numerous compared with the other color module genes, these key modules were selected for further analysis.
Figure 2.
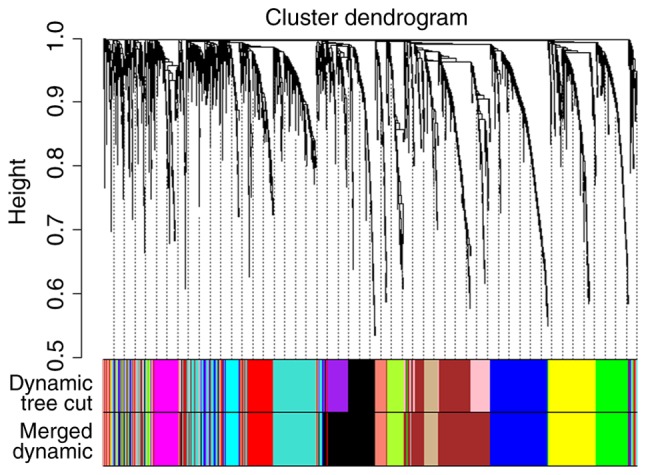
Hierarchical clustering dendrograms of identified co-expressed genes in modules. The dendrogram was generated by unsupervised hierarchical clustering of genes using topological overlap. The colored strips below each dendrogram indicate the module designation identified though the clusters of co-expressed genes, and assigned the merged module color to the original module color.
Table III.
Gene co-expression module sizes.
| Module color | Number of genes |
|---|---|
| Black | 320 |
| Blue | 380 |
| Brown | 152 |
| Cyan | 80 |
| Green | 204 |
| Green yellow | 111 |
| Grey | 106 |
| Magenta | 464 |
| Red | 292 |
| Salmon | 84 |
| Turquoise | 424 |
| Yellow | 274 |
PPI network and functional annotation for the modules
To better understand the associations between DEGs in the six different color modules, a PPI network was constructed using Cytoscape software (Fig. 3). The black module had 42 nodes and 58 interactions. There were 11 upregulated and 31 downregulated DEGs in this module. In total, six hub genes were identified in the center of the PPI network identified by the Cytohubb plugin, including epidermal growth factor (EGF), arginine vasopressin receptor 2, adrenoceptor β2, bradykinin receptor B2, endothelin receptor type A (ENDRA) and nerve growth factor receptor (NGFR). In addition, the KOBAS online tool was used to predict possible enrichment pathways (Table IV). There were 21 pathways enriched in this module, including ‘calcium signaling pathway’, ‘cGMP-PKG signaling pathway’ and ‘pathways in cancer’.
Figure 3.

Protein-protein interaction network visualization of modules identifies hub genes. Different colors represent the status of genes dysregulated, upregulated DEGs (red) and downregulated DEGs (green). The triangle nodes represent the hub genes in each module. The oval nodes represent genes and the lines represent interactions. DEGs, differentially expressed genes.
Table IV.
KEGG pathways of weighted gene co-expression network analysis modules.
| Module name | KEGG pathways | Input number | P-value |
|---|---|---|---|
| Black module | hsa04080:Neuroactive ligand-receptor interaction | 13 | 1.02×10−8 |
| hsa01100:Metabolic pathways | 21 | 3.37×10−7 | |
| hsa04020:Calcium signaling pathway | 8 | 7.78×10−6 | |
| hsa04810:Regulation of actin cytoskeleton | 7 | 1.05×10−5 | |
| hsa00830:Renin secretion | 4 | 2.61×10−5 | |
| hsa05202:Transcriptional misregulation in cancer | 6 | 2.84×10−5 | |
| hsa04110:Cell cycle | 5 | 7.34×10−5 | |
| hsa04151:PI3K-Akt signaling pathway | 8 | 1.23×10−4 | |
| hsa05200:Pathways in cancer | 8 | 2.45×10−4 | |
| hsa04014:Ras signaling pathway | 5 | 5.83×10−4 | |
| hsa04022:cGMP-PKG signaling pathway | 6 | 6.04×10−4 | |
| hsa04024:cAMP signaling pathway | 5 | 6.50×10−4 | |
| hsa04010:MAPK signaling pathway | 6 | 7.21×10−4 | |
| hsa05219:Bladder cancer | 4 | 7.37×10−4 | |
| hsa05218:Melanoma | 4 | 7.59×10−4 | |
| hsa04068:FoxO signaling pathway | 3 | 7.66×10−4 | |
| hsa04510:Focal adhesion | 5 | 8.40×10−3 | |
| hsa04923:Regulation of lipolysis in adipocytes | 4 | 4.38×10−3 | |
| hsa05160:Hepatitis C | 4 | 1.20×10−2 | |
| hsa04072:Phospholipase D signaling pathway | 3 | 3.34×10−2 | |
| hsa04144:Endocytosis | 4 | 4.93×10−2 | |
| Blue module | hsa01100:Metabolic pathways | 15 | 3.10×10−3 |
| hsa04020:Calcium signaling pathway | 5 | 3.17×10−3 | |
| hsa04080:Neuroactive ligand-receptor interaction | 6 | 4.24×10−3 | |
| hsa04060:Cytokine-cytokine receptor interaction | 8 | 3.67×10−2 | |
| hsa04080:Neuroactive ligand-receptor interaction | 6 | 3.69×10−2 | |
| hsa04068:FoxO signaling pathway | 3 | 4.02×10−2 | |
| hsa04922:Glucagon signaling pathway | 3 | 4.53×10−2 | |
| Magenta module | hsa04530:Tight junction | 6 | 3.27×10−6 |
| hsa04514:Cell adhesion molecule | 4 | 8.15×10−4 | |
| hsa05219:Bladder cancer | 3 | 6.33×10−3 | |
| hsa04390:Hippo signaling pathway | 3 | 9.48×10−3 | |
| hsa05218:Melanoma | 2 | 9.51×10−3 | |
| hsa05100:Bacterial invasion of epithelial cells | 3 | 1.75×10−2 | |
| hsa05200:Pathways in cancer | 4 | 1.88×10−2 | |
| hsa04670:Leukocyte transendothelial migration | 3 | 2.07×10−2 | |
| Red module | hsa04110:Cell cycle | 11 | 3.77×10−11 |
| hsa04114:Oocyte meiosis | 10 | 6.60×10−1° | |
| hsa04914:Progesterone-mediated oocyte maturation | 7 | 5.89×10−7 | |
| hsa04115:p53 signaling pathway | 4 | 3.63×10−4 | |
| hsa05200:Pathways in cancer | 6 | 7.66×10−4 | |
| hsa01100:Metabolic pathways | 12 | 2.08×10−3 | |
| hsa04152:AMPK signaling pathway | 3 | 4.54×10−3 | |
| hsa05166:HTLV-I infection | 4 | 7.30×10−3 | |
| hsa04060:Cytokine-cytokine receptor interaction | 4 | 1.10×10−2 | |
| hsa04080:Neuroactive ligand-receptor interaction | 4 | 1.33×10−2 | |
| hsa00982:Drug metabolism-cytochrome P450 | 2 | 1.36×10−2 | |
| hsa00980:Metabolism of xenobiotics by cytochrome P450 | 2 | 2.13×10−2 | |
| hsa01524:Platinum drug resistance | 2 | 2.13×10−2 | |
| hsa01230:Biosynthesis of amino acids | 2 | 3.38×10−2 | |
| hsa00430:Taurine and hypotaurine metabolism | 2 | 4.54×10−2 | |
| hsa05203:Viral carcinogenesis | 3 | 4.83×10−2 | |
| Turquoise module | hsa04110:Cell cycle | 17 | 3.25×10−15 |
| hsa05166:HTLV-I infection | 12 | 4.94×10−6 | |
| hsa04114:Oocyte meiosis | 8 | 5.99×10−6 | |
| hsa05200:Pathways in cancer | 13 | 2.23×10−5 | |
| hsa05202:Transcriptional misregulation in cancer | 8 | 2.94×10−5 | |
| hsa04914:Progesterone-mediated oocyte maturation | 6 | 8.26×10−4 | |
| hsa01040:Biosynthesis of unsaturated fatty acids | 3 | 1.36×10−2 | |
| hsa01212:Fatty acid metabolism | 3 | 4.05×10−2 | |
| Yellow module | hsa04668:TNF signaling pathway | 12 | 4.98×10−13 |
| hsa04380:Osteoclast differentiation | 9 | 2.27×10−10 | |
| hsa05166:HTLV-I infection | 11 | 2.03×10−8 | |
| hsa05323:Rheumatoid arthritis | 7 | 5.60×10−8 | |
| hsa04933:AGE-RAGE signaling pathway in diabetic complications | 7 | 3.68×10−7 | |
| hsa04010:MAPK signaling pathway | 7 | 7.13×10−7 | |
| hsa04210:Apoptosis | 5 | 2.20×10−4 | |
| hsa05161:Hepatitis B | 5 | 5.67×10−4 | |
| hsa05142:Chagas disease (American trypanosomiasis) | 4 | 6.82×10−4 | |
| hsa04620:Toll-like receptor signaling pathway | 4 | 1.58×10−3 | |
| hsa05168:Herpes simplex infection | 5 | 1.69×10−3 | |
| hsa01100:Metabolic pathways | 14 | 1.93×10−2 | |
| hsa04510:Focal adhesion | 5 | 4.47×10−2 |
KEGG, Kyoto Encyclopedia of Genes and Genomes; PI3K, phosphoinositide 3-kinase; Akt, protein kinase B; cGMP, cyclic guanosine monophosphate; PKG, protein kinase G; cAMP, adenosine monophosphate; MAPK, mitogen-activated protein kinase; AMPK, 5′ AMP-activated protein kinase; HTLV-1, human T-cell leukemia virus type 1; TNF, tumor necrosis factor; AGE, advanced glycation endproducts; RAGE, receptor for advanced glycation endproducts.
In the blue module, there were 40 nodes and 53 interactions. There were fewer upregulated genes than downregulated genes; 10 and 30, respectively. In total, three key dysregulated mRNAs, including glucagon receptor, prostaglandin E receptor 1 and SGK2, serine/threonine kinase 2, were identified as hub genes. There were seven enriched KEGG pathways in this module, which included ‘neuroactive ligand-receptor interaction’, ‘calcium signaling pathway’, ‘foxO signaling pathway’ and ‘glucagon signaling pathway’.
The magenta module had 22 nodes and 28 interactions. The number of upregulated genes was lower compared with the downregulated genes (five upregulated genes; 17 downregulated genes). Cadherin 1, claudin 7 and tight junction protein 3 were identified as hub genes. In total, eight enriched KEGG pathways were identified, which included ‘cell adhesion molecule’, ‘melanoma’ and ‘hippo signaling pathway’.
The red module had 70 nodes and 658 interactions. All the DEGs were upregulated in this module. Five hub genes were identified by the Cytohubb plugin, including mitotic arrest deficient 2 like 1, cyclin A2, cyclin dependent kinase 1 (CDK1), opa-interacting protein 5 (OIP5) and NUF2, NDC80 kinetochore complex component (NUF2). A total of 16 pathways were enriched in the red modules, including ‘progesterone-mediated oocyte maturation’, ‘HTLV-I infection’ and ‘viral carcinogenesis’, among others.
In the turquoise module, there were 116 nodes and 532 interactions. All the DEGs were upregulated in this module. Only three hub genes were identified, including BUB1 mitotic checkpoint serine/threonine kinase (BUB1) B, BUB1 and cell division cycle associated 8 (CDCA8). There were a total of eight pathways enriched in the turquoise module, including ‘cell cycle’, ‘oocyte meiosis’ and ‘progesterone-mediated oocyte maturation’, among others.
In the yellow module, there were 65 nodes and 293 interactions. All the DEGs were downregulated in this module. Five hub genes were identified, including FOS proto-oncogene, AP-1 transcription factor subunit, early growth response 1, activating transcription factor 3, dual specificity phosphatases and JunB proto-oncogene, AP-1 transcription factor subunit. A total of 13 pathways were identified, including ‘TNF signaling pathway’, ‘MAPK signaling pathway’ and ‘Toll-like receptor signaling pathway’, among others.
Co-expression network of the hub DEG-mediated pathways
According to KOBAS annotation, a co-expression network was constructed to exhibit the enrichment pathways and biological processes that may be activated by 25 hub DEGs (Fig. 4). Among these key DEGs, it was identified that seven had more interactions enriched in ‘cell cycle’ and ‘pathways in cancer’ compared with other regulators, including EGF, CDK1, ENDRA, NGFR, OIP5, NUF2 and CDCA8. These genes may represent key regulators associated with the progression of high-risk WT.
Figure 4.

Co-expression network of the hub DEGs mediated in the pathways. Each node represents a hub DEG, and the different colors represent the modules to which they belong. The number of connections indicates the association between the hub DEGs and signaling pathways. DEGs, differentially expressed genes.
Survival analysis of hub DEGs
The mRNAseq data and clinical information of all high-risk WT samples were combined for Kaplan-Meier univariate survival analysis. The seven most crucial hub genes were subjected to survival analysis. As a result, the high expression levels of three DEGs, including EGF, ENDRA and NGFR, were associated with a longer overall survival time compared with the low gene expression (P<0.05). On the contrary, four overexpressed DEGs (CDK1, OIP5, NUF2 and CDCA8) demonstrated that they were all associated with poor prognosis in patient overall survival (P<0.05). All survival analyses are plotted in Fig. 5.
Figure 5.


Survival curve analysis of hub DEGs for the overall survival in patients with high-risk Wilms tumor. In total, seven hub DEGs (CDK1, OIP5, NUF2, CDCA8, EGF, ENDRA and NGFR) are presented (P<0.05). DEGs, differentially expressed genes; CDK1, cyclin dependent kinase 1; OIP5, opa-interacting protein 5; NUF2, NUF2, NDC80 kinetochore complex component; CDCA8, cell division cycle associated 8; EGF, epidermal growth factor; ENDRA, endothelin receptor type A; NGFR, nerve growth factor receptor.
Discussion
WT is the most common genitourinary malignant tumor in children (1). High-risk WT, called anaplastic WT, and infants and children with rhabdoid tumor, were studied to determine why they have a much poorer prognosis. Even though numerous efforts have been made to investigate the key regulatory genes or molecules in malignant neoplasms, few studies have been able to predict the prognosis for high-risk WT, and no reliable biomarkers for the improvement of therapeutic decision-making have been identified.
In the present study, 2,921 overlapping DEGs, using the DESeq and edgR algorithms, were identified in gene expression profiling analysis. Furthermore, WGCNA was used to construct a free-scale gene co-expression network to investigate the associations between different sets of DEGs. To clarify the different clusters of WGCNA, PPI networks were built with Cytoscape software and the hub genes were identified by the Cytohubb plugin. Finally, using clinical information, it was identified that seven hub genes, including EGF, CDK1, ENDRA, NGFR, OIP5, NUF2 and CDCA8, may be potential biomarkers for prognosis prediction of patients with high-risk WT.
EGF is a member of the epidermal growth factor superfamily (25). The protein encoded by EGF acts as a potent mitogenic factor that serves a critical role in the proliferation, growth and differentiation of various cell types. Lanuti et al (26) identified that EGF is associated with patients with high-risk esophageal adenocarcinoma. Tanabe et al (27) identified that modulation of EGF gene expression levels is directly associated with the risk of developing hepatocellular carcinoma following liver cirrhosis. However, considering the survival analysis in the present study, it is noteworthy that the overexpression of EGF was demonstrated to be associated with better survival compared with a lower mean expression level in high-risk WT. In addition, KEGG pathway annotation demonstrated that EGF was associated with biological process and molecular function pathways, including ‘Rep1 signaling pathway’, ‘pathways in cancer’, and ‘PI3K-Akt signaling pathway’, which suggests that EGF may be the key regulator in the progression of WT to high-risk WT.
CDK1 is a member of the Ser/Thr protein kinase family, which serves a critical role in the G1/S and G2/M phase transitions of the eukaryotic cell cycle (28). In the present results, it was also identified that CDK1 is associated with significant cancer-associated pathways, including ‘oocyte meiosis’, ‘p53 signaling pathway’ and ‘viral carcinogenesis’. In addition, survival analysis demonstrated that high CDK1 expression decreased patient survival time. Although CDK1 was determined to be a prognostic predictor of high-risk WT, further research is required to verify this result.
ENDRA generally encodes the receptor for endothelin-1, a peptide that serves a role in potent and long-lasting vasoconstriction (29). The receptor for ENDRA is associated with guanine-nucleotide-binding proteins (30). In the present study, it was additionally identified that the ENDRA may activate specific pathways (‘cGMP-PKG signaling pathway’, ‘cAMP signaling pathway’ and ‘pathways in cancer’) to mediate high-risk WT. Combined with clinical data, survival analysis demonstrated that higher expression levels of ENDRA may prolong the overall survival time in patients with high-risk WT.
NGFR is a transmembrane receptor with intracellular tyrosine kinase activity (31). Through KEGG annotation, it was observed that NGF is enriched in ‘transcriptional dysregulation in cancer’, ‘PI3K-Akt signaling pathway’ and ‘Rap1 signaling pathway’. In 2008, Soland et al (32) verified that NGFR overexpression was associated with a pattern of invasion and a poor prognosis in oral squamous cell carcinoma. However in the present survival analysis, high NGFR expression was demonstrated to predict a longer survival time in patients with high-risk WT. Further investigations are required to confirm the impact of such gene expression on high-risk WT.
OIP5 is localized as adherent to centromeres (33). Expression of OIP5 is upregulated in a number of cancer types, making it a potential therapeutic target (34–37). Chun et al (37) reported that OIP5 is a highly expressed therapeutic target in colorectal and gastric carcinomas. Similarly, Koinuma et al (35) identified OIP5 as a molecular target in lung and esophageal carcinogenesis due to its biological functions. In the present study, survival analysis demonstrated that OIP5 overexpression was negatively associated with high-risk WT patient overall survival. Therefore, it was suggested that OIP5 may be a potential biomarker of high-risk WT, although the mechanism of OIP5-induced carcinogenesis should be further elucidated.
The protein encoded by NUF2 is generally a component protein that regulates chromosome segregation in the cell cycle (38). A previous study identified NUF2 to be associated with the malignant potential of colorectal cancer (39). In the present study, it was identified that, for the first time to the best of the authors' knowledge, NUF2 is associated with the clinical outcomes of patients with high-risk WT. Survival analysis demonstrated that NUF2 overexpression was negatively associated with patient overall survival. This result suggested that NUF2 expression knockdown may suppress the growth and progression of WT tumor cells. CDCA8 protein may additionally encode a component of the chromosomal passenger complex, which serves a regulatory role in mitosis and cell division (40). Additionally, higher expression of CDCA8 is associated with longer survival compared with lower expression.
However, the primary limitation of the present study was that these important DEGs remain to be verified by experiments; therefore, further analyses are required to determine the mechanisms underlying the process of malignant progression in high-risk WT. Future studies may aim to use polymerase chain reaction assays or western blotting to verify the expression levels of key genes between tumor and normal samples. Furthermore, conservative statistical methods with corrections for multiple testing at each level of analysis were applied; however, the present study still did not identify a better mathematical model to combine all eligible hub genes together for predicting the survival of WT. Therefore, future studies may aim for a better re-evaluation of the prognostic performance of the model for WT.
In conclusion, based on the gene expression profile analysis conducted in the present study, using TCGA database, DEmRNAs between high-risk WT samples and normal samples were identified. There were 2,921 DEGs identified by comprehensive bioinformatics analysis. The WGCNA method was used to identify hub genes involved in high-risk WT development. Specifically, EGF, CDK1, ENDRA, NGFR, OIP5, NUF2 and CDCA8 may serve a fundamental role in the development of high-risk WT and are predicted to be involved in carcinogenesis pathways. These findings may provide potential biomarkers for further study of WT mechanisms, and may be efficacious targets for therapeutic intervention or diagnosis in high-risk WT.
Acknowledgements
The authors gratefully acknowledge The Cancer Genome Atlas pilot project (established by the National Cancer Institute and NHGRI), which made available the genomic and clinical WT.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
CBX and CGH conceived and designed the study. XFW performed the bioinformatics analyses and was a major contributor in writing the manuscript. PS and NJY assisted in the bioinformatics analysis. CGH and XHZ selected and downloaded the datasets. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Amirian ES. The role of Hispanic ethnicity in pediatric Wilms' tumor survival. Pediatr Hematol Oncol. 2013;30:317–327. doi: 10.3109/08880018.2013.775618. [DOI] [PubMed] [Google Scholar]
- 2.Perme MP, Jereb B. Trends in survival after childhood cancer in Slovenia between 1957 and 2007. Pediatr Hematol Oncol. 2009;26:240–251. doi: 10.1080/08880010902900437. [DOI] [PubMed] [Google Scholar]
- 3.Pastore G, Znaor A, Spreafico F, Graf N, Pritchard-Jones K, Steliarova-Foucher E. Malignant renal tumours incidence and survival in European children (1978–1997): Report from the Automated Childhood Cancer Information System project. Eur J Cancer. 2006;42:2103–2114. doi: 10.1016/j.ejca.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 4.Geller JI. Current standards of care and future directions for ‘high-risk’ pediatric renal tumors: Anaplastic Wilms tumor and Rhabdoid tumor. Urol Oncol. 2016;34:50–56. doi: 10.1016/j.urolonc.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 5.PDQ Cancer Information Summaries. National Cancer Institute (US); Bethesda, MD: 2002. National Cancer Institute: Wilms Tumor and Other Childhood Kidney Tumors Treatment (PDQ®): Patient version. [Google Scholar]
- 6.Yuan N, Zhang G, Bie F, Ma M, Ma Y, Jiang X, Wang Y, Hao X. Integrative analysis of lncRNAs and miRNAs with coding RNAs associated with ceRNA crosstalk network in triple negative breast cancer. OncoTargets and Ther. 2017;10:5883–5897. doi: 10.2147/OTT.S149308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baltimore D. Our genome unveiled. Nature. 2001;409:814–816. doi: 10.1038/35057267. [DOI] [PubMed] [Google Scholar]
- 8.Huang C, Yuan N, Wu L, Wang X, Dai J, Song P, Li F, Xu C, Zhao X. An integrated analysis for long noncoding RNAs and microRNAs with the mediated competing endogenous RNA network in papillary renal cell carcinoma. Onco Targets and Ther. 2017;10:4037–4050. doi: 10.2147/OTT.S141951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang CG, Li FX, Pan S, Xu CB, Dai JQ, Zhao XH. Identification of genes associated with castration-resistant prostate cancer by gene expression profile analysis. Mol Med Rep. 2017;16:6803–6813. doi: 10.3892/mmr.2017.7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charlton J, Pavasovic V, Pritchard-Jones K. Biomarkers to detect Wilms tumors in pediatric patients: Where are we now? Future Oncol. 2015;11:2221–2234. doi: 10.2217/fon.15.136. [DOI] [PubMed] [Google Scholar]
- 11.Tomczak K, Czerwińska P, Wiznerowicz M. The cancer genome atlas (TCGA): An immeasurable source of knowledge. Contemp Oncol (Pozn) 2015;19:A68–A77. doi: 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JS. Exploring cancer genomic data from the cancer genome atlas project. BMB Rep. 2016;49:607–611. doi: 10.5483/BMBRep.2016.49.11.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rao Y, Lee Y, Jarjoura D, Ruppert AS, Liu CG, Hsu JC, Hagan JP. A comparison of normalization techniques for microRNA microarray data. Stat Appl Genet Mol Biol. 2008;7:Article22. doi: 10.2202/1544-6115.1287. [DOI] [PubMed] [Google Scholar]
- 14.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li D, Dye TD. Power and stability properties of resampling-based multiple testing procedures with applications to gene oncology studies. Comput Math Methods Med 2013. 2013:610297. doi: 10.1155/2013/610297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 18.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007;1:54. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39:W316–W322. doi: 10.1093/nar/gkr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. (Database Issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11. doi: 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goeman JJ, Oosting J, Cleton-Jansen AM, Anninga JK, van Houwelingen HC. Testing association of a pathway with survival using gene expression data. Bioinformatics. 2005;21:1950–1957. doi: 10.1093/bioinformatics/bti267. [DOI] [PubMed] [Google Scholar]
- 25.Jensen CH, Krogh TN, Støving RK, Holmskov U, Teisner B. Fetal antigen 1 (FA1), a circulating member of the epidermal growth factor (EGF) superfamily: ELISA development, physiology and metabolism in relation to renal function. Clin Chim Acta. 1997;268:1–20. doi: 10.1016/S0009-8981(97)00152-6. [DOI] [PubMed] [Google Scholar]
- 26.Lanuti M, Liu G, Goodwin JM, Zhai R, Fuchs BC, Asomaning K, Su L, Nishioka NS, Tanabe KK, Christiani DC. A functional epidermal growth factor (EGF) polymorphism, EGF serum levels, and esophageal adenocarcinoma risk and outcome. Clin Cancer Res. 2008;14:3216–3222. doi: 10.1158/1078-0432.CCR-07-4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanabe KK, Lemoine A, Finkelstein DM, Kawasaki H, Fujii T, Chung RT, Lauwers GY, Kulu Y, Muzikansky A, Kuruppu D, et al. Epidermal growth factor gene functional polymorphism and the risk of hepatocellular carcinoma in patients with cirrhosis. JAMA. 2008;299:53–60. doi: 10.1001/jama.2007.65. [DOI] [PubMed] [Google Scholar]
- 28.Marteil G, Gagne JP, Borsuk E, Richard-Parpaillon L, Poirier GG, Kubiak JZ. Proteomics reveals a switch in CDK1-associated proteins upon M-phase exit during the Xenopus laevis oocyte to embryo transition. Int J Biochem Cell Biol. 2012;44:53–64. doi: 10.1016/j.biocel.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 29.Russignan A, Spina C, Tamassia N, Cassaro A, Rigo A, Bagnato A, Rosanò L, Bonalumi A, Gottardi M, Zanatta L, et al. Endothelin-1 receptor blockade as new possible therapeutic approach in multiple myeloma. Br J Haematol. 2017;178:781–793. doi: 10.1111/bjh.14771. [DOI] [PubMed] [Google Scholar]
- 30.Zhu J, Cui L, Wang W, Hang XY, Xu AX, Yang SX, Dou JT, Mu YM, Zhang X, Gao JP. Whole exome sequencing identifies mutation of ENDRA involved in ACTH-independent macronodular adrenal hyperplasia. Fam Cancer. 2013;12:657–667. doi: 10.1007/s10689-013-9642-y. [DOI] [PubMed] [Google Scholar]
- 31.Canossa M, Twiss JL, Verity AN, Shooter EM. p75(NGFR) and TrkA receptors collaborate to rapidly activate a p75(NGFR)-associated protein kinase. EMBO J. 1996;15:3369–3376. doi: 10.1002/j.1460-2075.1996.tb00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soland TM, Brusevold IJ, Koppang HS, Schenck K, Bryne M. Nerve growth factor receptor (p75 NTR) and pattern of invasion predict poor prognosis in oral squamous cell carcinoma. Histopathology. 2008;53:62–72. doi: 10.1111/j.1365-2559.2008.03063.x. [DOI] [PubMed] [Google Scholar]
- 33.Williams JM, Chen GC, Zhu L, Rest RF. Using the yeast two-hybrid system to identify human epithelial cell proteins that bind gonococcal Opa proteins: Intracellular gonococci bind pyruvate kinase via their Opa proteins and require host pyruvate for growth. Mol Microbiol. 1998;27:171–186. doi: 10.1046/j.1365-2958.1998.00670.x. [DOI] [PubMed] [Google Scholar]
- 34.Li H, Zhang J, Lee MJ, Yu GR, Han X, Kim DG. OIP5, a target of miR-15b-5p, regulates hepatocellular carcinoma growth and metastasis through the AKT/mTORC1 and β-catenin signaling pathways. Oncotarget. 2017;8:18129–18144. doi: 10.18632/oncotarget.15185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koinuma J, Akiyama H, Fujita M, Hosokawa M, Tsuchiya E, Kondo S, Nakamura Y, Daigo Y. Characterization of an Opa interacting protein 5 involved in lung and esophageal carcinogenesis. Cancer Sci. 2012;103:577–586. doi: 10.1111/j.1349-7006.2011.02167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li HC, Chen YF, Feng W, Cai H, Mei Y, Jiang YM, Chen T, Xu K, Feng DX. Loss of the Opa interacting protein 5 inhibits breast cancer proliferation through miR-139-5p/NOTCH1 pathway. Gene. 2017;603:1–8. doi: 10.1016/j.gene.2016.11.046. [DOI] [PubMed] [Google Scholar]
- 37.Chun HK, Chung KS, Kim HC, Kang JE, Kang MA, Kim JT, Choi EH, Jung KE, Kim MH, Song EY, et al. OIP5 is a highly expressed potential therapeutic target for colorectal and gastric cancers. BMB Rep. 2010;43:349–354. doi: 10.5483/BMBRep.2010.43.5.349. [DOI] [PubMed] [Google Scholar]
- 38.Wigge PA, Kilmartin JV. The Ndc80p complex from Saccharomyces cerevisiae contains conserved centromere components and has a function in chromosome segregation. J Cell Biol. 2001;152:349–360. doi: 10.1083/jcb.152.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobayashi Y, Takano A, Miyagi Y, Tsuchiya E, Sonoda H, Shimizu T, Okabe H, Tani T, Fujiyama Y, Daigo Y. Cell division cycle-associated protein 1 overexpression is essential for the malignant potential of colorectal cancers. Int J Oncol. 2014;44:69–77. doi: 10.3892/ijo.2013.2177. [DOI] [PubMed] [Google Scholar]
- 40.Dai C, Miao CX, Xu XM, Liu LJ, Gu YF, Zhou D, Chen LS, Lin G, Lu GX. Transcriptional activation of human CDCA8 gene regulated by transcription factor NF-Y in embryonic stem cells and cancer cells. J Biol Chem. 2015;290:22423–22434. doi: 10.1074/jbc.M115.642710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.