

 Metabotropic glutamate type 7 (mGlu7) receptor is a member of the group III family of mGlu receptors.
Metabotropic glutamate type 7 (mGlu7) receptor is a member of the group III family of mGlu receptors.
Abstract
Metabotropic glutamate type 7 (mGlu7) receptor is a member of the group III family of mGlu receptors. It is widely distributed in the central nervous system (CNS) and is preferentially expressed on presynaptic nerve terminals where it is thought to play a critical role in modulating normal neuronal function and synaptic transmission, making it particularly relevant in neuropharmacology. The lack of small-molecule mGlu7 ligands with adequate potency, selectivity and drug-like properties has resulted in difficulties in the preclinical validation of mGlu7 modulation in disease models. In the last decade, allosteric modulators of mGlu7 receptors have emerged as valuable tools with good potency, selectivity and physicochemical properties to study and unleash the therapeutic potential of mGlu7 receptors. This review focusses on the medicinal chemistry of mGlu7 receptor allosteric ligands discovered since 2008.
Introduction
Metabotropic glutamate (mGlu) receptors are class C GPCRs that modulate synaptic transmission upon activation by l-glutamate, the most abundant excitatory neurotransmitter in the central nervous system (CNS) of vertebrates.1,2 To date, eight subtypes have been identified and classified into three groups based on sequence homology and signalling pharmacology: group I mGlu1,5, group II mGlu2,3 and group III mGlu4,6,7,8.3 Group I & II mGlu receptors have been the focus of most drug discovery programs so far, with numerous molecules advancing into clinical trials for neuropathological and psychiatric disorders.4,5 Group III receptors, despite their high therapeutic potential, have remained an area of relatively uncharted territory.6 Of the group III receptors, mGlu7 has attracted a great deal of interest; mGlu7 receptors are widely expressed in the CNS, with the highest density in brain regions associated with reward, cognition and emotion such as in the olfactory bulb, amygdala, nucleus accumbens, hypothalamus, hippocampus and sensory afferent neurons.7,8 mGlu7 receptors show the lowest affinity for glutamate compared to other mGlus, which has raised questions about the nature of its physiological role.9,10 Furthermore, mGlu7 receptor biology is predicted to be distinct from the other group III receptors due to its specific localization on presynaptic glutamatergic and GABAergic synapses.11
The fact that the glutamate binding site is highly conserved across group III receptors has hampered the development of sufficiently subtype-selective orthosteric ligands to elucidate the true contribution of each mGlu receptor subtype to the observed pharmacological effect. In the absence of mGlu7 ligands with good subtype selectivity and drug-like properties, mGlu7 receptor knock-out animals have been traditionally used as tools to investigate its physiological function and have provided valuable insights into the role of mGlu7 in neurological and psychiatric disorders including anxiety,12 depression,13 schizophrenia14 and epilepsy.15 Likewise numerous polymorphisms in the metabotropic glutamate receptor 7 gene (GRM7) have now been linked to autism,16 depression,17 attention deficit hyperactivity disorder (ADHD),18 bipolar disorder19 and schizophrenia.20
A breakthrough in the field came in 2005 when compound 1 (AMN082), the first selective mGlu7 receptor ligand, was reported.21 AMN082 activates mGlu7 receptor binding to an allosteric site in the transmembrane domain with an IC50 of 64 nM without affecting the affinity for orthosteric binders (Fig. 1). AMN082 is orally active and a brain penetrant, and since its discovery, it has been extensively used in vivo to study the role of the mGlu7 receptor, mainly in stress-related CNS disorders.22 However, AMN082 is rapidly metabolised in rat liver microsomes to N-benzhydrylethane-1,2-diamine (Met-1), a compound with significant binding affinity for dopamine, serotonin and norepinephrine transporters.23 This has raised concerns that some of the effects observed with AMN082 may not be purely mGlu7 receptor mediated and it cannot be discarded that other mechanisms could also play a role.24,25
Fig. 1. Structures of mGlu7 receptor allosteric ligands 1 and 2 described prior to 2008.

In 2007, scientists at Banyu Pharmaceutical reported the in vitro pharmacological characterization of a series of isoxazolopyridones, represented by compound 2 (MMPIP), as allosteric mGlu7 receptor antagonists (Fig. 1).26 MMPIP inhibited L-AP4-induced intracellular Ca2+ mobilization in CHO cells expressing the rat mGlu7 receptor with Gα15 (IC50 of 26 nM), but did not displace [3H]LY341495 bound to the mGlu7 receptor in a competition binding study. MMPIP is orally bioavailable and has a brain-to-plasma ratio of 1.0.27 MMPIP was labelled a suitable tool compound that has been used to study the pharmacology of mGlu7 receptors in rodents.28–30 A radioligand, [11C]MMPIP, has been reported and used to image mGlu7 receptors using in vitro autoradiography studies on rat brain sections.31 Recent studies with MMPIP have shown context-dependent pharmacology in cells and also a lack of activity in electrophysiology studies.
Since the discovery of AMN082 and MMPIP, significant progress has been made in the search for novel allosteric ligands of the mGlu7 receptor. This review article focusses on the progress made towards the identification of novel and selective mGlu7 receptor allosteric ligands in the period 2008–2018 with a focus on the medicinal chemistry aspects of the research.
Allosteric agonists
Recently, scientists at Janssen have used a virtual screen with proteochemometric modelling as a hit generation approach to identify allosteric modulators of the mGlu7 receptor.32 Among the virtual hits identified, compound 3 displayed low micromolar agonistic activity (pEC50 = 5.8, Emax = 76%) in CHO cells expressing the human mGlu7 receptor (Fig. 2). Compound 3 also showed agonistic activity at the mGlu3 and mGlu5 receptors with a pEC50 of 6.2 in both cases. The presence of the distal benzhydryl group in 3 shows a resemblance to the structure of AMN082 (1) and compound 3 was selected to perform a close-analogue search using the Janssen compound library with Tanimoto similarity and 2D ECFP6 circular topological fingerprints. The tetrahydroquinoline hit 4 was identified as a micromolar mGlu7 receptor allosteric agonist (pEC50 = 5.8, Emax = 58%) with sufficient chemical attractiveness to start a medicinal chemistry optimisation.33 The benzhydryl analogue 5 showed a close to 1 log unit increase in potency and confirmed the structural similarity of the tetrahydroquinoline series with AMN082. Modelling of compound 5 into a homology model of the mGlu7 receptor resulted in the identification of compound 6 with comparable potency (6: pEC50 = 6.96, Emax = 71% vs. AMN082: pEC50 = 6.8, Emax = 62%) and improved drug-like attributes over AMN082. Compound 6 showed a reduced lipophilicity with a Log P of 3 when compared to a Log P of 5.9 for compound 1. In general, compounds in this tetrahydroquinoline series suffered from high clearance in rat liver microsomes. Only selected examples were moderately stable in human liver microsomes. Although selectivity against other mGlu receptor subtypes was not fully evaluated, the authors suggest the compounds to be selective given the high structural similarity of the series with AMN082 and the shared binding mode.
Fig. 2. Structures and activity data of mGlu7 receptor allosteric agonists 3–6.

Three patent applications describing a series of mGlu7 receptor activators have been published by scientists at Takeda in 2017–2018. In the first patent application, 2,3,4,5-tetrahydro-1,5-benzoxazepines are claimed to activate the mGlu7 receptor with activities in a forskolin-stimulated cAMP production assay in CHO cells expressing the human mGlu7 gene.34 A representative example from this series is compound 7 (Fig. 3) with an mGlu7 EC50 of 1 nM. In the subsequent patent application, 1,4-dihydro-3(2H)-isoquinolinones bearing a carboxamide group at position C1 were reported as mGlu7 receptor modulators.35 The most potent compound described among the 27 examples given is compound 8 with an EC50 of 29.2 nM. No selectivity data was provided for the compounds in these two patent applications.
Fig. 3. Structures and activity data of mGlu7 receptor activators 7–10.

Finally, the third patent application claims chromane derivatives with mGlu7 receptor agonistic activity for the treatment of mGlu7 receptor regulated diseases, disorders or conditions.36 Compounds 9 and 10 are among the most potent examples given, with compound 10 being the most pharmacologically characterised from this chemical series. Thus compound 10 was found to be a full agonist at the human and mouse mGlu7 receptors (human mGlu7 EC50 = 6.9 ± 0.8 nM and mouse mGlu7 EC50 = 1.8 ± 3.5 nM) with no activity at the other group III mouse mGlu receptor subtypes. Compound 10 was classified as an mGlu7 receptor allosteric agonist after testing its activity in the presence of LY341495. Electrophysiology studies in mouse hippocampal slices showed that 10 modulates neurotransmitter release and synaptic activity with a similar profile to the non-selective mGlu7 orthosteric agonist L-AP4.37 The effect of compound 10 was not observed in slices from mGlu7 receptor knockout mice, suggesting that the observations in wild type mice are entirely mediated by the activation of the mGlu7 receptor.
Allosteric antagonists
Since the discovery of MMPIP in 2007 (ref. 25), only one report has been published describing the SAR around an isoxazolopyridone hit 11 (Fig. 4), leading to the identification of MMPIP.26 A HTS identified compound 11 as a 20 nM mGlu7 antagonist with no detectable activity at other mGlu receptor subtypes at 10 μM concentration. Compound 11 showed poor metabolic stability on rat hepatocytes and low aqueous solubility at pH 7.4. The medicinal chemistry strategy to explore hit 11 focussed on reducing its high lipophilicity (clog D7.4 = 3.5) while maintaining the good mGlu7 receptor activity. The strategy followed was based on modifying the substitution at positions 4, 5 and 7 of the isoxazolopyridone core.
Fig. 4. Structures and activity data of mGlu7 receptor activators 11 and 12.

The resulting structure–activity relationships (SARs) showed that alkyl substituents larger than a methyl group at position 5 were detrimental to the activity. Interestingly, a pyridyl-4-yl substituent at position 7 (12) reduced clog D7.4 to 2.3 while retaining acceptable mGlu7 receptor affinity (mGlu7 IC50 = 101 nM). Introduction of a 4-MeO-Ph substituent at position 4 in 12 resulted in MMPIP with a 4-fold increase in potency and a reduced clog D7.4 of 2.4 when compared with the HTS hit 11.
Positive allosteric modulators (PAMs)
The mGlu7 receptor positive allosteric modulator (mGlu7 PAM) avenue is paved with the discoveries of scientists at Vanderbilt University. The Vanderbilt team reported in 2014 the first examples of pan-group III positive allosteric modulators 13 (VU0155094) and 14 (VU0422288) (Fig. 5).38 Compound 13 was discovered in an HTS while looking for mGlu8 receptor PAMs. The compound potentiates mGlu4, 6, 7 and 8 but not the other mGlu receptor subtypes at 10 μM concentration. The compound was also clean in a Eurofins/PanLabs panel of 68 targets at 10 μM, showing only 50% inhibition for the norepinephrine transporter in a radioligand binding assay. Compound 14 was identified within an mGlu4 receptor PAM lead optimization program around a heterobiarylamide scaffold.39 Compound 14 is the most potent pan-group III allosteric modulator reported to date. It shows PAM activity for mGlu4, 7 and receptor subtypes in the range of 100 nM and it has a clean selectivity profile in the Ricerca/Eurofins Lead Profiler panel.
Fig. 5. Structures and activity data of mGlu7 receptor activators 13 and 14.

SAR studies by modification of the distal aromatic aryl groups in compounds 13 and 14 did not identify compounds with either increased activity or selectivity.
Despite their lack of selectivity for the mGlu7 receptor, both compounds have been proven to be valuable tools for ascertaining the role of mGlu7 in the modulation of synaptic transmission in the hippocampus. Thus, electrophysiology studies at Schaffer collateral-CA1 synapses in the hippocampus of adult rats, where mGlu7 is the only functional presynaptic group III mGlu, proved that both 13 and 14 (13: 30 μM and 14: 1 μM concentration) produced relevant and robust effects in modulating synaptic transmission via a presynaptic mechanism.
More recently, a novel mGlu7/8 PAM chemotype based on a series of pyrazolo[1,5-a]pyrimidines has been reported (Fig. 6).40 Compound 15 was selected after an HTS as an interesting hit with selectivity for mGlu7 versus mGlu4 and mGlu8 receptors and excellent CNS penetration in rats (plasma/brain Kp = 1.4, Kp,uu = 1.1). Furthermore, the chemical structure of 15 was amenable for an efficient parallel SAR exploration of the four substituents around the pyrazolopyrimidine central core. Screening of a set of 100 synthesis analogues revealed a steep SAR, with both 5-CH3 and 7-CF3 substituents being essential for retaining mGlu7 receptor PAM activity. A fluorine-walk strategy41 on the aromatic substituent proved to be successful, resulting in compounds 16–18 with sub-micromolar mGlu7 receptor PAM activities. Interestingly, compound 17 (VU6005649) is an mGlu7 receptor preferring PAM with no activity (EC50/IC50 > 10 mM) at mGlu1,2,3,4,5,6 receptors and 4-fold selectivity versus the mGlu8 receptor. Compound 17 was further evaluated in a mouse pharmacokinetic experiment (10 mg kg–1 intraperitoneal administration (i.p.)) where it was found to be CNS penetrant (Kp = 2.1 at 1 h post administration). In a mouse contextual fear conditioning model, compound 17 showed modest but significant efficacy, with pro-cognitive effects on associative learning at a dose of 50 mg kg–1 (i.p.).
Fig. 6. Structures and activity data of mGlu7 receptor PAMs 15–18.

Unfortunately, compound 17 also induced some level of sedation, which could contribute in diminishing mice capacity for associative learning. This sedation effect could be attributed to potential off-target effects. These include activity at neurokinin 1 receptors (NK1), where compound 17 behaves as a functional antagonist with a Ki of 650 nM. Nonetheless, this constituted the first example of mGlu7/8-mediated efficacy in this cognition model. Although the properties of compound 17 may not be optimal, it holds promise as an mGlu7/8 receptor PAM tool compound with CNS penetration and utility for interrogating mGlu7 and mGlu8 receptors in vivo.
Negative allosteric modulators (NAMs)
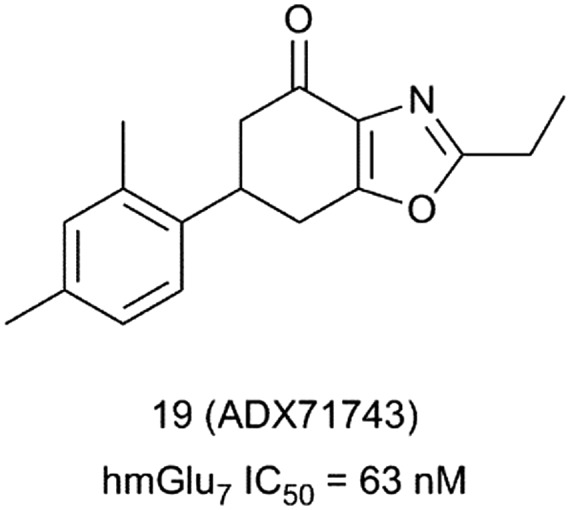
The first potent and selective mGlu7 receptor NAM was reported by Addex Therapeutics in 2013.42 Compound 19 (ADX71743) was developed from a hit molecule identified in a cell-based fluorescent Ca2+ mobilization assay HTS (Fig. 7). Compound 19 showed an IC50 of 63 nM at the human mGlu7 receptor. NAM activity was confirmed in Schild plot experiments, where 19 induced a concentration-dependent rightward shift of the L-AP4 concentration–response curve together with a decrease of L-AP4 efficacy. Compound 19 was found to be selective up to 30 μM concentration against the other members of the mGlu family and against the Cerep P27 panel.
Fig. 7. Structure and activity data of mGlu7 receptor NAM 19.
Despite being rapidly cleared, 19 showed good plasma exposures in rodents after subcutaneous administration with meaningful compound levels detected in cerebrospinal fluid at the high doses tested. The compound was characterised in several rodent behavioural models of psychosis, anxiety and depression. Compound 19 was active in the marble burying and elevated plus maze tests in mice, suggesting an anxiolytic-like effect after mGlu7 receptor inhibition.
In addition, compound 19 showed antipsychotic activity in the modified forced swim test and reversed MK-801-induced deficits in a social interaction test.43
In 2017, scientists at Vanderbilt University reported the identification of novel selective mGlu7 receptor NAMs after an HTS campaign.44 The screening found 156 compounds with antagonist/NAM activity, among which compound 20 was selected as an attractive hit with an IC50 of 5.8 μM at the mGlu7 receptor. 20 showed no affinity for mGlu4 and mGlu7 receptors up to 30 μM concentration (Fig. 8).
Fig. 8. Structures and activity data of mGlu7 receptor NAMs 20 and 21.

Medicinal chemistry optimization of compound 20 proved to be challenging. Analysis of ∼100 synthesis analogues showed a steep and flat SAR. The 1,2,4-triazole and meta-methoxy substituents were found to be essential for retaining mGlu7 receptor NAM activity. The only satisfactory replacement found for the chlorine atom was a trifluoromethoxy group. Compound 21 (VU6010608) emerged as the best mGlu7 receptor NAM from this exploration. Although it showed increased mGlu7 affinity and good selectivity over the other mGlu receptor subtypes, it suffered from poor pharmacokinetic properties.
Compound 21 was found to be rapidly cleared (CLp = 64.2 mL min–1 kg–1; t1/2 = 1.73 h) in rats with acceptable oral bioavailability (F (%) = 18.9). Disappointingly, mice free brain concentrations are below the in vitro mGlu7 receptor IC50 (59 nM vs. 759 nM) at a dose of 100 mg kg–1 (i.p.). This makes the compound unsuitable for in vivo experiments if efficacy is by free brain concentration. Nonetheless, 21 is a good tool compound for in vitro studies. In electrophysiology experiments, compound 21 showed a robust effect in blocking long-term potentiation at SC-CA1 brain slices after high frequency stimulation.
Conclusions
Considerable progress has been made over the last decade in the development of subtype-selective mGlu7 ligands. Allosteric binding sites are under less evolutionary pressure for conservation than orthosteric sites across a receptor family. Targeting these allosteric pockets in the mGlu7 receptor has been a key facet in addressing the selectivity issues encountered with orthosteric group III ligands. Although a good number of sub-type selective allosteric ligands of mGlu7 have been identified, allowing for an effective in vitro interrogation of the receptor, these tools still lack optimal pharmacokinetic properties for a thorough and unequivocal pharmacological validation of mGlu7 receptor modulation in preclinical animal disease models. The growing pool of allosteric ligands available to date will spur further research on mGlu7 receptors in the years to come and should serve as the launching platform to develop more potent and selective ligands suitable for a long-awaited in vivo preclinical target validation, and ultimately advancing a selective mGlu7 receptor ligand into clinical trials.
Conflicts of interest
There are no conflicts to declare.
Biographies

Henar Vázquez-Villa
Henar Vázquez-Villa obtained her Ph.D. in Chemistry in February 2005 from Universidad de Oviedo (Spain), under the guidance of Prof. J. Barluenga and Prof. J. M. González. She then carried out postdoctoral work with Prof. E. M. Carreira's group at ETH-Zürich (Switzerland). In 2007, she moved to the Faculty of Chemistry of Universidad de Alcalá (Spain), and a year later, she joined the Laboratory of Medicinal Chemistry of Universidad Complutense de Madrid, under the direction of Prof. M. L. López Rodríguez, to work in medicinal chemistry. Her current research interests include allosteric modulation of GPCRs, inhibition of the bacterial cell division protein FtsZ, and phenotypic drug discovery.
Andrés A. Trabanco was awarded his PhD degree by Universidad de Oviedo-Spain in 1999. He then moved to Imperial College of Science Technology and Medicine, London, UK for his postdoctoral studies. Andrés joined the Neuroscience MedChem Team at Janssen in 2000. Since 2008, he has led the Neuroscience Hit Generation Team in the search for novel hits/leads within the neuroscience therapeutic area. Andrés has been a chemistry leader and an active team member of several programs in the areas of schizophrenia, depression, anxiety, and cognition/Alzheimer's disease, which have delivered various clinical candidates. He is an inventor on 51 patent applications and an author of over 70 scientific publications.
References
- Conn P. J., Pin J.-P. Annu. Rev. Pharmacol. Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Niswender C. M., Conn P. J. Annu. Rev. Pharmacol. Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew J. N. C., Kemp J. A. Psychopharmacology. 2005;179:4–29. doi: 10.1007/s00213-005-2200-z. [DOI] [PubMed] [Google Scholar]
- Owen D. R. ACS Chem. Neurosci. 2011;2:394–401. doi: 10.1021/cn2000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C., Ma S. Med. Chem. Commun. 2017;8:501–515. doi: 10.1039/c6md00612d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavreysen H., Dautzenberg F. M. Curr. Med. Chem. 2008;15:671–684. doi: 10.2174/092986708783885246. [DOI] [PubMed] [Google Scholar]
- Kinzie J. M., Saugstad J. A., Westbrook G. L., Segerson T. P. Neuroscience. 1995;69:167–176. doi: 10.1016/0306-4522(95)00244-d. [DOI] [PubMed] [Google Scholar]
- Makoff A., Pilling C., Harrington K., Emson P. Brain Res. Mol. Brain Res. 1996;40:165–170. doi: 10.1016/0169-328x(96)00110-6. [DOI] [PubMed] [Google Scholar]
- Wright R. A., Arnold M. B., Wheeler W. J., Ornstein P. L., Schoepp D. D. Naunyn-Schmiedeberg's Arch. Pharmacol. 2000;362:546–554. doi: 10.1007/s002100000305. [DOI] [PubMed] [Google Scholar]
- Kammermeier P. J. BMC Neurosci. 2015;16:17. doi: 10.1186/s12868-015-0154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalezios Y., Lujan R., Shigemoto R., Roberts J. D., Somogyi P. Cereb. Cortex. 2002;12:961–974. doi: 10.1093/cercor/12.9.961. [DOI] [PubMed] [Google Scholar]
- Callaerts-Vegh Z., Beckers T., Ball S. M., Baeyens F., Callaerts P. F., Cryan J. F., Molnar E., D'Hooge R. J. Neurosci. 2006;26:6573–6582. doi: 10.1523/JNEUROSCI.1497-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan J. F., Kelly P. H., Neijt H. C., Sansig G., Flor P. J., van der Putten H. Eur. J. Neurosci. 2003;17:2409–2417. doi: 10.1046/j.1460-9568.2003.02667.x. [DOI] [PubMed] [Google Scholar]
- Hovelsø N., Sotty F., Montezinho L. P., Pinheiro P. S., Herrik K. F., Mrøk A. Curr. Neuropharmacol. 2012;10:12–48. doi: 10.2174/157015912799362805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansig G., Bushell T. J., Clarke V. R., Rozov A., Burnashev N., Portet C., Gasparini F., Schmutz M., Klebs K., Shigemoto R., Flor P. J., Kuhn R., Knoepfel T., Schroeder M., Hampson D. R., Collett V. J., Zhang C., Duvoisin R. M., Collingridge G. L., van der Putten H. J. Neurosci. 2001;21:8734–8745. doi: 10.1523/JNEUROSCI.21-22-08734.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhang Y., Zhao D., Dong R., Yang X., Tammimies K., Uddin M., Scherer S. W., Gai Z. Am. J. Med. Genet., Part B. 2015;168:258–264. doi: 10.1002/ajmg.b.32306. [DOI] [PubMed] [Google Scholar]
- Breen G., Webb B. T., Butler A. W., van den Oord E. J., Tozzi F., Craddock N., Gill M., Korszun A., Maier W., Middleton L., Mors O., Owen M. J., Cohen-Woods S., Perry J., Galwey N. W., Upmanyu R., Craig I., Lewis C. M., Ng M., Brewster S., Preisig M., Rietschel M., Jones L., Knight J., Rice J., Muglia P., Farmer A. E., McGuffin P. Am. J. Psychiatry. 2011;168:840–847. doi: 10.1176/appi.ajp.2011.10091342. [DOI] [PubMed] [Google Scholar]
- Mick E., Neale B., Middleton F. A., McGough J. J., Faraone S. V. Am. J. Med. Genet., Part B. 2008;147:1412–1418. doi: 10.1002/ajmg.b.30865. [DOI] [PubMed] [Google Scholar]
- Alliey-Rodriguez N., Zhang D., Badner J. A., Lahey B. B., Zhang X., Dinwiddie S., Romanos B., Plenys N., Liu C., Gershon E. S. Psychiatr. Genet. 2011;21:190–194. doi: 10.1097/YPG.0b013e3283457a31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuki T., Koga M., Ishigure H., Horiuchi Y., Arai M., Niizato K., Itokawa M., Inada T., Iwata N., Iritani S., Ozaki N., Kunugi H., Ujike H., Watanabe Y., Someya T., Arinami T. Schizophr. Res. 2008;101:9–16. doi: 10.1016/j.schres.2008.01.027. [DOI] [PubMed] [Google Scholar]
- Mitsukawa K., Yamamoto R., Ofner S., Nozulak J., Pescott O., Lukic S., Stoehr N., Mombereau C., Kuhn R., McAllister K. H., van der Putten H., Cryan J. F., Flor P. J. Proc. Natl. Acad. Sci. U. S. A. 2005;102:18712–18717. doi: 10.1073/pnas.0508063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. J., Niswender C. M. Proc. Natl. Acad. Sci. U. S. A. 2006;103:251–252. doi: 10.1073/pnas.0510051103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo S. J. S., Leonard S. K., Gilbert A., Dollings P., Smith D. L., Zhang M.-Y., Di L., Platt B. J., Neal S., Dwyer J. M., Bender C. N., Zhang J., Lock T., Kowal D., Kramer A., Randall A., Huselton C., Vishwanathan K., Tse S. Y., Butera J., Ring R. H., Rosenzweig-Lipson S., Hughes Z. A., Dunlop J. J. Pharmacol. Exp. Ther. 2011;338:345–352. doi: 10.1124/jpet.110.177378. [DOI] [PubMed] [Google Scholar]
- Ahnaou A., Raeyemaekers L., Huysmans H., Drinkenburg W. H. I. M. Behav. Brain Res. 2016;311:287–297. doi: 10.1016/j.bbr.2016.05.035. [DOI] [PubMed] [Google Scholar]
- Suzuki G., Tsukamoto N., Fushiki H., Kawagishi A., Nakamura M., Kurihara H., Mitsuya M., Ohkubo M., Ohta H. J. Pharmacol. Exp. Ther. 2007;323:147–156. doi: 10.1124/jpet.107.124701. [DOI] [PubMed] [Google Scholar]
- Nakamura M., Kurihara H., Suzuki G., Mitsuya M., Ohkubo M., Ohta H. Bioorg. Med. Chem. Lett. 2010;20:726–729. doi: 10.1016/j.bmcl.2009.11.070. [DOI] [PubMed] [Google Scholar]
- Hikichi H., Murai T., Okuda S., Maehara S., Satow A., Ise S., Nishino M., Suzuki G., Takehana H., Hata M., Ohta H. Eur. J. Pharmacol. 2010;639:106–114. doi: 10.1016/j.ejphar.2009.08.047. [DOI] [PubMed] [Google Scholar]
- Bahi A., Fizia K., Dietz M., Gasparini F., Flor P. J. Addict. Biol. 2012;101:193–200. doi: 10.1111/j.1369-1600.2010.00310.x. [DOI] [PubMed] [Google Scholar]
- Palazzo E., Marabese I., Luongo L., Boccella S., Bellini G., Giordano M. E., Rossi F., Scafuro M., de Novellis V., Maione S. Mol. Pain. 2013;9(44):1–12. doi: 10.1186/1744-8069-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo E., Romano R., Luongo L., Boccella S., De Gregorio D., Giordano M. E., Rossi F., Marabese I., Scafuro M. A., de Novellis V., Maione S. Pain. 2015;156:1060–1073. doi: 10.1097/j.pain.0000000000000150. [DOI] [PubMed] [Google Scholar]
- Yamasaki T., Kumata K., Yui J., Fujinaga M., Furutsuka K., Hatori A., Xie L., Ogawa M., Nengaki N., Kawamura K., Zhang M.-R. EJNMMI Res. 2013;3(54):1–10. doi: 10.1186/2191-219X-3-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresadern G., Trabanco A. A., Pérez-Benito L., Overington J. P., van Vlijmen H. T. W., van Westen G. J. P. J. Chem. Inf. Model. 2017;57:2976–2985. doi: 10.1021/acs.jcim.7b00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid J. M., Lavreysen H., Tresadern G., Pérez-Benito L., Tovar F., Fontana A., Trabanco A. A. ACS Chem. Neurosci. doi: 10.1021/acschemneuro.8b00331. [DOI] [PubMed] [Google Scholar]
- Mack S., Teall M. and Parmar A., WO Pat., 222083, 2017.
- Teall M., Stephenson A. and White K., WO Pat., 079862, 2018.
- Teall M., White K. and Mack S., WO Pat., 092921, 2018.
- Martín R., Ferrero J. J., Collado-Alsina A., Aguado C., Lujan R., Torres M., Sánchez-Prieto J. J. Physiol. 2018;596:921–940. doi: 10.1113/JP275371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalan-Sakrikar N., Field J. R., Klar R., Mattmann M. E., Gregory K. J., Zamorano R., Engers D. W., Bollinger S. R., Weaver C. D., Days E. L., Lewis L. M., Utley T. J., Hurtado M., Rigault D., Acher F., Walker A. G., Melancon B. J., Wood M. R., Lindsley C. W., Conn P. J., Xiang Z., Hopkins C. R., Niswender C. M. ACS Chem. Neurosci. 2014;5:1221–1237. doi: 10.1021/cn500153z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engers D. W., Niswender C. M., Weaver C. D., Jadhav S., Menon U. N., Zamorano R., Conn P. J., Lindsley C. W., Hopkins C. R. J. Med. Chem. 2009;52:4115–4118. doi: 10.1021/jm9005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe M., Seto M., Gogliotti R. G., Loch M. T., Bollinger K. A., Chang S., Engelberg E. M., Luscombe V. B., Harp J. M., Bubser M., Engers D. W., Jones C. K., Rodriguez A. L., Blobaum A. L., Conn P. J., Niswender C. M., Lindsley C. W. ACS Med. Chem. Lett. 2017;8:1110–1115. doi: 10.1021/acsmedchemlett.7b00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. J., Lindsley C. W., Meiler J., Niswender C. M. Nat. Rev. Drug Discovery. 2014;13:692–706. doi: 10.1038/nrd4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinichev M., Rouillier M., Girard F., Royer-Urios I., Bournique B., Finn T., Charvin D., Campo B., Le Poul E., Mutel V., Poli S., Neale S. A., Salt T. E., Lütjens R. J. Pharmacol. Exp. Ther. 2013;334:624–636. doi: 10.1124/jpet.112.200915. [DOI] [PubMed] [Google Scholar]
- Cieslik P., Wozniak M., Kaczorowska K., Branski P., Burnat G., Chocyk A., Bobula B., Gruca P., Litwa E., Palucha-Poniewiera A., Wasik A., Pilc A., Wieronska J. Front. Mol. Neurosci. doi: 10.3389/fnmol.2018.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed C. W., McGowan K. M., Spearing P. K., Stansley B. J., Roenfanz H. F., Engers D. W., Rodriguez A. L., Engelberg E. M., Luscombe V. B., Loch M. T., Remke D. H., Rook J. M., Blobaum A. L., Conn P. J., Niswender C. M., Lindsley C. W. ACS Med. Chem. Lett. 2017;8:1326. doi: 10.1021/acsmedchemlett.7b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]