

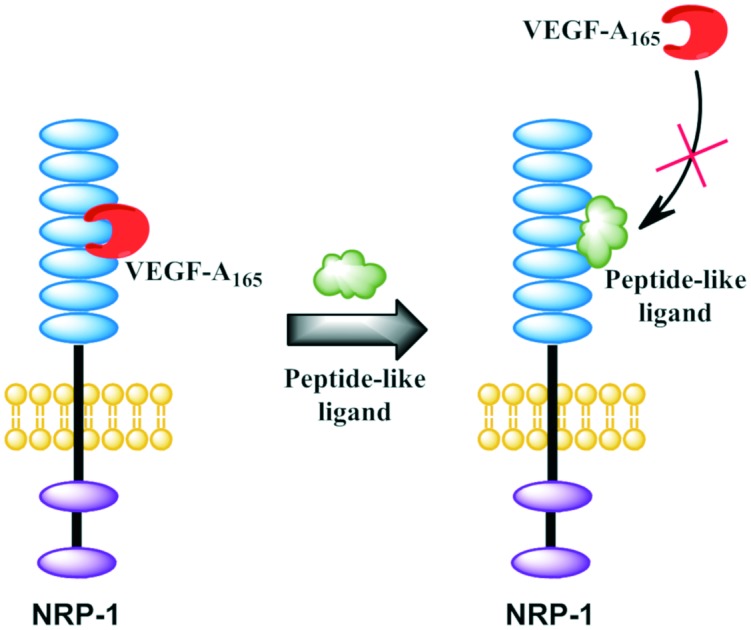
 We report a SAR study on branched peptide-like ligands, which gives an important insight into structural requirements for VEGF-A165/NRP-1 complex inhibitors.
We report a SAR study on branched peptide-like ligands, which gives an important insight into structural requirements for VEGF-A165/NRP-1 complex inhibitors.
Abstract
Many reports have suggested that NRP-1 acts as a co-receptor for VEGF-A165 and boosts tumour growth and metastasis. This NRP-1, due to its important role in tumour progression, triggered interest in the design of new molecules able to significantly inhibit NRP-1/VEGF-A165 interaction to suppress pathological angiogenesis. Our previous SAR studies of compounds, showing affinity for NRP-1, led us to develop branched peptides with general formula Lys(hArg)–AA2–AA3–Arg. Here, three series of analogues were synthesized, in which the middle fragment (AA2 and/or AA3) of initial sequences was substituted with unnatural Pro analogues with different rigidities and ring sizes. The synthesized compounds were screened for VEGF-A165 inhibitory activity on an improved assay (ELISA), which was selected based on our comparative inhibition study of the parent compounds, indicating that the method with chemiluminescence detection gives more accurate data. The results of affinity for NRP-1 and enzymatic stability of newly obtained compounds enabled the selection of new structures, showing a 2 and 4-fold lower IC50 value compared to parent peptides.
Introduction
Neuropilin-1 (NRP-1) belongs to the neuropilin family of relevant cell surface receptors, which are engaged in multiple important cellular signalling cascades. NRP-1 is a co-receptor for tyrosine kinase receptor VEGF-R2 (Flk-2)1,2 and its signalling is critical for VEGF-A/VEGF-R2-mediated angiogenesis,3 a complex biological process, fundamental in the transition of tumours from a benign to a malignant state. NRP-1 has five structured extracellular domains.4,5 Multiple reports indicate that a and b domains are essential for its ligand binding, including binding to vascular endothelial growth factor splice variant VEGF-A165.6–8 Evidence that NRP-1 might display separate functions, especially in cancer, through mechanisms that do not involve other receptors, is also described.9–13 NRP-1 may also interact with many other ligands, including Semaphorin class 3 (Sema3) and transforming growth factor β (TGF-β), which indicates the multiple functions of this receptor.4,14–17
NRP-1 directly binds the exon-8-encoded region of VEGF-A165via its b1b2 subdomains (FV/VIII coagulation factor domains). Heparin-mediated VEGF-R2/NRP-1 complex formation depends on heparin-binding regions located in both VEGF-A165 and NRP-1.18–20 Structural studies have revealed the crucial role of an interloop cleft at the b1 domain of human NRP-1, which forms a core conserved binding pocket.20 The binding pocket is specific for ligands with a C-terminal Arg, where the C-terminus and side chain (guanidine group) interact with the L3 and L5 loop of NRP-1.21
A number of anti-angiogenic strategies connected with the VEGF signalling pathway are used in current clinical treatment or in trial phase, but they either cause adverse effects or are not specific.22–24 Biologically active peptides are often considered as leading compounds in the drug development process. The requirement for the presence of the Lys/Arg–Xaa–Xbb–Arg/Lys motif at the C-terminus of peptides that bind to NRP-1 was described as the so-called C-end rule (CendR).25,26 The properties of the NRP-1 complex with various CendR peptides (e.g. RPAR, DKPPR, TKPRR, CDKPRR) have been intensively studied.27–31
Heptapeptide Ala–Thr–Trp–Leu–Pro–Pro–Arg (A7R) selectively binds to NRP-1 and exerts inhibitory effects on pro-metastatic human breast cancer growth.32–36 Based on A7R, we have recently developed shorter peptides which exhibit a significant VEGF-A165/NRP-1 binding inhibitory effect.31,37–40 The most active among them have the general sequence Lys(hArg)–AA2–AA3–Arg, where AA2 and/or AA3 are Pro residues and the ε-amine group of Lys forms an amide bond with homoarginine (hArg) carboxyl group.40 Pro is a unique amino acid, often considered to induce cis conformation in bioactive peptides. Additionally, peptides with Pro in the second position are susceptible to proteolitic degradation by dipeptidyl peptidase IV (DPP IV).
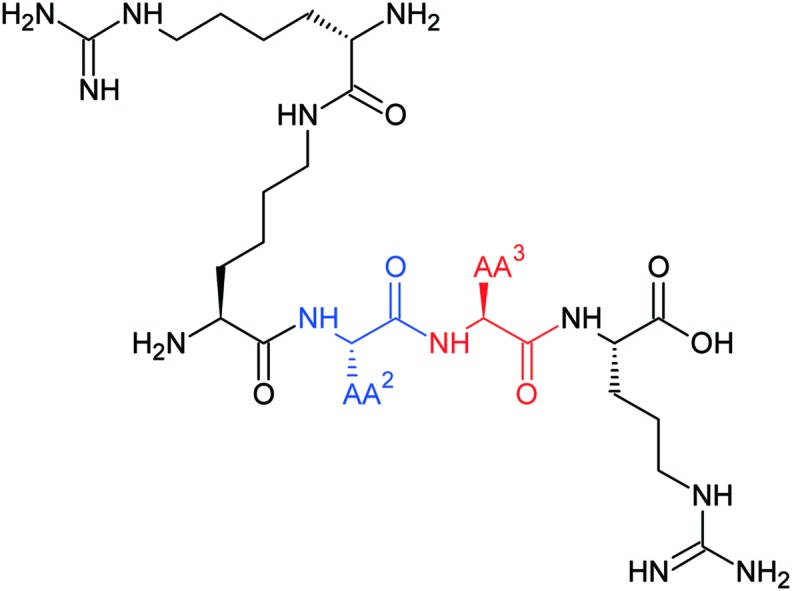
The previously obtained molecular dynamics simulation results suggest that our branched peptidomimetics may interact with NRP-1 by the Lys (hArg) fragment.40 We suspected that Pro might play a role in proper targeting of the N-terminal branching, limiting molecular lability. Here, we present affinity investigation of analogues derived from three previously described, promising peptides (Lys(hArg)–Pro–Pro–Arg, Lys(hArg)–Dab–Pro–Arg and Lys(hArg)–Pro–Ala–Arg),40 in which the middle fragment (AA2 and/or AA3) of initial sequences was substituted with unnatural Pro analogues with different rigidities and ring sizes.
Results and discussion
Design rationale
Proline is a unique proteinogenic amino acid containing a secondary amino group. Moreover, the characteristic cyclic structure of the Pro side chain gives it a unique configurational rigidity, compared to other proteinogenic amino acids. Pro plays an exceptional role in the structure of peptides and proteins through the conformational constraint introduced by its cyclic structure.
Pro probably plays a large role in targeting the N-terminal Lys(hArg) branched fragment of peptides, which allows them to interact with the NRP-1 receptor. Therefore, we decided to check how the size of the ring, its rigidness and possible substituents affect the activity of parent sequences: Lys(hArg)–Pro–Pro–Arg (2), Lys(hArg)–Dab–Pro–Arg (3) and Lys(hArg)–Pro–Ala–Arg (4). Three series of analogues were synthesized, in which Pro was substituted with its appropriate mimetics (Fig. 1).
Fig. 1. Proline mimetics used to optimize the sequence of compounds 2, 3 and 4.

First, we used trans-4-hydroxyproline (Hyp), which has a pyrrolidine ring with a hydroxyl-substituted group. Typically, the replacement of Pro in the peptide sequence on Hyp causes an increase enzymatic stability of the obtained compounds.41–43 In our research, an additional hydroxyl group in the pyrrolidine ring was intended to potentially form additional bonds with amino acid residues on the receptor surface. Another applied Pro mimetic was azetidine-2-carboxylic acid (Aze), which is a non-proteinogenic amino acid and has a 4-membered ring. Aze and Pro, due to the difference in ring size, vary in torsion angles and directions of turnover. Thus, peptides in which Pro has been substituted by Aze have a different tendency to cis-trans isomerization of the peptide bond.44 It was found that Aze containing peptides are more likely to adopt a cis conformation than those containing Pro.45,46 This effect is reasonably correlated with the lower elasticity of the 4-membered azetidine ring compared to the pyrrolidine ring. To obtain conformational rigidity of the peptide-like compounds we also used 3,4-dehydroproline (ΔPro). Compared to Pro, this amino acid has an almost flat and less elastic ring, which increases the population of cis conformers around the tertiary CO–N bond.47 ΔPro was previously used successfully for the synthesis of oxytocin,48 vasopressin,49 and even tuftsin50 analogues. Moreover, there was a possibility that the presence of a double bond will stabilize the peptide-like molecule complex with NRP-1 through π–π interaction, which was previously done for opioid peptidomimetics.51
We also examined the effect of replacing Pro with a larger and less rigid ring. For this purpose, we used piperazine-2-carboxylic acid (Piz).52 The 6-membered heterocyclic piperazine ring is a widespread structural motif in drug chemistry.53 We also used extended Pro analogues with fused rings – octahydroindole-2-carboxylic acid (Oic) and 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (Tic). Both the octahydroindole and the tetrahydroisoquinoline rings induce the specific structure of the peptide chain.54,55 Furthermore, the rigid aromatic ring of Tic is capable of π–π interaction with other aromatic rings, as well as π-cation interactions with positively charged protonated amino groups of Lys or Arg.56 All designed peptide-like structures are presented in Table 1.
Table 1. The general structure of designed peptide-like ligands. Fragments of parent peptides that are appropriate for a given series of compounds are underlined.
| |||||||
| N-Terminus | Compound | AA2–AA3 fragment (series I) | Compound | AA2–AA3 fragment (series II) | Compound | AA2–AA3 fragment (series III) | C-Terminus |
| H2N-Lys(hArg)- | 2 | P[combining low line]r[combining low line]o[combining low line]–P[combining low line]r[combining low line]o[combining low line] | 3 | Dab–P[combining low line]r[combining low line]o[combining low line] | 4 | P[combining low line]r[combining low line]o[combining low line]–Ala | –Arg–OH |
| 5 | Hyp–Hyp | 11 | Dab–Hyp | 17 | Hyp–Ala | ||
| 6 | Aze–Aze | 12 | Dab–Aze | 18 | Aze–Ala | ||
| 7 | ΔPro–ΔPro | 13 | Dab–ΔPro | 19 | ΔPro–Ala | ||
| 8 | Oic–Oic | 14 | Dab–Oic | 20 | Oic–Ala | ||
| 9 | Piz–Piz | 15 | Dab–Piz | 21 | Piz–Ala | ||
| 10 | Tic–Tic | 16 | Dab–Tic | 22 | Tic–Ala | ||
Comparison of assays for peptide binding studies on NRP-1
Affinity of peptides was first assessed in an assay of specific, high-affinity human (bt)-VEGF-A165 binding to recombinant rat NRP-1/Fc chimera.31,37–40,57 We observed 61% inhibition of rat NRP-1/VEGF-A165 binding with A7R peptide (1) at 10 μM, while Lys(hArg)–AA2–AA3–Arg branched peptides exerted more than 90% of inhibition (Table 2). Furthermore, values of inhibition decreased slower with compound concentration in the case of peptides 3 and 4. It was already shown that IC50 values of presented sequences with Lys(hArg) at the N-terminus were improved compared to A7R.40
Table 2. Inhibitory effects and IC50 of peptide binding rrNRP-1 (colorimetric detection) e .
| Compound | AA2–AA3 fragment | Inhibition of NRP-1/VEGF-A165 binding [%] |
IC50 [μM] | ||||
| 10 μM | 3 μM | 1 μM | 0.3 μM | 0.1 μM | |||
| 1 | n/a | 61.0 ± 0.4 | 38.1 ± 3.7 | 24.4 ± 3.5 | 14.6 ± 2.3 | 2.9 ± 2.1 | 5.9 ± 0.7 a |
| 2 | Pro–Pro | 92.2 ± 0.8 | 79.0 ± 1.5 | 41.4 ± 0.0 | 4.7 ± 0.1 | — | 1.0 ± 0.3 b |
| 3 | Dab–Pro | 99.6 ± 0.3 | 92.8 ± 1.1 | 83.7 ± 0.2 | 59.6 ± 0.1 | 35.8 ± 3.2 | 0.2 ± 0.4 c |
| 4 | Pro–Ala | 94.4 ± 0.4 | 89.4 ± 0.8 | 77.5 ± 0.1 | 54.2 ± 0.3 | 26.3 ± 2.1 | 0.3 ± 0.1 d |
alog IC50 = –5.23 ± 0.07.
blog IC50 = –5.99 ± 0.06.
clog IC50 = –6.70 ± 0.05.
dlog IC50 = –6.53 ± 0.05; R2 ranged between 0.98–0.99. Results are presented as the mean ± SEM. n/a means not applicable.
eA part of data was already published.40
We next assessed the second assay used in NRP-1/VEGF-A165 research.28,58,59 Despite the ostensible similarity between methods, we observed a great decrease in VEGF-A165 inhibition (Table 3). The affinity to human NRP-1 in this method is 2–2.5 times lower at 10 μM. The difference between the results of IC50 in both methods is significant.
Table 3. Inhibitory effects and IC50 of peptide binding rhNRP-1 (chemiluminescence detection).
| Compound | AA2–AA3 fragment | 100 μM | 50 μM | Inhibition of NRP-1/VEGF-A165 binding [%] |
IC50 [μM] | ||||
| 25 μM | 10 μM | 5 μM | 2.5 μM | 1 μM | |||||
| 1 | n/a | 58.2 ± 1.2 | 43.8 ± 1.3 | 35.0 ± 2.6 | 30.8 ± 3.8 | — | — | — | 84.1 ± 2.9 a |
| 2 | Pro–Pro | 77.0 ± 1.0 | 67.9 ± 1.4 | 54.1 ± 1.0 | 37.1 ± 1.0 | — | — | — | 20.9 ± 0.3 b |
| 3 | Dab–Pro | 79.8 ± 0.8 | 76.6 ± 0.9 | 70.4 ± 0.5 | 58.6 ± 1.4 | 43.7 ± 0.7 | 31.4 ± 0.5 | 13.8 ± 0.3 | 8.9 ± 0.8 c |
| 4 | Pro–Ala | 70.7 ± 0.5 | 62.1 ± 0.3 | 50.3 ± 1.0 | 35.5 ± 2.0 | — | — | — | 23.4 ± 7.9 d |
alog IC50 = –3.08 ± 1.18.
blog IC50 = –4.68 ± 0.04.
clog IC50 = –5.05 ± 0.13.
dlog IC50 = –4.63 ± 0.12; R2 ranged between 0.96–0.99. Results are presented as the mean ± SEM. n/a means not applicable.
Comparison of results acquired from two NRP-1 binding assays confirms that Lys(hArg)–AA2–AA3–Arg peptides show higher affinity for NRP-1 than the A7R peptide in both methods. We have also examined compound 2 affinity dependence on receptor species, rat or human, using the method with chemiluminescence detection. IC50 obtained for recombinant rat NRP-1 was only slightly lower (14.2 ± 0.8 μM) compared to human protein (20.9 ± 0.3 μM). These results of affinity evaluation suggest that receptor origin did not affect significantly the ligand affinity. Rat NRP-1 sequence shows a high percentage of homology to the sequence of the human receptor (93%). However, we suspect that a crucial difference between VEGF-A165/NRP-1 binding inhibition may have two main reasons. Firstly, the human NRP-1 peptide binding assay with chemiluminescence detection is a much more sensitive assay, compared to the rat NRP-1 assay with colorimetric detection. The used reagent based on luminol has lower detection limit than the colorimetric test with an ABTS substrate, while the amount of the receptor used for coating is also lower. Secondly, in the colorimetric assay anti-IgG was used to coat wells and receptors bound to Fab through its Fc fragment. Next, rat NRP-1 was added simultaneously with VEGF-A165 and the peptide. After incubation, excess reagents were removed during washing, including bound with the ligand and unbound rat NRP-1, thus making the colorimetric method not quantitative. A simplified scheme of assay procedure is illustrated in Fig. 2.
Fig. 2. Simplified scheme of assay procedure: A. colorimetric B. chemiluminescence.

Radioligand binding methods are the most sensitive and robust techniques to detect low levels of radioactively labelled ligands. Thus, the determination of ligand binding sites and affinity is very precise. High cost and hazards of handling elevated high levels of radioactivity are disadvantages. In the assay with iodinated protein, A7R inhibited [125I]-VEGF-A165 binding to NRP-1 in a concentration-dependent manner with an IC50 of 60 μM.34 This is similar to the result obtained using the assay with chemiluminescence detection for A7R (IC50 = 84 μM). IC50 calculated for the same peptide after the assay with colorimetric detection (IC50 = 6 μM) proved that this method is incomparable with other techniques and gives less reliable results.
Peptide-like ligand binding studies on NRP-1
The first synthesized series of compounds were analogues of branched peptide 2 (IC50 = 20.9 μM) (Table 4). The results obtained in the inhibition assay show that the hydroxyl group, attached to the 5-membered pyrrolidine ring of Hyp, does not affect the binding of compound 5 to the receptor (IC50 = 21.3 μM). Therefore, probably no additional interactions between a hydroxyl group present at the pyrrolidone ring and the surface of the protein have occurred. The use of a more rigid 4-membered ring of Aze in compound 6 reduced more than twice the ability of the molecule to inhibit the binding of VEGF-A165 to NRP-1 (IC50 = 48.6 μM). This is most likely due to changes in the geometry of the molecule, which reduced the ability of the C-terminal fragment Lys(hArg) to interact with the surface of the receptor. The analogue 7, in which the Pro–Pro sequence was changed to ΔPro–ΔPro, showed slightly better affinity for NRP-1 (IC50 = 14.0 μM) than the native sequence (IC50 = 20.9 μM). Pro and ΔPro have similar properties, but improving rigidness of the pyrrolidine ring by introducing a double bond, may facilitate optimal geometry and favour conformations that allow more receptor–ligand interactions. Peptide-like ligand 8, containing the Oic–Oic fragment in the central part of the chain, proved to be the most promising compound in this series. The additional 6-membered cyclohexyl ring attached to the pyrrolidine ring positively influenced the affinity of the molecule, reducing the IC50 by almost 3-fold (IC50 = 7.2 μM) in relation to the starting sequence. Compound 9 with a 6-membered piperazine ring showed only a small reduction in affinity for NRP-1 compared to 7. The use of Tic in analogue 10 as a Pro mimetic resulted in loss of inhibitory capacity (IC50 > 100 μM). The decrease in affinity for the NRP-1 receptor for compounds derived from sequence 2, depending on the Pro analogue used, can be ranked as follows: Oic > ΔPro > Piz > Pro ≈ Hyp > Aze ≫ Tic.
Table 4. Inhibitory effects and IC50 of series I peptide-like ligand binding rhNRP-1 (chemiluminescence detection).
| Compound | AA2–AA3 fragment | Inhibition of NRP-1/VEGF-A165 binding [%] |
IC50 [μM] | ||||||
| 100 μM | 50 μM | 25 μM | 10 μM | 5 μM | 2.5 μM | 1 μM | |||
| 2 | Pro–Pro | 77.0 ± 1.0 | 67.9 ± 1.4 | 54.1 ± 1.0 | 37.1 ± 1.0 | — | — | — | 20.9 ± 0.3 40 |
| 5 | Hyp–Hyp | 71.7 ± 0.6 | 63.1 ± 0.2 | 56.8 ± 0.7 | 36.7 ± 0.6 | — | — | — | 21.3 ± 0.6 a |
| 6 | Aze–Aze | 59.9 ± 0.3 | 53.6 ± 0.2 | 41.9 ± 1.8 | 30.0 ± 1.1 | — | — | — | 48.6 ± 1.7 b |
| 7 | ΔPro–ΔPro | 79.3 ± 0.8 | 72.2 ± 0.6 | 61.3 ± 0.5 | 45.4 ± 0.5 | — | — | — | 14.0 ± 0.1 c |
| 8 | Oic–Oic | 83.6 ± 0.3 | 77.6 ± 0.2 | 68.2 ± 0.2 | 53.4 ± 1.4 | 47.8 ± 2.3 | 42.1 ± 2.6 | 32.9 ± 2.1 | 7.2 ± 1.3 d |
| 9 | Piz–Piz | 70.8 ± 1.0 | 63.3 ± 1.7 | 58.7 ± 0.9 | 45.0 ± 1.9 | — | — | — | 16.0 ± 0.9 e |
| 10 | Tic–Tic | 45.2 ± 0.3 | 40.7 ± 1.1 | 31.4 ± 0.9 | 24.7 ± 1.1 | — | — | — | >100 |
alog IC50 = –4.67 ± 0.06.
blog IC50 = –4.31 ± 0.08.
clog IC50 = –4.85 ± 0.05.
dlog IC50 = –5.14 ± 0.10.
elog IC50 = –4.80 ± 0.09. R2 was between 0.93–0.98. Results are presented as the mean ± SEM.
The second examined series of synthesized compounds were analogues of branched peptide 3 (IC50 = 8.9 μM). The results are presented in Table 5. Due to the substitution of Pro in the second position by Dab (change from peptide 2), this structure is characterized by a higher number of free rotation bonds and a smaller rigidity of the central part of the molecule. In the case of analogue 11, in which Pro was substituted for Hyp, similar to compound 5 in series I, we did not observe significant changes in affinity to NRP-1 compared to the parent sequence. The substitution of Pro by Aze (12) resulted in a slight decrease in the inhibitory activity of the ligand (IC50 = 11.7 μM). The best compounds of series II were analogues 13 and 14. Increasing the rigidness of the ring by introducing a double bond (ΔPro) reduced the IC50 twice, while the octahydroindole ring Oic caused its nearly fourfold decrease. However, the presence of a six-membered, more flexible ring (Piz) caused a tenfold decrease in the inhibitory activity of the VEGF-A165/NRP-1 complex for compound 15 (IC50 = 80.5 μM). We also did not observe significant interaction between NRP-1 and analogue 16 with the Tic–Tic fragment. The decrease in affinity for the NRP-1 receptor for compounds derived from sequence 3, depending on the Pro analogue used, can be arranged in the following series: Oic ≈ ΔPro > Pro ≈ Hyp ≈ Aze ≫ Piz > Tic.
Table 5. Inhibitory effects and IC50 of series II peptide-like ligand binding rhNRP-1 (chemiluminescence detection).
| Compound | AA2–AA3 fragment | Inhibition of NRP-1/VEGF-A165 binding [%] |
IC50 [μM] | ||||||
| 100 μM | 50 μM | 25 μM | 10 μM | 5 μM | 2.5 μM | 1 μM | |||
| 3 | Dab–Pro | 79.8 ± 0.8 | 76.6 ± 0.9 | 70.4 ± 0.5 | 58.6 ± 1.4 | 43.7 ± 0.7 | 31.4 ± 0.5 | 13.8 ± 0.3 | 8.9 ± 0.8 (ref. 40) |
| 11 | Dab–Hyp | 78.6 ± 0.7 | 76.0 ± 0.6 | 67.3 ± 1.5 | 54.3 ± 2.0 | 36.0 ± 0.0 | 22.3 ± 1.5 | 22.5 ± 0.3 | 9.2 ± 0.7 a |
| 12 | Dab–Aze | 78.8 ± 0.6 | 75.8 ± 0.3 | 69.1 ± 0.3 | 49.6 ± 4.2 | 30.1 ± 1.8 | 12.9 ± 1.4 | 7.7 ± 1.0 | 11.7 ± 1.2 b |
| 13 | Dab–ΔPro | 87.1 ± 0.6 | 84.4 ± 0.3 | 78.1 ± 1.0 | 70.7 ± 0.3 | 58.0 ± 0.8 | 44.8 ± 0.4 | 29.9 ± 1.5 | 4.3 ± 0.2 c |
| 14 | Dab–Oic | 83.0 ± 0.3 | 79.5 ± 0.5 | 77.7 ± 1.3 | 63.4 ± 1.9 | 52.3 ± 3.3 | 50.9 ± 3.2 | 37.7 ± 2.1 | 2.3 ± 0.2 d |
| 15 | Dab–Piz | 51.8 ± 0.4 | 46.0 ± 0.6 | 34.8 ± 0.3 | 20.8 ± 0.6 | — | — | — | 80.5 ± 2.0 e |
| 16 | Dab–Tic | 0.0 ± 3.2 | 17.2 ± 1.4 | 35.0 ± 1.2 | 31.5 ± 2.5 | — | — | — | ≫100 |
alog IC50 = –5.03 ± 0.07.
blog IC50 = –4.97 ± 0.05.
clog IC50 = –5.54 ± 0.04.
dlog IC50 = –5.65 ± 0.08.
elog IC50 = –4.09 ± 0.07. R2 was between 0.92–0.98. Results are presented as the mean ± SEM.
The third series of synthesized compounds were branched peptide 4 analogues (IC50 = 23.4 μM). Analysis of compounds 17–21 showed no significant effect of the applied Pro mimetic in the second position on affinity for NRP-1 (Table 6). IC50 values were similar for all of these compounds and were in the range of 16–22 μM. The inhibitory activity of the peptide-like ligand 22 was twice as low relative to the parent sequence, suggesting that the presence of the tetrahydroisoquinoline ring negatively influenced the ligand interaction with the receptor. Thus, we did not significantly improve the receptor–ligand interaction with any synthesized analogue in the third series of compounds. The decrease in inhibition for compounds derived from sequence 4, depending on the Pro analogue used, can be arranged in the following series: Aze ≈ Oic > Hyp ≈ Pro ≈ ΔPro > Piz ≫ Tic.
Table 6. Inhibitory effect and IC50 of series III peptide-like ligand binding rhNRP-1 (chemiluminescence detection).
| Compound | AA2–AA3 fragment | Inhibition of NRP-1/VEGF-A165 binding [%] |
IC50 [μM] | |||
| 100 μM | 50 μM | 25 μM | 10 μM | |||
| 4 | Pro–Ala | 70.7 ± 0.5 | 62.1 ± 0.3 | 50.3 ± 1.0 | 35.5 ± 2.0 | 23.4 ± 7.9 40 |
| 17 | Hyp–Ala | 70.1 ± 1.1 | 65.0 ± 1.0 | 50.3 ± 0.2 | 43.6 ± 1.1 | 21.3 ± 1.0 a |
| 18 | Aze–Ala | 73.3 ± 1.4 | 72.2 ± 0.7 | 65.7 ± 0.5 | 41.9 ± 8.3 | 16.0 ± 3.7 b |
| 19 | ΔPro–Ala | 77.5 ± 0.6 | 66.2 ± 0.5 | 55.5 ± 0.8 | 36.3 ± 0.4 | 21.3 ± 0.7 c |
| 20 | Oic–Ala | 75.7 ± 0.7 | 65.6 ± 1.0 | 57.0 ± 1.1 | 46.3 ± 2.2 | 16.1 ± 1.8 d |
| 21 | Piz–Ala | 72.7 ± 0.5 | 64.8 ± 0.6 | 51.1 ± 1.5 | 48.5 ± 1.0 | 18.1 ± 1.6 e |
| 22 | Tic–Ala | 68.5 ± 0.2 | 52.1 ± 0.3 | 36.2 ± 0.3 | 22.5 ± 1.6 | 50.1 ± 0.2 f |
alog IC50 = –4.67 ± 0.09.
blog IC50 = –4.80 ± 0.08.
clog IC50 = –4.67 ± 0.04.
dlog IC50 = –4.79 ± 0.08.
elog IC50 = –4.74 ± 0.10.
flog IC50 = –4.30 ± 0.02. R2 was between 0.94–0.99. Results are presented as the mean ± SEM.
Stability studies in human plasma
The changes introduced in the parent sequences 2–4 were aimed, in addition to increasing the affinity for the receptor being studied, also to increase the stability of the compounds obtained in biological fluids. The rational design of the peptide-like ligands involves identifying the sites of hydrolysis of the compound by proteases, followed by the introduction of chemical modifications such as unnatural amino acids or amide bond mimetics. The limitation of the conformational liability induced by the Pro residues usually limits proteolysis, however, it depends on the position of this amino acid in the peptide chain. On the other hand, the presence of Pro, as well as the cationic amino acid residues Lys and Arg, in the appropriate position of the peptide sequence, may also enhance the susceptibility to enzymatic hydrolysis.60,61
While peptides containing a single Pro residue may be more susceptible to rapid enzymatic cleavage, the Pro–Pro motif shows remarkable stability in serum samples.61 Both the Pro–Pro motif (compound 2) and the Pro change in the AA2 position in the H2N–Lys(hArg)–AA2–AA3–Arg sequence for the unnatural amino acid with Dab (compound 3) cationic residue guaranteed high bond resistance for enzymatic hydrolysis.
The analysis of enzymatic stability in human blood plasma, with sodium citrate as an anticoagulant, of the two best peptide-like ligands with non-natural Pro analogues was performed. We observed increased stability of compounds with Pro mimetics compared to parent sequence 3 (t1/2 = 41.0 h)40 (Fig. 3).
Fig. 3. Comparison of the in vitro metabolic stability and half-life (t1/2) values of peptide-like ligands 13 and 14 incubated in human plasma.

Analysis of the metabolites of compounds 13 and 14 using LC-MS showed that enzymatic cleavage occurred on Lys-Dab and Lys-ε-hArg bonds, similar to branched peptide 3 (Fig. 4). However, we did not observe signals from the hydrolysis of the ΔPro/Oic–Arg amide bond. The presence of the Arg residue at the C-terminus of the sequence is crucial for the interaction of the peptide ligand with the receptor. Therefore, the results suggest that the replacement of Pro with the mimetic of this amino acid had a positive effect not only on the affinity of the compounds for NRP-1, but also on the stability of the key peptide bond.
Fig. 4. Proposed degradation pathways for peptide-like ligands A. 13 and B. 14.

Conclusions
In summary, three families of peptide-like ligands were synthesized, in which the positions AA2 and AA3 in the general sequence Lys(hArg)–AA2–AA3–Arg were substituted by Pro mimetics with different rigidities and ring sizes.
To select the best assay for the biological evaluation of synthesized compounds, we first conducted a detailed study on selection and optimization of the affinity assay. We compared the two previously described affinity assays for NRP-1 using A7R and Lys(hArg)–AA2–AA3–Arg parent peptides, which are already tested NRP-1/VEGF-A165 binding inhibitors. Our data show that these two assays are incomparable, as the difference between the results is significant. However, the assay with chemiluminescence detection seems to be much more proficient compared to the colorimetric method. Therefore, studies on the ability of the new compounds to inhibit VEGF-A165/NRP-1 complex formation were performed by means of an affinity assay with luminescence detection.
The results show that optimization of the central part of the parent sequence 3 (Lys(hArg)–Dab–Pro–Arg) led to compounds which show a 2 and a 4-fold decrease in the IC50. Based on these data, we conclude that replacing Pro with ΔPro or Oic in the third position of the Dab–AA3 fragment in our branched peptides, seems to increase interactions with NRP-1. This is probably due to the more rigid rings in ΔPro and Oic, which promote the optimal peptide-like chain position on the receptor surface.
Experimental section
General information
Unless otherwise specified, reagents were obtained from commercial sources and used without further purification. Resins, amino acids and coupling reagents were obtained from Iris Biotech. Solvents were obtained from Sigma-Aldrich. Synthesized compounds were purified using a Prominence preparative HPLC system (Phenomenex Jupiter Proteo C12 90 AXIA Å 10 μm 250 × 21.2 mm column). High resolution mass spectra (HRMS) were recorded on a Shimadzu LCMS-IT-TOF-MS with an ESI ionization source. The mass reported contains the most abundant isotopes with a mass error <5 ppm. LC–MS analysis was performed using a Prominence HPLC system (Phenomenex Jupiter Proteo C12 90 Å 10 μm 250 × 4.6 mm column) coupled with a Shimadzu LCMS-2020 single quadrupole mass spectrometer with an ESI ionization source. UV absorbance on 96-well plates was measured using a BioTek Cytation 3 reader. Luminescence was measured using a Tecan Infinite F200Pro microplate reader.
Peptide synthesis
The synthesis of all peptide-like ligands was carried out manually using a syringe on the pre-loaded Fmoc-Arg(Pbf) Wang resin (Activotec) with a capacity of 0.39 mmol g–1 (0.5 g) following the Fmoc chemistry. Coupling of 2 eq. amino acids (0.4 mmol) was done using 2 eq. HATU (152 mg, 0.4 mmol) and 5 eq. DIPEA (169 μL, 1 mmol) in DMF (5 mL). Completion of coupling was checked using the Kaiser or chloranil test. The deprotection step was done using 20% piperidine in DMF. Final compounds were cleaved from the resin with the use of 5 mL TFA : H2O : TIS (95 : 2.5 : 2.5, v : v : v) for 3 h and then precipitated by dropwise addition into cold diethyl ether. Crude peptides were collected by centrifugation and purified by preparative RP-HPLC on a C12 column with an H2O/ACN gradient containing 0.1% TFA. Peptide fractions were collected, lyophilized and analysed by LC-MS. Molecular weight and elemental composition were confirmed using HRMS. Yields, detailed RP-HPLC and LCMS data can be found in the ESI.†
Rat NRP-1 binding assay with colorimetric detection
This method was previously described.31,37–40,57 Briefly, the flat bottom surface of a 96-well plate was coated with 100 μL (2 μg mL–1) anti-human IgG (Fc-specific) (Sigma Aldrich) and incubated overnight at 4 °C. The plate was then washed thrice with PBS and treated with 2% BSA in PBS followed by incubation for 2 h at 37 °C to block non-specific binding. 50 μL (20 ng per well) of purified recombinant rat NRP-1/Fc chimera (R&D Systems) diluted in PBS containing 0.1% BSA and 0.005% Tween 80 (PBT) was added. Next, 50 μL of peptide dissolved in PBT in the range of concentrations (100–0.001 μM) and 50 μL of human (bt)-VEGF-A165 (R&D Systems) in PBT containing 2 μg mL–1 of heparin in the final concentration of 1 nM were added respectively to a final volume of 150 μL.
After overnight incubation at 4 °C, the plate was washed thrice in PBT and treated with a streptavidin–horseradish peroxidase (GE Healthcare) conjugate in PBS with 0.05% Tween 80 (1 : 8000). The plate was next incubated for 40 min at r.t. under obscurity. Washing with PBT was followed by 100 μL ABTS substrate (Sigma Aldrich) addition. As a positive control, only (bt)-VEGF-A165 was present in wells, while, as a negative control, only PBT was added. After 2 h, optical density was measured at 415 nm. Each peptide concentration was simultaneously tested in triplicate, and each experiment was repeated twice. Percentages of inhibition were calculated by the following formula: 100% – [[(S – SN)/(P – NS)] × 100%], where S is the signal intensity measured, NS is the signal measured in the negative control, and P is the maximum binding signal obtained with (bt)-VEGF-A165 without competitor.
Human NRP-1 binding assay with chemiluminescence detection
This method was previously described.28,58,59 Briefly, the flat bottom surface of a 96-well plate was coated with 100 μL (200 ng per well) recombinant human NRP-1 (R&D Systems) and incubated overnight at 4 °C. Non-specific binding was blocked by the incubation with 0.5% BSA in PBS. 50 μL of peptide dissolved in PBS in the range of concentrations and 50 μL (400 ng mL–1) of human (bt)-VEGF-A165 in PBS containing 4 μg mL–1 of heparin were added respectively. After 2 h incubation at r.t., the plate was washed and treated with a streptavidin–horseradish peroxidase conjugate in PBS (1 : 8000). Chemiluminescence was quantified immediately after addition of 100 μL chemiluminescent substrate (Thermo Scientific). In the positive control only (bt)-VEGF-A165 was present in wells, while, in the negative control, wells were not coated by NRP-1. Percentages of inhibition were calculated by the following formula: 100% – [[(S – SN)/(P – NS)] × 100%], where S is the signal intensity measured, NS is the signal measured in the negative control, and P is the signal measured in the positive control.
Blood collection and plasma preparation
All human procedures were performed in accordance with the ICH E6 (R2) Guideline for Good Clinical Practice and the Code of Good Customs in Science developed by the Polish Academy of Sciences, and approved by the Bioethics Committee of the Central Clinical Hospital of the Ministry of the Internal Affairs in Warsaw, Poland (Decision No. 64/2017). Human blood from four healthy donors (informed consent was obtained from all subjects), was directly drawn into evacuated tubes with sodium citrate as an anticoagulant agent to obtain plasma samples. Tubes were spun immediately and centrifuged at 2500g for 10 min at 4 °C to prevent possible platelet activation. Plasma was pipetted out of the blood collection tubes into 5 mL microtubes and pooled, collected in 2 mL microtubes and frozen at –80 °C until use.
Degradation assay in human plasma
A degradation assay was performed according to a method described previously.40 In a 1.5 mL microtube, human blood plasma was preactivated in Eppendorf ThermoMixer® Comfort (350 rpm) for 20 min at 37 °C. Afterwards aqueous peptide stock solution (6 mg mL–1; ∼5 μmol ml–1) was diluted in human plasma to give ∼1 μmol mL–1 final concentration of each peptide and incubation was continued. At specific time intervals (0–96 h), 100 μL of the mixture was collected in a 1.5 mL microtube and quenched by adding 100 μL of 98% ethanol. The obtained suspension was vortexed for 1 min (3000 min–1) and then centrifuged for 10 min in 4 °C (11 000g) to pellet proteins. 100 μL of supernatant was collected and lyophilized. The samples reconstituted in 100 μL of water were subjected to RP-HPLC analysis, followed by LC-MS analysis for selected samples. Before experiments with studied peptides, a pre-initial test to prove plasma activity was performed using endomorphin-2 (data not shown). Detailed RP-HPLC and LCMS data can be found in the ESI.†
Statistical analysis
Determination of IC50 and half-life of peptides in serum and plasma was conducted using the nonlinear regression function with Prism (Version-5.01, GraphPad software). All results are represented as an average with error bars indicating ±SD determined from the results of two or three independent experiments, each performed in duplicate or triplicate.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We thank Dr. Adam Mieczkowski (Institute of Biochemistry and Biophysics, Polish Academy of Sciences) for providing us with (S)-1-(Fmoc)-4-(Boc)piperazine-2-carboxylic acid. We are very grateful to Anna Laskowska (Mossakowski Medical Research Centre, Polish Academy of Sciences) for help with blood collection and experiments on human plasma. This publication is based upon work from COST Action CA15135: Multi-target paradigm for innovative ligand identification in the drug discovery process (MuTaLig), supported by COST (European Cooperation in Science and Technology, no. COST-STSM-CA15135-38226) and was supported by a grant from the University of Warsaw – Poland for young researchers, no. 12000-501/86-DSM-107500. LC and MS analyses were done in the Laboratory of Chemical Synthesis, CePT, Mossakowski Medical Research Centre. CePT laboratories are established within the project co-financed by EU from the European Regional Development Fund under the Operational Programme Innovative Economy, 2007–2013.
Footnotes
†Electronic supplementary information (ESI) available: Synthesis and analytical data. See DOI: 10.1039/c8md00537k
References
- Miao H. Q., Klagsbrun M. Cancer Metastasis Rev. 2000;19:29–37. doi: 10.1023/a:1026579711033. [DOI] [PubMed] [Google Scholar]
- Miao H. Q., Lee P., Lin H., Soker S., Klagsbrun M. FASEB J. 2000;14:2532–2539. doi: 10.1096/fj.00-0250com. [DOI] [PubMed] [Google Scholar]
- Soker S., Takashima S., Miao H. Q., Neufeld G., Klagsbrun M. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- Gu C., Limberg B. J., Whitaker G. B., Perman B., Leahy D. J., Rosenbaum J. S., Ginty D. D., Kolodkin A. L. J. Biol. Chem. 2002;277:18069–18076. doi: 10.1074/jbc.M201681200. [DOI] [PubMed] [Google Scholar]
- Lee C. C., Kreusch A., McMullan D., Ng K., Spraggon G. Structure. 2003;11:99–108. doi: 10.1016/s0969-2126(02)00941-3. [DOI] [PubMed] [Google Scholar]
- Whittle C., Gillespie K., Harrison R., Mathieson P. W., Harper S. J. Clin. Sci. 1999;97:303–312. [PubMed] [Google Scholar]
- Gluzman-Poltorak Z., Cohen T., Herzog Y., Neufeld G. J. Biol. Chem. 2000;275:18040–18045. doi: 10.1074/jbc.M909259199. [DOI] [PubMed] [Google Scholar]
- Hoeben A., Landuyt B., Highley M. S., Wildiers H., Van Oosterom A. T., De Bruijn E. A. Pharmacol. Rev. 2004;56:549–580. doi: 10.1124/pr.56.4.3. [DOI] [PubMed] [Google Scholar]
- Robinson S. D., Reynolds L. E., Kostourou V., Reynolds A. R., da Silva R. G., Tavora B., Baker M., Marshall J. F., Hodivala-Dilke K. M. J. Biol. Chem. 2009;284:33966–33981. doi: 10.1074/jbc.M109.030700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdembri D., Caswell P. T., Anderson K. I., Schwarz J. P., König I., Astanina E., Caccavari F., Norman J. C., Humphries M. J., Bussolino F., Serini G. PLoS Biol. 2009;7:e25. doi: 10.1371/journal.pbio.1000025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa M., Matsushita A., Korc M. Cancer Biol. Ther. 2007;6:1173–1180. doi: 10.4161/cbt.6.8.4363. [DOI] [PubMed] [Google Scholar]
- Li M., Yang H., Chai H., Fisher W. E., Wang X., Brunicardi F. C., Yao Q., Chen C. Cancer. 2004;101:2341–2350. doi: 10.1002/cncr.20634. [DOI] [PubMed] [Google Scholar]
- Grun D., Adhikary G., Eckert R. L. Oncogene. 2016;35:4379–4387. doi: 10.1038/onc.2015.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z., Tessier-Lavigne M. Cell. 1997;90:739–751. doi: 10.1016/s0092-8674(00)80534-6. [DOI] [PubMed] [Google Scholar]
- Kolodkin A. L., Levengood D. V., Rowe E. G., Tai U.-T., Giger R. J., Ginty D. D. Cell. 1997;90:753–762. doi: 10.1016/s0092-8674(00)80535-8. [DOI] [PubMed] [Google Scholar]
- Glinka Y., Prud'homme G. J. J. Leukocyte Biol. 2008;84:302–310. doi: 10.1189/jlb.0208090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinka Y., Stoilova S., Mohammed N., Prud'homme G. J. Carcinogenesis. 2011;32:613–621. doi: 10.1093/carcin/bgq281. [DOI] [PubMed] [Google Scholar]
- Soker S., Miao H. Q., Nomi M., Takashima S., Klagsbrun M. J. Cell. Biochem. 2002;85:357–368. doi: 10.1002/jcb.10140. [DOI] [PubMed] [Google Scholar]
- Koch S., Tugues S., Li X., Gualandi L., Claesson-Welsh L. Biochem. J. 2011;437:169–183. doi: 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- Vander Kooi C. W., Jusino M. A., Perman B., Neau D. B., Bellamy H. D., Leahy D. J. Proc. Natl. Acad. Sci. U. S. A. 2007;104:6152–6157. doi: 10.1073/pnas.0700043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H. F., Vander Kooi C. W. J. Biol. Chem. 2015;290:29120–29126. doi: 10.1074/jbc.R115.687327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N., Hillan K. J., Gerber H. P., Novotny W. Nat. Rev. Drug Discovery. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- Gross S., Rahal R., Stransky N., Lengauer C., Hoeflich K. P. J. Clin. Invest. 2015;125:1780–1789. doi: 10.1172/JCI76094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos N., Martin J., Ruan Q., Rafique A., Rosconi M. P., Shi E., Pyles E. A., Yancopoulos G. D., Stahl N., Wiegand S. J. Angiogenesis. 2012;15:171–185. doi: 10.1007/s10456-011-9249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teesalu T., Sugahara K. N., Kotamraju V. R., Ruoslahti E. Proc. Natl. Acad. Sci. U. S. A. 2009;106:16157–16162. doi: 10.1073/pnas.0908201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanuy D., Kotla R., Nussinov R., Teesalu T., Sugahara K. N., Alemán C., Haspel N. J. Struct. Biol. 2013;182:78–86. doi: 10.1016/j.jsb.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haspel N., Zanuy D., Nussinov R., Teesalu T., Ruoslahti E., Aleman C. Biochemistry. 2011;50:1755–1762. doi: 10.1021/bi101662j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. Q., Borriello L., Allain B., Pavoni S., Lopez N., Hermine O., Garbay Ch., Raynaud F., Lepelletier Y., Demange L. Int. J. Pept. Res. Ther. 2015;21:117–124. [Google Scholar]
- Kamarulzaman E. E., Gazzali A. M., Achard M., Frochot C., Barberi Heyob M., Boura C., Chaimbault P., Sibille E., Wahab H. A., Vanderesse R. Int. J. Mol. Sci. 2015;16:24059–24080. doi: 10.3390/ijms161024059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamarulzaman E. E., Vanderesse R., Gazzali A. M., Barberi-Heyob M., Boura C., Frochot C., Shawkataly O., Aubry A., Wahab H. A. J. Biomol. Struct. Dyn. 2017;35:26–45. doi: 10.1080/07391102.2015.1131196. [DOI] [PubMed] [Google Scholar]
- Fedorczyk B., Lipiński P. F. J., Tymecka D., Puszko A. K., Wileńska B., Perret G. Y., Misicka A. J. Pept. Sci. 2017;23:445–454. doi: 10.1002/psc.3009. [DOI] [PubMed] [Google Scholar]
- Binétruy-Tournaire R., Demangel C., Malavaud B., Vassy R., Rouyre S., Kraemer M., Plouët J., Derbin C., Perret G., Mazié J. C. EMBO J. 2000;19:1525–1533. doi: 10.1093/emboj/19.7.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perret G. Y., Starzec A., Hauet N., Vergote J., Le Pecheur M., Vassy R., Léger G., Verbeke K. A., Bormans G., Nicolas P., Verbruggen A. M., Moretti J. L. Nucl. Med. Biol. 2004;31:575–581. doi: 10.1016/j.nucmedbio.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Starzec A., Vassy R., Martin A., Lecouvey M., Di Benedetto M., Crépin M., Perret G. Y. Life Sci. 2006;79:2370–2381. doi: 10.1016/j.lfs.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Tirand L., Frochot C., Vanderesse R., Thomas N., Trinquet E., Pinel S., Viriot M. L., Guillemin F., Barberi-Heyob M. J. Controlled Release. 2006;111:153–164. doi: 10.1016/j.jconrel.2005.11.017. [DOI] [PubMed] [Google Scholar]
- Thomas N., Bechet D., Becuwe P., Tirand L., Vanderesse R., Frochot C., Guillemin F., Barberi-Heyob M. J. Photochem. Photobiol., B. 2009;96:101–108. doi: 10.1016/j.jphotobiol.2009.04.008. [DOI] [PubMed] [Google Scholar]
- Grabowska K., Puszko A. K., Lipiński P. F. J., Laskowska A. K., Wileńska B., Witkowska E., Misicka A. Bioorg. Med. Chem. Lett. 2016;26:2843–2846. doi: 10.1016/j.bmcl.2016.04.059. [DOI] [PubMed] [Google Scholar]
- Grabowska K., Puszko A. K., Lipiński P. F., Laskowska A. K., Wileńska B., Witkowska E., Perret G. Y., Misicka A. Bioorg. Med. Chem. 2017;25:597–602. doi: 10.1016/j.bmc.2016.11.024. [DOI] [PubMed] [Google Scholar]
- Tymecka D., Lipiński P. F. J., Fedorczyk B., Puszko A., Wileńska B., Perret G. Y., Misicka A. Peptides. 2017;94:25–32. doi: 10.1016/j.peptides.2017.06.003. [DOI] [PubMed] [Google Scholar]
- Tymecka D., Puszko A. K., Lipiński P. F., Fedorczyk B., Wileńska B., Sura K., Perret G. Y., Misicka A. Eur. J. Med. Chem. 2018;158:453–462. doi: 10.1016/j.ejmech.2018.08.083. [DOI] [PubMed] [Google Scholar]
- Vermeirssen V., Van Camp J., Verstraete W. J. Geophys. Res. Oceans. 2004;92:357–366. doi: 10.1079/bjn20041189. [DOI] [PubMed] [Google Scholar]
- Knappe D., Zahn M., Sauer U., Schiffer G., Sträter N., Hoffmann R. ChemBioChem. 2011;12:874–876. doi: 10.1002/cbic.201000792. [DOI] [PubMed] [Google Scholar]
- Knappe D., Cassone M., Nollmann F. I., Otvos L., Hoffmann R. Protein Pept. Lett. 2014;21:321–329. doi: 10.2174/09298665113206660105. [DOI] [PubMed] [Google Scholar]
- Rubenstein E. J. Neuropathol. Exp. Neurol. 2008;67:1035–1040. doi: 10.1097/NEN.0b013e31818add4a. [DOI] [PubMed] [Google Scholar]
- Tsai F. H., Overberger C. G., Zand R. Biopolymers. 1990;30:1039–1049. doi: 10.1002/bip.360301105. [DOI] [PubMed] [Google Scholar]
- Bessonov K., Vassall K. A., Harauz G. J. Mol. Graphics Modell. 2013;39:118–125. doi: 10.1016/j.jmgm.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Flores-Ortega A., Casanovas J., Zanuy D., Nussinov R., Alemán C. J. Phys. Chem. B. 2007;111:5475–5482. doi: 10.1021/jp0712001. [DOI] [PubMed] [Google Scholar]
- Moore S., Felix A. M., Meienhofer J., Smith C. W., Walter R. J. Med. Chem. 1977;20:495–500. doi: 10.1021/jm00214a007. [DOI] [PubMed] [Google Scholar]
- Smith C. W., Walter R. Science. 1978;199:297–299. doi: 10.1126/science.619455. [DOI] [PubMed] [Google Scholar]
- Amoscato A. A., Babcock G. F., Nishioka K. Peptides. 1984;5:489–494. doi: 10.1016/0196-9781(84)90075-5. [DOI] [PubMed] [Google Scholar]
- Torino D., Mollica A., Pinnen F., Lucente G., Feliciani F., Davis P., Lai J., Ma S. W., Porreca F., Hruby V. J. Bioorg. Med. Chem. Lett. 2009;19:4115–4118. doi: 10.1016/j.bmcl.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieczkowski A., Koźmiński W., Jurczak J. Synthesis. 2010;2:221–232. [Google Scholar]
- Al-Ghorbani M., Bushara B. A., Zabiulla S., Mamatha S. V., Khanum S. A. J. Chem. Pharm. Res. 2015;7:281–301. [Google Scholar]
- Hicks R. P., Abercrombie J. J., Wong R. K., Leung K. P. Bioorg. Med. Chem. 2013;21:205–214. doi: 10.1016/j.bmc.2012.10.039. [DOI] [PubMed] [Google Scholar]
- Haney E. F., Hancock R. E. Biopolymers. 2013;100:572–583. doi: 10.1002/bip.22250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham P. L., An introduction to drug synthesis, Oxford UniversityOxford University PressPress, Oxford, UK, 2015. [Google Scholar]
- Starzec A., Miteva M. A., Ladam P., Villoutreix B. O., Perret G. Y. Bioorg. Med. Chem. 2014;22:4042–4048. doi: 10.1016/j.bmc.2014.05.068. [DOI] [PubMed] [Google Scholar]
- Allain B., Jarray R., Borriello L., Leforban B., Dufour S., Liu W. Q., Pamonsinlapatham P., Bianco S., Larghero J., Hadj-Slimane R., Garbay C., Raynaud F., Lepelletier Y. Cell Signal. 2012;24:214–223. doi: 10.1016/j.cellsig.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Borriello L., Montès M., Lepelletier Y., Leforban B., Liu W. Q., Demange L., Delhomme B., Pavoni S., Jarray R., Boucher J. L., Dufour S., Hermine O., Garbay C., Hadj-Slimane R., Raynaud F. Cancer Lett. 2014;349:120–127. doi: 10.1016/j.canlet.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Vanhoof G., Goossens F., De Meester I., Hendriks D., Scharpé S. FASEB J. 1995;9:736–744. [PubMed] [Google Scholar]
- Jambunathan K., Galande A. K. Protein Pept. Lett. 2014;21:32–38. doi: 10.2174/09298665113209990069. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.