

 Bioisosteric [7]annulenothiophenes are well tolerated by GluN2B subunit containing NMDA receptors, but do not require a benzylic OH moiety.
Bioisosteric [7]annulenothiophenes are well tolerated by GluN2B subunit containing NMDA receptors, but do not require a benzylic OH moiety.
Abstract
The involvement of NMDA receptors containing the GluN2B subunit in neurodegenerative disorders including Alzheimer's and Parkinson's disease renders this NMDA receptor subtype an interesting pharmacological target. The aim of this study was the bioisosteric replacement of benzene, methoxybenzene and aniline moieties of known potent GluN2B selective NMDA receptor antagonists by a thiophene ring. In a nine-step synthesis starting from commercially available propionic acid 9 the thiophene derivative 7a was obtained as a bioisostere of the potent GluN2B ligands cis-3 and trans-3. [7]Annuleno[b]thiophene 8a without a benzylic OH moiety was prepared in a six-step synthesis starting from carboxylic acid 18. 8a represents a bioisostere of potent GluN2B ligands 4 and 5. [7]Annulenothiophene 8a without a benzylic OH moiety reveals approx. 8-fold higher GluN2B affinity (Ki = 26 nM) than the analogous thiophene derivative 7a with a benzylic OH moiety (Ki = 204 nM). Both thiophene bioisosteres show a slight preference for GluN2B receptors over both σ receptors. The data indicate that the bioisosteric replacement of benzene or substituted benzene rings by a thiophene ring is well tolerated by the NMDA receptor. Furthermore, the benzylic OH moiety seems not to be essential for high GluN2B affinity.
1. Introduction
Glutamate receptors are transmembrane proteins in neuronal cells, which specifically bind the excitatory neurotransmitter (S)-glutamate. The class of glutamate receptors can be divided into two types depending on their intracellular signal transduction pathways. Whereas metabotropic glutamate receptors labelled mGluR1 to mGluR8 activate G-proteins, ionotropic glutamate receptors represent ligand gated ion channels and control the passage of cations. Ionotropic glutamate receptors are subdivided into three types named after their selective agonists 2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl)propionic acid (AMPA), kainic acid (kainate) and N-methyl-d-aspartate (NMDA) receptor.
The NMDA receptor (NMDAR) is a heterotetrameric, ligand-gated ion channel. Activation leads to Ca2+ and Na+ influx and K+ efflux. Furthermore, the receptor can interact with multiple intracellular proteins via different subunits.1 The receptor is mainly localized on the postsynaptic side of the synaptic cleft but can also be present in a smaller extent on the presynaptic side.2 Seven different genes are known encoding for NMDAR subunits. Four different genes encode for the four GluN2 subunits GluN2A-D.3 The GluN3A and GluN3B subunits are encoded by two different genes.4 Additionally, eight splice variants of the GluN1 subunit (GluN1a-h) result from alternative splicing. The presence of different GluN2 subunits and thus the composition of the heterotetrameric NMDA receptors depend on the developmental stage of the particular organism and the regions of the central nervous system. The functional diversity regarding NMDAR's biophysical and pharmacological properties is predominantly determined by the subunit composition.5
A functional NMDAR consists of at least one GluN1 and one GluN2 subunit. The GluN1 subunit contains the binding site for glycine, whereas the GluN2 subunit presents the binding site for (S)-glutamate. Each subunit is characterized by four domains. The amino-terminal domain (ATD) and the agonist-binding domain (ABD) are located extracellularly. The transmembrane domain (TMD) forms the ion-pore. The C-terminal domain (CTD) on the intracellular side is the access point for different protein–protein interactions.6,7
The NMDAR exhibits some unique features. At first, in its resting state, the NMDAR pore is blocked by Mg2+ ions. This blockade is voltage dependent and is removed by depolarization of neighboring receptors, such as AMPA or kainate receptors.8 Secondly, for the activation of the NMDAR two agonists glycine and (S)-glutamate have to be present simultaneously. This phenomenon termed coagonism is unique for the NMDA receptor.9 These properties lead to an effect called synaptic coincidence, which is responsible for synaptic plasticity. A special form of this plasticity is the long-term potentiation (LTP), which is associated with neurological phenomena such as learning and memory. Several studies demonstrated the involvement of the GluN2B subunit in these processes.10–12
The NMDAR is also of pathophysiological interest. An overstimulation of the NMDAR leads to an excessive Ca2+ influx, which leads via several signal cascades to cell death by apoptosis.13 This phenomenon, known as excitotoxicity, is believed to be the major reason for neuronal cell death after acute brain damage (e.g. stroke, injury) but also within neurodegenerative diseases. Several studies indicated the unique role of the GluN2B subunit in neurodegenerative diseases such as Parkinson's disease,14,15 Huntington's disease,16,17 but also hypoxia and ischemia,18,19 ethanol20,21 and opioid abuse.22 Alzheimer's disease is also associated with the NMDAR, but various GluN2 subunits are involved in this process.23 While non-selective NMDAR antagonists such as open channel blockers usually exhibit a broad range of side effects, including psychotomimetic, behavioral and cardiovascular effects, GluN2B selective antagonists are well tolerated.24 Therefore, antagonists addressing selectively NMDARs containing the GluN2B subunit are envisaged as promising new pharmacological drug candidates.
The NMDAR possesses binding sites for different ions and molecules, such as Zn2+ ions, H+, polyamines and NO (ATD), the negative allosteric modulator ifenprodil (1) (ATD), glycine and (S)-glutamate (LBD) as well as the open channel blockers MK-801, phencyclidine and memantine (TMD).25 The ifenprodil binding site is located in the ATD26 at the interface between the GluN1 and GluN2B subunits.27 Addressing this binding site with positive and negative allosteric modulators allows a selective modulation of NMDARs containing the GluN2B subunit.
The benzylpiperidine ifenprodil (1) originally designed as an α1 receptor antagonist28 was the first compound binding at a particular region of the GluN2B subunit.29 (Fig. 1) It binds with high affinity towards GluN2B subunit containing NMDARs (IC50 = 13.3 nM)30 and prefers the activated and desensitized state of the NMDAR compared to the resting state.31 However, some major drawbacks have inhibited ifenprodil to become a drug for clinical use. Undesired side effects, such as memory deficits, psychotomimetic effects and hypertension resulting from interaction with other receptors, such as 5-HT1A, α1, σ1 and σ2 receptors, prevented further clinical trials with ifenprodil. Moreover, the rapid biotransformation leads to very low bioavailability of ifenprodil.32
Fig. 1. Lead compounds 1–5 with high GluN2B affinity. #Only one enantiomer of the racemic mixtures 1, cis-3, trans-3, and 5 is depicted in the figure, respectively.

An X-ray crystal structure reported by Furukawa et al.27 displayed for the first time that ifenprodil binds at the interface between the GluN1b and GluN2B subunits. Two H-bonds between the C( O)NH2 moiety of Gln110 of the GluN2B protein and the protonated amino moiety and the benzylic OH moiety of ifenprodil stabilize the ligand in the binding pocket. A further H-bond between the carboxy moiety of Glu236 (GluN2B) and the phenolic OH moiety are seen in the X-ray crystal structure. Another promising GluN2B antagonist is Ro 25-6981 (2) revealing an even higher affinity (IC50 = 9 nM) towards GluN2B subunit containing NMDARs than ifenprodil (IC50 = 13.3 nM). (Fig. 1) Ro 25-6981 (2) reveals the same selectivity over the other GluN2 subtypes as ifenprodil.33
In order to improve the selectivity and metabolic stability a conformational restriction approach was pursued. The selectivity of a ligand results from its adaptability to binding pockets of different receptors, which depends on the flexibility of the ligand. Therefore, a ligand with a more rigid structure should show increased selectivity for a particular binding pocket of a receptor.
A formal connection of the CH2 moiety at the piperidine ring with the benzene ring of Ro 25-6981 (2) leads to benzo[7]annulenamines of type 6. A series of amines 6 has been synthesized including compounds cis-3 and trans-3 (Fig. 1) showing high GluN2B affinity and high selectivity over σ1 and σ2 receptors.34 Removal of the benzylic OH moiety and the aromatic substituent X resulted in the “naked” benzo[7]annulenamine 4 with unexpectedly high GluN2B affinity (Ki = 57 nM) and selectivity over both σ receptor types.35 Introduction of an amino moiety as an alternative H-bond donating and accepting functional group led to reduction of GluN2B affinity of amine 536 (Fig. 1).
Due to the fast biotransformation of phenols and anilines these structural elements do not represent the first line structural elements of innovative drugs. Therefore, a bioisosteric replacement of the donor-substituted benzene ring of 6 by a thiophene ring (compounds 7 and 8) was envisaged.37 With respect to size and electron density, the thiophene ring is regarded as bioisostere of phenols and anilines. In this manuscript, synthesis and pharmacological properties of thiophene bioisosteres 7 with benzylic OH moiety and 8 without benzylic OH moiety are reported (Fig. 2).
Fig. 2. Conformational restriction approach and bioisosteric benzene/thiophene replacement.

2. Synthesis
The synthesis of thiophene analog 7a started from commercially available carboxylic acid 9. At first carboxylic acid 9 was transformed into Weinreb amide 10 in 89% yield.38 Afterwards, Weinreb amide 10 was reduced with DIBAL-H to afford the aldehyde 11, which was reacted with the Wittig reagent Ph3P CHCO2Et to give the α,β-unsaturated ester 12 (Scheme 1). In literature, an alternative synthetic route to obtain the aldehyde 11 was reported using Pd-catalyzed Heck reaction of 2-iodothiophene with allylic alcohol. Aldehyde 11 was not isolated, but converted directly into the α,β-unsaturated ester 12.39 Both routes gave high yields of α,β-unsaturated ester 12. Due to our experience with the formation and reduction of Weinreb amides and the availability of acid 9 in our lab, we preferred the Weinreb route.
Scheme 1. Synthesis of aminoalcohol 7a. Reagents and reaction conditions: (a) 1. CDI, CH2Cl2, rt, 45 min; 2. CH3NHOCH3, CH2Cl2, rt, 24 h, 89%. (b) DIBAL-H, toluene, –78 °C, 2 h. (c) (C6H5)3P CHCO2Et, CH2Cl2, rt, 24 h, 80%. (d) Benzylamine, EtOH, 80 °C, 20 h, 42%. (e) TosCl, DIPEA, CH2Cl2, rt, 24 h, 87%. (f) 0.1 M NaOH, THF/H2O, rt, 26 h, 69%. (g) P4O10, CH2Cl2, –10 °C, 23 h, 27%. (h) NaBH4, MeOH, 0 °C to rt, 5.5 h, 78%. (i) Mg0, MeOH, rt, ultra-sonic, 5 h, 64%.

Conjugate addition of benzylamine at α,β-unsaturated ester 12 provided β-aminoester 13, which was protected with tosyl chloride to yield sulfonamide 14. For the following intramolecular Friedel–Crafts acylation ester 14 was hydrolyzed with NaOH to afford carboxylic acid 15 in 69% yield.
The cyclization of carboxylic acid 15 was the key step of the planned synthesis. A Friedel–Crafts acylation was envisaged to obtain the desired ketone 16. Conversion of the carboxylic acid 15 into its carboxylic acid chloride and subsequent cyclization with Lewis acids such as AlCl3 or ZnCl2 failed to give the ketone 16. Usually, a conversion could not be observed, since the carboxylic acid chloride was not formed. However, the cyclization of a carboxylic acid with a similar structure was performed with P4O10.40 Therefore, carboxylic acid 15 was treated with P4O10, which led to the ketone 16 in only 10% yield. Finally, a yield of 27% was achieved performing the reaction at low temperature (–10 °C) in CH2Cl2 at a very low concentration of the carboxylic acid 15 (0.01 mol L–1). At higher temperature the carboxylic acid 15 decomposed into a black solid.
The bicyclic ketone 16 was reduced with NaBH4 to provide the secondary alcohol 17 as 6 : 4 mixture of two diastereomers. Mg0 in methanol41 was able to remove the tosyl group leading to the desired final secondary amine 7a as 6 : 4 mixture of diastereomers. The mixture of diastereomeric alcohols 17 and 7a could not be separated by fc. Overall, this synthetic pathway was very challenging, since some key reaction steps were not reproducible and gave variable yields (e.g. intramolecular Friedel–Crafts acylation of 15, detosylation of 17). Therefore, only one test compound 7a was prepared and a novel synthetic strategy was developed to obtain further analogs.
In order to analyze the effect of the benzylic OH moiety [7]annulenothiophene 8a without benzylic OH moiety should be synthesized and tested. A double nucleophilic substitution with dimethyl 3-oxoglutarate was planned as key step of the synthesis (Scheme 2). The preparation of dibromide 19 was performed as described in the literature.42 Dibromide 19 reacted twice with dimethyl 3-oxoglutarate to form the seven-membered keto-diester 20. According to the NMR spectra, 20 exists as mixture of four products: two regioisomeric enols and two diastereomeric β-ketodiesters. Ester hydrolysis and decarboxylation of 20 was performed upon heating a methanolic solution of 20 with 7 M HCl to obtain ketone 21 in 37% yield. Finally, reductive amination of ketone 21 with benzylamine and NaBH(OAc)3 43 provided the benzylamine 8a in 57% yield.
Scheme 2. Synthesis of benzylamine 8a. Reagents and reaction conditions: (a) three steps (metalation/carboxylation, reduction, bromination) according to ref. 42. (b) K2CO3, THF, 76 °C, 46 h, 40%. (c) 7 M HCl, MeOH, 96 °C, 6 h, 37%. (d): Benzylamine, NaBH(OAc)3, CH2Cl2, rt, 48 h, 57%.

3. GluN2B affinity and selectivity over σ1 and σ2 receptors
To determine the affinity of [7]annulenothiophenamines 7a and 8a towards GluN2B subunit containing NDMARs competitive receptor binding assays using the radioligand [3H]ifenprodil were performed. A cell membrane preparation from stably transfected L(tk-) cells was used as receptor material.30 In order to test the selectivity of the new ligands, the affinity at σ1 and σ2 receptors was also recorded. The σ1 and σ2 receptor competitive binding assays were performed with the radioligands [3H]-(+)-pentazocine and [3H]di-o-tolylguanidine, respectively. Membrane preparations from guinea pig brain and rat liver served as receptor source, respectively. Since the radioligand [3H]di-o-tolylguanidine also labels σ1 receptors, an excess of non-radioactive (+)-pentazocine was added in the σ2 assay to block the σ1 receptors (Table 1).44–46
Table 1. Affinity of [7]annulenothiophenamines 7a and 8a and reference compounds towards NMDAR and related receptors.
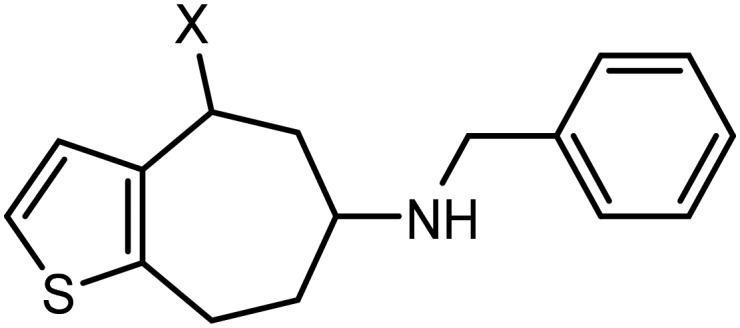
[7]Annulenothiophenamine 7a with benzylic OH moiety exhibits a moderate GluN2B affinity with a Ki value of 204 nM. Affinities towards σ1 as well as σ2 receptors are lower with Ki values greater than 500 nM. A slight preference of 7a for the GluN2B receptor over both σ receptor subtypes can be concluded (Table 1).
The corresponding [7]annulenothiophenamine 8a without benzylic OH moiety shows 8-fold higher GluN2B affinity than 7a. Its Ki value is 26 nM. The σ1 (Ki = 79 nM) and σ2 affinities (Ki = 50 nM) of 8a are in the same range as the GluN2B affinity. Nevertheless, a slight preference for the GluN2B receptor over both σ receptor subtypes can be observed.
Compared to the naked benzo[7]annulenamine 4 (Ki = 57 nM) the [7]annulenothiophene bioisostere 8a (Ki = 26 nM) reveals two-fold higher GluN2B affinity. The selectivity of 4 and 8a over both σ receptor subtypes is very similar. Although the distance between the basic amino moiety and the terminal phenyl ring in 8a is far from the optimal length of 3–4 methylene moieties, the bioisosteric replacement of the benzene ring of 4 by the thiophene ring of 8a increased the GluN2B affinity slightly.
The diastereomeric benzo[7]annulenamines cis-3 and trans-3 show considerably higher GluN2B affinity than the thiophene analog 7a. Structurally, 7a contains the bioisosteric thiophene ring instead of the methoxybenzene ring of 3 and a shorter phenylalkyl side chain at the exocyclic amino moiety. It remains to be analyzed, whether the bioisosteric replacement of the methoxybenzene ring or the reduction of the side chain length are responsible for the reduced GluN2B affinity of 7a.
4. Conclusion
The pharmacological data obtained with the thiophene derivatives 7a and 8a indicate that the bioisosteric replacement of the benzene, methoxybenzene or aniline ring of GluN2B ligands 3, 4, or 5 by the thiophene ring is well tolerated. In the case of the not further substituted [7]annulenothiophene 8a even an increased GluN2B affinity without loss of selectivity over both σ receptor subtypes was observed. Since the [7]annulenothiophene 8a without a benzylic OH moiety is even more potent than the analog 7a with a benzylic OH moiety it can be concluded that this OH moiety is not essential for high GluN2B affinity.
5. Experimental part
5.1. Chemistry, general
Unless otherwise noted, moisture sensitive reactions were conducted under dry nitrogen. CH2Cl2 was distilled over CaH2. THF was distilled over sodium/benzophenone. Et2O and toluene were dried over molecular sieve 0.4 Å. Thin layer chromatography (tlc): silica gel 60 F254 plates (Merck). Flash chromatography (fc): silica gel 60, 40–64 μm (Merck); parentheses include: diameter of the column (d), length of the stationary phase, fraction size (V), eluent. Melting point: melting point apparatus Mettler Toledo MP50 Melting Point System, uncorrected. MS: microOTOF-Q II (Bruker Daltonics); APCI, atmospheric pressure chemical ionization. IR: FT-IR spectrophotometer MIRacle 10 (Shimadzu) equipped with ATR technique. Nuclear magnetic resonance (NMR) spectra were recorded on Agilent 600-MR (600 MHz for 1H, 151 MHz for 13C) or Agilent 400-MR spectrometer (400 MHz for 1H, 101 MHz for 13C); δ in ppm related to tetramethylsilane and measured referring to CHCl3 (δ = 7.26 ppm (1H NMR) and δ = 77.2 ppm (13C NMR)), CHD2OD (δ = 3.31 ppm (1H NMR) and δ = 49.0 ppm (13C NMR)) and DMSO-d6 (δ = 2.54 ppm (1H NMR) and δ = 39.5 ppm (13C NMR)); coupling constants are given with 0.5 Hz resolution; the assignments of 13C and 1H NMR signals were supported by 2-D NMR techniques where necessary.
5.2. HPLC method for the determination of purity
Pump: LPG-3400SD, degasser: DG-1210, autosampler: ACC-3000T, UV-detector: VWD-3400RS, interface: DIONEX UltiMate 3000, data acquisition: Chromeleon 7 (Thermo Fisher Scientific); column: LiChrospher® 60 RP-select B (5 μm), LiChroCART® 250–4 mm cartridge; guard column: LiChrospher® 60 RP-select B (5 μm), LiChroCART® 4–4 mm cartridge (No.: 1.50963.0001), manu-CART® NT cartridge holder; flow rate: 1.0 mL min–1; injection volume: 5.0 μL; detection at λ = 210 nm; solvents: A: method 1: water with 0.05% (v/v) trifluoroacetic acid; method 2: water; B: method 1: acetonitrile with 0.05% (v/v) trifluoroacetic acid; method 2: acetonitrile: gradient elution: (A%): 0–4 min: 90%, 4–29 min: 90 → 0%, 29–31 min: 0%, 31–31.5 min: 0 → 90%, 31.5–40 min: 90%. The purity of all compounds was determined by this method. The purity of all test compounds is higher than 95% (unless otherwise noted).
5.3. Synthetic procedures
5.3.1. N-Methoxy-N-methyl-3-(thiophen-2-yl)propanamide (10)38
Under N2, carbonyldiimidazole (5.8 g, 36 mmol) was added to a solution of 3-(thiophen-2-yl)propanoic acid (9, 5 g, 32 mmol) in dry CH2Cl2 (100 mL) at room temperature. After the gas evolution stopped, the solution was stirred for another 45 min. Subsequently, N,O-dimethylhydroxylammonium chloride (3.4 mg, 35 mmol) was added. The resulting solution was stirred at room temperature overnight. An aqueous HCl solution (1 M, 50 mL) was added and vigorous stirring was continued for another 10 min. The aqueous layer was extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were washed with a saturated NaHCO3 solution (aq., 2 × 40 mL), brine (30 mL) and water (30 mL), dried (Na2SO4) and concentrated in vacuo. Rf = 0.25 (cyclohexane/ethyl acetate 7 : 3). Brown oil, yield 5.7 g (90%), purity (HPLC): 85.8%, (tR = 17.2 min), C9H13NO2S (199.3 g mol–1). 1H NMR (600 MHz, CDCl3): δ (ppm) = 2.81 (t, J = 7.7 Hz, 2H, 2 × 2-H), 3.16–3.20 (m, 5H, 2 × 3-H, NCH3), 3.64 (s, 3H, OCH3), 6.84 (dd, J = 3.3/1.0 Hz, 1H, 3-Hthioph), 6.92 (dd, J = 5.2/3.3 Hz, 1H, 4-Hthioph), 7.12 (dd, J = 5.1/1.3 Hz, 1H, 5-Hthioph). 13C NMR (151 MHz, CDCl3): δ (ppm) = 24.9 (C-3), 32.3 (NCH3), 34.1 (C-2), 61.4 (OCH3), 123.5 (C-5thioph), 124.8 (C-3thioph), 126.9 (C-4thioph), 144.1 (C-2thioph), 176.6 (C O). Exact MS (APCI): m/z = 200.0740 (calcd. 200.0740 for C9H14NO2S+ [M + H]+). FT-IR (neat): ṽ (cm–1) = 1728 (C Oamide).
5.3.2. 3-(Thiophen-2-yl)propanal (11)
A solution of Weinreb amide 10 (1 g, 5 mmol) in toluene was cooled to –78 °C. Subsequently, a solution of diisobutylaluminum hydride (20 mL, 24 mmol) in toluene was added dropwise. Within this addition, it was necessary that the reaction temperature remains below –70 °C. Ethyl acetate (10 mL) and aqueous HCl (1 M, 70 mL) were added at –78 °C. The mixture was warmed up to room temperature overnight. The aqueous layer was extracted with CH2Cl2 (5 × 20 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. The crude product was used for the following reaction without further purification. Rf = 0.41 (cyclohexane/ethyl acetate 9 : 1). Colorless oil, yield 0.81 g (100%), C7H8OS (140.2 g mol–1).
5.3.3. Ethyl 5-(thiophen-2-yl)pent-2-enoate (12)39
(Ethoxycarbonylmethylene)triphenylphosphorane (Ph3P CHCO2Et, 11 g, 32 mmol) was added to the mixture of aldehyde 11 (5.8 g, 29 mmol) in freshly distilled CH2Cl2 (100 mL). After 96 h at room temperature, the mixture was concentrated in vacuo and the residue was purified by flash column chromatography (ø = 6 cm, l = 12 cm, fraction size = 30 mL, cyclohexane/ethyl acetate 9 : 1, Rf = 0.51). Yellow oil, yield 4.3 g (80%), purity (HPLC): 92.6%, (tR = 22.3 min), C11H14O2S (210.3 g mol–1). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.28 (t, J = 7.1 Hz, 2.7H, OCH2CH3trans) 1.29 (t, J = 7.2 Hz, 0.3H, OCH2CH3cis), 2.58 (m, 2H, 2 × 4-Htrans&cis), 2.94–3.04 (m, 2H, 2 × 5-Htrans&cis), 4.17 (q, J = 7.1 Hz, 0.2H, OCH2CH3cis) 4.18 (q, J = 7.1 Hz, 1.8H, OCH2CH3trans), 5.82 (dt, J = 11.6/1.5 Hz, 0.1H, 2-Hcis), 5.87 (dt, J = 15.7/1.6 Hz, 0.9H, 2-Htrans), 6.81 (dd, J = 3.5/1.2 Hz, 1H, 3-Hthioph), 6.24 (dt, J = 11.5/7.0 Hz, 0.1H, 3-Hcis), 6.92 (dd, J = 5.1/3.4 Hz, 1H, 4-Hthioph), 6.99 (dt, J = 15.7/6.8 Hz, 0.9H, 3-Htrans), 7.13 (dd, J = 5.1/1.2 Hz, 1H, 5-Hthioph). Ratio trans : cis is 9 : 1. 13C NMR (101 MHz, CDCl3): δ (ppm) = 14.4 (OCH2CH3), 28.6 (C-5), 34.2 (C-4), 60.4 (OCH2CH3), 122.4 (C-2), 123.5 (C-5thioph), 124.7 (C-3thioph), 127.0 (C-4thioph), 143.6 (C-2thioph), 147.3 (C-3), 166.6 (C O). Exact MS (APCI): m/z = 211.0779 (calcd. 211.0787 for C11H15O2S+ [M + H]+). FT-IR (neat): ṽ (cm–1) = 2932 (C–H), 1717 (C Oα,β-unsat. ester).
5.3.4. Ethyl 3-(benzylamino)-5-(thiophen-2-yl)pentanoate (13)
Freshly distilled benzylamine (2.7 mL, 25 mmol) was added to α,β-unsaturated ester 12 (4.5 g, 22 mmol) in absolute ethanol (50 mL). The mixture was heated to reflux for 20 h under N2. Afterwards the mixture was concentrated in vacuo and the residue was purified by flash column chromatography (ø = 6 cm, l = 12 cm, fraction size = 30 mL, cyclohexane/ethyl acetate 9 : 1, Rf = 0.1). Yellow oil, yield 2.9 g (42%), purity (HPLC): 94.4%, (tR = 18.1 min), C18H23NO2S (317.45 g mol–1). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.69 (d, J = 35.0 Hz, 1H, NH), 1.82–1.98 (m, 2H, 2 × 4-H), 2.52 (d, J = 6.1 Hz, 2H, PhCH2N), 2.94 (td, J = 8.0/0.9 Hz, 2H, 2 × 2-H), 3.05–3.15 (m, 1H, 3-H), 3.75–3.86 (m, 2H, 2 × 5-H), 4.14 (qd, J = 7.1/0.6 Hz, 2H, OCH2CH3), 6.77 (dq, J = 3.3/1.0 Hz, 1H, 3-Hthioph), 6.91 (dd, J = 5.1/3.4 Hz, 1H, 4-Hthioph), 7.11 (dd, J = 5.1/1.2 Hz, 1H, 5-Hthioph), 7.23–7.27 (m, 1H, 4-HPh), 7.29–7.36 (m, 4H, 2,3,5,6-HPh). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.4 (OCH2CH3), 26.3 (C-5), 36.3 (C-4), 39.0 (C-2), 50.9 (PhCH2N), 53.7 (C-3), 60.6 (OCH2CH3), 123.2 (C-5thioph), 124.4 (C-3thioph), 126.9 (C-4thioph), 127.2 (C-4Ph), 128.4 (2C, C-3Ph, C-5Ph), 128.6 (2C, C-2Ph, 6-CPh), 140.2 (C-1Ph), 144.9 (C-2thioph), 172.4 (C O). A signal for the NH proton is not seen in the spectrum. Exact MS (APCI): m/z = 318.1528 (calcd. 318.1522 for C18H24NO2S+ [M + H]+). FT-IR (neat): ṽ (cm–1) = 2978 (C–H), 1728 (C O).
5.3.5. Ethyl 3-[N-benzyl-N-(4-methylphenylsulfonyl)amino]-5-(thiophen-2-yl)pentanoate (14)
N-Ethyl-N-isopropylpropan-2-amine (2.1 mL, 10 mmol) was added to a solution of benzylamine 13 (2.9 g, 9.1 mmol) in freshly distilled CH2Cl2 (100 mL). Subsequently 4-methylbenzenesulfonyl chloride (2.1 g, 11 mmol) was added and everything was stirred for 24 h under N2 at room temperature. The mixture was concentrated in vacuo and the residue was purified by flash column chromatography (ø = 6 cm, l = 12 cm, fraction size = 30 mL, cyclohexane/ethyl acetate 9 : 1, Rf = 0.08). Yellow solid, mp 86–87 °C yield 3.4 g (87%), purity (HPLC): 99.8%, (tR = 25.7 min), C25H29NO4S2 (471.6 g mol–1), melting point: 82.6 °C. 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.18 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.67–1.72 (m, 2H, 2 × 4-H), 2.31 (d, J = 7.0 Hz, 2H, 2 × 2-H), 2.43 (s, 3H, tosyl-CH3), 2.52–2.64 (m, 2H, 2 × 5-H), 4.01 (q, J = 7.2 Hz, 2H, OCH2CH3), 4.23 (d, J = 15.4 Hz, 1H, PhCH2N), 4.28 (quint, J = 6.9 Hz, 1H, 3-H), 4.50 (d, J = 15.4 Hz, 1H, PhCH2N), 6.51–6.52 (m, 1H, 3-Hthioph), 6.83 (dd, J = 5.1/3.4 Hz, 1H, 4-Hthioph), 7.04 (dd, J = 5.1/1.2 Hz, 1H, 5-Hthioph), 7.28–7.35 (m, 5H, 2,3,4,5,6-HPh), 7.38–7.41 (m, 2H, 3,5-Htosyl), 7.75 (d, J = 8.3 Hz, 2H, 2,6-Htosyl). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.2 (OCH2CH3), 21.7 (tosyl-CH3), 26.9 (C-5), 35.8 (C-4), 39.0 (C-2), 48.8 (PhCH2N), 55.5 (C-3), 60.9 (OCH2CH3), 123.1 (C-5thioph), 124.3 (C-3thioph), 126.8 (C-4thioph), 127.4 (2C, C-2tosyl, C-6tosyl), 128.1 (C-4tosyl), 128.7 (2C, C-2Ph, C-6Ph), 128.8 (2C, C-3tosyl,C-5tosyl), 129.9 (2C, C-3Ph, C-5Ph), 137.6 (C-4Ph), 137.9 (C-1Ph), 143.6 (C-1tosyl), 144.0 (C-2thioph), 170.9 (C O). Exact MS (APCI): m/z = 472.1622 (calcd. 472.1611 for C25H30NO4S2+ [M + H]+). FT-IR (neat): ṽ (cm–1) = 2978 (C–H), 1728 (C O), 1339 (S Osulfonamide).
5.3.6. 3-[N-Benzyl-N-(4-methylphenylsulfonyl)amino]-5-(thiophen-2-yl)pentanoic acid (15)
An aqueous NaOH solution (0.1 M, 15 mL) was added to a solution of ester 14 (0.64 g, 1.4 mmol) in THF. The mixture was stirred for 24 h at room temperature. The mixture was concentrated in vacuo and HCl (1 M, 20 mL) was added. Diethyl ether (30 mL) was added and the layers were separated. The aqueous layer was extracted with diethyl ether (3 × 30 mL) again. The combined organic layers were dried (Na2SO4) and concentrated in vacuo. The residue was purified by flash column chromatography (ø = 2 cm, l = 15 cm, fraction size 10 mL, CH2Cl2/MeOH 98 : 2 + 2% HCOOH, Rf = 0.47). Colorless solid, mp 82–83 °C yield 0.49 g (82%), purity (HPLC): 98.5%, (tR = 22.2 min), C23H25NO4S2 (443,6 g mol–1). 1H NMR (600 MHz, CDCl3-d): δ (ppm) = 1.73 (ddt, J = 11.4/9.3/4.1 Hz, 2H, 2 × 4-H), 2.23–2.39 (m, 2H, 2 × 2-H), 2.42 (s, 3H, p-tosyl-CH3), 2.50–2.64 (m, 2H, 2 × 5-H), 4.25 (d, J = 15.5 Hz, 2H, 3-H, PhCH2N), 4.48 (d, J = 15.4 Hz, 1H, PhCH2N), 6.48–6.53 (m, 1H, 3-Hthioph), 6.82 (dd, J = 5.1/3.4 Hz, 1H, 4-Hthioph), 7.04 (dd, J = 5.1/1.3 Hz, 1H, 5-Hthioph), 7.27–7.34 (m, 5H, 2,3,4,5,6-HPh), 7.36–7.42 (m, 2H, 3,5-Htosyl), 7.71–7.77 (m, 2H, 2,6-Htosyl). 13C NMR (151 MHz, CDCl3-d): δ (ppm) = 21.7 (p-tosyl-CH3), 27.1 (C-5), 35.6 (C-4), 38.6 (C-2), 48.8 (PhCH2N), 55.2 (C-3), 123.2 (C-5thioph), 126.8 (C-3thioph), 127.4 (C-4thioph), 127.4 (2C, C-2tosyl, C-6tosyl), 128.1 (C-4Ph), 128.7 (2C, C-3tosyl, C-5tosyl), 128.8 (2C, C-2Ph, C-6Ph), 129.9 (2C, C-3Ph, C-5Ph), 130.0 (C-1tosyl), 137.4 (C-1Ph), 143.7 (C-4tosyl), 143.8 (C-2thioph), 174.9 (C O). Exact MS (APCI): m/z = 426.1183 (calcd. 426.1192 for C23H24NO3S2+ [M-H2O + H]+). FT-IR (neat): ṽ (cm–1) = 3063 (O–Hacid), 1709 (C Oacid), 1335 (S Osulfonamide).
5.3.7. N-Benzyl-4-methyl-N-(4-oxo-5,6,7,8-tetrahydro-4H-[7]annuleno[b]thiophen-6-yl)benzenesulfonamide (16)
Under N2, P4O10 (0.18 mg, 1.3 mmol) was added to a solution carboxylic acid 15 (0.01 M, 97 mg, 0.22 mmol) in freshly distilled CH2Cl2 (22 mL) at –10 °C. The mixture was stirred overnight under N2. NaOH (aq., 0.1 M, 30 mL) was added. The mixture was filtered, the filter was washed with ethyl acetate (3 × 30 mL) and CH2Cl2 (2 × 15 mL). The remaining solid was mixed with water and stirred extensively overnight. This mixture was extracted with ethyl acetate (3 × 10 mL) and CH2Cl2 (2 × 5 mL). The filtrate was extracted with ethyl acetate (10 mL) and CH2Cl2 (5 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. The crude product was purified by flash column chromatography (ø = 1 cm, l = 15 cm, fraction size = 3 mL, CH2Cl2/MeOH 99 : 1 + 1% HCOOH, Rf = 0.41). Colorless oil, yield 24 mg (25%), purity (HPLC): 83%, (tR = 23.3 min), C23H23NO3S2 (425.6 g mol–1). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.89 (dddd, J = 13.8/9.3/7.3/4.2 Hz, 1H, 7-H), 2.12 (dddd, J = 13.2/9.4/6.7/3.7 Hz, 1H, 7-H), 2.45 (s, 3H, p-tosyl-CH3), 2.63 (dd, J = 14.7/3.7 Hz, 1H, 5-H), 2.94–2.84 (m, 2H, 8-H, 5-H), 3.02 (ddd, J = 16.7/9.4/4.1 Hz, 8-H), 4.24 (d, J = 16.1 Hz, 1H, PhCH2N), 4.30–4.39 (m, 1H, 6-H), 4.55 (d, J = 16.1 Hz, 1H, PhCH2N), 6.96 (d, J = 5.4 Hz, 1H, 2-Hthioph), 7.27–7.37 (m, 8H, 2,3,4,5,6-HPh, 3,5-Htosyl, 3-Hthioph), 7.68–7.72 (m, 2H, 2,6-Htosyl). 13C NMR (101 MHz, CDCl3): δ (ppm) = 21.7 (p-tosyl-CH3), 26.0 (C-7), 32.9 (C-8), 47.2 (C-5), 48.7 (PhCH2N), 54.2 (C-6), 122.4 (C-2thioph), 127.4 (2C, C-2tosyl, C-6tosyl), 127.8 (C-4Ph), 128.8 (C-3thioph), 128.8 (2C, C-2Ph, C-6Ph), 129.9 (2C, C-3Ph, C-5Ph), 130.1 (2C, C-3tosyl, C-5tosyl), 139.0 (C-3athioph), 143.8 (C-4tosyl), 152.0 (C-8athioph), 193.1 (C-8). Exact MS (APCI): m/z = 426.1208 (calcd. 426.1192 for C23H24NO3S2+ [M + H]+). FT-IR (neat): ṽ (cm–1) = 1663 (C Oaryl ketone), 1331 (S Osulfonamide).
5.3.8. N-Benzyl-N-(4-hydroxy-5,6,7,8-tetrahydro-4H-[7]annuleno[b]thiophen-6-yl)-4 methylbenzenesulfonamide (17)
Under N2, NaBH4 (9 mg, 0.5 mmol) was added to a solution of ketone 16 (100 mg, 0.24 mmol) in freshly distilled MeOH (15 mL) at 0 °C. After 45 min, the mixture was warmed up to room temperature. After another 2 h the solvent was removed in vacuo. Water (5 mL) was added to the residue. The mixture was extracted with CH2Cl2 (2 × 10 mL). The organic layer was washed with water, dried (Na2SO4) and concentrated in vacuo. The crude product is purified by flash column chromatography (ø = 2 cm, l = 15 cm, fraction size = 10 mL, cyclohexane/ethyl acetate 1 : 1, Rf = 0.48). Yellow oil, yield 54 mg (54%), C23H25NO3S2 (427.6 g mol–1). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.44 (m, 1H, 7-H), 1.68–1.81 (m, 2H, 5-H, 7-H), 1.87–1.93 (m, 1H, 5-H), 2.45 (s, 3H, p-tosyl-CH3), 2.50 (ddd, J = 15.5/11.9/2.4 Hz, 1H, 8-H), 2.82 (ddd, J = 15.8/6.4/2.3 Hz, 1H, 8-H), 4.04–4.16 (m, 1H, 6-H), 4.33 (d, J = 16.0 Hz, 0.85H, PhCH2N), 4.34 (d, J = 16.0 Hz, 0.15H, PhCH2N), 4.43 (d, J = 15.9 Hz, 0.85H, PhCH2N), 4.44 (d, J = 16.0 Hz, 0.15H, PhCH2N), 4.64 (dd, J = 10.6/2.8 Hz, 0.85H, 4-H), 4.87 (dd, J = 4.6/1.4 Hz, 0.15H, 4-H), 6.93 (d, J = 5.2 Hz, 1H, 3-Hthioph), 7.01 (d, J = 5.3 Hz, 1H, 2-Hthioph), 7.27–7.37 (m, 7H, 2,3,4,5,6-HPh, 3,5-Htosyl), 7.70–7.75 (m, 2H, 2,6-Htosyl). Ratio of the diastereomers is 85 : 15. A signal for the OH proton is not seen in the spectrum.
5.3.9. 6-(Benzylamino)-5,6,7,8-tetrahydro-4H-[7]annuleno[b]thiophen-4-ol (7a)
Under N2, Mg (322 mg, 13.3 mmol) was added to a slurry of alcohol 17 (354 mg, 0.83 mmol) in freshly distilled MeOH (10 mL). After 4 h in the ultrasonic bath, another portion of elemental Mg (294 mg, 12.1 mmol) was added. After another 3 h, acetic acid (20 mL) was added (pH: 5). Subsequently a NH3/NH4+ buffer was added, and the mixture was extracted with CH2Cl2 (3 × 15 mL). The organic layer was concentrated in vacuo. The residue was purified by flash column chromatography (ø = 2 cm, l = 15 cm, fraction size = 15 mL, cyclohexane/ethyl acetate 1 : 1 + 1% N,N-dimethylethanamine, Rf = 0.15). Colorless solid, mp 105–106 °C, yield 146 mg (64%), purity (HPLC): 96.4%, (tR = 14.2 min), C16H19NOS (273.4 g mol–1). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.92 (ddd, J = 14.7/3.0/1.9 Hz, 1H, 5-H), 1.96–2.13 (m, 2H, 2 × 7-H), 2.44 (ddd, J = 14.6/6.4/5.2 Hz, 1H, 5-H), 2.74 (ddd, J = 16.0/7.9/2.4 Hz, 1H, 8-H), 3.24 (ddd, J = 16.1/10.3/2.4 Hz, 1H, 8-H), 3.36–3.43 (m, 1H, 6-H), 3.85 (d, J = 12.9 Hz, 0.85H, PhCH2N), 3.88 (d, J = 13.1 Hz, 0.15H, PhCH2N), 3.91 (d, J = 13.1 Hz, 0.15H, PhCH2N), 4.02 (d, J = 12.8 Hz, 0.85H, PhCH2N), 4.96 (dd, J = 6.5/1.9 Hz, 0.85H, 4-H), 5.09 (dd, J = 7.3/1.8 Hz, 0.15H, 4-H), 6.93 (d, J = 5.2 Hz, 1H, 3-Hthioph), 6.94 (d, J = 5.2 Hz, 1H, 2-Hthioph), 7.26–7.42 (m, 5H, 2,3,4,5,6-HPh). Signals for the NH and OH protons are not seen in the spectrum. Ratio of the diastereomeres is 85 : 15. 13C NMR (151 MHz, CDCl3): δ (ppm) = 23.6 (C-8), 33.5 (C-7), 35.4 (C-5), 51.6 (PhCH2N), 56.8 (C-6), 70.1 (C-4), 120.8 (C-4thioph), 127.6 (C-4Ph), 128.5 (2C, C-2Ph, C-4Ph), 128.8 (2C, C-3Ph, C-5Ph), 129.9 (C-5thioph), 138.9 (C-1Ph) 139.3 (C-2thioph), 142.7 (C-3thioph). Exact MS (APCI): m/z = 274.1266 (calcd. 274.1260 for C16H20NOS [M + H]+). FT-IR (neat): ṽ (cm–1) = 2978 (C–H), 2947 (O–H), 2916 (N–H).
5.3.10. Dimethyl 6-oxo-5,6,7,8-tetrahydro-4H-[7]annuleno[b]thiophene-5,7-dicarboxylate (20)
Dibromide 1942 (0.8 g, 3 mmol) was dissolved in dry THF (30 mL) and anhydrous K2CO3 (1 g, 7.4 mmol) was added. Subsequently, dimethyl 3-oxoglutarate (0.5 mL, 3.3 mmol) was added. The mixture was heated to reflux for 18 h under N2. Afterwards, the mixture was filtered through Celite®, washed with ethyl acetate (3 × 20 mL), the organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by flash column chromatography (ø = 5 cm, l = 12 cm, fraction size = 15 mL, cyclohexane/ethyl acetate 1 : 1, Rf = 0.48). Brown oil, yield 333 mg (40%), purity (HPLC): 40/53%, (tR = 18.5/18.8 min), C13H14O5S (282.3 g mol–1). 1H NMR (600 MHz, CDCl3): δ (ppm) = 2.98–3.29 (m, 6H, 2 × 4-H, 2 × 8-H, 5-H, 7-H), 3.66–3.82 (m, 6H, 5,7-CO2CH3), 6.89 (d, J = 5.1 Hz, 1H, 3-Hthioph), 7.06 (d, J = 5.0 Hz, 1H, 2-Hthioph). The shifts in the 1H-NMR spectrum represent the sum of all possible isomers, including cis/trans-diastereomers as well as keto/enol-tautomers. The 1H-NMR spectrum is rather complicated, since compound 20 exists as a mixture of two regioisomeric enols and two diastereomeric β-ketodiesters. Exact MS (APCI): m/z = 283.0608 (calcd. 283.0635 for C13H15O5S+ [M + H]+). FT-IR (neat): ṽ (cm–1) = 1736 (C Oester).
5.3.11. 4,5,7,8-Tetrahydro[7]annuleno[b]thiophen-6-one (21)
Diester 20 (511 mg, 1.81 mmol) was dissolved in methanol (20 mL). Aqueous HCl (6 M, 10 mL) was added and the mixture was heated to reflux for 8 h. Once the excessive gas evolution stopped, the mixture was extracted with ethyl acetate (3 × 20 mL) and washed with saturated NaHCO3 solution (aq., 20 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. The residue was purified by flash column chromatography (ø = 3 cm, l = 15 cm, fraction size = 15 ml, cyclohexane/ethyl acetate 5 : 1, Rf = 0.27). Colorless oil, yield 160 mg (53%), purity (HPLC): 85.3%, (tR = 17.4 min), C9H10OS (166.2 g mol–1). 1H NMR (400 MHz, CDCl3): δ (ppm) = 2.72–2.78 (m, 4H, 2 × 5-H, 2 × 7-H), 2.90–2.96 (m, 2H, 2 × 4-H), 3.02–3.08 (m, 2H, 2 × 8-H), 6.78–6.81 (d, 5.1 Hz, 1H, 3-Hthioph), 7.00 (d, J = 5.1 Hz, 1H, 2-Hthioph). 13C NMR (101 MHz, CDCl3): δ (ppm) = 24.5 (C-8), 25.7 (C-4), 42.8 (C-7), 43.6 (C-5), 121.5 (C-2thioph), 130.2 (C-3thioph), 136.7 (C-3athioph), 137.3 (C-8athioph), 212.6 (C-6). Exact MS (APCI): m/z = 167.0528 (calcd. 167.0525 for C9H11OS+ [M + H+]). FT-IR (neat): ṽ (cm–1) = 1735 (C O).
5.3.12. N-Benzyl-5,6,7,8-tetrahydro-4H-[7]annuleno[b]thiophen-6-amine (8a)
Ketone 21 (196 mg, 1.2 mmol) was dissolved in CH2Cl2 (10 mL). Under N2, benzylamine (0.21 mL, 1.9 mmol) was added. Afterwards, NaBH(OAc)3 (468 mg, 2.2 mmol) was added and the mixture was stirred overnight at room temperature. A saturated NaHCO3 solution (aq., 20 mL) was added. The mixture was extracted with CH2Cl2 (3 × 20 mL), the organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by flash chromatography (ø = 3 cm, l = 15 cm, fraction size = 10 mL, cyclohexane/ethyl acetate/2 : 1 + 0.5% N,N-dimethylethanamine, Rf = 0.24). Colorless oil, yield 191 mg (63%), purity (HPLC): 93%, (tR = 17 min), C16H19NS (257.4 g mol–1). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.49–1.54 (m, 1H, 7-H), 1.54–1.60 (m, 1H, 7-H), 2.01–2.10 (m, 2H, 5-H, 7-H), 2.53 (ddd, J = 15.4/11.0/2.0 Hz, 1H, 4-H), 2.68 (ddd, J = 15.9/10.8/2.0 Hz, 1H, 8-H), 2.87 (tt, J = 9.1/3.0 Hz, 1H, 6-H), 2.92 (ddd, J = 15.4/7.9/2.0 Hz, 1H, 4-H), 3.00 (ddd, J = 15.8/7.9/2.0 Hz, 1H, 8-H), 3.87 (s, 2H, PhCH2N), 6.76 (d, J = 5.1 Hz, 1H, 3-Hthioph), 6.88 (d, J = 5.0 Hz, 1H, 2-Hthioph), 7.25–7.28 (m, 1H, 4-HPh), 7.32–7.37 (m, 4H, 2,3,5,6-HPh). A signal for the NH proton is not seen in the spectrum. 13C NMR (151 MHz, CDCl3): δ (ppm) = 25.4 (C-8), 26.2 (C-4), 33.9 (C-7), 34.3 (C-5), 51.3 (PhCH2N), 60.2 (C-6), 119.8 (C-2thioph), 127.0 (2C, C-3Ph, C-5Ph), 128.2 (C-4Ph), 128.6 (2C, C-2Ph, C-6Ph), 130.4 (C-3thioph), 138.8 (C-3athioph), 140.0 (C-8athioph), 140.9 (C-1Ph). Exact MS (APCI): m/z = 258.1297 (calcd. 258.1311 for C16H20NS+ [M + H]+). FT-IR (neat): ṽ (cm–1): 3024 (N–H), 2920 (C–H).
5.4. Receptor binding studies
5.4.1. Materials
Guinea pig brains, rat brains and rat livers were commercially available (Harlan-Winkelmann, Borchen, Germany). Pig brains were a donation of the local slaughterhouse (Coesfeld, Germany). The recombinant L(tk-) cells stably expressing the GluN2B receptor were obtained from Prof. Dr. Dieter Steinhilber (Frankfurt, Germany). Homogenizers: Elvehjem Potter (B. Braun Biotech International, Melsungen, Germany) and Soniprep® 150 (MSE, London, UK). Centrifuges: cooling centrifuge Eppendorf 5427R (Eppendorf, Hamburg, Germany) and high-speed cooling centrifuge model Sorvall® RC-5C plus (Thermo Fisher Scientific, Langenselbold, Germany). Multiplates: standard 96 well multiplates (Diagonal, Muenster, Germany). Shaker: self-made device with adjustable temperature and tumbling speed (scientific workshop of the institute). Harvester: MicroBeta® FilterMate 96 Harvester. Filter: Printed Filtermat Type A and B. Scintillator: Meltilex® (Typ A or B) solid state scintillator. Scintillation analyzer: MicroBeta® Trilux (all Perkin Elmer LAS, Rodgau-Jügesheim, Germany).
5.4.2. Cell culture and preparation of membrane homogenates from GluN2B cells
Mouse L(tk-) cells stably transfected with the dexamethasone-inducible eukaryotic expression vectors pMSG GluN1a, pMSG GluN2B (1 : 5 ratio) were grown in modified Earl's medium (MEM) containing 10% of standardized FCS (Biochrom AG, Berlin, Germany). The expression of the NMDA receptor at the cell surface was induced after the cell density of the adherent growing cells had reached approximately 90% of confluency. For the induction, the original growth medium was replaced by growth medium containing 4 μM dexamethasone and 4 μM ketamine (final concentration). After 24 h, the cells were rinsed with phosphate buffered saline solution (PBS, Biochrom AG, Berlin, Germany), harvested by mechanical detachment and pelleted (10 min, 1200 × g).
For the binding assay, the cell pellet was resuspended in PBS solution and the number of cells was determined using a Scepter® cell counter (MERCK Millipore, Darmstadt, Germany). Subsequently, the cells were lysed by sonication (4 °C, 6 × 10 s cycles with breaks of 10 s). The resulting cell fragments were centrifuged with a high performance cool centrifuge (23 500 × g, 4 °C). The supernatant was discarded and the pellet was resuspended in a defined volume of PBS yielding cell fragments of approximately 500 000 cells per mL. The suspension of membrane homogenates was sonicated again (4 °C, 2 × 10 s cycles with a break of 10 s) and stored at –80 °C.
5.4.3. Preparation of membrane homogenates from Guinea pig brain
5 guinea pig brains were homogenized with the potter (500–800 rpm, 10 up and down strokes) in 6 volumes of cold 0.32 M sucrose. The suspension was centrifuged at 1200 × g for 10 min at 4 °C. The supernatant was separated and centrifuged at 23 500 × g for 20 min at 4 °C. The pellet was resuspended in 5–6 volumes of buffer (50 mM TRIS, pH 7.4) and centrifuged again at 23 500 × g (20 min, 4 °C). This procedure was repeated twice. The final pellet was resuspended in 5–6 volumes of buffer and frozen (–80 °C) in 1.5 mL portions containing about 1.5 mg protein per mL.
5.4.4. Preparation of membrane homogenates from rat liver
Two rat livers were cut into small pieces and homogenized with the potter (500–800 rpm, 10 up and down strokes) in 6 volumes of cold 0.32 M sucrose. The suspension was centrifuged at 1200 × g for 10 min at 4 °C. The supernatant was separated and centrifuged at 31 000 × g for 20 min at 4 °C. The pellet was resuspended in 5–6 volumes of buffer (50 mM TRIS, pH 8.0) and incubated at rt for 30 min. After the incubation, the suspension was centrifuged again at 31 000 × g for 20 min at 4 °C. The final pellet was resuspended in 5–6 volumes of buffer and stored at –80 °C in 1.5 mL portions containing about 2 mg protein per mL.
5.4.5. Protein determination
The protein concentration was determined by the method of Bradford,47 modified by Stoscheck.48 The Bradford solution was prepared by dissolving 5 mg of Coomassie Brilliant Blue G 250 in 2.5 mL of EtOH (95%, v/v). 10 mL deionized H2O and 5 mL phosphoric acid (85%, m/v) were added to this solution, the mixture was stirred and filled to a total volume of 50 mL with deionized water. The calibration was carried out using bovine serum albumin as a standard in 9 concentrations (0.1, 0.2, 0.4, 0.6, 0.8, 1.0, 1.5, 2.0 and 4.0 mg mL–1). In a 96 well standard multiplate, 10 μL of the calibration solution or 10 μL of the membrane receptor preparation were mixed with 190 μL of the Bradford solution, respectively. After 5 min, the UV absorption of the protein–dye complex at λ = 595 nm was measured with a plate reader (Tecan Genios®, Tecan, Crailsheim, Germany).
5.4.6. General procedures for the binding assays
The test compound solutions were prepared by dissolving approximately 110 μmol (usually 2–4 mg) of test compound in DMSO so that a 10 mM stock solution was obtained. To obtain the required test solutions for the assay, the DMSO stock solution was diluted with the respective assay buffer. The filtermats were presoaked in 0.5% aqueous polyethylenimine solution for 2 h at rt before use. All binding experiments were carried out in duplicates in the 96 well multiplates. The concentrations given are the final concentration in the assay. Generally, the assays were performed by addition of 50 μL of the respective assay buffer, 50 μL of test compound solution in various concentrations (10–5, 10–6, 10–7, 10–8, 10–9 and 10–10 mol L–1), 50 μL of the corresponding radioligand solution and 50 μL of the respective receptor preparation into each well of the multiplate (total volume 200 μL). The receptor preparation was always added last. During the incubation, the multiplates were shaken at a speed of 500–600 rpm at the specified temperature. Unless otherwise noted, the assays were terminated after 120 min by rapid filtration using the harvester. During the filtration, each well was washed five times with 300 μL of water. Subsequently, the filtermats were dried at 95 °C. The solid scintillator was melted on the dried filtermats at a temperature of 95 °C for 5 min. After solidifying of the scintillator at rt, the trapped radioactivity in the filtermats was measured with the scintillation analyzer. Each position on the filtermat corresponding to one well of the multiplate was measured for 5 min with the [3H]-counting protocol. The overall counting efficiency was 20%. The IC50 values were calculated with the program GraphPad Prism® 3.0 (GraphPad Software, San Diego, CA, USA) by non-linear regression analysis. Subsequently, the IC50 values were transformed into Ki values using the equation of Cheng and Prusoff.49 The Ki values are given as mean value ± SEM from three independent experiments.
5.4.7. Ifenprodil binding site of the NMDA receptor30
The competitive binding assay was performed with the radioligand [3H]ifenprodil (60 Ci mmol–1; BIOTREND, Cologne, Germany). The thawed cell membrane preparation from the transfected L(tk-) cells (about 20 μg protein) was incubated with various concentrations of test compounds, 5 nM [3H]-ifenprodil, and TRIS/EDTA-buffer (5 mM TRIS/1 mM EDTA, pH 7.5) at 37 °C. The non-specific binding was determined with 10 μM unlabeled ifenprodil. The Kd value of ifenprodil is 7.6 nM.30
5.4.8. Affinity towards the σ1 receptor44–46
The assay was performed with the radioligand [3H]-(+)-pentazocine (22.0 Ci mmol–1; Perkin Elmer). The thawed membrane preparation of guinea pig brain cortex (about 100 μg of the protein) was incubated with various concentrations of test compounds, 2 nM [3H]-(+)-pentazocine, and TRIS buffer (50 mM, pH 7.4) at 37 °C. The non-specific binding was determined with 10 μM unlabeled (+)-pentazocine. The Kd value of (+)-pentazocine is 2.9 nM.50
5.4.9. Affinity towards the σ2 receptor44–46
The assays were performed with the radioligand [3H]di-o-tolylguanidine (specific activity 50 Ci mmol–1; ARC, St. Louis, MO, USA). The thawed rat liver membrane preparation (about 100 μg protein) was incubated with various concentrations of the test compound, 3 nM [3H]di-o-tolylguanidine and buffer containing (+)-pentazocine (500 nM (+)-pentazocine in TRIS buffer (50 mM TRIS, pH 8.0)) at rt. The non-specific binding was determined with 10 μM non-labeled di-o-tolylguanidine. The Kd value of di-o-tolylguanidine is 17.9 nM.51
Conflicts of interest
The authors declare no conflict of interest.
Supplementary Material
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) which is gratefully acknowledged. Moreover, we are grateful to Cells-in-Motion (CiM) Cluster of Excellence for supporting this project.
Footnotes
†Electronic supplementary information (ESI) available: Contains the NMR spectra of synthesized compounds. See DOI: 10.1039/c8md00545a
References
- McBain C. J., Mayer M. L. Physiol. Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- Liu H., Wang H., Sheng M., Jan L. Y., Jan Y. N., Basbaum A. I. Proc. Natl. Acad. Sci. U. S. A. 1994;91:8383–8387. doi: 10.1073/pnas.91.18.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R., Borges K., Bowie D., Traynelis S. F. Pharmacol. Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Eriksson M., Nilsson A., Froelich-Fabre S., Åkesson E., Dunker J., Seiger Å., Folkesson R., Benedikz E., Sundström E. Neurosci. Lett. 2002;321:177–181. doi: 10.1016/s0304-3940(01)02524-1. [DOI] [PubMed] [Google Scholar]
- Paoletti P., Neyton J. Curr. Opin. Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Loftis J. M., Janowsky A. Pharmacol. Ther. 2003;97:55–85. doi: 10.1016/s0163-7258(02)00302-9. [DOI] [PubMed] [Google Scholar]
- Paoletti P. Eur. J. Neurosci. 2011;33:1351–1365. doi: 10.1111/j.1460-9568.2011.07628.x. [DOI] [PubMed] [Google Scholar]
- Nowak L., Bregestovski P., Ascher P., Herbet A., Prochiantz A. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Kleckner N. W., Dingledine R. Science. 1988;241:835–837. doi: 10.1126/science.2841759. [DOI] [PubMed] [Google Scholar]
- Clayton D. A., Mesches M. H., Alvarez E., Bickford P. C., Browning M. D. J. Neurosci. 2002;22:3628–3637. doi: 10.1523/JNEUROSCI.22-09-03628.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y. P., Shimizu E., Dube G. R., Rampon C., Kerchner G. A., Zhuo M., Liu G., Tsien J. Z. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Thomas K. L., Davis S., Hunt S. P., Laroche S. Learn. Mem. 1996;3:197–208. doi: 10.1101/lm.3.2-3.197. [DOI] [PubMed] [Google Scholar]
- Mody I. Trends Pharmacol. Sci. 1995;16:356–359. doi: 10.1016/s0165-6147(00)89070-7. [DOI] [PubMed] [Google Scholar]
- Dunah A. W., Wang Y., Yasuda R. P., Kameyama K., Huganir R. L., Wolfe B. B., Standaert D. G. Mol. Pharmacol. 2000:342–352. [PubMed] [Google Scholar]
- Oh J. D., Russell D., Vaughan C. L., Chase T. N. Brain Res. 1998;813:150–159. doi: 10.1016/s0006-8993(98)01049-x. [DOI] [PubMed] [Google Scholar]
- Chen N., Luo T., Wellington C., Metzler M., McCutcheon K., Hayden M. R., Raymond L. A. J. Neurochem. 1999;72:1890–1898. doi: 10.1046/j.1471-4159.1999.0721890.x. [DOI] [PubMed] [Google Scholar]
- Zeron M. M., Chen N., Moshaver A., Lee A. T., Wellington C. L., Hayden M. R., Raymond L. A. Mol. Cell. Neurosci. 2001;17:41–53. doi: 10.1006/mcne.2000.0909. [DOI] [PubMed] [Google Scholar]
- Cheng C., Fass D. M., Reynolds I. J. Brain Res. 1999;849:97–108. doi: 10.1016/s0006-8993(99)01995-2. [DOI] [PubMed] [Google Scholar]
- Kim W.-T., Kuo M.-F., Mishra O. P., Delivoria-Papadopoulos M. Brain Res. 1998;799:49–54. doi: 10.1016/s0006-8993(98)00464-8. [DOI] [PubMed] [Google Scholar]
- Kalluri H. S. G., Mehta A. K., Ticku M. K. Mol. Brain Res. 1998;58:221–224. doi: 10.1016/s0169-328x(98)00112-0. [DOI] [PubMed] [Google Scholar]
- Narita M., Soma M., Narita M., Mizoguchi H., Tseng L. F., Suzuki T. Eur. J. Pharmacol. 2000;401:191–195. doi: 10.1016/s0014-2999(00)00428-3. [DOI] [PubMed] [Google Scholar]
- Narita M., Aoki T., Suzuki T. Neuroscience. 2000;101:601–606. doi: 10.1016/s0306-4522(00)00405-x. [DOI] [PubMed] [Google Scholar]
- Sze C.-I., Bi H., Kleinschmidt-DeMasters B. K., Filley C. M., Martin L. J. J. Neurol. Sci. 2001;182:151–159. doi: 10.1016/s0022-510x(00)00467-6. [DOI] [PubMed] [Google Scholar]
- Kemp J. A. and Kew J. N. C., Gill R. NMDA receptor antagonists and their potential as neuroprotective agents, Springer-Verlag, Berlin Heidelberg, 1999. [Google Scholar]
- Stark H., Graßmann S., Reichert U. Pharm. Unserer Zeit. 2000;29:159–166. doi: 10.1002/(SICI)1615-1003(200005)29:3<159::AID-PAUZ159>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Masuko T., Kashiwagi K., Kuno T., Nguyen N. D., Pahk A. J., Fukuchi J., Igarashi K., Williams K. Mol. Pharmacol. 1999;55:957–969. doi: 10.1124/mol.55.6.957. [DOI] [PubMed] [Google Scholar]
- Karakas E., Simorowski N., Furukawa H. Nature. 2011;475:249–253. doi: 10.1038/nature10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carron C., Jullien A., Bucher B. Arzneim. Forsch. 1971;21:1992–1998. [PubMed] [Google Scholar]
- Williams K. Mol. Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Schepmann D., Frehland B., Lehmkuhl K., Tewes B., Wünsch B. J. Pharm. Biomed. Anal. 2010;53:603–608. doi: 10.1016/j.jpba.2010.04.014. [DOI] [PubMed] [Google Scholar]
- Kew J. N., Trube G., Kemp J. A. J. Physiol. 1996;497:761–772. doi: 10.1113/jphysiol.1996.sp021807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck E., Begrow F., Verspohl E., Wünsch B. J. Pharm. Biomed. Anal. 2014;88:96–105. doi: 10.1016/j.jpba.2013.08.014. [DOI] [PubMed] [Google Scholar]
- Fischer G., Mutel V., Trube G., Malherbe P., Kew J. N., Mohacsi E., Heitz M. P., Kemp J. A. J. Pharmacol. Exp. Ther. 1997;283:1285–1292. [PubMed] [Google Scholar]
- Benner A., Bonifazi A., Shirataki C., Temme L., Schepmann D., Quaglia W., Shoji O., Watanabe Y., Daniliuc C., Wünsch B. ChemMedChem. 2014;9:741–751. doi: 10.1002/cmdc.201300547. [DOI] [PubMed] [Google Scholar]
- Gawaskar S., Schepmann D., Bonifazi A., Wünsch B. Bioorg. Med. Chem. 2014;22:6638–6646. doi: 10.1016/j.bmc.2014.10.004. [DOI] [PubMed] [Google Scholar]
- Gawaskar S., Temme L., Schreiber J. A., Schepmann D., Bonifazi A., Robaa D., Sippl W., Strutz-Seebohm N., Seebohm G., Wünsch B. ChemMedChem. 2017;12:1212–1222. doi: 10.1002/cmdc.201700311. [DOI] [PubMed] [Google Scholar]
- Brown N. Mol. Inf. 2014;33:458–462. doi: 10.1002/minf.201400037. [DOI] [PubMed] [Google Scholar]
- Rudzinski D. M., Kelly C. B., Leadbeater N. E. Chem. Commun. 2012;48:9610–9612. doi: 10.1039/c2cc35037h. [DOI] [PubMed] [Google Scholar]
- Panther J., Röhrich A., Müller T. J. J. ARKIVOC. 2011;2012:297. [Google Scholar]
- Junker A., Yamaguchi J., Itami K., Wünsch B. J. Org. Chem. 2013;78:5579–5586. doi: 10.1021/jo400692p. [DOI] [PubMed] [Google Scholar]
- Tewes B., Frehland B., Schepmann D., Schmidtke K.-U., Winckler T., Wünsch B. Bioorg. Med. Chem. 2010;18:8005–8015. doi: 10.1016/j.bmc.2010.09.026. [DOI] [PubMed] [Google Scholar]
- Dey T., Navarathne D., Invernale M. A., Berghorn I. D., Sotzing G. A. Tetrahedron Lett. 2010;51:2089–2091. [Google Scholar]
- Abdel-Magid A. F., Mehrman S. J. Org. Process Res. Dev. 2006;10:971–1031. [Google Scholar]
- Hasebein P., Frehland B., Lehmkuhl K., Fröhlich R., Schepmann D., Wünsch B. Org. Biomol. Chem. 2014;12:5407–5426. doi: 10.1039/c4ob00510d. [DOI] [PubMed] [Google Scholar]
- Meyer C., Neue B., Schepmann D., Yanagisawa S., Yamaguchi J., Würthwein E.-U., Itami K., Wünsch B. Bioorg. Med. Chem. 2013;21:1844–1856. doi: 10.1016/j.bmc.2013.01.038. [DOI] [PubMed] [Google Scholar]
- Miyata K., Schepmann D., Wünsch B. Eur. J. Med. Chem. 2014;83:709–716. doi: 10.1016/j.ejmech.2014.06.073. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Stoscheck C. M. Methods Enzymol. 1990;182:50–68. doi: 10.1016/0076-6879(90)82008-p. [DOI] [PubMed] [Google Scholar]
- Cheng Y., Prusoff W. H. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- DeHaven-Hudkins D. L., Fleissner L. C., Ford-Rice F. Y. Eur. J. Pharmacol. 1992;227:371–378. doi: 10.1016/0922-4106(92)90153-m. [DOI] [PubMed] [Google Scholar]
- Mach R. H., Smith C. R., Childers S. R. Life Sci. 1995;57:PL57–PL62. doi: 10.1016/0024-3205(95)00301-l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.