

Abstract
We report Mycobacterium chimaera pulmonary disease in 4 patients given a diagnosis of cystic fibrosis in a university hospital in Montpellier, France. All patients had M. chimaera–positive expectorated sputum specimens, clinical symptoms of pulmonary exacerbation, or a decrease in spirometry test results that improved after specific treatment.
Keywords: Mycobacterium chimaera, bacteria, Mycobacterium avium complex, tuberculosis and other mycobacteria, nontuberculosis mycobacteria, NTM, respiratory infections, cystic fibrosis, pulmonary disease, forced expiratory volume, forced vital capacity, France
Mycobacterium chimaera is a member of the Mycobacterium avium complex, which was elevated to species rank in 2004. M. chimaera was reported by Tortoli et al. (1) as a cause of human lung disease but has been widely known as the bacteria responsible for an outbreak of endocarditis and disseminated infection after cardiac surgery in 2013 (2).
Although virulence and pathogenicity of M. chimaera in lung disease are currently debated, several cases of M. chimaera lung infections have been reported in settings of chronic obstructive pulmonary disease, malignancy, or immunosuppression (3–5). We found 1 case of M. chimaera infection in a patient with cystic fibrosis (6). Other nontuberculous mycobacteria (NTM), especially M. abscessus and M. intracellulare, are well-known pathogens in such a setting (7). We report M. chimaera pulmonary disease in 4 patients with cystic fibrosis.
After reviewing data for 248 patients who were examined at the Cystic Fibrosis Center of Montpellier, France, during 2010–2017, we observed that 24 (9.7%) of 248 patients had >1 respiratory smear sample positive for NTM; for 4 (16.7%) of 24, the sample was positive for M. chimaera. The 4 case-patients were Caucasian, age 8–21 years, and who had a newborn diagnosis of cystic fibrosis, preexisting respiratory impairment, and digestive malabsorption. The association of an increased cough and sputum production, breathlessness, and fatigue with a reduction in forced expiratory volume in 1 s (FEV1) or forced vital capacity (FVC) was diagnosed as pulmonary exacerbation (7) for all patients (Figure). Diagnosis was confirmed by computed tomography by the presence of bronchiectasis and nodules (tree-in-bud pattern) for case-patients 1 and 3.
Figure.
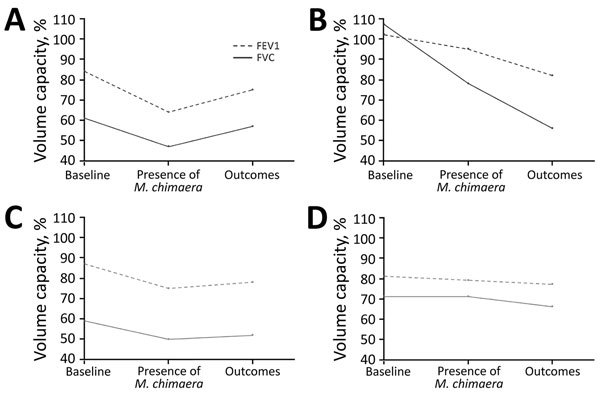
Evolution of lung function for 4 cystic fibrosis patients with Mycobacterium chimaera pulmonary disease, France, 2010–2017. A) Case-patient 1, B) case-patient 2, C) case-patient 3, D) case-patient 4. Case-patients 1 and 3 were given specific treatment for M. chimaera disease for 3 months; case-patient 2 was not given specific treatment; case-patient 4 was given only partial treatment. FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity.
Respiratory specimens collected every 3–6 months for 1 year were digested and decontaminated by using the sodium dodecyl sulfate–NaOH method and then centrifuged using fluorescence microscopy. Sputum samples from all patients were negative for acid-fast bacilli. Samples were cultured on solid and liquid media (BACTEC MGIT 960 System; Becton Dickinson Diagnostic Systems, https://www.bd.com) which identified, after 11–41 days, mycobacteria from >2 separate sputum samples.
We performed species identification by using a commercial kit (GenoType NTM-DR assay; Hain Lifescience, https://www.hain-lifescience.de) and identified isolates as M. chimaera. We also performed molecular identification of isolates as M. chimaera as described (1,8). All isolates were susceptible to macrolides and aminoglycosides. We also isolated several other microorganisms: Pseudomonas aeruginosa and methicillin-resistant Staphylococcus aureus from case-patient 1; Haemophilus influenzae from case-patient 2; M. avium and Stenotrophomonas maltophila from case-patient 3; and M. abscessus, methicillin-resistant S. epidermidis, and P. aeruginosa from case-patient 4.
Azithromycin, rifampin, and ethambutol (in combination) and ceftazidime, tobramycin, and inhaled colistin were given to case-patient 1. No antimicrobial drugs were given to case-patient 2. Azithromycin and rifampin (in combination) and inhaled colistin were given to case-patient 3. Clarithromycin and linezolid were given to case-patient 4. All 4 case-patients required physiotherapy.
All case-patients were followed up for >1 year after the first positive smears for M. chimaera were obtained. We found a substantial reduction in symptoms of pulmonary exacerbations and sterilization of sputum specimens for patients given macrolides and rifampin with or without aminoglycosides after 3 months, as well as improvement in FEV1 and FVC after 6 months. In contrast, patients not given treatment (case-patient 2) or given only partial treatment with an anti-NTM antimicrobial drug regimen (case-patient 4) showed a decrease in FEV1 and FVC after 6 months (Figure) and slight recovery or no change after 1 year.
Therefore, we hypothesized that M. chimaera showed virulence and pathogenicity in our patients because of their clinical picture and evolution. We are aware that the diagnosis of NTM diseases according to American Thoracic Society criteria (9) might not be made with complete certainty because definitive exclusion of other diagnoses was often difficult for cystic fibrosis patients. Several confounding factors, such as co-infection with conventional pathogens, were observed, which could explain the observed favorable outcome. However, specific treatment against NTM improved outcome, which strengthens our presumption of the potential pathogenic role of M. chimaera in lung disease of patients with cystic fibrosis.
Cystic fibrosis transmembrane conductance regulator disorder results in mucus retention and bronchiectasis that favor repeated respiratory tract infection, including NTM diseases (7). In these circumstances, cystic fibrosis might promote M. chimaera infection in a similar manner to that in patients with chronic obstructive pulmonary disease. The lack of improvement of respiratory function for case-patient 4, who had been given treatment for infection with other pathogens, but only partially for M. chimaera, supports our hypothesis. However, whether M. chimaera is only a surrogate of respiratory impairment without any virulence or a real pathogenic microorganism remains unknown. In conclusion, M. chimaera lung disease should prompt physicians to consider this bacteria as an emergent pathogen in cystic fibrosis patients.
Acknowledgments
We thank the patients at the Cystic Fibrosis Center of Montpellier and all members of the Bacteriology Department of Montpellier University Hospital for participating in this study.
Biography
Dr. Larcher is a clinical fellow in the Intensive Care Medicine Department at the University Hospital of Montpellier. He is also a PhD student in the PhyMedEx Laboratory at the University of Montpellier, in association with the French Institute of Health and Medical Research and the French National Center for Scientific Research. His primary research interests are sepsis and infectious diseases in critically ill patients.
Footnotes
Suggested citation for this article: Larcher R, Lounnas M, Dumont Y, Michon A-R, Bonzon L, Chiron R, et al. Mycobacterium chimaera pulmonary disease in cystic fibrosis patients, France, 2010–2017. Emerg Infect Dis. 2019 Mar [date cited]. https://doi.org/10.3201/eid2503.181590
References
- 1.Tortoli E, Rindi L, Garcia MJ, Chiaradonna P, Dei R, Garzelli C, et al. Proposal to elevate the genetic variant MAC-A, included in the Mycobacterium avium complex, to species rank as Mycobacterium chimaera sp. nov. Int J Syst Evol Microbiol. 2004;54:1277–85. 10.1099/ijs.0.02777-0 [DOI] [PubMed] [Google Scholar]
- 2.van Ingen J, Kohl TA, Kranzer K, Hasse B, Keller PM, Katarzyna Szafrańska A, et al. Global outbreak of severe Mycobacterium chimaera disease after cardiac surgery: a molecular epidemiological study. Lancet Infect Dis. 2017;17:1033–41. 10.1016/S1473-3099(17)30324-9 [DOI] [PubMed] [Google Scholar]
- 3.Bills ND, Hinrichs SH, Aden TA, Wickert RS, Iwen PC. Molecular identification of Mycobacterium chimaera as a cause of infection in a patient with chronic obstructive pulmonary disease. Diagn Microbiol Infect Dis. 2009;63:292–5. 10.1016/j.diagmicrobio.2008.12.002 [DOI] [PubMed] [Google Scholar]
- 4.Boyle DP, Zembower TR, Reddy S, Qi C. Comparison of clinical features, virulence, and relapse among Mycobacterium avium complex species. Am J Respir Crit Care Med. 2015;191:1310–7. 10.1164/rccm.201501-0067OC [DOI] [PubMed] [Google Scholar]
- 5.Moon SM, Kim S-Y, Jhun BW, Lee H, Park HY, Jeon K, et al. Clinical characteristics and treatment outcomes of pulmonary disease caused by Mycobacterium chimaera. Diagn Microbiol Infect Dis. 2016;86:382–4. 10.1016/j.diagmicrobio.2016.09.016 [DOI] [PubMed] [Google Scholar]
- 6.Cohen-Bacrie S, David M, Stremler N, Dubus J-C, Rolain J-M, Drancourt M. Mycobacterium chimaera pulmonary infection complicating cystic fibrosis: a case report. J Med Case Reports. 2011;5:473. 10.1186/1752-1947-5-473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elborn JS. Cystic fibrosis. Lancet. 2016;388:2519–31. 10.1016/S0140-6736(16)00576-6 [DOI] [PubMed] [Google Scholar]
- 8.Tortoli E. The new mycobacteria: an update. FEMS Immunol Med Microbiol. 2006;48:159–78. 10.1111/j.1574-695X.2006.00123.x [DOI] [PubMed] [Google Scholar]
- 9.Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, et al. ; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367–416. 10.1164/rccm.200604-571ST [DOI] [PubMed] [Google Scholar]