

 Methoxy substituted 2-benzylidene-1-indanones possess improved adenosine A1 and A2A receptor affinity in the nanomolar range.
Methoxy substituted 2-benzylidene-1-indanones possess improved adenosine A1 and A2A receptor affinity in the nanomolar range.
Abstract
A prior study reported on hydroxy substituted 2-benzylidene-1-indanone derivatives as A1 and/or A2A antagonists for the potential treatment of neurological conditions. A lead compound (1a) was identified with both A1 and A2A affinity in the micromolar range. The current study explored the structurally related methoxy substituted 2-benzylidene-1-indanone derivatives with various substitutions on ring A and B of the benzylidene indanone scaffold in order to enhance A1 and A2A affinity. This led to compounds with both A1 and A2A affinity in the nanomolar range, namely 2c (A1Ki (rat) = 41 nM; A2AKi (rat) = 97 nM) with C4-OCH3 substitution on ring A together with meta (3′) hydroxy substitution on ring B and 2e (A1Ki (rat) = 42 nM; A2AKi (rat) = 78 nM) with C4-OCH3 substitution on ring A together with meta (3′) and para (4′) dihydroxy substitution on ring B. Additionally, 2c is an A1 antagonist. Consequently, the methoxy substituted 2-benzylidene-1-indanone scaffold is highly promising for the design of novel A1 and A2A antagonists.
Introduction
The adenosine system – specifically adenosine A1 and A2A receptors – is a promising drug target for the non-dopaminergic treatment of the neurodegenerative disorder Parkinson's disease (PD).1 These adenosine receptors (AR's), together with A2B and A3 AR's, are either inhibitory (A1 and A3) or stimulatory (A2A and A2B) G-protein coupled receptors.2
The spotlight first fell on the A1 and A2A AR's when an epidemiological study found that consumption of the non-selective AR antagonist caffeine – present in coffee and tea – is associated with a reduced risk of developing PD.3 Since then, numerous preclinical studies in rodents and non-human primate models of PD have supported the potential of A1 and A2A AR antagonists for the treatment of PD.4
The structure of the xanthine derivative caffeine is similar to that of adenosine.5 Caffeine and other A1 and A2A AR antagonists exert effects contrary to endogenous adenosine and, in so doing, affects various neurotransmitters, receptors and signalling pathways.4,6
There is growing evidence that selective A2A AR antagonists, like KW-6002 (istradefylline), may be novel non-dopaminergic treatment for PD; KW-6002 is approved in Japan since 2013 for the treatment of wearing-off phenomenon associated with l-dopa treatment in PD.7,8 Selective A2A AR antagonists may possibly alleviate parkinsonian motor symptoms due to the close anatomical and functional relationship between A2A AR's and dopamine D2 receptors on the indirect striatopallidal GABAergic pathway.7,8 In animal studies, agonists and antagonists of A2A AR's produce behavioural effects similar to antagonists and agonists of dopamine D2 receptors, respectively.9 Thus, blockade of A2A AR's on the indirect striatopallidal GABAergic pathway reduces postsynaptic effects of dopamine depletion (a pathological hallmark of PD) and, subsequently, reduces motor symptoms associated with PD.10 Additionally, KW-6002 may address a non-motor symptom of PD, namely depression, evidenced by a decrease in immobility time during the forced swim test and tail suspension test in rodents (animal models of depression) when the said drug was administered.11,12 Neurodegeneration may be stopped or, at least, slowed by A2A AR antagonists as KW-6002 attenuated striatal dopamine depletion in the MPTP animal model of PD.13
Synaptic plasticity; the ability of synapses to strengthen or weaken in response to increases or decreases in their activity, is the basis for learning and memory.14 Brain areas associated with cognition are the prefrontal cortex and hippocampus.14 Endogenous adenosine via A1 AR's—which are abundantly expressed in the prefrontal cortex and hippocampus—modulates synaptic plasticity phenomena long-term depression and long-term potentiation and, in so doing, inhibits learning and memory.15 A1 AR antagonists block A1 AR mediated inhibitory modulation of synaptic plasticity in the prefrontal cortex and hippocampus: neurotransmitter release and synaptic transmission is increased, facilitating synaptic plasticity and, thus, learning and memory.14 Pharmacological studies support the above, by means of the selective A1 AR antagonists BIIP20 and FR194921 which are active in animal models of cognitive deficits.16,17
It proved to date challenging to translate findings on AR function into clinical studies and no other AR antagonists have been approved.18
The investigation of dual A1/A2A AR antagonists for the potential treatment of PD has been investigated in the past.19–23 For example, 5-[5-amino-3-(4-fluorophenyl)pyrazin-2-yl]-1-isopropylpyridine-2(1H)-one (ASP5854) (see Fig. 1) showed promise in animal models of PD as well as cognition and has Ki values of 9.03 nM at the human A1 AR and 1.76 at the human A2A AR.19 Additionally, 2-amino-8-[2-(4-morpholinyl)ethoxy]-4-phenyl-5H-indeno-[1,2-d]pyrimidin-5-one (JNJ-40255293) (see Fig. 1) is also a dual A1/A2A AR antagonist with efficacy in animal models of PD (A1Ki (human) = 48 nM; A2AKi (human) = 6.5 nM).20,21 Of note, this compound is structurally related to the benzylidene indanones currently under investigation. 8-Substituted 1,3-dimethyltetrahydropyrazino[2,1-f]purinedione derivatives were evaluated as dual A1/A2A AR antagonist in a multitarget approach for the treatment of neurodegenerative disorders.22 The 7-aminopyrazolo[4,3-d]pyrimidine derivatives were also explored as dual A1/A2A AR antagonists with several of these compounds possessing nanomolar affinity for the human A2A AR and slightly lower human A1 AR.23
Fig. 1. The structures of ASP-5854 and JNJ-40255293.

Therefore, dual A1 and A2A AR antagonists may be non-dopaminergic drugs for the symptomatic treatment of both PD motor symptoms (for example bradykinesia, rigidity, resting tremor and postural instability) and PD non-motor symptoms (for example cognitive deficits such as cognitive dysfunction and depression) as well as exhibit neuroprotective properties.4
Benzopyrones are a class of compounds with significantly diverse biological activities24 (including antiparkinsonian and neuroprotective properties)25–30 and are, thus, considered a privileged scaffold in medicinal chemistry.24 Benzopyrones constitute the basic framework of flavonoids,31,32 and the structurally related isocoumarins33 and coumarins.24 It is reported that the flavonoid derivative 5,3′-dihydroxyflavone possess an A1Ki (rat) value of 0.956 μM and an A2AKi (rat) value of 1.44 μM (see Fig. 2).34 In fact, the aurone derivatives (specifically hispidol (A1Ki (rat) = 0.352 μM) and maritimetin (A1Ki (rat) = 3.47 μM and A2AKi (rat) = 9.35 μM) (see Fig. 2)), which are members of the flavonoid family,28 served as inspiration for previous work on the benzylidene tetralones35,36 as well as benzylidene indanones37 – leading to compounds with A1 and A2A AR affinity in the micromolar range. For example, the benzylidene tetralone derivative (E)-5-hydroxy-2-(3-hydroxybenzylidene)-3,4-dihydronaphthalen-1(2H)-one has an A1Ki (rat) value of 1.62 μM and an A2AKi (rat) value of 5.46 μM (see Fig. 2).36
Fig. 2. The structures and Ki values of compounds structurally related to benzylidene indanones and the structural modifications to lead compound 1a to determine features essential for dual A1/A2A AR affinity.

In recent times, hydroxy substituted 2-benzylidene-1-indanone analogues were explored as A1 and/or A2A AR antagonists for the potential treatment of neurological conditions.37 Of note, is compound 1a with Ki values for both the A1 and A2A AR below 1 μM (A1Ki (rat) = 0.435 μM; A2AKi (rat) = 0.903 μM). It was found that C4 hydroxy substitution on ring A of the benzylidene indanones in combination with meta (C3′) and para (C4′) dihydroxy substitution on ring B is preferable for both A1 and A2A AR binding.
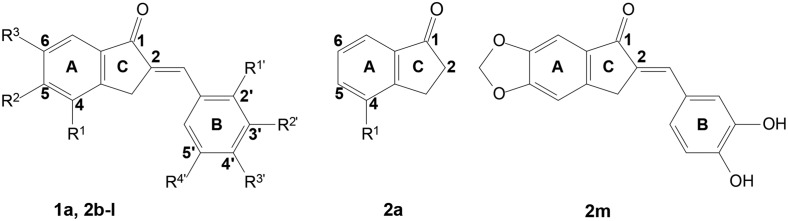
These compounds comprise of a benzylidene indanone scaffold (i.e. fused 6- and 5-membered rings, namely ring A and ring C), where ring C bears a C2-phenyl substituted sidechain (namely ring B). Theoretically, these compounds may be either E- or Z-isomers.38 The E-configuration is favourable for thermodynamic reasons because of steric interaction between the aryl and carbonyl groups in case of the Z-isomers.38,39
Based on the structure of 1a, we investigated the structurally related methoxy substituted 2-benzylidene-1-indanone derivatives as potent A1 and A2A AR antagonists.
The 2-benzylidene-1-indanone scaffold was modified to include substituent changes to ring A and ring B (see Fig. 2). Firstly, C4-, C5- and/or C6-methoxy substitution on ring A was made, in order to determine whether hydroxy or methoxy substitution is preferable for A1 and A2A AR affinity and, secondly, the optimal position of the methoxy group for both A1 and A2A AR affinity was determined. Also, an unsubstituted ring A and methylenedioxy substitution on ring A was incorporated. Substitution at the ortho (C2′), meta (C3′), para (C4′) and/or C5 position(s) of phenyl ring B with hydroxy-, methoxy- or dimethylamino group(s) was investigated.
Accordingly, the 2-benzylidene-1-indanones was evaluated to ascertain which structure activity relationships govern A1 and A2A AR affinity.
Results and discussion
Chemistry
Reagents and test compounds were synthesised as depicted in Scheme 1. The key starting material for 2b–g, namely 2a was synthesised by methylation of 4-hydroxy-1-indanone, as described,40 whereas, reagents for 2j, 2l and 2m were commercially available. In turn, test compounds (2b–g, 2i, 2k & 2m) were prepared via an acid catalysed aldol condensation reaction.35–37 Test compounds 2h, 2j and 2l were available from a previous study.41 The novel compounds 2b–g, 2i, 2k and 2m (fair yields) were purified by recrystallization from a suitable solvent (either MeOH or EtOH) and, in each instance, the structure, molecular mass and purity of these compounds were verified by 1H NMR, 13C NMR, MS and/or HPLC analysis.
Scheme 1. Synthesis of 2a–g, 2i, 2k & 2m. Reagents and conditions: a) acetone, K2CO3, MeI, 50 °C (18 h); b) MeOH, HCl (32%), 120 °C (24 h).

Biology
Radioligand binding assays
The affinities of the 2-benzylidene-1-indanone analogues (2a–m) at rat A1 and A2A AR subtypes were determined with radioligand competition experiments as described previously.42,43 The A1 and A2A AR radioligand binding assay results are summarized in Table 1. Two reference compounds, namely CPA and DPCPX, were included in the study and the results are in accordance with literature values (see Table 1).
Table 1. The dissociation constant (Ki) values for the binding of the 2-benyzlidene-1-indanone analogues at rat A1 and A2A AR's.
| ||||||||||
| # | Ring A |
Ring B |
K
i ± SEM (μM)
a
(% displacement)
b
|
SI d | ||||||
| R 1 | R 2 | R 3 | R 1′ | R 2′ | R 3′ | R 4′ | A1 c vs. [3H]DPCPX | A2A c vs. [3H]NECA | (A1/A2A) | |
| Lead compound | ||||||||||
| 1a | OH | H | H | H | OH | OH | H | 0.435 ± 0.050 a , e | 0.903 ± 0.081 a , e | 0.5 d , e |
| Methoxy substituted 2-benzylidene-1-indanones | ||||||||||
| 2a | OCH3 | H | H | — | — | — | — | >100 (78%) b | >100 (72%) b | — |
| 2b | OCH3 | H | H | H | H | H | H | 1.65 ± 0.15 a | >100 (62%) b | — |
| 2c | OCH3 | H | H | H | OH | H | H | 0.041 ± 0.002 a | 0.097 ± 0.009 a | 0.4 d |
| 2d | OCH3 | H | H | H | H | OH | H | >100 (31%) b | >100 (65%) b | — |
| 2e | OCH3 | H | H | H | OH | OH | H | 0.042 ± 0.009 a | 0.078 ± 0.002 a | 0.5 d |
| 2f | OCH3 | H | H | OCH3 | H | OCH3 | OCH3 | >100 (46%) b | >100 (29%) b | — |
| 2g | OCH3 | H | H | H | H | N(CH3)2 | H | >100 (52%) b | >100 (23%) b | — |
| 2h | H | OCH3 | H | H | H | H | H | >100 (42%) b | >100 (82%) b | — |
| 2i | H | OCH3 | H | H | OH | OH | H | 3.28 ± 0.31 a | 6.32 ± 0.32 a | 0.5 d |
| 2j | H | OCH3 | H | H | H | N(CH3)2 | H | >100 (64%) b | >100 (47%) b | — |
| 2k | H | OCH3 | OCH3 | H | OH | OH | H | 4.29 ± 0.18 a | 18.02 ± 1.39 a | 0.2 d |
| 2l | H | H | H | H | H | N(CH3)2 | H | >100 (51%) b | >100 (65%) b | — |
| 2m | — | — | — | — | — | — | — | >100 (70%) b | 1.07 ± 0.10 a | — |
| Reference compounds | ||||||||||
| CPA (A1 agonist) | 0.0068 ± 0.0001 a | 0.163 ± 0.001 a | 24 d | |||||||
| (0.0079); f | (0.331) g | (22) g | ||||||||
| (0.015) g | ||||||||||
| DPCPX (A1 antagonist) | 0.0004 ± 0.0002 a | 0.545 ± 0.204 a | 1363 d | |||||||
| (0.0005); g | (0.530); g | (958) g | ||||||||
| (0.0003) h | (0.340) h | (1130) h | ||||||||
aAll Ki values determined in triplicate and expressed as mean ± SEM.
bPercentage displacement of the radioligand at a maximum tested concentration (100 μM).
cRat receptors were used (A1: rat whole brain membranes; A2A: rat striatal membranes).
dSelectivity index (SI) for the A2A receptor isoform calculated as the ratio of A1Ki/A2AKi.
eLiterature value obtained from reference.37
fLiterature value obtained from reference.49
gLiterature value obtained from reference.42
hLiterature value obtained from reference.50
A prior study identified the 4-hydroxy substituted 2-benzylidene-1-indanone analogue 1a as a lead compound to design novel and potent A1 and A2A AR antagonists.37 This study's parent scaffold is the 4-methoxy substituted compound 2a, which is unsubstituted at position 2 and lacks both A1 and A2A AR affinity.
Structural modification to ring A
Firstly, in analogy to previous work on the 2-benzylidene-1-tetralones (which determined whether OH- or OCH3-group substitution on ring A was preferable to attain both A1 and A2A AR affinity),36 the current study investigated C4-OH substitution versus C4-OCH3 substitution on ring A, as well as an unsubstituted ring A and 1,3-dioxolane substitution on ring A by comparing the Ki values of analogous compounds. Secondly, similar to a previous study of the 2-benzylidene-1-indanones – which determined optimal OH-substitution on ring A together with meta (3′) and para (4′) diOH-substitution on ring B37 – the impact of OCH3-substitution at either position C4, C5 and/or C6 of ring A, in combination with various ring B substitutions were evaluated by comparing the Ki values of analogous compounds.
C4-OH substitution vs. C4-OCH3 substitution
Comparison of lead compound 1a (A1Ki (rat) = 0.435 μM; A2AKi (rat) = 0.903 μM)37 to its methoxy substituted counterpart 2e (A1Ki (rat) = 0.042 μM; A2AKi (rat) = 0.078 μM) showed that in the 2-benzylidene-1-indanone scaffold C4-OCH3 substitution is favoured over C4-OH substitution on ring A; where a tenfold increase in A1 AR affinity and a twelvefold increase in A2A AR affinity were observed.
Interestingly, the structurally related 2-benzylidene-1-tetralone analogues generally prefer OH-substitution over OCH3-substitution on ring A.35,36
An unsubstituted ring A in combination with a dimethylamino group in the para (4′) position (2g) diminished both A1 and A2A AR affinity as seen with comparison of 2g to unsubstituted 2l.
Methylenedioxy substitution on ring A (2m) reduced both A1 AR affinity (A1Ki (rat) = >100 μM) and A2A AR affinity (A2AKi (rat) = >1.07 μM) when compared to 2e, however the said compound retained relatively good A2A AR affinity.
Optimal position of OCH3-group
Similar to the hydroxy substituted 2-benzylidene-1-indanone analogues, the position of the OCH3-group on ring A modulates A1 and A2A AR binding affinity and C4-OCH3 substitution on ring A is preferred over either C5-OCH3 substitution or C5- and C6-diOCH3 substitution, as evidenced when C4-OCH3 substitution on ring A (2e; A1Ki (rat) = 0.042 μM and A2AKi (rat) = 0.078 μM) is compared to C5-OCH3 substitution (2i; A1Ki (rat) = 3.28 μM and A2AKi (rat) = 6.32 μM) and C5 and C6-diOCH3 substitution (2k A1Ki (rat) = 4.29 μM and A2AKi (rat) = 18.02 μM).
Comparison of 4-methoxy substituted 2b (A1Ki (rat) = 1.65 μM; A2AKi (rat) = >100 μM) to 5-methoxy substituted 2h (A1Ki & A2AKi (rat) = >100 μM), showed that C4-OCH3 substitution on ring A, instead of C5-OCH3 substitution, lead to increased A1 AR affinity – yet, A2A AR affinity was unaffected.
Additionally, both the A1 and A2A AR's favour C4-OCH3 substitution on ring A to either C5-OCH3 substitution or C5 and C6-diOCH3 substitution when 4-methoxy substituted 2e (A1Ki (rat) = 0.042 μM; A2AKi (rat) = 0.078 μM) was compared to 5-methoxy substituted 2i (A1Ki (rat) = 3.28 μM; A2AKi (rat) = 6.32 μM) and 5,6-dimethoxy substituted 2k (A1Ki (rat) = 4.29 μM; A2AKi (rat) = 18.02 μM); a dramatic increase in both A1 and A2A AR affinity was observed – seeing as 2e possess A1 and A2A AR Ki values in the nanomolar range. In fact, the A1 AR affinity of 2e is 78 times better than that of 2i and 102 times better than that of 2k, whereas as the A2A AR affinity of 2e is 81 times better than that of 2i and 231 times better than that of 2k. However, comparison of 4-methoxy substituted 2g, 5-methoxy substituted 2j and unsubstituted 2l – all with Ki values >100 μM – such a trend could not be observed. All in all, it may be said that 4-methoxy substitution on ring A is preferred over either 5-methoxy substitution, 5,6-dimethoxy substitution or no substitution on ring A.
Additionally, when compound 2m – containing a methylenedioxy on ring A – was compared to compounds 2e, 2i and 2k, all with meta (3′) and para (4′) diOH-substitution on ring B, it was seen that A1 AR affinity (>100 μM) was diminished, yet the compound retained relatively good A2A AR affinity (A2AKi (rat) = 1.07 μM).
An unsubstituted ring A in combination with para (4′) N(CH3)2-substitution on ring B (2l) was compared to C4-OCH3 substitution on ring A (2g) and C5-OCH3 substitution on ring A (2j) in combination with para (4′) N(CH3)2-substitution on ring B of the 2-benzylidene-1-indanones and, although, none of these compounds possess micromolar affinity for either the A1 or A2A AR, it seems that the A1 AR prefers no substitution on ring A in combination with N(CH3)2-substitution (2l) on ring B, rather than OCH3-substitution at position C4 or C5 of ring A. The A2A AR again favours C4-OCH3 substitution over C5-OCH3 and then no substitution on ring A.
Structural modification to ring B
Comparison of compound 2a (A1Ki & A2AKi (rat) = >100 μM) to compound 2b (A1Ki (rat) = 1.65 μM; A2AKi (rat) = >100 μM) showed that the benzylidene linked at position C2 on ring C increased both A1 and A2A AR affinity – conveying the necessity of the benzylidene at position C2 on ring C – this trend was also observed with regards to 5-substituted 2-benzylidene-1-tetralones and hydroxy substituted 2-benzylidene-1-indanones.36,37
The A1Ki (rat) value of compound 2b (1.65 μM) when compared to the A1Ki (rat) value of compound 2a (>100 μM) suggests that phenyl ring B is valuable to A1 AR affinity.
In correlation with previous studies35–37 OH-group substitution on ring B favours A1 and A2A AR binding, especially meta (3′) hydroxy-group substitution (2c) and meta (3′) and para (4′) diOH-group substitution (2e).
It seems that both A1 and A2A AR's prefer either meta (3′) OH-group substitution (giving an A1Ki (rat) value of 0.041 μM and an A2AKi (rat) value of 0.097 μM) or meta (3′) and para (4′) diOH-group substitution (giving an A1Ki (rat) value of 0.042 μM and an A2AKi (rat) value of 0.078 μM); seeing as these Ki values are quite similar.
Interestingly, para (4′) hydroxy-substitution diminished both A1 and A2A AR affinity (2d: A1 & A2A (rat) Ki = >100 μM).
Other substitutions that diminish both A1 and A2A AR affinity include 2′,4′,5′-trimethoxy substitution on ring B (2f), as well as dimethylamino substitution (2g & 2j) when compared to its unsubstituted counterpart 2b (in the case of 2f and 2g) and 2h (in the case of 2j) (Fig. 3).
Fig. 3. Structural requirements of the 2-benzylidene-1-indanone scaffold for dual A1/A2A affinity.

These compounds were found to be E-isomers. The E-configuration is favourable for thermodynamic reasons because of steric interaction between the aryl and carbonyl groups in case of the Z-isomers.38,39
The benzylidene indanone scaffold (1a, 2b–m) contains an α,β-unsaturated ketone group perceived as a potential Michael acceptor.44,45 Although, compounds that act as Michael acceptors are generally biologically active,44,45 Michael acceptors are also notoriously reactive compound substructures.46,47 These compound substructures, containing an electrophile, might show reactivity towards nucleophiles such as thiols.47 Various biologically-relevant nucleophiles are thiols, for example glutathione, coenzyme A and protein cysteines.47 Off-target effects are often due to such compound substructures reactivity.47 Yet, a compound that acts as a Michael acceptor (and contains an electrophile) may still be useful after on-reaction with a suitable nucleophile to yield a lower energy compound.48
Compounds containing a catechol group, like 2e, 2i, 2k & 2m, are widespread in the literature as potential starting points to further explore structure activity relationships – yet, compounds that contain known reactive moieties, such as a catechol group, are also considered a liability;46–48 as catechols potentially are chelators, redox-active and oxidizes to form protein-reactive quinones.47,48 For example, the activity of 2e, with relatively good A1 and A2A AR affinity, may be due to oxidation of the ortho-hydroquinone compound substructure (or catechol group) to ortho-quinone – a protein-reactive quinone.
Reactive compound substructures are a risk factor in early drug discovery and development; as the results of biological assays are subject to interference from reactive moieties,47,48 like the α,β-unsaturated ketone group (present in all compounds in the current study, except 2a) or the catechol group (present in 2e, 2i, 2k & 2m). Failure to identify these moieties may lead to wasted resources, as such, compounds have poor development potential.47,48 Therefore a lack of reactivity interference, must be demonstrated,47 before further development.
GTP shift assay
GTP shift experiments are performed to determine whether the test compounds that exhibit A1 AR affinity function as agonists or antagonists. For this purpose, compound 2c was selected as it exhibited the highest A1 AR binding affinity among the investigated compounds. The affinities of the reference (CPA and DPCPX) and test compound 2c were determined in the absence and presence of 100 μM GTP and are reported with the calculated GTP shifts in Table 2. The calculated GTP shift results for CPA and DPCPX (see Table 2) were found to correspond with literature values, where CPA acts as an agonist (see Fig. 4) and DPCPX as an antagonist. Generally, a rightward shift of the binding curve in the presence of GTP (due to an uncoupling of the A1 AR from its Gi protein) is expected for an A1 AR agonist.42,51 In the case of an A1 AR antagonist no significant shift is anticipated in the presence of GTP.42,51 The results suggest that compound 2c act as A1 AR antagonist – as no significant rightward shift of the binding curve was observed in the presence of GTP (see Fig. 4).
Table 2. The A1 AR affinities (in the absence and presence of GTP) and calculated GTP shifts of selected 2-benyzlidene-1-indanone analogues.
| # |
K
i ± SEM (μM)
a
|
GTP shift d | |
| A1 b vs. [3H]DPCPX | A1 b + GTP c vs. [3H]DPCPX | ||
| Lead compound | |||
| 1a | 0.435 ± 0.050 e | 0.339 ± 0.071 e | 0.90 e |
| Methoxy substituted 2-benzylidene-1-indanones | |||
| 2c | 0.041 ± 0.002 a , b | 0.060 ± 0.002 a , c | 1.46 d |
| Reference compounds | |||
| CPA (A1 agonist) | 0.0068 ± 0.0001 a | 0.099 ± 0.015 a | 15 |
| (0.0079); f | (0.099) g | (14) g | |
| (0.015) g | |||
| DPCPX (A1 antagonist) | 0.0004 ± 0.0002 a | 0.0004 ± 0.0002 a | 1.0 |
| (0.0005); g | (0.0004) g | (1.0) g | |
| (0.0003) h | |||
aAll Ki values determined in triplicate and expressed as mean ± SEM.
bRat receptors were used (A1: rat whole brain membranes).
cGTP shift assay, where the 100 μM GTP was added to the A1 AR radioligand binding assay.
dGTP shifts calculated by dividing the Ki in the presence of GTP by the Ki in the absence of GTP.
eLiterature value obtained from reference.37
fLiterature value obtained from reference.49
gLiterature value obtained from reference.42
hLiterature value obtained from reference.50
Fig. 4. The binding curves of compounds 2c and CPA (reference compound) are examples of A1 AR antagonistic action (A) and A1 AR agonistic action (B), respectively, determined via a GTP shift assays (with and without 100 μM GTP) in rat whole brain membranes expressing A1 ARs with [3H]DPCPX as radioligand. (A) GTP shift of 1.46 calculated for compound 2c, (B) GTP shift of 15 calculated for compound CPA.

Conclusions
In summary, this study involved the synthesis, characterization and evaluation of methoxy substituted 2-benzylidene-1-indanone derivatives to understand the importance of structural modifications to ring A and B of the 2-benzylidene-1-indanone scaffold in gaining or even losing A1 and/or A2A AR affinity. Upon analysis, it was found that C4-OCH3 substitution on ring A (2e) is preferred to C4-OH substitution (1a). Additionally, meta (3′) OH-group substitution (2c; A1Ki (rat) = 41 nM; A2AKi (rat) = 97 nM) and meta (3′) and para (4′) diOH-group substitution (2e; A1Ki (rat) = 42 nM; A2AKi (rat) = 78 nM) on ring B along with C4 OCH3-substitution on ring A is complimentary to A1 and A2A AR affinity, affording these non-selective compounds Ki values in the nanomolar range for both the A1 and A2A AR. To reiterate the significance of the aforementioned, 2c and 2e showed an approximately tenfold increase in A1 and A2A AR affinity when compared to lead compound 1a. Other compounds showed A1 affinity (2b) and A2A affinity (2m) in the micromolar range, whereas compounds 2i and 2k showed dual A1 and A2A AR affinity. Yet, none of the other target compounds come close to 2c and 2e with regards to A1 and A2A AR affinity. Functional characterization of 2c proved this compound to be an A1 AR antagonist. In view of these findings, compounds 2c and 2e present the potential starting points to further explore the structure activity relationships for affinities of this class of compounds as ligands for A1 and A2A AR. However, since both 2c and 2e contain potential reactive compound substructures, experiments that demonstrate the absence of reactivity interference using a variety of methods must be performed. If necessary, the undesirable reactive compound substructures may be removed or modified during medicinal chemistry optimization.
Experimental
Chemistry
General remarks
Unless otherwise noted, all starting materials and solvents were procured from Sigma-Aldrich and used without further purification. Proton (1H) and carbon (13C) nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III 600 spectrometer at frequencies of 600 MHz and 151 MHz, respectively, with deuterated dimethylsulfoxide (DMSO-d6) as solvent. Chemical shifts are reported in parts per million (δ) in relation to the signal of tetramethylsilane (Si(CH3)4). Spin multiplicities are indicated as: s (singlet), d (doublet), dd (doublet of doublets), td (triplet of doublets), t (triplet), q (quartet) and m (multiplet). High resolution mass spectra (HRMS) were recorded on a Bruker micrOTOF-Q II mass spectrometer in atmospheric pressure chemical ionisation (APCI) mode. High performance liquid chromatography (HPLC) analyses were determined on an Agilent 1100 HPLC system. Melting points (mp) for 2a–g were measured with a Buchi B545 melting point apparatus, whereas mp's for 2i, 2k & 2m were measured by means of differential scanning calorimetry (DSC) with a Mettler DSC 3 Star System (Mettler Toledo, Greifensee, Switzerland) and are uncorrected and are uncorrected. Thin layer chromatography (TLC) was done using silica gel 60 (Merck) with UV254 fluorescent indicator.
Synthesis of 2a
4-Methoxy-2,3-dihydro-1H-inden-1-one (2a)
To a solution of 4-hydroxy-1-indanone (2.00 g, 13.5 mmol) in acetone (70.0 mL), K2CO3 (7.46 g, 54.0 mmol) and then MeI (11.5 g, 81.0 mmol) were added and mechanically stirred at 50 °C under reflux for 18 h. Next, the reaction mixture was concentrated, extracted with EtOAc (3 × 100 mL), combined organic extracts dried (MgSO4), filtered and concentrated to yield compound 2a as brown crystals (2.00 g, 91%): Rf: 0.51 (PE/EtOAc 4 : 1); mp: 105.3–107.1 °C; 1H NMR (600 MHz, DMSO) δ 2.61 (dd, J = 6.7, 4.6 Hz, 2H), 2.97–2.92 (m, 2H), 3.87 (s, 3H), 7.22 (dd, J = 18.8, 7.7 Hz, 2H), 7.40 (t, J = 7.7 Hz, 1H); 13C NMR (151 MHz, DMSO) δ 22.31, 35.75, 55.52, 114.48, 115.48, 129.09, 138.20, 143.52, 156.89, 206.39. APCI-HRMS m/z calculated for C10H10O2 (MH+): 163.0753, found: 163.0754. Purity (HPLC): 98.6%.
Synthesis of 2b–g, 2i, 2k and 2m
(E)-2-Benzylidene-4-methoxy-2,3-dihydro-1H-inden-1-one (2b)
Compound 2a (0.200 g, 1.233 mmol) and benzaldehyde (0.131 g, 1.233 mmol) were suspended in MeOH (4 mL) and HCl (32%, 6 mL) and mechanically stirred at 120 °C under reflux for 24 h. Thereafter, the reaction mixture was cooled to room temperature, ice (20 g) was added and the resulting precipitate was filtered, dried (60 °C) and recrystallized from a suitable solvent (either MeOH or EtOH) to yield 2b as light brown crystals (0.22 g, 71%): Rf: 0.68 (PE/EtOAc 3 : 1); mp: 385.3–385.5 °C; 1H NMR (600 MHz, DMSO) δ 3.96–3.90 (m, 5H), 7.29 (d, J = 7.9 Hz, 1H), 7.36 (d, J = 7.5 Hz, 1H), 7.57–7.43 (m, 5H), 7.78 (d, J = 7.4 Hz, 2H); 13C NMR (151 MHz, DMSO) δ 28.90, 55.56, 115.20, 116.00, 129.05, 129.44, 129.86, 130.79, 133.08, 134.71, 134.78, 138.05, 138.59, 156.50, 193.30. APCI-HRMS m/z calculated for C17H14O2 (MH+): 251.1067, found: 251.1088. Purity (HPLC): 98.1%.
(E)-2-(3-Hydroxybenzylidene)-4-methoxy-2,3-dihydro-1H-inden-1-one (2c)
Prepared as for 2b from compound 2a (0.300 g, 1.850 mmol) and 3-hydroxybenzaldehyde (0.226 g, 1.850 mmol) to yield compound 2c as light brown crystals (0.44 g, 90%): Rf: 0.29 (PE/EtOAc 3 : 1); mp: 104.3–104.4 °C; 1H NMR (600 MHz, DMSO) δ 3.90 (d, J = 14.8 Hz, 5H), 6.86 (dd, J = 8.0, 1.9 Hz, 1H), 7.20 (dd, J = 10.1, 4.7 Hz, 2H), 7.38–7.26 (m, 3H), 7.45 (dd, J = 15.6, 7.9 Hz, 2H), 9.73 (s, 1H); 13C NMR (151 MHz, DMSO) δ 29.01, 55.56, 115.20, 115.98, 116.59, 117.24, 122.39, 129.45, 130.07, 133.30, 134.43, 135.95, 137.97, 138.65, 156.51, 157.73, 193.32. APCI-HRMS m/z calculated for C17H14O3 (MH+): 267.1016, found: 267.1017. Purity (HPLC): 100%.
(E)-2-(4-Hydroxybenzylidene)-4-methoxy-2,3-dihydro-1H-inden-1-one (2d)
Prepared as for 2b from compound 2a (0.300 g, 1.850 mmol) and 4-hydroxybenzaldehyde (0.226 g, 1.850 mmol) to yield compound 2d as mustard powder (0.45 g, 92%): Rf: 0.14 (PE : EtOAc 3 : 1); mp: 275.3–396.2 °C; 1H NMR (600 MHz, DMSO) δ 3.86 (d, J = 0.4 Hz, 2H), 3.91 (s, 3H), 6.91 (d, J = 8.6 Hz, 2H), 7.27 (d, J = 7.9 Hz, 1H), 7.33 (d, J = 7.5 Hz, 1H), 7.44 (dd, J = 12.7, 4.8 Hz, 2H), 7.64 (d, J = 8.7 Hz, 2H), 10.15 (s, 1H); 13C NMR (151 MHz, DMSO) δ 28.97, 55.53, 115.06, 115.63, 116.10, 125.89, 129.30, 131.13, 133.03, 133.61, 137.69, 139.03, 156.48, 159.50, 193.19. APCI-HRMS m/z calculated for C17H14O3 (MH+): 267.1016, found: 267.1020. Purity (HPLC): 100%.
(E)-2-(3,4-Dihydroxybenzylidene)-4-methoxy-2,3-dihydro-1H-inden-1-one (2e)
Prepared as for 2b from compound 2a (0.300 g, 1.850 mmol) and 3,4-dihydroxybenzaldehyde (0.255 g, 1.850 mmol) to yield compound 2e as light yellow crystals (0.47 g, 90%): Rf: 0.29 (PE/EtOAc 3 : 1); mp: 294.0–295.1 °C; 1H NMR (600 MHz, DMSO) δ 3.84 (s, 2H), 3.92 (s, 3H), 6.87 (d, J = 8.2 Hz, 1H), 7.10 (dd, J = 8.3, 1.9 Hz, 1H), 7.27 (t, J = 5.1 Hz, 2H), 7.33 (d, J = 7.5 Hz, 1H), 7.37 (s, 1H), 7.44 (t, J = 7.7 Hz, 1H); 13C NMR (151 MHz, DMSO) δ 29.05, 55.54, 115.07, 115.63, 116.13, 117.08, 124.73, 126.32, 129.33, 130.89, 134.08, 137.62, 139.11, 145.69, 148.19, 156.49, 193.17. APCI-HRMS m/z calculated for C17H14O4 (MH+): 283.0965, found: 283.0959. Purity (HPLC): 100%.
(E)-2-(2,4,5-Trimethoxybenzylidene)-4-methoxy-2,3-dihydro-1H-inden-1-one (2f)
Prepared as for 2b from compound 2a (0.300 g, 1.850 mmol) and 2,4,5-trimethoxybenzaldehyde (0.363 g, 1.850 mmol) to yield compound 2f as green powder (0.34 g, 54%): Rf: 0.14 (PE/EtOAc 4 : 1); mp: 217.6–218.0 °C; 1H NMR (600 MHz, DMSO) δ 3.82 (s, 3H), 3.94–3.87 (m, 9H), 3.95 (d, J = 1.3 Hz, 2H), 6.79 (s, 1H), 7.37–7.27 (m, 3H), 7.45 (t, J = 7.8 Hz, 1H), 7.45 (t, J = 7.8 Hz, 1H), 7.89 (t, J = 1.9 Hz, 1H); 13C NMR (151 MHz, DMSO) δ 28.52, 55.60, 55.86, 56.47, 97.64, 113.20, 114.49, 115.08, 115.66, 127.30, 129.29, 131.31, 137.70, 139.08, 142.73, 152.39, 154.91, 156.45, 167.00, 193.20. APCI-HRMS m/z calculated for C20H20O5 (MH+): 341.1384, found: 341.1393. Purity (HPLC): 95.1%.
(E)-2-(4-(Dimethylamino)benzylidene)-4-methoxy-2,3-dihydro-1H-inden-1-one (2g)
Prepared as for 2b from compound 2a (0.300 g, 1.850 mmol) and 4-(dimethylamino)benzaldehyde (0.276 g, 1.850 mmol) to yield compound 2g as brown crystals (0.33 g, 61%): Rf: 0.46 (PE/EtOAc 4 : 1); mp: 190.7–191.7 °C; 1H NMR (600 MHz, DMSO) δ 3.01 (s, 6H), 3.85 (s, 2H), 3.92 (s, 3H), 6.85–6.77 (m, 2H), 7.26 (d, J = 7.9 Hz, 1H), 7.32 (d, J = 7.4 Hz, 1H), 7.44 (dd, J = 9.6, 5.0 Hz, 2H), 7.62 (d, J = 8.9 Hz, 2H); 13C NMR (151 MHz, DMSO) δ 29.16, 55.53, 112.01, 114.93, 115.32, 122.13, 129.09, 129.20, 132.72, 134.31, 137.35, 139.45, 151.23, 156.43, 192.86. APCI-HRMS m/z calculated for C19H19NO2 (MH+): 294.1489, found: 294.1499 Purity (HPLC): 97.9%.
(E)-2-(3,4-Dihydroxybenzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (2i)
Prepared as for 2b from 5-methoxy-1-indanone (0.300 g, 1.850 mmol) and 3,4-dihydroxybenzaldehyde (0.255 g, 1.850 mmol) to yield compound 2i as brown powder (0.33 g, 63%): Rf: 0.13 (DCM/EtOAc 10 : 1); mp: 281.26 °C; 1H NMR (600 MHz, DMSO) δ 3.88 (s, 3H), 3.96 (s, 2H), 6.85 (d, J = 8.2 Hz, 1H), 7.01 (dd, J = 8.5, 2.2 Hz, 1H), 7.08 (dd, J = 8.3, 2.0 Hz, 1H), 7.17 (dd, J = 12.5, 2.0 Hz, 2H), 7.29 (t, J = 1.7 Hz, 1H), 7.69 (d, J = 8.5 Hz, 1H), 9.24 (s, 1H), 9.64 (s, 1H); 13C NMR (151 MHz, DMSO) δ 32.09, 55.76, 110.23, 115.15, 116.02, 117.44, 123.87, 125.20, 126.57, 130.91, 131.91, 132.43, 145.58, 147.75, 152.55, 164.61, 191.59. APCI-HRMS m/z calculated for C17H14O4 (MH+): 283.0965, found: 283.0972. Purity (HPLC): 95.4%.
(E)-2-(3,4-Dihydroxybenzylidene)-5,6-dimethoxy-2,3-dihydro-1H-inden-1-one (2k)
Prepared as for 2b from 5,6-dimethoxy-1-indanone (0.300 g, 1.561 mmol) and 3,4-dihydroxybenzaldehyde (0.216 g, 1.561 mmol) to yield compound 2k as dark green powder (0.41 g, 84%): Rf: 0.12 (DCM/EtOAc/PE 8 : 1 : 1); mp: 262.66 °C; 1H NMR (600 MHz, DMSO) δ 3.83 (s, 12H), 3.90 (s, 20H), 6.85 (d, J = 8.2 Hz, 4H), 7.07 (dd, J = 8.3, 2.0 Hz, 4H), 7.18 (dd, J = 12.3, 4.1 Hz, 12H), 7.27 (s, 4H), 9.43 (s, 7H); 13C NMR (151 MHz, DMSO) δ 31.69, 55.65, 55.96, 104.50, 108.07, 116.01, 117.41, 123.75, 126.62, 130.32, 131.98, 132.19, 144.61, 145.58, 147.67, 149.25, 154.94, 191.88. APCI-HRMS m/z calculated for C18H16O5 (MH+): 313.1071, found: 313.0993. Purity (HPLC): 98.6%.
(E)-6-(3,4-Dihydroxybenzylidene)-6,7-dihydro-5H-indeno[5,6-d][1,3]dioxol-5-one (2m)
Prepared as for 2b from 5,6-methylenedioxy-1-indanone (0.300 g, 1.703 mmol) and 3,4-dihydroxybenzaldehyde (0.235 g, 1.703 mmol) to yield compound 2m as green powder (0.45 g, 90%): Rf: 0.09 (DCM/EtOAc 10 : 1); mp: 317.96 °C; 1H NMR (600 MHz, DMSO) δ 3.88 (s, 2H), 6.17 (s, 2H), 6.84 (d, J = 8.2 Hz, 1H), 7.06 (dd, J = 8.2, 1.7 Hz, 1H), 7.15 (dd, J = 9.0, 2.4 Hz, 3H), 7.25 (s, 1H), 9.25 (s, 1H), 9.63 (s, 1H); 13C NMR (151 MHz, DMSO) δ 31.97, 101.97, 102.37, 105.92, 116.01, 117.33, 123.93, 126.49, 131.95, 132.18, 132.28, 145.59, 146.99, 147.77, 148.03, 153.48, 191.34. APCI-HRMS m/z calculated for C17H12O5 (MH+): 297.0758, found: 297.0755. Purity (HPLC): 87.2%.
Biology
General remarks
All commercially available reagents were obtained from various manufacturers: radioligands [3H]NECA (specific activity 27.1 Ci mmol–1) procured from PerkinElmer and [3H]DPCPX (specific activity 120 Ci mmol–1) from Amersham Biosciences, filter-count from PerkinElmer and Whatman GF/B 25 mm diameter filters from Merck. Radio activity was calculated by a Packard Tri-CARB 2810 TR liquid scintillation counter.
Radioligand binding assays
The collection of tissue samples for the A1 and A2A AR binding studies were approved by the Research Ethics Committee of the North-West University (application number NWU-0035-10-A5). The affinities of the 2-benzylidene-1-indanone analogues (2a–m) at rat A1 and A2A AR subtypes were determined with radioligand competition experiments as described previously.22,23 The competition experiments were carried out in the presence of the radioligands [3H]-8-cylcopentyl-1,3-dipropylxanthine ([3H]DPCPX; 0.1 nM; Kd = 0.36 nM) and 5′-N-[3H]-ethylcarboxamideadenosine ([3H]NECA; 4 nM; Kd = 15.3 nM) for the A1 and A2A AR radioligand binding assays, respectively.22,24 In addition, the A2A AR binding studies were determined in the presence of N6-cyclopentyladenosine (CPA) to minimize the binding of [3H]NECA to A1 AR's. Non-specific binding was defined by the addition of a final concentration of 100 μM CPA. The sigmoidal-dose response curves, via Graphpad Software Inc. package, were obtained by plotting the specific binding versus the logarithm of the test compound's concentrations. Subsequently, the Ki values were obtained by using the IC50 values that were determined from sigmoidal-dose response curves. All incubations were carried out in triplicate and the Ki values are expressed as the mean ± standard error of mean (SEM). CPA and DPCPX (unlabelled) were used as reference compounds and their assay results confirmed validity of the radioligand binding assays (see Table 1).
GTP shift assays
The GTP shift assay was performed as described previously with rat whole brain membranes and [3H]DPCPX (0.1 nM; Kd = 0.36 nM) in the absence and presence of a final concentration of 100 μM GTP (see Table 2).22,23 Non-specific binding was defined by the addition of 10 μM DPCPX (unlabelled). If a calculated GTP shift of approximately 1 is obtained, that compound is considered to function as an antagonist. On the other hand, the presence of GTP affects the competition curve of an agonist and shifts the curve to the right, as previously demonstrated by the A1 AR agonist CPA.22 The sigmoidal-dose response curves were obtained via the Graphpad Software Inc. package and the Ki values determined as described above. The GTP shift was calculated by dividing the Ki value of a compound reported in the presence of GTP by the Ki value obtained in the absence of GTP.22
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
Financial support for this work was provided by the North-West University (NWU), the National Research Foundation (96135) and the Medical Research Council, South Africa. We are grateful to Dr. J. Jordaan of Chemical Research Beneficiation, NWU for NMR and MS analysis, as well as Prof. J. Du Preez for HPLC analysis and Prof. W. Liebenberg for mp analysis of 2i, 2k & 2m – both of Pharmaceutics, School of Pharmacy, NWU. We also thank Miss M. S. Nel for the use of compounds 2h, 2j & 2l.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00540k
References
- Soliman A. M., Fathalla A. M., Moustafa A. A. Pharmacol. Rep. 2018;70(4):661–667. doi: 10.1016/j.pharep.2018.02.003. [DOI] [PubMed] [Google Scholar]
- Palmer T. M., Stiles G. L. Neuropharmacology. 1995;34(7):683–694. doi: 10.1016/0028-3908(95)00044-7. [DOI] [PubMed] [Google Scholar]
- Ross G. W., Abbot R. D., Petrovitch H., Morens D. M., Grandinetti A., Tung K. H., Tanner C. M., Masaki K. H., Blanchette P. L., Curb J. D., Popper J. S. JAMA, J. Am. Med. Assoc. 2000;283(20):2674–2679. doi: 10.1001/jama.283.20.2674. [DOI] [PubMed] [Google Scholar]
- Prediger R. D. S., Ribeiro J. A., Sebastião A. M. J. Alzheimers Dis. J. Alzheimer's Dis. 2010;2010;2020(S1)(S1):S205–S2205. S3–S15. [Google Scholar]
- Ribeiro J. A., Sebastião A. M. J. Alzheimer's Dis. 2010;20(S1):S3–S15. [Google Scholar]
- Chen J. F., Lee C. and Chern Y., in International Review of Neurobiology, ed. R. A. Harris and P. Jenner, Academic Press, London, 2014, vol. 119, pp. 1–49. [DOI] [PubMed] [Google Scholar]
- Pinna A. CNS Drugs. 2014;28(5):455–474. doi: 10.1007/s40263-014-0161-7. [DOI] [PubMed] [Google Scholar]
- Ferré S., Popoli P., Giménez-Llort L., Rimondini R., Müller C. E., Strömberg I., Örgen S. O., Fuxe K. Parkinsonism Relat. Disord. 2001;7:235–241. doi: 10.1016/s1353-8020(00)00063-8. [DOI] [PubMed] [Google Scholar]
- Ferré S., Von Euler G., Johansson B., Fredholm B. B., Fuxe K. Neuroscience. 1992;51:501–512. doi: 10.1016/0306-4522(92)90291-9. [DOI] [PubMed] [Google Scholar]
- Schwarzschild M. A., Agnati L., Fuxe K., Chen J., Morelli M. Trends Neurosci. 2006;29:647–654. doi: 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Yacoubi M. E., Ledent C., Parmentier M., Bertorelli R., Onginni E., Costentin J., Vaugeois J. Br. J. Pharmacol. 2001;134:68–77. doi: 10.1038/sj.bjp.0704240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K., Kobayashi M., Mori A., Jenner A., Kanda T. Pharmacol., Biochem. Behav. 2013;114–115:23–30. doi: 10.1016/j.pbb.2013.10.022. [DOI] [PubMed] [Google Scholar]
- Chen J. F., Xu K., Petzer J. P., Staal R., Xu Y. H., Beilstein M., Sonsalla P. K., Castagnoli K., Castagnoli N., Schwarzschild M. A., J. Neurosci., 2001, 21 , RC143 , , 1 of 6 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi R. N., Pamplona F. A., Prediger R. D. S. Front. Biosci. 2008;13:2614–2632. doi: 10.2741/2870. [DOI] [PubMed] [Google Scholar]
- Ribeiro J. A., Sebastião A. M., De Mendonca A. Drug News Perspect. 2003;16:80–86. doi: 10.1358/dnp.2003.16.2.740246. [DOI] [PubMed] [Google Scholar]
- Pitsikas N., Borsini F. Eur. J. Pharmacol. 1997;328:19–22. doi: 10.1016/s0014-2999(97)83021-x. [DOI] [PubMed] [Google Scholar]
- Maemoto T., Tada M., Mihara T., Ueyama N., Matsuoka H., Harada K., Yamaji T., Shirakawa K., Kuroda S., Akahane A., Iwashita A. J. Pharmacol. Sci. 2004;96:42–52. doi: 10.1254/jphs.fp0040359. [DOI] [PubMed] [Google Scholar]
- Baraldi P. G., Tabrizi M. A., Gessi S., Borea P. A. Chem. Rev. 2008;108(1):238–263. doi: 10.1021/cr0682195. [DOI] [PubMed] [Google Scholar]
- Mihara T., Mihara K., Yarimizu J., Mitani Y., Matsuda R., Yamamoto H., Aoki S., Akahane A., Iwashita A., Matsuoka N. J. Pharmacol. Exp. Ther. 2007;223(2):708–719. doi: 10.1124/jpet.107.121962. [DOI] [PubMed] [Google Scholar]
- Shook B. C., Rassnick S., Wallace N., Crooke J., Ault M., Chakravarty D., Barbay J. K., Wang A., Powell M. T., Leonard K., Alford V. J. Med. Chem. 2012;55(3):1402–1417. doi: 10.1021/jm201640m. [DOI] [PubMed] [Google Scholar]
- Atack J. R., Shook B. C., Rassnick S., Jackson P. F., Rhodes K., Drinkenburg W. H., Ahnaou A., Te Riele P., Langlois X., Hrupka B., De Haes P. ACS Chem. Neurosci. 2014;5(10):1005–1019. doi: 10.1021/cn5001606. [DOI] [PubMed] [Google Scholar]
- Brunschweiger A., Koch P., Schlenk M., Rafehi M., Radjainia H., Küppers P., Hinz S., Pineda F., Wiese M., Hockemeyer J., Heer J., Denonne F., Müller C. E. Bioorg. Med. Chem. 2016;24(21):5462–5480. doi: 10.1016/j.bmc.2016.09.003. [DOI] [PubMed] [Google Scholar]
- Squarcialupi L., Falsini M., Catarzi D., Varano F., Betti M., Varani K., Vincenzi F., Dal Ben D., Lambertucci C., Volpini R., Colotta V. Bioorg. Med. Chem. 2016;24(12):2794–2808. doi: 10.1016/j.bmc.2016.04.048. [DOI] [PubMed] [Google Scholar]
- Gaspar A., Reis J., Matos M. J., Uriarte E., Borges F. Eur. J. Med. Chem. 2012;54:914–918. doi: 10.1016/j.ejmech.2012.05.033. [DOI] [PubMed] [Google Scholar]
- Moro S., van Rhee A. M., Sanders L. H., Jacobson K. A. J. Med. Chem. 1998;41:46–52. doi: 10.1021/jm970446z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander S. P. Phytother. Res. 2006;20:1009–1012. doi: 10.1002/ptr.1975. [DOI] [PubMed] [Google Scholar]
- Karton Y., Jiang J. L., Ji X. D., Melman N., Olah M. E., Stiles G. L., Jacobson K. A. J. Med. Chem. 1996;39:2293–2301. doi: 10.1021/jm950923i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A., Moro S., Manthey J. A., West P. L., Ji X. D. Adv. Exp. Med. Biol. 2002;505:163–171. doi: 10.1007/978-1-4757-5235-9_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X., Melman N., Jacobson K. A. J. Med. Chem. 1996;39:781–788. doi: 10.1021/jm950661k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalingam K. B., Radhakrishnan A. K., Haleagrahara N. Oxid. Med. Cell. Longevity. 2015;2015:1–14. doi: 10.1155/2015/314560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim K. E., Tagliaferro A. R., Bobilya D. J. J. Nutr. Biochem. 2002;13:572–584. doi: 10.1016/s0955-2863(02)00208-5. [DOI] [PubMed] [Google Scholar]
- Kumar S., Pandey A. K. Sci. World J. 2013;2013:1–16. [Google Scholar]
- Matosiuk D., Gralak D., Ryznar M. Annales Universitatis Mariae Curie-Sklodowska, Sectio DDD: Pharmacia. 2011;24:19–25. [Google Scholar]
- Van der Walt M. M., Terre'Blanche G. Bioorg. Chem. 2018;77:136–143. doi: 10.1016/j.bioorg.2018.01.004. [DOI] [PubMed] [Google Scholar]
- Legoabe L. J., Van der Walt M. M., Terre'Blanche G. Chem. Biol. Drug Des. 2018;91:234–244. doi: 10.1111/cbdd.13074. [DOI] [PubMed] [Google Scholar]
- Janse van Rensburg H. D., Terre'Blanche G., Van der Walt M. M., Legoabe L. J. Bioorg. Chem. 2017;74:251–259. doi: 10.1016/j.bioorg.2017.08.013. [DOI] [PubMed] [Google Scholar]
- Janse van Rensburg H. D., Legoabe L. J., Terre'Blanche G. and Van der Walt M. M., Drug Res., 10.1055/a-0808-3993. [DOI] [PubMed]
- Hallgas B., Dobos Z., Eosz E., Hollósy F., Schwab R. E., Szabó E. Z., Erõs D., Idei M., Keri G., Lóránd T. J. Chromatogr., B. 2005;819(2):283–291. doi: 10.1016/j.jchromb.2005.02.014. [DOI] [PubMed] [Google Scholar]
- Van der Wenden E. M., Hartog-Witte H. R., Roelen H. C., von Frijtag Drabbe Künzel J. K., Pirovano M., Mathôt R. A., Danhof M., Van Aerschot A., Lidaks M. J., Ijzerman A. P. Eur. J. Pharmacol. 1995;290(3):189–199. doi: 10.1016/0922-4106(95)00064-x. [DOI] [PubMed] [Google Scholar]
- Cantin L. D., Choi S., Clark R. B., Hentemann M. F., Ma X., Rudolph J., Liang S. X., Akuche C., Lavoie R. C., Chen L. and Majumdar D., US Pat., 7,714,004B2, 2010.
- Nel M. S., Petzer A., Petzer J. P., Legoabe L. J. Bioorg. Med. Chem. Lett. 2016;26(19):4599–4605. doi: 10.1016/j.bmcl.2016.08.067. [DOI] [PubMed] [Google Scholar]
- Van der Walt M. M., Terre'Blanche G. Bioorg. Med. Chem. 2015;23:6641–6649. doi: 10.1016/j.bmc.2015.09.012. [DOI] [PubMed] [Google Scholar]
- Van der Walt M. M., Terre'Blanche G. Eur. J. Med. Chem. 2016;125:652–656. doi: 10.1016/j.ejmech.2016.09.072. [DOI] [PubMed] [Google Scholar]
- Zhuang C., Zhang W., Sheng C., Zhang W., Xing C., Miao Z. Chem. Rev. 2017;117:7762–7810. doi: 10.1021/acs.chemrev.7b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes J. C. J. M. D. S. RSC Adv. 2017;7:9357–9372. [Google Scholar]
- Bruns R. F., Watson I. A. J. Med. Chem. 2012;55:9763–9772. doi: 10.1021/jm301008n. [DOI] [PubMed] [Google Scholar]
- Dahlin J. L., Baell J. B. and Walters M. A., in Assay Guidance Manual, ed. G. S. Sittampalam, N. P. Coussens, K. Brimacombe, A. Grossman, M. Arkin, D. Auld, C. Austin, J. Baell, B. Bejcek, J. M. M. Caaveiro, T. D. Y. Chung, J. L. Dahlin, V. Devanaryan, T. L. Foley, M. Glicksman, M. D. Hall, J. V. Haas, J. Inglese, P. W. Iversen, S. D. Kahl, S. C. Kales, M. Lal-Nag, Z. Li, J. McGee, O. McManus, T. Riss, O. J. Trask Jr, J. R. Weidner, M. J. Wildey, M. Xia and X. Xu, Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda (MD), 2004, pp. 1–43. [Google Scholar]
- Baell J. B., Holloway G. A. J. Med. Chem. 2010;53(7):2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Bruns R. F., Fergus J. H., Badger E. W., Bristol J. A., Santay L. A., Hartman J. D., Hays S. J., Huang C. C. Naunyn-Schmiedeberg's Arch. Pharmacol. 1987;335:59–63. doi: 10.1007/BF00165037. [DOI] [PubMed] [Google Scholar]
- Lohse M. J., Klotz K., Lindenborn-Fotinos J., Reddington M., Schwabe U., Olsson R. A. Naunyn-Schmiedeberg's Arch. Pharmacol. 1987;336:204–210. doi: 10.1007/BF00165806. [DOI] [PubMed] [Google Scholar]
- Gutschow M., Schlenk M., Gäb J., Paskaleva M., Wessam Alnouri M., Scolari S., Igbal J., Müller C. E. J. Med. Chem. 2012;55:3331–3341. doi: 10.1021/jm300029s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.