

Abstract
The major human pathogens Trypanosoma cruzi, Trypanosoma brucei, and Leishmania major are collectively known as the Tritryps. The initial comparative analysis of their genomes has uncovered that Tritryps share a great number of genes, but repetitive DNA seems to be extremely variable between them. However, the in-depth characterization of repetitive DNA in these pathogens has been in part neglected, mainly due to the well-known technical challenges of studying repetitive sequences from de novo assemblies using short reads. Here, we compared the repetitive DNA repertories between the Tritryps genomes using genome-wide, low-coverage Illumina sequencing coupled to RepeatExplorer analysis. Our work demonstrates that this extensively implemented approach for studying higher eukaryote repeatomes is also useful for protozoan parasites like trypanosomatids, as we recovered previously observed differences in the presence and amount of repetitive DNA families. Additionally, our estimations of repetitive DNA abundance were comparable to those obtained from enhanced-quality assemblies using longer reads. Importantly, our methodology allowed us to describe a previously undescribed transposable element in Leishmania major (TATE element), highlighting its potential to accurately recover distinctive features from poorly characterized repeatomes. Together, our results support the application of this low-cost, low-coverage sequencing approach for the extensive characterization of repetitive DNA evolutionary dynamics in trypanosomatid and other protozoan genomes.
Keywords: trypanosomatids, Tritryps, repetitive DNA, RepeatExplorer, RepeatExplorer, transposable elements
Main Text
Collectively known as the “Tritryps,” the unicellular mono-flagellated protozoan parasites Trypanosoma cruzi, Trypanosoma brucei, and Leishmania major are the causative agents of American trypanosomiasis, cutaneous leishmaniasis, and African tryrpanosomiasis, respectively. These dixenous parasites belong to the family Trypanosomatidae, within the order Kinetoplastida (Votýpka et al. 2015). Despite Tritryps share many general characteristics which are used as distinctive taxonomic markers (i.e., their unique mitochondria known as kinetoplast), each species has its own insect vector, particular life-cycle features, different target tissues, and distinct disease pathogenesis in mammalian hosts (Jackson 2015).
The genomes of T. cruzi, T. brucei, and L. major have been initially sequenced and compared with better understand gene evolution and genetic variation in these related pathogens (Ghedin et al. 2004; El-Sayed, Myler, Blandin, et al. 2005). A remarkable finding derived from the comparative analysis of Tritryps genomes was the great number of shared genes (El-Sayed, Myler, Blandin, et al. 2005). However, the repetitive DNA was extremely different in these species. Repetitive DNA sequences are scarce in the L. major genome, but comprises up to half of the T. cruzi genome. Moreover, L. major is believed to be devoid of active transposable elements (TEs) (Ghedin et al. 2004; Ivens et al. 2005; Bringaud et al. 2006), but both T. cruzi and T. brucei genomes harbor intact and autonomous TEs (Wickstead et al. 2003; El-Sayed, Myler, Bartholomeu, et al. 2005; Bringaud et al. 2008; Thomas et al. 2010; Berná et al. 2018). Caused by this intrinsic genome complexity—abundance of repetitive sequences and genes organized in tandem—the T. cruzi genome remained fragmented even through long-read sequencing (1,142 and 599 scaffolds in hybrid and nonhybrid strains, respectively; Berná et al. 2018), and all of the T. cruzi sequencing projects based on short reads have demonstrated that genome assembly and downstream comparative analyses are extremely challenging in this species.
Genome annotation procedures are mainly focused on standard genetic elements, frequently neglecting repetitive sequences due to their hard-achieving de novo assembly (Treangen and Salzberg 2012). As a consequence, repetitive DNA is poorly described and studied (Altemose et al. 2014). In this context, RepeatExplorer has emerged as a widely used approach to comprehensively evaluate the nature of repetitive sequences. This bioinformatic tool attempts to cluster low-coverage high-throughput sequencing reads using a graph-based algorithm to characterize and quantify the complete repetitive DNA fraction of a genome (Novák et al. 2010, 2013, 2017), which nowadays is known as the “repeatome” (Maumus and Quesneville 2014). Low-coverage sequencing is a cost-effective approach that does not require having previous information about the target genome and avoids dealing with whole-genome assemblies. Beyond RepeatExplorer was originally conceived to analyze plant repeatomes, it has been successfully applied in mammals (Pagán et al. 2012), insects (Ruiz-Ruano et al. 2016; Palacios-Gimenez et al. 2017; Pita et al. 2017), and fishes (Utsunomia et al. 2017). Here, we used low-coverage sequencing and the RepeatExplorer approach to compare the repeatomes of T. cruzi, T. brucei, and L. major to reference genomes. Four genomes of T. cruzi—with different sequencing technology approaches—were compared. CL Brenner strain with BAC-end Sanger sequencing (El-Sayed, Myler, Bartholomeu, et al. 2005), Sylvio X10 strain with 454 technology (Franzén et al. 2011), and the newly less collapsed PacBio sequenced strains Dm28c and TCC (Berná et al. 2018). The extensive amplification of repeated DNA is the main reason why the T. cruzi genomes were very poorly assembled. Since firsts genomes of T. brucei and L. major are considered as high quality assemblies, those were used as reference (Berriman et al. 2005; Ivens et al. 2005).
First, kinetoplast DNA (mini- and maxicircles) was removed from raw Illumina reads and after quality filtering a random subsampling was performed to obtain ∼1× coverage in each genome. This resulted in 353,334 reads from T. cruzi, 173,836 reads from T. brucei, and 218,778 reads from L. major that were subsequently used in the RepeatExplorer analyses. The software initially identified 293, 203, and 199 clusters for T. cruzi, T. brucei, and L.major, respectively. In T. cruzi, we estimated that 51.25% of the genome corresponds to repetitive DNA sequences. Out of them, 28.81% were annotated as coding sequences belonging to multigenic families, 8.85% as LINEs (Long Interspersed Elements), 3.73% as DIRS-like or tyrosin recombinase (YR) elements (mostly VIPER element), 3.48% as satellite DNA, 0.31% represented ribosomal DNA (rDNA), and 5.07% remained as undetermined repeats. Conversely, in T. brucei, only 20.69% of the genome harbors repetitive DNA sequences. Out of them, we were able to determine that 9.53% belong to coding sequences from multigenic families, 5.67% to LINE TEs, 3.59% were satellite DNA repeats, 0.33% as rDNA, and 1.57% of the genome remained as undetermined repeats. Finally, the repetitive DNA fraction in L. major was smaller than in the genus Trypanosoma, corresponding only to 8.80% of the genome. The vast majority of this repetitive DNA consisted in multigenic families (see details later), which reached the 3.93% of the genome. Additionally, 1.32% was identified as TEs named telomere-associated mobile elements (TATEs), 0.29% as LINE TEs, and 0.27% assigned to satellite DNA repeats. In addition, several clusters belonged to rDNA genes and snoRNA regions, which accounted for the 0.57% and 0.34% of the genome, respectively. The remaining 2.09% of the genome was annotated as undetermined repeats (fig. 1). In terms of quantitative comparison, figure 2 is representing the total amount of genome content in mega base pairs (Mbp), depicting the repetitive and nonrepetitive sequences. Although difference in genome size on Tritryps is highly influenced by the repetitive DNA content, it does not the only responsible, since nonrepetitive DNA fraction abundance is quite different in each genome.
Fig. 1.

—Comparison of Trypanosoma cruzi, T. brucei, and Leishmania major repeatomes. Bar plots show the relative amount of each repetitive DNA fraction on the (A) T. cruzi, (B) T. brucei, and (C) L. major genome. Pie charts represent the relative amount of repetitive and nonrepetitive DNA on each genome.
Fig. 2.
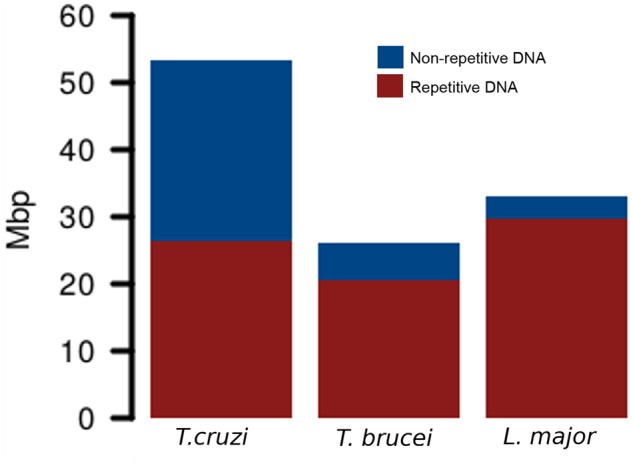
—Comparison of Trypanosoma cruzi, T. brucei, and Leishmania major genomes. Bar plots represent the total genome content in Mega base pairs (Mbp). In each genome, repetitive and nonrepetitive DNA is depicted.
In agreement to previous quantification of repetitive DNA in the genomes of T. cruzi CL Brener (El-Sayed, Myler, Bartholomeu, et al. 2005), Sylvio X-10 (Franzén et al. 2011), Dm28c and TCC (Berná et al. 2018) strains, our current analysis showed that almost half of the genome is composed by these sequences. Within the repetitive fraction, the most abundant sequences correspond to multigenic families as previously described on reference genomes. However, the relative abundance of each family remained uncertain and probably underestimated (El-Sayed, Myler, Bartholomeu, et al. 2005). It was only with the upcoming of the new referenced genomes (Berná et al. 2018), that we were able to compare our measure data with a noncollapsed genome, rendering quite similar quantification of the multigenic families as a whole. Still, it seems that our methodology deals with pseudogenes better that the classical methods for annotating multigenic families in an assembled genome. Since RepeatExplorer clusterization merge them together, more pseudogenes are annotated. On the other hand, we are not able to separate between proper genes and pseudogenes, but this can not be the objective using Illumina reads. Here, we were able to quantify the relative abundance of trans-sialidase (TS), retrotransposon hot spot (RHS), mucins, mucin-associated surface proteins (MASP), surface protein dispersed gene family-1 (DGF-1) and GP63 multigenic families (fig. 1). The amount of these families vary among different T. cruzi strains, as has been shown between CLBrener and X-10 Sylvio, which could be related to their infection capacity, since these protein-coding genes are involved in the parasite–host interactions (Franzén et al. 2011). The application of low-coverage sequencing and RepeatExplorer analysis over multiple T. cruzi strains with differential infectivity may uncover the relationship between multigenic dynamics and pathogenesis. TEs ranked second in terms of abundance being almost 13% of the genome. LINEs (such as the L1Tc, NARTc, CZAR, and TcTREZO elements) where more abundant than YR elements (VIPER element and their nonautonomous derivative SIRE). These repetitive DNA sequences were also underestimated on previous analyses (El-Sayed, Myler, Bartholomeu, et al. 2005; Franzén et al. 2011; Berná et al. 2018). As explained for multigenic families, TE fragments are not usually identified when classical procedures are done. Nevertheless, it must been taken into account that TEs richness differences between strains has been already described (Vargas et al. 2004). It could be attributed to natural variations between T. cruzi strains, but considering the remarkable disparity (5% estimated for CL Brener) this must deserve further attention, since additional factors than strain diversification may be explaining TEs dynamics. Moreover, TEs quantification on both newly generated reference genomes showed almost the same TEs abundance for TCC strain—closely related to CL Brenner—and Dm28c strain (Berná et al. 2018), evidencing that RepeatExplorer is a valid tool for TEs recognition and quantification. Lastly, satellite DNA sequences encompass >3% of the genome, being vastly dominated by the 195-nt satellite. Previous 195-nt satellite quantification on CL Brenner estimated that 5% of the genome is composed by this repeat (Martins et al. 2008). However, variation of 195-nt abundance has been reported to be 4- to 6-fold between DTU TcI and DTU TcII strains (Elias et al. 2003; Vargas et al. 2004). This difference is also observed in the new reference genomes from TCC and Dm28c. Actually, the Dm28c quantification is close to that reported here, reinforcing that low-coverage sequencing provides reproducible estimations of repeat element abundances. Several other tandem repeats have been recently described (Berná et al. 2018) but only a few of them were retrieved by RepeatExplorer, indicating that their abundances are below the threshold set for a standard analysis. However, we aimed to render a coarse-grain, genome-wide overview rather than a meticulous description of all repeats.
Trypanosoma brucei genome is composed by ∼20% of repetitive DNA. Similar to T. cruzi, multigenic families were the most abundant repeats reaching ∼10% of the genome, with RHS and VGS/ESAG as the most representative families. TEs in T. brucei represented 5.67% of the genome, but in this case is only composed by LINE sequences, such as the Tbingi elements, its related nonautonomous RIMEs, and a few SLAC elements. Although VIPER elements are described in T. brucei (Lorenzi et al. 2006), these repeats are known to be in very low copy number, hence undetectable under our approach. The first draft genome of T. brucei presented in 2005 (Berriman et al. 2005) only reported that subtelomeric genes were just over 20% and that TEs represented 2% of the genome (El-Sayed, Myler, Bartholomeu, et al. 2005), however, nothing is said about the satellite DNA. Our results showed two prominent satellite DNA families, the 177-bp repeat described to be part of intermediate and minicromosomes which are enriched by VSG genes (Sloof et al. 1983; Wickstead et al. 2004; Obado et al. 2005), and the 147-bp repeat (named CIR147) present in the centromere form the majority of macrochromosomes (Obado et al. 2007).
The most surprising results came along with the TEs analysis in L. major. Repetitive DNA comprehends <10% of the genome, as was expected since former genome analyses described smaller subtelomeric regions than in Trypanosoma species (Ivens et al. 2005; Peacock et al. 2007). Furthermore, the closely related L. braziliensis and L. infantum have also ∼10% of the genome composed by DNA repeats (Peacock et al. 2007). Although the reference genome for L. infantum has been resequenced using long-reads technology, revealing an expansion of coding genes copy number, the amount of repetitive DNA was not cited (González-De La Fuente et al. 2017). As observed in Trypanosoma, the majority of the repeated genome was represented by gene-coding sequences, being GP-63 and the Leucin-rich repeats among the most abundant elements. Remarkably, as in L. braziliensis genome (Peacock et al. 2007) but not described so far for L. major, we found traces of a the LINE element related to CRE2 (from Crithidia fasciculata), which is also related with CZAR and SLACs TEs from T. cruzi and T. brucei, respectively. Another interesting finding was the presence of a truncated element bearing a reverse transcriptase domain, from the LINE order. This probably corresponds to the LmDIRE elements, which are included in the ingi2 clade (Bringaud et al. 2009). By far, an exceptional finding was the recovering of TATE copies representing 1.32% of the genome. These elements were previously reported in other Leishmania species from the subgenus Viannia, such as L. braziliensis (Peacock et al. 2007) and L. panamensis (Llanes et al. 2015), but not from the subgenus Leishmania, as L. major. Sequence similarity searches on the L. major genome available on TriTrypdb (https://www.TriTrypdb.org) did not retrieve any positive results. Currently, TATEs are not classified within any of the TEs families, nor even as a concrete class. However, the presence of a tyrosine recombinase suggests that possibly TATEs are DIRS-like TEs (Peacock et al. 2007). Here, we were able to reconstruct a consensus sequence for the retrotranscriptase domain of the L. major TATE element, and determine that all kinetoplastid TATEs described hitherto form a separated clade from other DIRS-like elements (fig. 3). Further analysis on these elements would be of major interest for better understanding the dynamics of Leishmania genomes. Beyond the already know impact of TEs in trypanosomatid genomes (Bringaud et al. 2008; Thomas et al. 2010), our finding that TATE elements account for a considerable part of the L. major genome, could change the evolutionary paradigm of a genome that was believed to be almost TE-free. Actually, it has been already suggested that TATEs are not restricted to telomeric regions in L. panamensis genome, and that they could be playing a central role in gene regulation and structuring (Llanes et al. 2015). For example, being candidates to participate on recombinational events leading to genetic amplification (Ubeda et al. 2014). Genome localization of TATE elements within L. major genome could not be determined by our methodology; new assemblies from long reads would be the best approach on this issue.
Fig. 3.
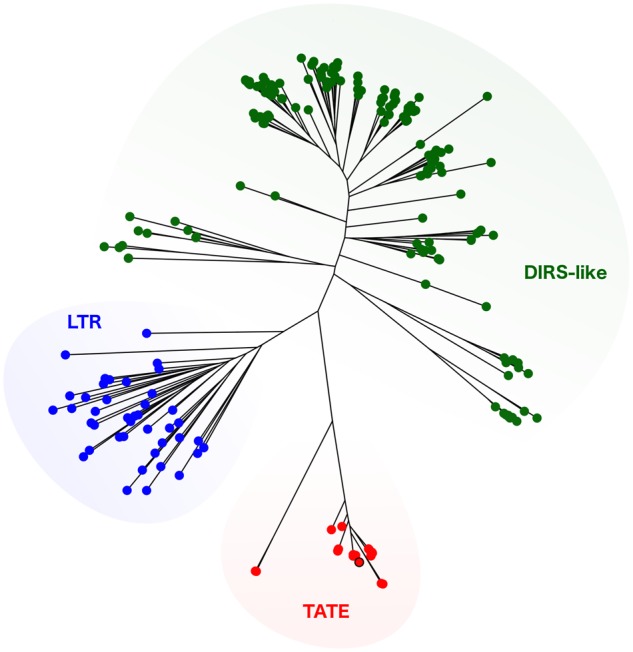
—Phylogenetic characterization of TATE elements. Maximum-likelihood phylogenetic tree using full retrotranscriptase domain sequences from all Trypanosomatidae TATE reported hitherto, and several DIRS-like elements retrieved from databases. Other LTR elements were used as outgroup. The Leishmania major TATE consensus sequence is marked by a black edge.
In conclusion, we have shown that our results are comparable to those obtained for other Tritryps strains and implementing different sequencing strategies, such as high-coverage and long-read genomic assemblies. This supports that our method using low-coverage, Illumina short reads is useful for a genome-wide characterization of trypanosomatid repeatomes, and could be useful to perform comparative analyses of the repetitive DNA repertories in other protozoan species. Noteworthy, our strategy allowed to identify genetic features that were not described so far, such as TATEs elements in the L. major genome.
Materials and Methods
Strains and DNA Purification
Trypanosoma cruzi Dm28c (Contreras et al. 1988) epimastigotes were cultured axenically in liver infusion tryptose medium supplemented with 10% (v/v) inactivated fetal bovine serum (GIBCO, Gaithersburg, MD) at 28 °C. Leishmania major (FRIEDLIN strain) and T. brucei (TREU927 strain) were cultured in modified RPMI medium containing 10% (v/v) inactivated fetal bovine serum (GIBCO, Gaithersburg, MD) at 28 °C. Quick-DNA Universal kit (Zymo Research, Irvine, CA) was used according to the manufacturer’s specifications for isolation of genomic DNA in logarithmic growth phase. The DNA was resuspended in sterile distilled water and stored at 4 °C until use. Quantification was performed using Qubit™ dsDNA HS Assay Kit (Invitrogen by Thermo Fisher Scientific, San Jose, CA).
Illumina Sequencing and Bioinformatic Analyses
Genomic libraries were prepared with the Nextera XT DNA Sample Preparation Kit (Illumina, San Diego, CA), analyzed using 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA), and then sequenced using a MiSeq Illumina platform, which produced 540831, 1790895, and 1322286 pair-end reads (2 ×150 cycles) for T. cruzi, T. brucei, and L.major, respectively. Low-coverage sequencing data results are available on SRP155233.
Kinetoplast DNA (mini- and maxicircles) was removed from raw Illumina reads, using DeconSeq (Schmieder and Edwards 2011) with a custom database made from several kinetoplastid maxicircles sequences deposited in GenBank. Quality filtering was performed with TRIMMOMATIC (Bolger et al. 2014) under LEADING: 3 TRAILING: 3 SLIDINGWINDOW: 4 : 20 MINLEN: 149 parameters. The random subsampling was performed to obtain ∼1× coverage in each genome with the shuf bash command. Graph-based clustering analyses were carried on separately using RepeatExplorer default options, implemented within the Galaxy environment (http://repeatexplorer.org/) (Novák et al. 2010, 2013, 2017). Cluster annotation was supported with a custom database of repeated gene families, retroelements, and satDNA, based on the newly PacBio sequenced reference genome annotation (Berná et al. 2018).
Leishmania major TATE consensus sequence was determined assembling the raw reads which belonged to RepeatExplorer clusters annotated as TATEs. Assembly of these reads was performed using CAP3 (Huang and Madan 1999), following a hand curation of the sequence alignment using SeaView (Gouy et al. 2010). TATE elements from other kinetoplastid genomes (Bodo saltans, Leptomonas pyrrhocoris, T. theileri, L. braziliensis, Angomonas deanei) were retrieved from NCBI, using BLAST search using the L. major TATE consensus sequence as query. DIRS-like sequences were downloaded from Repbase (https://www.girinst.org/repbase/) and only those with complete retrotranscriptase domains were used. DIRS-1 retrotranscriptase domain sequences were also recovered from the GenBank cd03714 sequence cluster, and retrotranscriptase domain sequences from other LTR elements were used as outgroup (cd01647 sequence cluster). Alignment of amino acid sequences was performed using MAFFT software (Katoh et al. 2002) under the G-INS-i method. Phylogenetic reconstruction was performed with PhyML (Guindon et al. 2010) under the WAG substitution model and the aLRT (Shimodaria–Hasegawa-like) test was employed for internal node support.
Acknowledgments
This work was funded by Agencia Nacional de Investigación e Innovación (UY) DCI-ALA/2011/023–502, ‘Contrato de apoyo a las políticas de innovación y cohesión territorial’, Fondo para la Convergencia Estructural del Mercado Común del Sur (FOCEM) 03/11, and by Research Council United Kingdom Grand Challenges Research Funder ‘A Global Network for Neglected Tropical Diseases’ grant number MR/P027989/1. FDV has a ANII doctoral fellowship (No. POS_NAC_2016_1_129916). SP, GI and CR are members of the Sistema Nacional de Investigadores (SNI-ANII, UY).
Footnotes
Data deposition: This project has been deposited at NCBI under the accesion SRP155233
Literature Cited
- Altemose N, Miga KH, Maggioni M, Willard HF.. 2014. Genomic characterization of large heterochromatic gaps in the human genome assembly. PLoS Comput Biol. 10:e10036.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berná L, et al. 2018. Expanding an expanded genome: long-read sequencing of Trypanosoma cruzi. Microb Genomics. 4(5):e0001:1–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriman M, Ghedin E, Hertz-fowler C.. 2005. The genome of the African trypanosome, Trypanosoma brucei. Science 309(5733):416–422. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringaud F, Berriman M, Hertz-Fowler C.. 2009. Trypanosomatid genomes contain several subfamilies of ingi-related retroposons. Eukaryot Cell. 8(10):1532–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringaud F, et al. 2006. Evolution of non-LTR retrotransposons in the trypanosomatid genomes: Leishmania major has lost the active elements. Mol Biochem Parasitol. 145(2):158–170. [DOI] [PubMed] [Google Scholar]
- Bringaud F, Ghedin E, El-Sayed NMA, Papadopoulou B.. 2008. Role of transposable elements in trypanosomatids. Microbes Infect. 10(6):575–581. [DOI] [PubMed] [Google Scholar]
- Contreras VT, et al. 1988. Biological aspects of the Dm 28c clone of Trypanosoma cruzi after metacyclogenesis in chemically defined media. Mem Inst Oswaldo Cruz. 83(1):123–133. [DOI] [PubMed] [Google Scholar]
- Elias MCQB, Vargas NS, Zingales B, Schenkman S.. 2003. Organization of satellite DNA in the genome of Trypanosoma cruzi. Mol Biochem Parasitol. 129(1):1–9. [DOI] [PubMed] [Google Scholar]
- El-Sayed NM, Myler PJ, Blandin G, et al. 2005. Comparative genomics of trypanosomatid parasitic protozoa. Science 309(5733):404–409. [DOI] [PubMed] [Google Scholar]
- El-Sayed NM, Myler PJ, Bartholomeu DC, et al. 2005. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309(5733):409–415. [DOI] [PubMed] [Google Scholar]
- Franzén O, et al. 2011. Shotgun sequencing analysis of Trypanosoma cruzi i Sylvio X10/1 and comparison with T. cruzi VI CL Brener. PLoS Negl Trop Dis. 5:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghedin E, et al. 2004. Gene synteny and evolution of genome architecture in trypanosomatids. Mol Biochem Parasitol. 134(2):183–191. [DOI] [PubMed] [Google Scholar]
- González-De La Fuente S, et al. 2017. Resequencing of the Leishmania infantum (strain JPCM5) genome and de novo assembly into 36 contigs. Sci Rep. 7(1):18050:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O.. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 27(2):221–224. [DOI] [PubMed] [Google Scholar]
- Guindon S, et al. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59(3):307–321. [DOI] [PubMed] [Google Scholar]
- Huang XQ, Madan A.. 1999. SymBioSys\rCAP3: a DNA sequence assembly program. Genome Res. 9(9):868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivens AC, et al. 2005. The genome of the kinetoplastid parasite, Leishmania major. Science 309(5733):436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AP. 2015. Genome evolution in trypanosomatid parasites. Parasitology 142(S1):S40–S56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T.. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llanes A, Restrepo CM, Del Vecchio G, Anguizola FJ, Lleonart R.. 2015. The genome of Leishmania panamensis: insights into genomics of the L. (Viannia) subgenus. Sci Rep. 5:8550.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi HA, Robledo G, Levin MJ.. 2006. The VIPER elements of trypanosomes constitute a novel group of tyrosine recombinase-enconding retrotransposons. Mol Biochem Parasitol. 145(2):184–194. [DOI] [PubMed] [Google Scholar]
- Martins C, et al. 2008. Genomic organization and transcription analysis of the 195-bp satellite DNA in Trypanosoma cruzi. Mol Biochem Parasitol. 160(1):60–64. [DOI] [PubMed] [Google Scholar]
- Maumus F, Quesneville H.. 2014. Deep investigation of Arabidopsis thaliana junk DNA reveals a continuum between repetitive elements and genomic dark matter. PLoS One 9(4):e94101.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák P, et al. 2017. TAREAN: a computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 45(12):e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák P, Neumann P, Macas J.. 2010. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics 11(1):378.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák P, Neumann P, Pech J, Steinhaisl J, Macas J.. 2013. RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29:792–793. [DOI] [PubMed] [Google Scholar]
- Obado SO, Bot C, Nilsson D, Andersson B, Kelly JM.. 2007. Repetitive DNA is associated with centromeric domains in Trypanosoma brucei but not Trypanosoma cruzi. Genome Biol. 8(3):R37.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obado SO, Taylor MC, Wilkinson SR, Bromley EV, Kelly JM.. 2005. Functional mapping of a trypanosome centromere by chromosome fragmentation identifies a 16-kb GC-rich transcriptional ‘strand-switch’ domain as a major feature. Genome Res. 15(1):36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagán HJT, et al. 2012. Survey sequencing reveals elevated DNA transposon activity, novel elements, and variation in repetitive landscapes among vesper bats. Genome Biol Evol. 4:575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios-Gimenez OM, et al. 2017. High-throughput analysis of the satellitome revealed enormous diversity of satellite DNAs in the neo-Y chromosome of the cricket Eneoptera surinamensis. Sci Rep. 7(1):6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock CS, et al. 2007. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet. 39(7):839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pita S, et al. 2017. Comparative repeatome analysis on Triatoma infestans Andean and Non-Andean lineages, main vector of Chagas disease. PLoS One 12(7):e0181635.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Ruano FJ, López-León MD, Cabrero J, Camacho JPM.. 2016. High-throughput analysis of the satellitome illuminates satellite DNA evolution. Sci Rep. 6:28333.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R.. 2011. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One 6(3):e17288.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloof P, et al. 1983. Characterization of satellite DNA in Trypanosoma brucei and Trypanosoma cruzi. J Mol Biol. 167(1):1–21. [DOI] [PubMed] [Google Scholar]
- Thomas MC, Macias F, Alonso C, López MC.. 2010. The biology and evolution of transposable elements in parasites. Trends Parasitol. 26(7):350–362. [DOI] [PubMed] [Google Scholar]
- Treangen TJ, Salzberg SL.. 2012. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 13(1):36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubeda JM, et al. 2014. Genome-wide stochastic adaptive DNA amplification at direct and inverted DNA repeats in the parasite Leishmania. PLoS Biol. 12(5):e1001868.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utsunomia R, et al. 2017. A glimpse into the satellite DNA library in characidae fish (Teleostei, Characiformes). Front Genet. 8:103:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas N, Pedroso A, Zingales B.. 2004. Chromosomal polymorphism, gene synteny and genome size in T. cruzi I and T. cruzi II groups. Mol Biochem Parasitol. 138(1):131–141. [DOI] [PubMed] [Google Scholar]
- Votýpka J, et al. 2015. New approaches to systematics of trypanosomatidae: criteria for taxonomic (re)description. Trends Parasitol. 31(10):460–469. [DOI] [PubMed] [Google Scholar]
- Wickstead B, Ersfeld K, Gull K.. 2003. Repetitive elements in genomes of parasitic protozoa. Microbiol Mol Biol Rev. 67(3):360–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstead B, Ersfeld K, Gull K.. 2004. The small chromosomes of Trypanosoma brucei involved in antigenic variation are constructed around repetitive palindromes. Genome Res. 14(6):1014–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]