

Abstract
The development and function of stem and progenitor cells that produce blood cells are vital in physiology. GATA-binding protein 2 (GATA2) mutations cause GATA-2 deficiency syndrome involving immunodeficiency, myelodysplastic syndrome, and acute myeloid leukemia. GATA-2 physiological activities necessitate that it be strictly regulated, and cell type–specific enhancers fulfill this role. The +9.5 intronic enhancer harbors multiple conserved cis-elements, and germline mutations of these cis-elements are pathogenic in humans. Since mechanisms underlying how GATA2 enhancer disease mutations impact hematopoiesis and pathology are unclear, we generated mouse models of the enhancer mutations. While a multi-motif mutant was embryonically lethal, a single-nucleotide Ets motif mutant was viable, and steady-state hematopoiesis was normal. However, the Ets motif mutation abrogated stem/progenitor cell regeneration following stress. These results reveal a new mechanism in human genetics, in which a disease predisposition mutation inactivates enhancer regenerative activity, while sparing developmental activity. Mutational sensitization to stress that instigates hematopoietic failure constitutes a paradigm for GATA-2 deficiency syndrome and other contexts of GATA-2–dependent pathogenesis.
Keywords: Hematology, Stem cells
Keywords: Bone marrow differentiation, Hematopoietic stem cells, Mouse models

Introduction
Development of the hematopoietic system involves massive genome remodeling and the establishment of complex genetic networks. The transcription factor GATA-binding protein 2 (GATA-2) establishes and maintains genetic networks governing hematopoietic stem and progenitor cell (HSPC) genesis and function (1–5) and is a major determinant of all blood cell lineages (6, 7). Human GATA2 deficiency syndrome resulting from germline coding or enhancer (+9.5) mutations includes monocytopenia and mycobacterial infection syndrome (2, 8, 9); DC, monocyte, B, and NK lymphoid deficiency (10); Emberger syndrome (primary lymphedema with myelodysplastic syndromes [MDS]) (8, 10–12); and familial MDS/acute myeloid leukemia (AML) (11). These pathologies might extend beyond germline mutations, as a patient with a somatic GATA2 mutation had similar phenotypes (13). Though mechanisms underlying GATA-2 pathologies and their unpredictable onset are unresolved, a GATA-2 growth–promoting circuit in AML (14) with clinical relevance (15) has been described. The variable onset and multicomponent phenotypes characterizing patients with GATA-2 deficiency syndrome (16–19) suggest that additional genetic and/or environment factors contribute to disease progression. Consistent with this inference, symptomatic patients often harbor additional mutations, e.g., in ASXL transcriptional regulator 1 (ASXL1) (20), or cytogenetic abnormalities such as monosomy 7 (18).
Since murine Gata2 ablation is lethal at approximately E10.5 (6), conditional (21, 22), heterozygous (23, 24), and enhancer-mutant mice (1, 2, 4, 5, 25) have been used to elucidate GATA-2 regulation and function. Transcriptional control of GATA-2 requires cell type–specific enhancers 9.5 kb downstream and 77 kb upstream of the Gata2 start site (2, 5, 26). Whether these enhancers function and are essential in regenerative contexts is unknown. The +9.5 triggers hematopoietic stem cell (HSC) emergence from hemogenic endothelium and confers vascular integrity (1, 2) (Figure 1). While Gata2-mutant mice have not previously been found to have malignancies, GATA-2 mediates leukemogenesis in a Tet2 ablation and FLT3–internal tandem duplication–overexpression (FLT3-ITD–overexpression) AML mouse model (27). Thus, normal hematopoiesis requires stringent GATA-2 regulation.
Figure 1. The +9.5 enhancer WGATAR motif is insufficient for HSC genesis and function.

(A) Mouse +9.5 mutations. Deletion of the E-box spacer GATA composite elements abrogates +9.5 function (2). The question marks denote uncertainty regarding whether the mutation impacts protein occupancy at adjacent sites. (B) Human +9.5 mutations found in patients with GATA-2 deficiency syndrome (2, 9).
The +9.5 enhancer contains multiple transcription factor motifs, including an E-box–spacer–WGATAR and Ets motif (Figure 1). These include canonical binding motifs for the heptad of transcription factors and cofactors governing the generation of HSPCs (28), including stem cell leukemia/T cell acute lymphoblastic leukemia (SCL/TAL1) (E-box: CANNTG), GATA-2 (WGATAR), and the Ets factors ETS-related gene (ERG) and Fli-1 proto-oncogene (FLI1) (GGAW). The Gata2 +9.5 E-box, GATA, and downstream motifs (including the Ets motif) are essential for +9.5 site enhancer activity in reporter assays (29, 30), and human patients with heterozygous disruptions of the +9.5 E-box or Ets motifs exhibit decreased GATA2 expression (2, 9). Deletion of the E-box–spacer–WGATAR (+9.5–/–) composite element reduced chromatin accessibility and abrogated occupancy of SCL/TAL1 and its cofactor LIM domain–binding 1 (LDB1) in fetal liver cells (31), revealing +9.5 enhancer–dependent chromatin accessibility and regulatory complex assembly at the Gata2 locus.
Patients with GATA-2 deficiency syndrome with enhancer mutations can lack the E-box or harbor single-nucleotide Ets mutations (2, 9, 32) that disrupt the GGAW motif, which is essential for high-affinity DNA binding (33). Both the E-box and Ets motif mutations reduce GATA2 expression (2, 9). The Ets 1017+572C>T transition was detected in at least 6 families and is the most common noncoding GATA2 mutation described to date (9, 34–36). Neither of these mutations is predicted to introduce known transcription factor–binding motifs (37). Since E-box– and Ets motif–mutant patients share phenotypes, +9.5 cis-elements might contribute equivalently to enhancer activity, including transcriptional complex assembly. Alternatively, different +9.5 cis-elements may exert qualitatively or quantitatively distinct functions, with essentially any corruption of +9.5 enhancer activity being pathogenic.
Here, we generated mouse strains to test these models and found differential cis-element contributions to +9.5 enhancer function. Mutation of E-box and Ets motifs, with retention of WGATAR, abrogated HSC emergence and was embryonically lethal, resembling mice lacking the E-box–spacer–WGATAR composite element (2). Though the Ets motif was dispensable for embryogenesis, it enhanced Gata2 upregulation and HSPC regeneration in response to myeloablation. These results revealed the human genetics concept that disease mutation segregates regenerative versus developmental functions of an enhancer. Furthermore, our analyses with the unique in vivo models provided evidence for a paradigm of GATA-2–dependent pathogenesis.
Results
Multiple cis-elements within the Gata2 +9.5 enhancer collectively support HSPC genesis.
The +9.5 deletion is embryonically lethal at approximately E14 (1, 2). It was unclear whether individual motifs within the +9.5 are essential or dispensable for enhancer activity. To test models to determine how distinct cis-elements contribute to +9.5 enhancer activity, we generated a mouse strain lacking the E-box and Ets, but retaining WGATAR [+9.5(E-box Ets)–/–] (Figure 2, A–C). We asked whether WGATAR confers any +9.5 activities in diverse cellular and development contexts, if it recapitulates the defects of +9.5–/– embryos, or if unique phenotypes emerge. By E13.5, +9.5(E-box Ets)–/– embryos showed a decrease of approximately 90% in fetal liver cellularity and reduced fetal liver cellularity and size (Figure 2, D and E), resembling +9.5–/– embryos. We found that homozygous mutants had severe hemorrhages (100% penetrance) and variable edema (Figure 2F) and died by E14.5 (Figure 2F and Table 1). Gata2 mRNA levels in +9.5(E-box Ets)–/– fetal liver from live embryos were 23-fold lower than those in WT littermates (Figure 2G), also resembling +9.5–/– embryos.
Figure 2. The +9.5 enhancer WGATAR motif is insufficient for embryogenesis.

(A) Sequences of WT (+9.5+/+) and E-box Ets–mutant [+9.5(E-box Ets)–/–] mice. (B) Sequence of mutant mice. (C) PCR genotyping. (D) Quantification of E13.5 liver cells from +9.5(E-box Ets+/+) (n = 11), +9.5(E-box Ets)+/– (n = 30), and +9.5(E-box Ets)–/– (n = 6) mice. Data are from 5 experiments. (E) E13.5 fetal livers from littermate mice. (F) E13.5 littermates with anemia, hemorrhage, and edema. (G) Gata2 mRNA quantification of fetal liver cells from +9.5(E-box Ets)+/+ (n = 5), +9.5(E-box Ets)+/– (n = 9), and +9.5(E-box Ets)–/– (n = 4) mice. Data are from 3 experiments. Quantitative data are represented as box-and-whisker plots, with bounds from the 25th to 75th percentiles, the median line, and whiskers ranging from minimum to maximum values. *P < 0.05, by 2-tailed, unpaired Student’s t test with Benjamini-Hochberg correction.
Table 1. Genotypes of embryos from +9.5(E-box Ets) heterozygous matings at developmental stages and weaning.

HSC genesis in the +9.5–/– aorta-gonad-mesonephros (AGM) is defective, given the lack of HSC-containing clusters and depletion of long-term repopulating HSCs (LT-HSCs) (1). To determine whether the +9.5(E-box Ets)–/– mutation impacts the endothelial-to-hematopoietic transition, we performed 3D confocal analysis of embryos to quantify emerging hematopoietic cells. Endothelial and hematopoietic cells express CD31, and hematopoietic, but not endothelial, cells, express c-Kit (38). While CD31+c-Kit+ clusters were abundant in E10.5 WT AGM, +9.5(E-box Ets)–/– embryos lacked clusters (Figure 3, A and B), indicating an HSC emergence defect.
Figure 3. The +9.5 enhancer WGATAR motif is insufficient for HSC genesis and function.

(A) Whole-mount immunostaining of E10.5 dorsal aorta (DA). CD31+ cells are shown in magenta and c-Kit+ cells in green. Scale bars: 100 μm. (B) c-Kit+ cell quantification within the dorsal aorta from +9.5(E-box Ets)+/+ (n = 3), +9.5(E-box Ets)+/– (n = 4), and +9.5(E-box Ets)–/– (n = 4) mice. Data are from 2 experiments. (C) Flow cytometric analysis of E13.5 fetal liver HSCs (Lin−CD41−CD48−Mac1+Sca1+Kit+CD150+) and MPPs (Lin−CD41−CD48−Mac1+Sca1+Kit+CD150–). FSC, forward scatter. (D and E) HSC and MPP quantification (percentage of live fetal liver cells from +9.5(E-box Ets)+/+ (n = 12), +9.5(E-box Ets)+/– (n = 19), and +9.5(E-box Ets)–/– (n = 4) mice. Data are from 3 experiments. Quantitative data are represented as box-and-whisker plots, with bounds from the 25th to 75th percentiles, the median line, and whiskers ranging from minimum to maximum values. *P < 0.05, **P < 0.01, and ***P < 0.001, by 2-tailed, unpaired Student’s t test with Benjamini-Hochberg correction.
At E13.5, the +9.5–/– fetal liver is devoid of immunophenotypic and LT-HSCs (Lin–CD48–CD41–Sca1+Kit+Mac1+CD150+) (2). We found that E13.5 +9.5(E-box Ets)–/– fetal liver was depleted of immunophenotypic HSCs (Figure 3, C and D). The percentage of multipotent progenitors (MPPs) (Lin–CD48–CD41–Sca1+Kit+Mac1+CD150–) was 5-fold lower than in WT littermates (Figure 3E). At this stage, the percentage of LT-HSCs was approximately 40% lower in +9.5(E-box Ets)+/– embryos than in WT embryos. MPP percentages were 2-fold higher in +9.5(E-box Ets)+/– embryos (Figure 3E), which may reflect compensation for the LT-HSC reduction. These results indicate that WGATAR is insufficient to support developmental hematopoiesis without the neighboring E-box and Ets motifs.
The Gata2+9.5 enhancer Ets motif is dispensable for HSPC genesis. To test models for how the human disease Ets mutation (1017+572C>T) impacts GATA-2 regulation and function, we generated a mouse strain harboring the disease mutation (Figure 4, A and B). Whereas +9.5–/– and +9.5(E-box Ets)–/– embryos exhibited severe anemia and hemorrhaging at E12.5 and died by E14.5 (Figure 2F and Table 1) (2), +9.5(Ets)–/– mice lacked hemorrhages (Figure 4C) and were born at Mendelian ratios (Table 2). The cellularity of WT and +9.5(Ets)–/– fetal livers was not significantly different (Figure 4D), although Gata2 expression was approximately 50% lower in the mutants (Figure 4E). Importantly, the lack of an Ets requirement for embryogenesis distinguishes the +9.5(Ets)–/– mutant from all other +9.5 mutants described (Figure 2 and Figure 3) (2) .
Figure 4. The +9.5 enhancer Ets motif is dispensable for HSC emergence.

(A) Sequences of WT [+9.5(Ets)+/+] and Ets motif-mutant [+9.5(Ets)–/–] embryos. Asterisk indicates C>T transition. (B) Sequences of +9.5+/+ and +9.5(Ets)–/– animals. (C) E15.5 littermates. (D) Quantification of E15.5 liver cells from +9.5+/+ (n = 18), +9.5(Ets)+/– (n = 20), and +9.5(Ets)–/– (n = 10) embryos. Data are from 4 experiments. (E) Gata2 mRNA quantification in fetal liver cells from +9.5(Ets)+/+ (n = 4), +9.5(Ets)+/– (n = 7), and +9.5(Ets)–/– (n = 5) embryos. Data are from 2 experiments. Quantitative data are represented as box-and-whisker plots, with bounds from the 25th to 75th percentiles, the median line, and whiskers ranging from minimum to maximum values. *P < 0.05 and **P < 0.01, by 2-tailed, unpaired Student’s t test with Benjamini-Hochberg correction.
Table 2. Genotypes of embryos from +9.5(Ets) heterozygous matings at weaning.

While +9.5+/– embryos are morphologically normal (2), HSC emergence (4), immunophenotypic and long-term–repopulating fetal liver HSCs, and Gata2 expression are approximately 50% lower than in WT mice (1, 2). To determine whether +9.5(Ets)–/– embryos resemble +9.5+/– embryos, we analyzed HSC emergence. CD31+c-Kit+ cell numbers decreased by approximately 50% in +9.5(Ets)–/– embryos (Figure 5, A and B). To assess whether this defect persisted throughout embryogenesis, we quantified immunophenotypic HSCs and MPPs in E15.5 fetal liver (Figure 5, C–E). We found that the percentages of +9.5(Ets)–/– fetal liver HSCs and MPPs were approximately 70% and 50% lower, respectively, than in WT mice. While the percentage of +9.5(Ets)+/– fetal liver HSCs did not differ significantly from that in WT embryos, the percentage of MPPs was approximately 2-fold higher in +9.5(Ets)+/– embryos. Thus, unlike the E-box–spacer–WGATAR composite element, which was essential for developmental hematopoiesis and embryogenesis, the Ets motif was dispensable, yet it contributed to select +9.5 functions.
Figure 5. The +9.5 enhancer Ets motif is dispensable for HSC emergence.

(A) Whole-mount immunostaining of E10.5 dorsal aorta. CD31+ cells are shown in magenta and c-Kit+ cells in green. Scale bars: 100 μm. (B) c-Kit+ cell quantification within the dorsal aorta from +9.5+/+ (n = 6), +9.5(Ets)+/– (n = 8), and +9.5(Ets)–/– (n = 6) mice. Data are from 4 experiments. (C) Flow cytometric analysis of E15.5 fetal liver HSCs (Lin−CD41−CD48−Mac1+Sca1+Kit+CD150+) and MPPs (Lin−CD41−CD48−Mac1+Sca1+Kit+CD150–). (D and E) Quantification of HSCs and MPPs (percentage of live fetal liver cells) from +9.5(Ets)+/+ (n = 9), +9.5(Ets)+/– (n = 11), and +9.5(Ets)–/– (n = 8) mice. Data are from 2 experiments. Quantitative data are represented as box-and-whisker plots, with bounds from the 25th to 75th percentiles, the median line, and whiskers ranging from minimum to maximum values; *P < 0.05, **P < 0.01, and ***P < 0.001, by 2-tailed, unpaired Student’s t test with Benjamini-Hochberg correction.
Human disease GATA2–mutant mouse model: combinatorial impact of a predisposition mutation and myeloablative stress on pathogenesis.
Since the Ets motif was dispensable for embryogenesis, we tested whether the Ets motif mutation affected adult hematopoiesis. Peripheral WBC, RBC, and platelet numbers were comparable between +9.5(Ets)–/– and WT mice (Figure 6A).
Figure 6. The +9.5 enhancer Ets motif confers hematopoietic regeneration and survival following stress.

(A) Steady-state peripheral blood parameters (n = 18 per genotype). (B) MA plot summarizing RNA-Seq analysis of transcripts from BM-derived WT and +9.5(Ets)–/– LSK cells in the steady state. (C) Kaplan-Meier survival curve for mice following two 5-FU doses (250 mg/kg, days 0 and 11): +9.5+/+ (n = 17), +9.5(Ets)+/– (n = 23), and +9.5(Ets)–/– (n = 10). Data are from 2 experiments. Significance was determined using the log-rank test. (D) qRT-PCR quantitation of mRNA from BM 9 days after vehicle (PBS) or 5-FU (250 mg/kg) treatment of +9.5+/+ (n = 8) and +9.5(Ets)–/– (n = 8) mice. Data are from 4 experiments. Quantitative data are represented as box-and-whisker plots, with bounds from the 25th to 75th percentiles, the median line, and whiskers ranging from minimum to maximum values. **P < 0.01, by Tukey’s multiple comparisons test. (E) Hematologic parameters following 5-FU treatment (150 mg/kg). n = 9 per genotype; data are from 3 experiments. (F) MA plot summarizing RNA-Seq analysis of transcripts from BM-derived WT and +9.5(Ets)–/– LSK cells on day 10 after treatment with 5-FU. (G) Relationships derived from the RNA-Seq data in F. Venn diagram compares 5-FU–regulated (total upregulated and downregulated) transcripts in BM-derived WT and +9.5(Ets)–/– LSK cells on day 10 after 5-FU treatment. These relationships were further deconvoluted by separating 5-FU–upregulated transcripts from the downregulated transcripts. PLTs, platelets.
In the steady state, a predisposition mutation might generate measurable but functionally “silent” molecular alterations. In this scenario, a secondary genetic aberration(s) or environmental insult might exacerbate such alterations and trigger phenotypic alterations that do not arise with either the predisposition mutation alone or the secondary aberration or insult alone. Alternatively, the predisposition mutation might be insufficient to derail molecular processes in the steady state, and only after a secondary aberration or insult would measurable molecular alterations be manifested. To distinguish between these mechanisms, we used RNA-Seq to compare transcriptomes of Lin–Sca1+c-Kit+ (LSK) cell populations isolated from WT and +9.5(Ets)–/– mice. After filtering out the outlying genes, the data were processed with DEseq at a FDR of 0.05 and a fold-change minimum of 2. Strikingly, of the 53,342 transcripts quantitated, only 11 were differentially expressed (Figure 6B). Thus, we determined that the predisposition mutation had little to no impact on the LSK cell transcriptome, consistent with a model in which the predisposition mutation creates a “silent” defect that is manifested only upon a secondary aberration or insult.
The silent nature of the predisposition mutation led us to question what triggers the emergence of pathogenic phenotypes. We reasoned that an increased demand on the hematopoietic system involving the transition of quiescent-to-proliferating HSCs and HSPC regeneration might unveil deleterious phenotypes in mutants harboring the predisposition mutation. To test whether myeloablation-induced stress reveals Ets activity, we used 5-fluorouracil (5-FU) to kill cycling cells and promote HSC proliferation, thereby regenerating the hematopoietic system (39). Two doses of 5-FU (250 mg/kg) were administered with an eleven-day interval for maximal stimulation of HSPC expansion (40). The +9.5(Ets)–/– mice had a reduced median survival of 14 days versus 22 days for WT mice (Figure 6C).
During regeneration, Gata2 transcripts are induced in the BM LSK cell population (40). We performed quantitative reverse transcription PCR (qRT-PCR) of BM and RNA-Seq analysis of LSK cells and found that Gata2 transcript levels did not differ significantly in BM from untreated +9.5(Ets)–/– mice compared with that from WT mice (Figure 6D). In contrast, 9 days after 5-FU treatment, Gata2 expression increased by approximately 15-fold in BM from WT mice, but not in that from +9.5(Ets)–/– mice (Figure 6D). The Ets motif was therefore essential for myeloablation-induced Gata2 expression and function. In principle, the Ets motif–mutant phenotypes might reflect hypersensitivity to 5-FU–mediated cell death and/or defective regeneration. Using a sublethal 5-FU dose (150 mg/kg), RBC, WBC, and platelet regeneration was delayed in +9.5(Ets)–/– mice (Figure 6E).
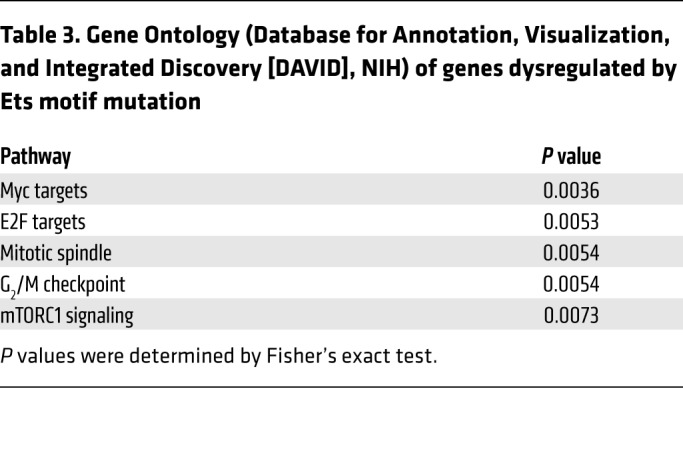
Since the predisposition mutation had little to no impact on the LSK transcriptome in the steady state, we used RNA-Seq to globally analyze whether the mutation affects 5-FU–induced gene expression changes. Differential expression analysis between vehicle-treated WT and 5-FU–treated LSK cells detected 2974 genes (754 upregulated, 2220 downregulated) at a FDR of 0.05 and a minimum fold-change of 2 (Figure 6F). Of these genes, only 423 were differentially expressed between vehicle-treated and 5-FU–treated mutant LSK cells (Figure 6G). Thus, the predisposition mutation altered the responsiveness of 85.8% of the genes that were 5-FU responsive in WT LSK cells. Gene Ontology analysis indicated that genes dysregulated by the Ets motif mutation included those linked to cell-cycle regulation (e.g., G2/M checkpoint) and cellular proliferation (e.g., Myc and E2F targets) (Table 3). The analysis detected 630 differentially expressed genes between vehicle-treated mutant and 5-FU–treated mutant LSK cells. While 423 of these were also differentially expressed between vehicle-treated WT and 5-FU–treated WT LSK cells, the remaining 32.8% were unresponsive to 5-FU treatment in the WT cells. The predisposition mutation corrupted the LSK transcriptome in the context of hematopoietic injury followed by regeneration, but not in the steady state. These results provide strong evidence that the Ets motif mutation generates a disease predisposition.
Table 3. Gene Ontology (Database for Annotation, Visualization, and Integrated Discovery [DAVID], NIH) of genes dysregulated by Ets motif mutation.
To establish how defective Gata2 induction impacts BM, we analyzed histological sections 9 and 11 days after 5-FU treatment (Figure 7A). No gross differences between untreated samples were apparent, consistent with the peripheral blood counts. While both WT and +9.5(Ets)–/– mice were hypocellular 9 days after treatment, we detected regeneration 11 days after treatment in WT sections (Figure 7, A and B). In contrast, there was still considerable hypocellularity 11 days after treatment of +9.5(Ets)–/– animals, confirming the delayed regeneration in the mutants.
Figure 7. The +9.5 enhancer Ets motif promotes hematopoietic regeneration after stress.

(A) H&E staining of BM after treatment with vehicle (PBS) or 9 or 11 days after treatment with 5-FU (250 mg/kg). Scale bars: 50 μm. (B) Quantification of BM cells as shown in A. n = 4 per genotype and condition; data are from 4 experiments. Quantitative data are represented as box-and-whisker plots, with bounds from the 25th to 75th percentiles, the median line, and whiskers ranging from minimum to maximum values. *P < 0.05, by 2-tailed, unpaired Student’s t test with Benjamini-Hochberg correction.
To analyze +9.5(Ets)–/– BM cellularity, we quantified immunophenotypic HSPCs in untreated and treated (9 and 11 days after 5-FU) mice. In the untreated mice, we observed that +9.5(Ets)–/– and WT HSCs (Lin−Sca1+Kit+CD48–CD150+, common myeloid progenitors (CMPs) (Lin−Sca1–Kit+FcγR–CD34+), granulocyte macrophage progenitors (GMPs) (Lin−Sca1–Kit+FcγR+CD34+), and megakaryocyte-erythrocyte progenitors (MEPs) (Lin−Sca1–Kit+FcγR–CD34–) were unaffected (Figure 8, A and B). MPP (Lin–Sca1+Kit+CD48–CD150–) percentages were 9-fold lower in mutants (Figure 8, A and B). Nine days after 5-FU treatment, HSC percentages were 12-fold higher in WT animals and were maintained at 11 days after treatment (Figure 8, A and B). The disease mutation abrogated the HSC increase. While all immunophenotypic progenitor cell populations were severely depleted by 5-FU after 9 days, 11 days after 5-FU treatment, WT progenitor percentages were 3.6- to 5.9-fold higher than those in untreated animals (Figure 8, A and B). In contrast, mutant progenitor percentages were restored only to the steady-state level. Thus, Ets confers Gata2 expression as a vital step in HSPC regeneration. To assess HSC function, BM from untreated animals was competitively transplanted into lethally irradiated mice. Sixteen weeks after transplantation, we observed that the +9.5(Ets)–/– BM multilineage repopulating activity was 3.0-fold lower (P < 0.0001) than that of WT BM (71 ± 4.6% and 23 ± 6.0% for mutant vs. WT BM, respectively) (Figure 9A). Analysis of donor-derived hematopoietic precursors in BM reconstituted with +9.5(Ets)–/– cells revealed significant reductions in LSK, HSC, LS–K, CMP, GMP, and MEP percentages relative to those in WT donor BM (Figure 9B).
Figure 8. +9.5 enhancer Ets motif–dependent HSPC regeneration.

BM was harvested 9 or 11 days after vehicle (PBS) or 5-FU (250 mg/kg) treatment. (A) Flow cytometric analysis of LSK cells (Lin−CD48−Sca1+Kit+), HSCs (Lin−CD48−Sca1+Kit+CD150+), MPPs (Lin−CD48−Sca1+Kit+CD150–), LS–K cells (Lin−Sca1–Kit+), MEPs (Lin−Sca1–Kit+FcγR–CD34–), CMPs (Lin−Sca1–Kit+FcγR–CD34+), and GMPs (Lin−Sca1–Kit+FcγR+CD34+). (B) LSK, HSC, MPP, LS–K, MEP, CMP, and GMP quantification (percentage of live BM cells). n = 8–10 per genotype and treatment; data are from 5 experiments. Quantitative data are represented as box-and-whisker plots, with bounds from the 25th to 75th percentiles, the median line, and whiskers ranging from minimum to maximum values. *P < 0.05, **P < 0.01, and ***P < 0.001, by Tukey’s multiple comparisons test.
Figure 9. +9.5 enhancer Ets motif–dependent regeneration of LT-HSCs.

(A) Multilineage repopulating activity 16 weeks after competitive transplantation (n = 10 per genotype). *P < 0.05 and ***P < 0.001, by 2-tailed, unpaired Student’s t test. (B) Analysis of donor-derived LSK cells, HSCs, MPPs, LS–K cells, CMPs, GMPs, and MEPs in BM 16 weeks after competitive transplantation (n = 10 per genotype). Statistical significance was determined by 2-tailed, unpaired Student’s t test.
Regeneration after hematopoietic injury, such as that caused by 5-FU–induced myeloablation, involves increased expression of the HSC-regulatory genes Scl, Gata2, and Ets variant 2 (Etv2) (40), each of which contains binding motif(s) within the +9.5 enhancer. ETV2 occupies the +9.5 enhancer in mouse embryonic stem cell–derived embryoid bodies (Figure 10A, irreproducible discovery rate [IDR] <0.01), and the +9.5 Ets motif mutant phenocopied the conditional deletion of Etv2 encoding a hematopoietic/vascular-regulatory ETS factor (40, 41). While ETV2 deficiency does not affect progenitors or blood cells in the steady state, vascular (42) or hematopoietic (40) injury induces ETV2, which then functions to promote recovery. Given the hundreds of ETV2 chromatin occupancy sites detected by ChIP-Seq, it is striking that the single-nucleotide mutation in the +9.5 enhancer disrupts hematopoietic regeneration in a manner analogous to the conditional deletion of ETV2 throughout the hematopoietic system. To test whether there is a hierarchical relationship between ETV2 and Gata2 in the hematopoietic injury response, we quantified Gata2 expression in regenerating HSPCs from WT and Tie2-Cre Etv2fl/fl conditional–knockout mice after 5-FU injury. We found that Gata2 expression was 1.5-fold lower (P = 0.001) in Etv2fl/fl LSK cells than in WT cells after 5-FU treatment (Figure 10B), suggesting that ETV2 functions upstream of GATA-2 in the injury response.
Figure 10. Evidence for an ETV2/GATA-2 regenerative axis.

(A) ETV2 ChIP-Seq. ETV2 occupancy was detected at the +9.5 enhancer in mouse embryonic stem cell–derived embryoid bodies (GEO GSE59402) (41). (B) Sorted LSK cells from WT (n = 6) or Tie2-Cre Etv2 conditional–knockout mice (n = 6) 10 days after 5-FU injection (250 mg/kg). Gata2 mRNA levels were normalized to Actb. **P < 0.01, by 2-tailed, unpaired Student’s t test.
Discussion
Recurring GATA2 mutations in human pathologies (11, 14, 43) highlight vital GATA-2 functions (18, 32, 44), some of which were predictable on the basis of prior studies of Gata2-mutant mouse models. As decreased (2, 6, 9) or elevated GATA-2 (45, 46) levels disrupt hematopoiesis, establishing and maintaining GATA-2 expression within a physiological window is crucial. Though this control is accomplished, in part, via +9.5 and –77 enhancer–dependent transcriptional induction of GATA-2 expression, many questions remain unanswered regarding the underlying mechanisms, including the relative contributions (qualitative and quantitative) of the individual cis-elements constituting the enhancers to enhancer function in diverse developmental and adult contexts, as well as the constitution of regulatory factors conferring or suppressing enhancer activity.
Mechanistic and pathological insights have emerged from analyses of Gata2 mouse models, including Gata2 enhancer mutants with impaired HSPC levels and functions (1, 2, 4, 5, 21, 23, 24). Here we dissected models governing +9.5 enhancer function and discovered that the WGATAR motif, without E-box and Ets motifs, was insufficient for embryogenesis and developmental hematopoiesis; WGATAR required additional cis-elements to confer the critical +9.5 enhancer activity and the control of developmental hematopoiesis (Figure 11). While the Ets motif was dispensable in embryos and adults in the steady state, the Ets motif disease mutation abrogated myeloablation-dependent GATA-2 induction and hematopoietic regeneration in BM. Myeloablation increased GATA-2 expression and HSC numbers in the BM of WT, but not +9.5(Ets)–/–, mice. Combined with our comparison of WT and +9.5(Ets)–/– LSK cell transcriptomes, these results establish a pathogenesis paradigm, in which the Ets motif disease mutation is, in effect, “silent” and singularly does not corrupt the LSK transcriptome or steady-state hematopoiesis. Because the mutation altered the capacity of 5-FU to induce transcriptomic changes, the mutation predisposed the cells to a secondary insult that instigated hematopoietic failure.
Figure 11. Human disease predisposition mutation segregates developmental versus regenerative enhancer functions.

Model depicts the +9.5(E-box Ets)–/– HSC emergence and fetal liver hematopoietic defects, which phenocopy the previously described +9.5–/– phenotype. Both mutations are embryonically lethal. In contrast, the human disease +9.5 Ets motif mutant is not an embryonically lethal mutation. In adult mice, the mutation impedes HSPC regeneration. We propose that the human mutation renders the hematopoietic system vulnerable to subsequent insults that demand HSPC regeneration to reestablish the steady state. The intimate connection between the predisposition mutation and mechanisms elicited by secondary insults may underlie the variable penetrance of disease onset in GATA-2 deficiency syndrome.
It is instructive to consider what qualitative insults and quantitative changes initiate and/or accelerate pathogenesis. While we used mild or severe myeloablation, does a damage threshold constitute a binary switch to trigger pathogenesis, or does modest stress induce phenotypes that are reversible or compensated for? Our model will enable this problem to be rigorously dissected in the contexts of environmental insults and genetic aberrations. As mimicking viral infection with the type I IFN inducer polyinosinic:polycytidylic acid (pI:pC) in Fanca–/– mice resulted in HSC functional defects (47), high levels of DNA damage, and BM failure, there may be mechanistic parallels between this system and the defective hematopoietic regeneration caused by the Gata2 Ets motif predisposition mutation.
We provide evidence for a new mechanism in human genetics, in which a disease mutation of a cis-element inactivates the regenerative activity of an enhancer, while sparing its developmental activities, which require multiple cis-elements. Dissecting this mechanism in the unique mouse model described here established a paradigm in which the cis-element predisposition mutation, combined with a secondary insult, corrupts hematopoietic-regulatory mechanisms, leading to hematopoietic failure, a feature of GATA-2-deficiency syndrome in humans. Furthermore, these studies illustrate the power of microdissecting enhancers to unveil physiological and pathological mechanisms.
Methods
Statistics.
Results are presented as either the mean ± SEM or as box-and-whisker plots, with bounds from the 25th to the 75th percentiles, the median line, and whiskers ranging from minimum to maximum values. Multiple independent cohorts were used in each experiment. Statistical comparisons were performed using 2-tailed Students t tests (significance cutoff of P < 0.05) , with correction of statistical overrepresentation of functions calculated using the Benjamini-Hochberg multiple tests correction procedure or Tukey’s multiple comparisons test. A log-rank test was performed on the Kaplan-Meier survival curve. GraphPad Prism (GraphPad Software) was used to perform the statistical analyses.
Generation of mutant mice.
Pronuclear injection was conducted on a C57BL/6J background at the 1-cell zygote stage at the UW-Madison Biotechnology Center using recombinant Cas9, guide RNA (gRNA) (5′-TCGTGTATCTGTTTCCGGACCGG-3′), and repair oligonucleotide (5′-TTTCAAAACAGCCCAGCAAGAGGCAGGACTGAGTCGAGGTGGCTCTGAAAACTTGCCGGTCCAGAAACAGATACACGAAGTTTCCTTATCTACCGGCTGCAGATGTCCGGATAGGAAACTCCGGC-3′) The C>T transition, without additional mutations, was detected in 4 of 25 live pups by DNA sequencing at and surrounding the mutation site. One additional founder contained the Ets point mutation and a 28-bp deletion corrupting the E-box motif. Further genotyping was performed using a TaqMan Assay (ABI, 6253771_1) or the primers Gata2 –78 forward: CACACAGCGGCCACCAA and Gata2 +120 reverse: AAAGCGGGTGAACGATTTAAAC.
Mouse embryo isolation.
Embryos were obtained from timed matings between heterozygous males and females, with the day of vaginal plug detection considered day 0.5. Pregnant females were euthanized with CO2, and fresh embryos were transferred into ice-cold PBS for dissection.
qRT-PCR.
Total RNA was purified with TRIzol (Invitrogen, Thermo Fisher Scientific). DNAse (Invitrogen, Thermo Fisher Scientific) treatment was performed on 0.1 to 1 μg RNA at 25°C for 15 minutes, followed by addition of 2.5 mM EDTA at 65°C for 10 minutes. cDNA was prepared by annealing with 250 ng of a 1:5 mixture of random hexamer and oligonucleotide (dT) primers incubated with m-MLV Reverse Transcriptase (Invitrogen, Thermo Fisher Scientific) with 10 mM DTT, RNasin (Promega), and 0.5 mM dNTPs at 42°C for 1 hour, followed by heat inactivation at 95°C for 5 minutes. cDNA was analyzed in reactions (20 μl) containing 2 μl cDNA, primers, and 10 μl Power SYBR Green (Applied Biosystems) by qRT-PCR with a Viia7 Real-Time RT-PCR Cycler (Applied Biosystems). Standard curves of serial 1:5 dilutions of cDNAs were prepared from control cDNA with the highest predicted gene expression. Values were normalized to the standard curve and 18S control.
Whole-embryo confocal microscopy.
E10.5 embryos were fixed, stained, and analyzed as described previously (1, 38). Embryos were stained with biotinylated anti–PECAM-1 (BD Biosciences, 553371) and anti–c-Kit (BD Biosciences, 553352) antibodies. Samples were mounted in a 1:2 mixture of benzyl alcohol (Sigma-Aldrich, 402834) and benzyl benzoate (Acros Organics, 105862500) to increase tissue transparency and then visualized with a Nikon A1RS Confocal Microscope. 3D reconstructions were generated from Z-stacks (80–200 optical sections) using Fiji software.
5-FU myeloablation model.
Myeloablation was induced by a single dose (150 mg/kg) or 2 doses (250 mg/kg) of 5-FU (MilliporeSigma, F6627) administered intraperitoneally on days 0 and 11. Blood samples were collected by retro-orbital bleeding (~10 μl per collection), and hematologic parameters were quantified on a HemaVet complete blood count (CBC) instrument (Drew Scientific).
BM isolation.
BM from femurs and tibiae of 8- to 14-week-old mice was collected by removing epiphyses and flushing the BM with a 25-gauge needle and syringe containing IMDM plus 2% FBS.
Histological analysis.
Femurs were fixed with 10% neutral-buffered formalin (Dot Scientific, DSF10800-500) and embedded in paraffin using standard procedures. Sections (10-μm) were stained with H&E. The hematopoietic content of serial images along the entire length of the femurs was quantified.
Flow cytometric analysis.
Fetal liver or BM was dissociated and resuspended in IMDM with 2% FBS and passed through 25-μm cell strainers to obtain single-cell suspensions prior to antibody staining. All antibodies were purchased from eBioscience (Thermo Fisher Scientific) unless otherwise stated. Lineage markers for the LSK cell populations were stained with the FITC-conjugated antibodies B220 (11-0452), CD3 (11-0031), CD4 (11-0041), CD5 (11-0051), CD8 (11-0081), CD41 (11-0411) (for fetal liver only), CD48 (11-0481), Gr-1 (11-5931), and TER-119 (11-5921). Other surface proteins were detected with PE-conjugated CD150 (BioLegend, 115904; PE-Cy7–conjugated Mac1, 25-0112 [for fetal liver only]); PerCP-Cy5.5–conjugated Sca1, and allophycocyanin-conjugated (APC-conjugated) c-Kit (2B8, 17-1171) antibodies. Analysis of myeloid progenitors was conducted as described in Johnson et al. (5). Lineage markers were stained with FITC-conjugated B220, CD3, CD4, CD5, CD8, CD19 (11-0193), IgM (11-5890), Il7Ra (11-1271), AA4.1 (11-5892), and TER-119 antibodies. Other surface proteins were detected with PE-conjugated FcγR (12-0161), eFluor 660–conjugated CD34 (50-0341), PerCP-Cy5.5–conjugated Sca1 (45-5981), and PE-Cy7–conjugated c-Kit (BioLegend, 105814). After staining, cells were washed with PBS and resuspended in IMDM, 2% FBS, and DAPI and analyzed on a LSR II Flow Cytometer (BD Biosciences).
RNA-Seq.
BM from 5-FU– or PBS-treated animals (10 days after treatment) was dissociated, resuspended in IMDM with 10% FBS and passed through 25-μm cell strainers to obtain single-cell suspensions prior to antibody staining. LSK cells (B220–CD3–CD4–CD5–CD8–Sca1+c-Kit+; 3,000–10,000 cells per replicate) were collected on a FACSAria II cell sorter (BD Biosciences). RNA was purified using an RNAeasy Micro Kit (QIAGEN, 74004). RNA libraries were prepared for sequencing using a standard NuGEN Ovation protocol. Library sequencing was conducted at the UW-Madison Biotechnology Center using an Illumina HiSeq 2500 (1 × 100). Data processing was conducted as described previously (48), and data were deposited in the NCBI’s Gene Expression Omnibus (GEO) database (GEO GSE123080).
BM transplantation.
Adult C57BL/6 recipient mice (CD45.1+, 6 to 8 weeks old; stock no. 002014, The Jackson Laboratory) were lethally irradiated using an XRAD 320 irradiator for a single dose of 8.5 Gy. BM cells were harvested from individual 8-week-old animals (CD45.2+). A total of 106 BM cells were mixed with the same number of CD45.1+ BM cells and injected into individual irradiated CD45.1+ recipients. The transplanted recipient mice were maintained on a Irradiated Uniprim Diet (Envigo; TD.06596) for 2 weeks. Blood obtained from the retro-orbital venous sinus or BM was isolated after transplantation and analyzed using flow cytometry for donor-derived hematopoiesis.
Study approval.
All animal protocols were approved by the UW-Madison IACUC in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC International) regulations.
Author contributions
AAS and EHB conceived and designed the research. AAS, CM, JW, MC, and KDJ conducted experiments. YZ and SK analyzed RNA-Seq data. IH analyzed histology. YZ and JZ facilitated the transplantations. KC generated ETV2 ChIP-Seq data. PL analyzed the ChIP-Seq data. AAS and EHB wrote the manuscript. AAS, EHB, CM, KDJ, SK, KC, and JZ edited the manuscript.
Acknowledgments
This work was supported by the NIH (DK68634 and DK50107, to EHB); the Evans MDS Foundation (to EHB and IH); Midwest Athletes Against Childhood Cancer (to EHB and IH); a National Heart, Lung, and Blood Institute (NHLBI) Training Grant in Hematology (T32 HL07899, to AAS); and the Carbone Cancer Center (P30CA014520).
Version 1. 01/08/2019
In-Press Preview
Version 2. 02/11/2019
Electronic publication
Version 3. 03/01/2019
Print issue publication
Funding Statement
DK50107
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
License: Copyright 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(3):1180–1192.https://doi.org/10.1172/JCI122694.
Contributor Information
Alexandra A. Soukup, Email: soukup@wisc.edu.
Ye Zheng, Email: yzheng74@wisc.edu.
Charu Mehta, Email: cmehta3@wisc.edu.
Jun Wu, Email: 377545520wustl@gmail.com.
Peng Liu, Email: pliu55@wisc.edu.
Miao Cao, Email: mcao36@wisc.edu.
Inga Hofmann, Email: ihofmann@wisc.edu.
Yun Zhou, Email: yzhou255@wisc.edu.
Jing Zhang, Email: zhang@oncology.wisc.edu.
Kirby D. Johnson, Email: kjohns25@wisc.edu.
Kyunghee Choi, Email: kchoi@wustl.edu.
Sunduz Keles, Email: keles@stat.wisc.edu.
Emery H. Bresnick, Email: ehbresni@wisc.edu.
References
- 1.Gao X, et al. Gata2 cis-element is required for hematopoietic stem cell generation in the mammalian embryo. J Exp Med. 2013;210(13):2833–2842. doi: 10.1084/jem.20130733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson KD, et al. Cis-element mutated in GATA2-dependent immunodeficiency governs hematopoiesis and vascular integrity. J Clin Invest. 2012;122(10):3692–3704. doi: 10.1172/JCI61623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linnemann AK, O’Geen H, Keles S, Farnham PJ, Bresnick EH. Genetic framework for GATA factor function in vascular biology. Proc Natl Acad Sci U S A. 2011;108(33):13641–13646. doi: 10.1073/pnas.1108440108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehta C, et al. Integrating Enhancer Mechanisms to Establish a Hierarchical Blood Development Program. Cell Rep. 2017;20(12):2966–2979. doi: 10.1016/j.celrep.2017.08.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson KD, et al. Cis-regulatory mechanisms governing stem and progenitor cell transitions. Sci Adv. 2015;1(8):e1500503. doi: 10.1126/sciadv.1500503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai FY, et al. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature. 1994;371(6494):221–226. doi: 10.1038/371221a0. [DOI] [PubMed] [Google Scholar]
- 7.Tsai FY, Orkin SH. Transcription factor GATA-2 is required for proliferation/survival of early hematopoietic cells and mast cell formation, but not for erythroid and myeloid terminal differentiation. Blood. 1997;89(10):3636–3643. [PubMed] [Google Scholar]
- 8.Hsu AP, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118(10):2653–2655. doi: 10.1182/blood-2011-05-356352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu AP, et al. GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood. 2013;121(19):3830–3837, S1. doi: 10.1182/blood-2012-08-452763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickinson RE, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118(10):2656–2658. doi: 10.1182/blood-2011-06-360313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hahn CN, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43(10):1012–1017. doi: 10.1038/ng.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ostergaard P, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome) Nat Genet. 2011;43(10):929–931. doi: 10.1038/ng.923. [DOI] [PubMed] [Google Scholar]
- 13.Sekhar M, et al. Can somatic GATA2 mutation mimic germ line GATA2 mutation? Blood Adv. 2018;2(8):904–908. doi: 10.1182/bloodadvances.2017012617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katsumura KR, Ong IM, DeVilbiss AW, Sanalkumar R, Bresnick EH. GATA Factor-Dependent Positive-Feedback Circuit in Acute Myeloid Leukemia Cells. Cell Rep. 2016;16(9):2428–2441. doi: 10.1016/j.celrep.2016.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carey A, et al. Identification of Interleukin-1 by Functional Screening as a Key Mediator of Cellular Expansion and Disease Progression in Acute Myeloid Leukemia. Cell Rep. 2017;18(13):3204–3218. doi: 10.1016/j.celrep.2017.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wlodarski MW, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127(11):1387–1397; quiz 1518. doi: 10.1182/blood-2015-09-669937. [DOI] [PubMed] [Google Scholar]
- 17.Hsu AP, McReynolds LJ, Holland SM. GATA2 deficiency. Curr Opin Allergy Clin Immunol. 2015;15(1):104–109. doi: 10.1097/ACI.0000000000000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spinner MA, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123(6):809–821. doi: 10.1182/blood-2013-07-515528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McReynolds LJ, Calvo KR, Holland SM. Germline GATA2 mutation and bone marrow failure. Hematol Oncol Clin North Am. 2018;32(4):713–728. doi: 10.1016/j.hoc.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.West RR, Hsu AP, Holland SM, Cuellar-Rodriguez J, Hickstein DD. Acquired ASXL1 mutations are common in patients with inherited GATA2 mutations and correlate with myeloid transformation. Haematologica. 2014;99(2):276–281. doi: 10.3324/haematol.2013.090217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim KC, et al. Conditional Gata2 inactivation results in HSC loss and lymphatic mispatterning. J Clin Invest. 2012;122(10):3705–3717. doi: 10.1172/JCI61619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Pater E, et al. Gata2 is required for HSC generation and survival. J Exp Med. 2013;210(13):2843–2850. doi: 10.1084/jem.20130751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ling KW, et al. GATA-2 plays two functionally distinct roles during the ontogeny of hematopoietic stem cells. J Exp Med. 2004;200(7):871–882. doi: 10.1084/jem.20031556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodrigues NP, et al. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood. 2005;106(2):477–484. doi: 10.1182/blood-2004-08-2989. [DOI] [PubMed] [Google Scholar]
- 25.Katsumura KR, et al. Human leukemia mutations corrupt but do not abrogate GATA-2 function. Proc Natl Acad Sci U S A. 2018;115(43):E10109–E10118. doi: 10.1073/pnas.1813015115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grass JA, et al. Distinct functions of dispersed GATA factor complexes at an endogenous gene locus. Mol Cell Biol. 2006;26(19):7056–7067. doi: 10.1128/MCB.01033-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shih AH, et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell. 2015;27(4):502–515. doi: 10.1016/j.ccell.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson NK, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7(4):532–544. doi: 10.1016/j.stem.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 29.Wozniak RJ, et al. Molecular hallmarks of endogenous chromatin complexes containing master regulators of hematopoiesis. Mol Cell Biol. 2008;28(21):6681–6694. doi: 10.1128/MCB.01061-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wozniak RJ, Boyer ME, Grass JA, Lee Y, Bresnick EH. Context-dependent GATA factor function: combinatorial requirements for transcriptional control in hematopoietic and endothelial cells. J Biol Chem. 2007;282(19):14665–14674. doi: 10.1074/jbc.M700792200. [DOI] [PubMed] [Google Scholar]
- 31.Sanalkumar R, et al. Mechanism governing a stem cell-generating cis-regulatory element. Proc Natl Acad Sci U S A. 2014;111(12):E1091–E1100. doi: 10.1073/pnas.1400065111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katsumura KR, Bresnick EH, GATA Factor Mechanisms Group The GATA factor revolution in hematology. Blood. 2017;129(15):2092–2102. doi: 10.1182/blood-2016-09-687871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hollenhorst PC, McIntosh LP, Graves BJ. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu Rev Biochem. 2011;80:437–471. doi: 10.1146/annurev.biochem.79.081507.103945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ganapathi KA, et al. GATA2 deficiency-associated bone marrow disorder differs from idiopathic aplastic anemia. Blood. 2015;125(1):56–70. doi: 10.1182/blood-2014-06-580340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koegel AK, Hofmann I, Moffitt K, Degar B, Duncan C, Tubman VN. Acute lymphoblastic leukemia in a patient with MonoMAC syndrome/GATA2 haploinsufficiency. Pediatr Blood Cancer. 2016;63(10):1844–1847. doi: 10.1002/pbc.26084. [DOI] [PubMed] [Google Scholar]
- 36.Wlodarski MW, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127(11):1387–1397; quiz 1518. doi: 10.1182/blood-2015-09-669937. [DOI] [PubMed] [Google Scholar]
- 37.Messeguer X, Escudero R, Farré D, Núñez O, Martínez J, Albà MM. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics. 2002;18(2):333–334. doi: 10.1093/bioinformatics/18.2.333. [DOI] [PubMed] [Google Scholar]
- 38.Yokomizo T, Yamada-Inagawa T, Yzaguirre AD, Chen MJ, Speck NA, Dzierzak E. Whole-mount three-dimensional imaging of internally localized immunostained cells within mouse embryos. Nat Protoc. 2012;7(3):421–431. doi: 10.1038/nprot.2011.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lerner C, Harrison DE. 5-Fluorouracil spares hemopoietic stem cells responsible for long-term repopulation. Exp Hematol. 1990;18(2):114–118. [PubMed] [Google Scholar]
- 40.Xu CX, et al. ETV2/ER71 regulates hematopoietic regeneration by promoting hematopoietic stem cell proliferation. J Exp Med. 2017;214(6):1643–1653. doi: 10.1084/jem.20160923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu F, et al. Induction of hematopoietic and endothelial cell program orchestrated by ETS transcription factor ER71/ETV2. EMBO Rep. 2015;16(5):654–669. doi: 10.15252/embr.201439939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park C, et al. Injury-Mediated Vascular Regeneration Requires Endothelial ER71/ETV2. Arterioscler Thromb Vasc Biol. 2016;36(1):86–96. doi: 10.1161/ATVBAHA.115.306430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katsumura KR, Yang C, Boyer ME, Li L, Bresnick EH. Molecular basis of crosstalk between oncogenic Ras and the master regulator of hematopoiesis GATA-2. EMBO Rep. 2014;15(9):938–947. doi: 10.15252/embr.201438808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dickinson RE, et al. The evolution of cellular deficiency in GATA2 mutation. Blood. 2014;123(6):863–874. doi: 10.1182/blood-2013-07-517151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vicente C, et al. Overexpression of GATA2 predicts an adverse prognosis for patients with acute myeloid leukemia and it is associated with distinct molecular abnormalities. Leukemia. 2012;26(3):550–554. doi: 10.1038/leu.2011.235. [DOI] [PubMed] [Google Scholar]
- 46.Persons DA, et al. Enforced expression of the GATA-2 transcription factor blocks normal hematopoiesis. Blood. 1999;93(2):488–499. [PubMed] [Google Scholar]
- 47.Walter D, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015;520(7548):549–552. doi: 10.1038/nature14131. [DOI] [PubMed] [Google Scholar]
- 48.Tanimura N, et al. GATA/Heme Multi-omics Reveals a Trace Metal-Dependent Cellular Differentiation Mechanism. Dev Cell. 2018;46(5):581–594.e4. doi: 10.1016/j.devcel.2018.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]