

Abstract
ARHGEF1 is a RhoA-specific guanine nucleotide exchange factor expressed in hematopoietic cells. We used whole-exome sequencing to identify compound heterozygous mutations in ARHGEF1, resulting in the loss of ARHGEF1 protein expression in 2 primary antibody–deficient siblings presenting with recurrent severe respiratory tract infections and bronchiectasis. Both ARHGEF1-deficient patients showed an abnormal B cell immunophenotype, with a deficiency in marginal zone and memory B cells and an increased frequency of transitional B cells. Furthermore, the patients’ blood contained immature myeloid cells. Analysis of a mediastinal lymph node from one patient highlighted the small size of the germinal centers and an abnormally high plasma cell content. On the molecular level, T and B lymphocytes from both patients displayed low RhoA activity and low steady-state actin polymerization (even after stimulation of lysophospholipid receptors). As a consequence of disturbed regulation of the RhoA downstream target Rho-associated kinase I/II (ROCK), the patients’ lymphocytes failed to efficiently restrain AKT phosphorylation. Enforced ARHGEF1 expression or drug-induced activation of RhoA in the patients’ cells corrected the impaired actin polymerization and AKT regulation. Our results indicate that ARHGEF1 activity in human lymphocytes is involved in controlling actin cytoskeleton dynamics, restraining PI3K/AKT signaling, and confining B lymphocytes and myelocytes within their dedicated functional environment.
Keywords: Immunology
Keywords: Adaptive immunity, B cells, Genetic diseases
Introduction
ARHGEF1 (also known as GEF1, P115-RHOGEF, and LSC) belongs to the dbl-homology guanine nucleotide exchange factor family (1–3) of proteins that activate monomeric GTPases by stimulating the release of guanosine diphosphate (GDP) and thus allowing the binding of guanosine triphosphate (GTP). ARHGEF1 is specific for the GTPase RhoA (4). The protein is predominantly expressed in hematopoietic cells and is involved in the signaling of G protein–coupled receptors associated with Gα12/13-containing heterotrimeric G proteins (5). Studies of Arhgef1–/– mice have revealed a lack of marginal zone B cells and impaired antibody responses to T-independent and T-dependent antigens (3, 6). A recent study suggested that somatic ARHGEF1 mutations are involved in the pathogenesis of germinal center (GC) B cell–like diffuse large B cell lymphoma (GCB-DLBCL), since this malignant disease is frequently associated with loss-of-function ARHGEF1 mutations (7). As seen in the context of GCB-DLBCL, B cells from Arhgef1–/– mice failed to activate sphingosine-1-phosphate (S1P) signaling. Furthermore, excessive egress of GC B cells into the lymphatic system and blood was observed in Arhgef1–/– mice, indicating the loss of a retention signal (7). Although ARHGEF1 has been analyzed in the mouse and in cancer-related conditions, less is known about the protein’s physiological in vivo contribution to the human immune system.
Primary antibody deficiencies (PADs) are the most common primary immunodeficiencies (PIDs) in humans (8). These deficiencies can result from intrinsic or extrinsic defects in B cell development, terminal B cell differentiation, antibody maturation, and/or T cell development (8). Although the genetic characterization of PAD patients is improving rapidly, most patients with PAD do not have a defined molecular diagnosis (8).
Using a whole-exome sequencing (WES) approach, we identified compound heterozygous germline mutations in ARHGEF1 in 2 PAD patients from the same family. These mutations led to ARHGEF1 deficiency, impaired RhoA activity, disturbed cytoskeleton dynamics, and impaired regulation of AKT signaling in both patients’ T and B lymphocytes. Our findings suggest that ARHGEF1 has a critical role in B lymphocyte homeostasis and function and in the confinement of the different hematopoietic cells to their respective dedicated functional environments.
Results
Clinical and immunology presentation.
Two female siblings (P1 and P2) born to healthy, nonconsanguineous parents presented during childhood with recurrent upper and lower respiratory tract infections; this included episodes of pneumonia from the age of 7 and 11 years onwards, respectively. The sisters were diagnosed with bronchiectasis and evaluated for PID at the age of 10 and 18 years, respectively. Antibody production (including T cell–dependent and –independent vaccine responses to poliovirus, tetanus, diphtheria toxoids, and pneumococcal immunizations) was defective in both patients (Table 1). P1 also presented with a low isohemagglutinin titer. Polyvalent IgG replacement therapy was initiated, and a lung lobectomy was performed on P1 at the age of 12 because of persistent suppuration associated with localized bronchiectasis (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI120572DS1). At 13 years of age, P1 developed immune thrombocytopenia. At last follow-up, P1 was aged 30 and was doing well on subcutaneous IgG replacement therapy.
Table 1. Clinical and immunological features of the 2 patients with PAD.
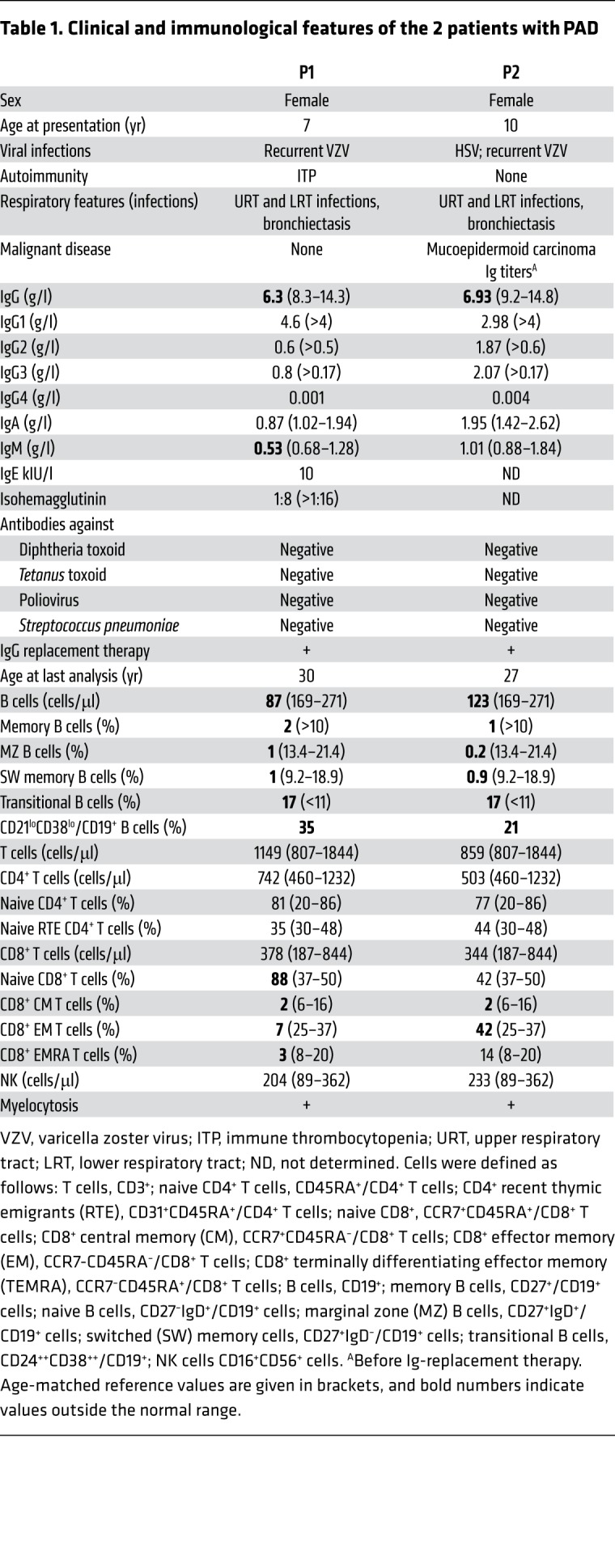
P2 experienced 3 episodes of herpes zoster, a severe, acute, oral herpes simplex virus 1 (HSV-1) primary infection, and recurrent lung infections; at 21 years of age, she was diagnosed with bronchial mucoepidermoid carcinoma and underwent a lung lobectomy. At last follow-up, P2 was aged 27 and doing well on subcutaneous Ig replacement therapy.
Blood samples from both patients repeatedly contained myelocytes (Figure 1, A and B). Consequently, a bone marrow examination of P2 was performed, but did not provide any evidence of a myeloproliferative or myelodysplastic syndrome. Both patients presented with low CD19+ B cell blood counts, an elevated frequency of transitional B cells (identified as CD19+/CD21+CD24++ [Figure 1C] or CD19+/ CD24++CD38++ cells), and an expansion of the CD21loCD38lo B cell subset (Table 1). Switched memory (CD19+/CD27+IgD–) and marginal zone (CD19+/CD27+IgD+) B cells were almost undetectable in both patients (Figure 1D). Cell counts, percentages of natural killer cells, and CD3+, CD4+, and CD8+ T cells were within the normal range (Table 1). An increased frequency of naive CD8+ T cells (CD8+/CCR7+CD45RA+) and a decreased frequency of all CD8+ memory subsets were observed in P1 but not P2 (Table 1). Both patients presented with a decreased frequency of CD8+ central memory and effector memory T cell subsets (Table 1). Remarkably, expression of the chemokine receptor CCR7 was higher on the patients’ CD8+ naive T cells than on controls (Supplemental Figure 1). Both parents had normal serum immunoglobulin levels, and the mother exhibited normal lymphocyte subsets.
Figure 1. Myelocytosis, an increase in transitional B cells, and the absence of marginal zone and memory B cells are hallmarks of the patients’ phenotype.

(A) Pictures of blood smears from P1 and P2 after staining with May-Grunwald-Giemsa reagent, showing the abnormal presence of myelocytes. Original magnification, ×100. (B) Distribution of the different myeloid cell populations in the blood of both affected siblings. Each circle (P1) or square (P2) denotes an independent blood sample. n = 2. Pro., promyelocytes; My., myelocytes; Meta., metamyelocytes. (C and D) Representative FACS plots analyzing the frequency of transitional B lymphocytes (C), marginal zone, memory, and naive B lymphocytes (D) in the blood of 2 healthy donors (HD1, HD2) and both patients. These experiments were performed 3 times. transi, transitional; Me., memory; MZ, marginal zone; N, naive.
Overall, the patients’ clinical and immunological characteristics were indicative of a PAD due primarily to disturbed B lymphocyte functions. However, a contribution from other affected cell types (including T lymphocytes) could not be ruled out.
A disturbed GC reaction.
In view of the occurrence of a bronchial mucoepidermoid carcinoma in P2, mediastinal lymph node biopsies were available. The lymph nodes were free of malignant cells. As shown in Figure 2, the GCs were smaller in P2 than in controls. It is noteworthy that the GCs were round and their structure was not disrupted. The GC mantle zone was somewhat less thick (according to IgD and CD79a staining), and the follicular dendritic cell meshwork was smaller (according to CD21 staining) than in controls. Strikingly, very few B cells were present in the GC in general and in its center in particular (according to CD20, CD79a, BCL6, and CD10 staining), in contrast with controls, in which GC B cells were evenly distributed. Proliferating (Ki67 stained) cells were mostly localized at the GC margin and were less frequent than in controls. The localization of PD-1–positive T follicular helper cells within the GCs was not altered. A high frequency of CD138-positive (syndecan-1) plasma cells was observed within the GCs, along with a higher number of interfollicular intracellular IgM- and IgA-positive cells compared with controls. Overall, the immunohistochemical analysis suggests that a disturbed GC reaction was combined with a relatively intense plasma cell development.
Figure 2. Histological analysis of mediastinal lymph node biopsies of P2, indicating a disturbed GC reaction.

Pictures of mediastinal lymph nodes of P2 and a control lymph node stained with the indicated antibodies, highlighting the GC structures. Original magnification, ×200; scale bar: 50 μm (H&E, CD3, CD5, CD20, CD79a, CD10, BCL6, PD1, Ki67, IgD, CD138, IgA, and IgM).Original magnification, ×100; scale bar: 100 μm (CD4 and CD8). Original magnification, ×50; scale bar: 200 μm (CD21).
Identification of an ARHGEF1 deficiency.
WES of DNA from total blood samples from both patients was performed with a view to identifying the underlying genetic cause of their disease. The WES results of both siblings were compared, leading to the identification of compound heterozygous variants in ARHGEF1: a nonsense variant on Chr19: 42398710: C>T (hg19 build 137) (NM_199002.1, exon 12, c. 898 C>T, p.R300X) and a splice acceptor site variant on Chr19: 42406933: G>T (Figure 3). Both variants (confirmed by Sanger sequencing; Figure 3B) were predicted to be highly damaging for the corresponding protein function, with combined annotation-dependent depletion scores of 41 and 23.3 for the nonsense and splice acceptor site variants, respectively. These variants were not annotated in our in-house database or in several open-access human genetic variation databases, including the Exome Aggregation Consortium (ExAC), the Exome Sequencing Project, the Short Genetic Variations Database (dbSNP), and the Swiss-Prot Variant database (SwissVar). Sanger sequencing of ARHGEF1 in the patients’ healthy parents confirmed the inheritance of the nonsense mutation from the father and the splice acceptor site mutation from the mother (Figure 3, A–C). To assess the impact of the ARHGEF1 splice acceptor site variant, mRNA processing of ARHGEF1 transcripts was analyzed by reverse-transcriptase PCR (RT-PCR) in RNA extracted from PBMCs collected from the patients, their mother, and a healthy donor. An aberrant ARHGEF1 transcript was detected in cells from both patients and their mother (Figure 3D), although it was expressed at lower levels than the main transcript, suggesting degradation. Sequencing of the transcript evidenced abnormal exon skipping of exon 19, introducing a frame shift with creation of a premature stop codon (E557Kfs34X).
Figure 3. Compound heterozygous mutations in ARHGEF1 lead to protein deficiency in the patients’ lymphocytes.

(A) Pedigree of the 2 siblings presenting with PAD. (B) Sanger sequencing of ARHGEF1 in P1, P2, and their parents. A nonsense mutation c.898C>T (Chr19: 42398710: C>T; hg19 build 137) was inherited from the father, and a splice mutation c.1669-1G>T (Chr19: 42406933: G>T) was inherited from the mother. F, father; M, mother. (C) Impact of ARHGEF1 mutations on the protein sequence. (D) RT-PCR analysis of the presence of ARHGEF1 transcripts lacking exon 19 (Δ exon 19) in the patients’ PBMCs, their mother, and a healthy donor (HD). L, ladder; H2O, water control. The schema for PCR analyses is depicted here; arrows designate primers used to analyze the effect of the c.1669-1G>T mutation on ARHGEF1 exon 19 splicing. The absence of the transcript lacking exon 19 was verified in 2 blood samples from independent healthy donors. (E and F) Western blots showing the expression of ARHGEF1 and RhoA in protein lysates of (E) B-EBV and (F) T cell blasts derived from patients. GAPDH was included as a loading control. (G and H) Enzyme-linked immunosorbent assay of the level of active RhoA (RhoA-GTP) in (G) B-EBV cells and (H) T cell blasts derived from patients. The level of active RhoA in the patients’ cells was compared with that found in HD-derived cells. In G, each symbol indicates an independent measure. Two independent healthy donor–derived B-EBV cell lines (HD1, circles, n = 3; HD2, white squares, n = 1), 2 independent B-EBV cell lines from P1 (P1-1, black squares, n = 3; P1-2, crosses, n = 2), and 1 B-EBV cell line from P2 (P2, triangles, n = 3) were analyzed. *P < 0.05; **P < 0.01, 1-sample, 2-tailed t test on normalized log2-transformed measurements. The experiment presented in H was performed only once.
To assess the effect of the compound heterozygous variants on ARHGEF1 protein expression, an immunoblot analysis with an antibody against the N-terminal part of ARHGEF1 was performed on patients’ lymphoblasts. No immunoreactive bands for ARHGEF1 protein, i.e., neither full-length nor truncated forms, were detected in total cell lysates from patient-derived, EBV-transformed lymphoblastoid cells or IL-2–propagated T cell blasts — in contrast with healthy donor cells (Figure 3, E and F, and Supplemental Figure 2). Taken as a whole, these data indicate that the compound heterozygous variants in ARHGEF1 in both patients resulted in ARHGEF1-deficient expression in lymphocytes.
Impaired RhoA activity and disturbed actin cytoskeleton dynamics in B and T lymphocytes from ARHGEF1-deficient patients.
ARHGEF1 is a specific guanine nucleotide exchange factor for RhoA GTPase, but not for RAC, CDC42, or RAS (4, 9). We therefore used an enzyme-linked immunosorbent assay to measure RhoA activity in lymphocytes from both patients. Although a normal amount of total RhoA protein was detected in a Western blot analysis (Figure 3, E and F), RhoA activity in B-EBV–transformed lymphoblastoid cells and T cell blasts derived from both patients was 2-to 3-fold lower than in cells from healthy donors (Figure 3, G and H). Given that RhoA is a key regulator of actin cytoskeleton dynamics, we next used FACS to analyze the amount of polymerized actin (F-actin) in blood lymphocyte subsets from both patients and healthy donors. We found the F-actin content to be abnormally low in all T and B lymphocyte subsets from the ARHGEF1-deficient patients when compared with that of healthy donors (Figure 4, A and B). Similar results were observed for T cell blasts from the patients and healthy donors (Supplemental Figure 3).
Figure 4. Reduced polymerized actin in lymphocytes is a signature of ARHGEF1 deficiency.

(A and B) Representative polymerized actin (F-actin) levels measured by FACS in (A) CD4+ and CD8+ naive T cells (CD4+CD45RA+CD31+, CD8+CD45RA+CCR7+) and (B) B cells (CD19+) in blood samples from patients and a healthy donor. (C) Representative FACS analyses showing the induction of actin polymerization in the naive CD4+ T cell compartment of PBMCs treated with various lysophospholipids. Cells were stimulated with S1P (10 μM for 1 minute), the thromboxane analogue U46619 (1 μM for 1 minute), and LPA (7.7 μM for 15 minutes). Stimulation with SDF1 (3 μg/ml for 1 minute) was assessed as a lysophospholipid-independent means of inducing actin polymerization. Experiments presented in A, B, and C were performed twice. It is noteworthy that the F-actin content was higher in memory T cells than in naive T cells (Supplemental Figure 4). (D) Enzyme-linked immunosorbent assay showing the induction of active RhoA (RhoA-GTP) after LPA stimulation (15 μM for 15 minutes) of T cell blasts from patients (P1, n = 2, square; P2, n = 2, triangle) and healthy donors (n = 4). Results are expressed as fold induction of unstimulated conditions. *P < 0.05, 1-sample, 2-tailed t test on normalized log2-transformed measurements.
ARHGEF1 possesses a regulator of G protein signaling domain (Figure 3C) and is thought to be involved in Gα 12/13–mediated signaling (3, 5). To analyze the impact of ARHGEF1 deficiency on this signaling, we investigated the stimulation of actin polymerization by ligands reported to signal via Gα 12/13–coupled receptors (S1P, lysophosphatidic acid [LPA], and the thromboxane A2 analog U46619) (3, 10–12) in PBMCs. In response to S1P, LPA, and U46619, patient-sourced CD4+ and CD8+ naive T cells displayed low or null levels of actin polymerization relative to healthy donor cells. In contrast, induction of the actin polymerization by chemokine stromal-derived factor 1α (SDF1), a ligand that is not strictly dependent on Gα 12/13 (13), was detectable in patients’ cells. However, the F-actin content after SDF1 stimulation remained lower in patients’ cells compared with controls (Figure 4C and Supplemental Figure 4). LPA-induced actin polymerization was also disturbed in patients’ B-EBV cell lines relative to a control cell line (Supplemental Figure 5A). The possibility that diminished expression of CXCR4 and/or lysophospholipid receptors in patient cells was responsible for the impaired ligand-induced actin polymerization was excluded by surface and RNA expression analysis in T cell blasts (Supplemental Figure 6). To confirm that impaired RhoA activation was responsible for the actin polymerization defect in ARHGEF1-deficient cells, we analyzed RhoA activity of T cell blasts stimulated with LPA. Induction of RhoA activity upon LPA stimulation was impaired in T cell blasts from both patients as compared with healthy donor cells (Figure 4D). Incubation of T cell blasts with the RhoA activator II, a commercially available drug that blocks Rho GTPase activity (14), resulted in a 2-fold increase in RhoA activity in P1 and a 3-fold increase in healthy control cells (Supplemental Figure 3D). In order to bypass potential problems of expression and signaling properties of lysophospholipid receptors, we analyzed the effect of enforced Gα 13 expression on RhoA activity in healthy donor– and patient-derived B-EBV cells. Increased RhoA activity was only found in healthy donor cells, although both control and patient cells exhibited increased expression of GNA13 RNA (Supplemental Figure 5B).
Next, we investigated whether RhoA activation could rescue the actin polymerization defect observed in patients’ cells. PBMCs and T cell blasts from the patients were treated for 1 hour with RhoA activator II. This treatment almost completely rescued actin polymerization in naive CD4 T lymphocytes and IL-2–propagated T cell blasts from the patients (Figure 5A and Supplemental Figure 3C). Actin polymerization promoted by RhoA involves the activation of its downstream target, the Rho-associated kinase I/II (ROCK) (15). In order to test ROCK’s functionality in patients’ cells, we treated them with the ROCK inhibitor Y27632. We observed that the patients’ cells did not modulate the F-actin content in response to Y27632 treatment, whereas the F-actin content was drastically diminished in T cell blasts derived from healthy donors (Figure 5B). In particular, the level of F-actin in Y27632-treated healthy donor cells was similar to that observed in untreated patient cells (Figure 5B). Next, we used fluorescence microscopy to analyze the cellular distribution of polymerized actin. A lower amount of cortical F-actin was observed in patients’ T cell blasts relative to that in healthy donor cells (Supplemental Figure 3A). To determine whether ARHGEF1 deficiency was responsible for the low level of actin polymerization, the patients’ T cell blasts were transduced using a retrovirus encoding WT ARHGEF1. Retrovirus-mediated expression of ARHGEF1 normalized cortical F-actin levels in P1’s cells (Figure 5, C and D, and Supplemental Figure 7). Taken as a whole, these data indicate that patients’ lymphocytes had a constitutive defect in RhoA/ROCK-mediated actin polymerization and impaired lysophospholipid receptor signaling as a consequence of ARHGEF1 deficiency.
Figure 5. Rescue of the actin polymerization defect in patients’ lymphocytes by drug-induced activation of the RhoA/ROCK pathway and by retroviral correction of ARHGEF1 expression.

(A) Representative FACS plots showing the effect of the RhoA activator II (RhoA act., 32 μg/ml for 1 hour) on actin polymerization in lymphocytes from PBMCs from a healthy donor and the patients (P1, P2). The experiment was performed 3 times. (B) Representative FACS analyses highlighting the effect of the ROCK inhibitor Y27632 (0.6 mg/ml for 1 hour) on the level of polymerized actin (F-actin) in T cell blasts from the patients and a healthy donor. The experiment was performed 3 times for P1 and twice for P2. (C) Representative confocal microscopy images showing the effect of the retroviral transduction of an ARHGEF1 construction (ARHGEF1-IRES-Thy1.1) or an empty vector (IRES-Thy1.1) on the level of F-actin in T cell blasts from P1 and a healthy donor. Original magnification, ×40 (48 hours after transduction). The experiment was performed twice. (D) Single-cell quantification of the fluorescence intensity of F-actin after ARHGEF1 expression in the confocal photographs shown in C. Between 80 and 130 cells were evaluated for each condition. ***P < 0.001, 2- tailed unpaired Student’s t test.
Impaired migration and formation of extended trailing edges in lymphocytes from ARHGEF1–deficient patients.
We compared the ability of PBMCs from patients and healthy donors to migrate toward SDF1. ARHGEF1-deficient B and T lymphocytes migrated less efficiently toward SDF1 (Figure 6A). ARHGEF1 has been implicated in regulation of RhoA activity downstream of adhesion to fibronectin (16). On fibronectin-coated Boyden chambers, we observed a strongly reduced Transwell migration of patients’ T cell blasts compared with those of controls (Figure 6B). Time-lapse microscopy of patients’ T cell blasts on fibronectin-coated surfaces revealed increased uropods (trailing edges) depicted by increased maximum tail length and decreased migration (mean displacement) (Figure 6, C–E). The decreased Transwell migration, mean displacement, and elongated trailing edges could reflect increased adhesion to fibronectin or defective deadhesion. Against increased adhesion, we showed that expression levels of the different integrin α and β chains on T cell blasts of P1 and P2 were either reduced or comparable to those of control cells (Supplemental Figure 8A). In addition, expression of the high-affinity conformation of lymphocyte function–associated antigen-1 (LFA-1) (integrin αLβ2; CD11a/CD18) was not increased in SDF1-activated ARHGEF1-deficient CD4+ and CD8+ memory T cell blasts, as compared with controls (Supplemental Figure 8B). Taken as a whole, these data suggest that human ARHGEF1 deficiency is associated with migration defects possibly caused by impaired deadhesion.
Figure 6. ARHGEF1-deficient lymphocytes exhibit impaired motility.

(A) SDF1-directed chemotaxis (Boyden chambers) analysis of PBMCs from patients (P1, n = 2; P2, n = 2) and healthy donors (n = 4). The migration index was defined as ratio of the number of migrating cells from the patient sample divided by the number of migrating cells from the healthy donor sample. *P < 0.05, 1-sample, 2-tailed t test on ratio. (B) Effects of fibronectin (Fn) coating on the passive migration of T cell blasts from healthy donors (n = 2) and patients (P1, n = 1; P2, n = 1). (C) Representative photographs showing the displacement of patient and healthy donor T cell blasts. Live cell imaging was performed during a minimum of 3 hours with photographs acquired every 50 seconds. Cropped areas are depicted here, and complete videos are available as Supplemental Videos 1–3. Stars show uropod anchoring and detachment point. Original magnification, ×20. (D) Dot plot showing the mean displacement of healthy donor and patient T cell blasts determined by live cell imaging as described in C. ****P < 0.0001, 2-tailed Welch’s t test on log2-transformed data. (E) Dot plot of the measure of the length of cell’s tails at the maximum of their elongation. *P < 0.05; **P < 0.01, 2-tailed Welch’s t test on log2-transformed data. For D and E, each symbol represents a single cell measurement. Measurements were performed with T cell blasts from independent healthy donors (circle;s n = 3), P1 (squares; n = 1), and P2 (triangles; n = 2).
Impaired regulation of AKT signaling.
One important function of the RhoA/ROCK pathway is the control of PI3K/AKT signaling via the regulation of phosphatase and tensin homolog (PTEN) (17). We thus compared the ability of T cell blasts from patients and healthy donors to repress AKT activation after SDF1/CXCR4-mediated PI3K activation. By measuring AKT phosphorylation (Ser473) with FACS, we found that repression of the AKT signal was less efficient in patients’ T cell blasts than in healthy donors’ blasts (Figure 7A). Treatment with RhoA activator II reduced SDF1-mediated AKT phosphorylation of patients and control T cell blasts similarly, indicating that the reduced repression of AKT phosphorylation in patients’ cells resulted from a defect upstream of RhoA. Of note, treatment of T cell blasts with the ROCK inhibitor Y27632 potentiated the phosphorylation of AKT after SDF1 stimulation (Figure 7A and Supplemental Figure 9A). Upon enforced ARHGEF1 expression in patients’ cells, a diminished AKT (Ser473) phosphorylation was observed as compared with cells transduced with an empty vector (Figure 7B). These results indicate that ARHGEF1’s ability to restrain AKT activation in lymphocytes via the modulation of RhoA/ROCK activity was abnormally low in patients’ cells.
Figure 7. ARHGEF1-deficient lymphocytes are less able to repress AKT activation.

(A) FACS analyses showing the level of AKT Ser473 phosphorylation after stimulation with SDF1 (6 μg/ml) for the indicated times with or without pretreatment with the RhoA act.II (32 μg/ml, 1 hour) or the ROCK inhibitor Y27632 (0.6 mg/ml, 1 hour) in T cell blasts derived from the 2 patients (P1, P2) and from a healthy donor. The experiment was performed twice, except for under ROCK inhibitor Y27632 conditions (once). (B) Representative histograms of the level of AKT Ser473 after ARHGEF1 forced expression in patient and healthy donor T cell blasts. The experiment was conducted 2 times. (C) Representative FACS plot showing the level of AKT phosphorylation after stimulation with SDF1 alone (6 μg/ml, 10 minutes) or in combination with S1P (20 μM, 10 minutes) in patient- or healthy donor–derived T cell blasts. (D) Inhibitory effect of S1P on SDF1-mediated AKT phosphorylation in patient- and healthy donor–derived T cell blasts. Each value represents the pAKT inhibition in independent T cell culture established from 6 different healthy donors (n = 6), P1 (n = 4), and P2 (n = 3). **P < 0.01, Mann-Whitney U test. Nonstim, nonstimulated.
It has been suggested that the activation of RhoA/ROCK/PTEN signaling by lysophospholipids (including S1P) promotes lymphocyte niche confinement by counterbalancing the PI3K/AKT signaling induced by chemokines, including SDF1 or CXCL13 (10, 18). Since this could not be tested in patients’ GC B cells, we investigated this signaling crosstalk in T cell blasts as a surrogate assay. Costimulation of control T cell blasts with SDF1 and S1P reduced the level of phosphorylated Ser473 AKT by about 40%, suggesting that S1PR-mediated inhibition of AKT phosphorylation operates in T lymphocytes (Supplemental Figure 9, A and B). Inhibition of AKT phosphorylation could also be achieved by substituting S1P with RhoA activator II, which highlighted the importance of RhoA activity in AKT regulation (Supplemental Figure 9B). Furthermore, the restriction of AKT activation by S1P-mediated signaling was found to involve ROCK activity, since pretreating healthy donor T cells with the Y27632 inhibitor completely abolished S1P’s repressive effect on AKT phosphorylation (Supplemental Figure 9, A and B). These results confirmed ROCK’s role in S1P-mediated AKT inhibition in human T cells. We next tested to determine whether patients’ lymphocytes could restrict AKT phosphorylation following SDF1 and S1P costimulation as efficiently as healthy donor cells could. Patients’ cells were significantly less efficient than those of controls (by 30%–40%) in dampening AKT phosphorylation (Figure 7, C and D). An elevated frequency of ribosomal protein S6 phosphorylation in B lymphocytes (as a consequence of increased PI3K/AKT/mTOR activation) has been observed in activated PI3Kδ syndromes (APDS1 and -2) caused by a gain-of-function PI3Kδ-signaling mutation (19). We also observed a higher frequency of phosphorylated ribosomal protein S6 in ARHGEF1-deficient B cells than in healthy donor cells (Supplemental Figure 10). Taken as a whole, these data suggest that the control of AKT signaling is impaired in ARHGEF1-deficient cells.
Discussion
We have identified human autosomal recessive ARHGEF1 deficiency as a cause of PAD. Compound heterozygous mutations in ARHGEF1 led to the absence of ARHGEF1 protein expression in 2 siblings. Neither the nonsense mutation nor the splice acceptor site mutation we found in patients was annotated in several major human genetics databases. It is noteworthy that at least 7 other rare variants resulting in a premature stop codon have been annotated in public access databases (Supplemental Table 1) for an extremely low number of cases. We assume that any combination of these variations will lead to ARHGEF1 deficiency.
We found that ARGHEF1-deficient patients’ lymphocytes had low RhoA activity and low cortical F-actin polymerization and that F-actin polymerization was restored by a RhoA activator and by the retroviral expression of ARHGEF1, indicating the causal nature of the ARHGEF1 defect. Several features reminiscent of the ARHGEF1 deficiency phenotype — including impaired antibody responses to T-dependent and T-independent antigens, the absence of blood marginal zone B cells, impaired signaling by lymphocytes in response to the phospholipids S1P, LPA, and U46619, impaired actin polymerization, and impaired control of AKT phosphorylation — have been observed in Arhgef1-deficient murine models (6, 7, 20). Thus, the similarity of the patients’ phenotype with the murine profile strengthens the hypothesis whereby ARHGEF1 deficiency causes PAD.
We observed a defect in RhoA/ROCK activation and actin polymerization in ARHGEF1-deficient lymphocytes cultured in the absence of lysophospholipid stimulation. These data suggest that ARHGEF1 functions by maintaining the intrinsic “tonic” activity of the RhoA/ROCK signaling pathway in human lymphocytes. The tonic activity of RhoA mediated by ARHGEF1 not only regulates the actin cytoskeleton, but also helps to dampen AKT phosphorylation following PI3K/AKT activation. Indeed, we showed that restriction of SDF1-induced AKT activation over time, without lysophospholipid costimulation, is less efficient in ARHGEF1-deficient lymphocytes. In addition, we showed that enforced ARHGEF1 expression restored control of AKT phosphorylation in patients’ cells. These results indicate that ARHGEF1/RhoA/ROCK activity functions as a constitutive break in the PI3K/AKT pathway. The observation of an elevated frequency of phosphorylated ribosomal protein S6 in ARHGEF1-deficient B cells further supports the notion of impaired AKT/mTOR regulation in ARHGEF1-deficient cells.
Impaired migration, especially of marginal zone B cells and the development of elongated trailing edges, has been described in Arhgef1-deficient mice (6). We showed that patient B and T cells migrated less efficiently toward SDF1 compared with controls. This observation might be explained by differences in T and B cell subset composition within the blood samples of the patients and healthy donor or by an intrinsic cellular defect present in the ARHGEF1–deficient patient lymphocytes. The latter hypothesis was further suggested by the observation of reduced Transwell migration of patient lymphocytes on a fibronectin-coated Boyden chamber. ARHGEF1-deficient T cell blasts exhibited a reduced cellular motility and extended trailing edges on fibronectin-coated surfaces, an observation reminiscent of the reported migration defect of Arhgef1-deficient marginal zone B cells on ICAM-1/VCAM-1–coated surfaces (6). We showed that ARHGEF1-deficient CD4+ and CD8+ memory T cell blasts exhibited no increased expression levels of the high-affinity conformation of LFA-1 after activation compared with controls, making it unlikely that hyperactivation of β2 integrins mediates the excessive adhesion of AHRGEF1-deficient cells. Our data rather suggest that the migration defect in human ARHGEF1 deficiency is associated with impaired deadhesion. A role of AHRGEF1 in integrin trafficking has been recently suggested based on the observation that Ahrgef1 deletion in murine leukocytes resulted in an increased pool of membrane β2 integrins and prevented angiotensin II–induced β2 integrin activation (21). Further exploration will be necessary to decipher the mechanisms underlying a possible connection of ARHGEF1 with integrin functioning.
Disruption of the mesenteric lymph node GC architecture associated with GC B cell detection in lymph and blood has been described in Arhgef1-deficient mice and mixed bone marrow chimeras (Arhgef1-deficient bone marrow transferred into Ly5.1 congenic host) (7). However, disruption of the GC structure associated with extensive egress of GC B cells (as observed in these murine models) was not clearly present in P2’s mediastinal lymph nodes. The presence of GC B cells at the GC’s margin only and not within the middle of the GC might reflect several non–mutually exclusive defects, such as (a) impaired proliferation of GC B cells, (b) disturbance of either the entrance, retention, or egress of GC B cells, (c) increased differentiation of GC B cells into plasmablasts/plasma cells, and (d) impaired survival of GC B cells (although there is no evidence of increased apoptosis). The absence of B cells within the middle of the GC could be explained by disturbed signaling of Gα 12/13–coupled receptors, such as S1PR2, which are reportedly involved in the maintenance and guidance of activated B cells in GCs (10). However, our observation of round, nondisrupted GC structures may also suggest that activation of a particular receptor is required for egress of ARHGEF1-deficient human GC B cells. It is noteworthy that S1PR3 has recently been described as an egress-promoting receptor for murine GC B cells, although S1PR3 is not upregulated in human tonsillar GC B cells (22). We also observed a higher frequency of CD138-positive plasma cells within the GC, suggesting a relative increased differentiation of GC B lymphocytes into plasma cells. The regulation of AKT/mTOR signaling in ARHGEF1-deficient T lymphocytes was found to be impaired. If one assumes that a similar defect is present in ARHGEF1-deficient B cells (as reported in ARHGEF1-deficient human diffuse large B cell lymphoma cell lines; ref. 7), increased PI3K/AKT signaling in human GC B lymphocytes (because of impaired regulation of ARHGEF1/RhoA/ROCK signaling) might promote the excessive differentiation of GC B lymphocytes into plasma cells.
Collectively, these data strongly suggest that ARGHEF1 deficiency is a new cause of inherited immune deficiency. Although ARHGEF1 is expressed in both B and T lymphocytes and abnormalities are found in both compartments in the absence of ARGHEF1 expression, the clinical and immunological presentation of the ARHGEF1-deficient patients (e.g., absence of marginal zone, increased numbers of transitional B cells in blood, disrupted distribution of B cells within the GC, and impaired T cell–independent antibody responses) strongly suggests that AHRGEF1 deficiency causes an intrinsic B cell defect.
The constant presence of immature myeloid cells in the blood of both patients (independently of inflammation, infection, hemolyses, or evidence for myeloid malignancy) highlights ARHGEF1’s function in retaining myelocytes in the bone marrow. This appears to be a human-specific ARHGEF1 function, as an increased frequency of myelocytes in the blood was not observed in an Arhgef1-deficient murine model (Jason Cyster, Dan Liu, and Scott Kogan, unpublished observations).
P2 was diagnosed with bronchial mucoepidermoid carcinoma at the age of 21. This raises the possibility that ARHGEF1 deficiency might predispose to certain types of cancer. Although ARHGEF1 expression is reported to be predominant in hematopoietic cells and ARHGEF1 loss-of-function mutations were observed in GCB-DLBCL, the expression of ARHGEF1 in other cells has also been reported (23), suggesting that other tumors could occur in the context of ARHGEF1 deficiency.
ARHGEF1 deficiency is a member of the growing group of PIDs, which includes Wiskott-Aldrich syndrome, Wiskott-Aldrich syndrome protein–interacting protein deficiency (24), ARPC1B deficiency (25), DOCK2 deficiency (26), DOCK8 deficiency (27), Ras homologue family member H deficiency (28), moesin deficiency (29), macrophage-stimulating 1 growth factor deficiency (30, 31), coronin-1A deficiency (32), RASGRP1-deficiency (33) and tetratricopeptide repeat domain 7A (TTC7A) deficiency (34) presenting with disturbed actin cytoskeleton dynamics. Intriguingly, several features in ARHGEF1-deficient lymphocytes (including diminished RhoA activity, diminished F-actin polymerization, and elevated CCR7 expression) contrast with those observed in TTC7A deficiency — highlighting the important of the tight regulation of RhoA activity for human lymphocytes (34). Clinically, patients with TTC7A deficiency present with early onset inflammatory bowel disease and progressive immune deficiency caused by the impairment of epithelial cells and lymphocytes (34).
The presence of circulating immature myeloid cells, the elevated frequency of transitional B cells, the absence of marginal zone B cells in the blood, and the disturbed distribution of B cells within the GC of ARHGEF1-deficient patients indicate the likely requirement of ARHGEF1 signaling for the retention and/or localization of these cells in their dedicated environment. ARHGEF1 deficiency could be seen as a mirror to warts, hypogammaglobulinemia, immunodeficiency, and myelokathexis (WHIM) syndrome, caused by a gain-of-function mutation in CXCR4. The peripheral neutropenia and profound B cell lymphopenia observed in patients with WHIM syndrome is explained (at least to a substantial extent) by a trafficking defect that disturbs the cells’ egress from the bone marrow (35).
In conclusion, our description of a immune deficiency caused by loss of ARHGEF1 function paves the way for further molecular investigations of the mechanisms underlying the trafficking and localization of B lymphocyte and myeloid cells.
Methods
WES.
Exome capture was performed using the SureSelect Human All Exon Kit (Agilent Technologies). Agilent SureSelect Human All Exon (58 Mb, V6) libraries were prepared from 3 μg of genomic DNA sheared with an ultrasonicator (Covaris), as recommended by the manufacturer. Barcoded exome libraries were pooled and sequenced using a HiSeq 2500 system (Illumina), generating paired-end reads. After demultiplexing, sequences were mapped against the human genome reference (NCBI build37/hg19 version) with the Burrows-Wheeler Aligner (BWA). The mean depth of coverage obtained for the 2 exome libraries was greater than ×115, with 97% or more and 93% or more of the targeted exonic bases covered by at least 15 and 30 independent sequencing reads, respectively (≥97% at ×15 and ≥93% at ×30). Variant calling was carried out with the Genome Analysis Toolkit (GATK), SAMtools, and Picard Tools. Single nucleotide variants were called with the GATK Unified Genotyper, whereas indel calls were made with the GATK IndelGenotyper, version 2. All variants with a read coverage of ×2 or less and a Phred-scaled quality of 20 or less were filtered out. All the variants were annotated and filtered using PolyWeb (our in-house annotation software).
Sanger sequencing was performed with the primers forward 5′-GAAGTCGGGGAGGAACTTCT-3′ and reverse 5′-CCCCCAGTATGGATGCTATG-3′ for the stop mutation and forward 5′-GAAAATCTCCTCCCGCTTCT-3′ and reverse 5′-CTGCAGTGAGCTGTGATGGT-3′ for the splice mutation. For mRNA sequencing, RT-PCRs were performed with 100 ng of total RNA, using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher). Exon 19 splicing was analyzed and sequenced using the following primers: forward, 5′-CCAGAAAATCTCCTCCCGCT-3′; reverse, 5′-GGTCCTCCATGTCACGCA-3′.
Peripheral blood lymphocyte phenotyping.
Blood collected in heparin tubes was directly stained for B cell and T cell surface markers with the following fluorescent-conjugated antibodies: CD19 (clone HIB19, catalog 2111030, Sony), IgM (clone MHM-88, catalog 2172580, Sony), CD21 (clone B-ly4, catalog 561374, BD), CD24 (clone ML5, catalog 555428, BD), CD31 (clone M89D3, catalog 558094, BD), CD45RA (clone HI100, catalog 560675, BD), CD57 (clone NK-1, catalog 555619, BD), CD3 (clone UCHT1, catalog 25-0038-42, BD), IgD (clone IA6-2, catalog 555779, BD), CCR7 (clone 150503, catalog FAB197F, RD system), CD27 (clone O323, catalog 302810, BioLegend), CXCR4 (clone 12G5, catalog 306528, BioLegend), CD4 (clone VIT4, catalog 130-092-373, Miltenyi Biotec), CD8 (clone BW135/80, catalog 130-096-902, Miltenyi Biotec), CD1c (clone AD5-8E7, catalog 130-090-507, Miltenyi Biotec), and CD23 (clone M-L23.4, catalog 130-099-986, Miltenyi Biotec). Red blood cell lysis was achieved either with the lysis buffer provided in the PerFix EXPOSE Kit (Beckman Coulter) or with BD FACS lysing solution. Cells washed in PBS were then analyzed with a MACSQuant analyzer (Miltenyi Biotec) or an SP6800 spectral analyzer (Sony).
Cell culture.
PBMCs were obtained by Ficoll-Plaque density gradient centrifugation. T cell blasts were established by activating PBMCs with phorbol myristate acetate (20 ng/ml, MilliporeSigma) and ionomycin (1 μM) in a RPMI 1640 GlutaMAX medium (Thermo Fisher Scientific) supplemented with 1% penicillin/streptomycin and 10% human AB serum (complete medium) for 3 days. After a second Ficoll-Plaque density gradient centrifugation, cells were expanded in complete medium containing 100 U/ml IL-2.
The Necker Imagine Centre de Resources Biologiques (CRB) (Paris France) generated EBV-transformed lymphoblastoid cell lines (B-EBV). B-EBV cells were cultured in RPMI 1640 GlutaMAX, 1% penicillin/streptomycin medium supplemented with 10% FCS (Invitrogen).
RhoA activation and ROCK inhibition assays.
The RhoA activation assay was performed after a 1-hour treatment with 16–32 μg/ml of the RhoA activator II (Cytoskeleton) in a complete medium for PBMCs or in a RPMI 1640 GlutaMAX, 1% penicillin/streptomycin medium supplemented with 0.5% fatty acid–free BSA (lipid-free medium) for T cell blasts. For ROCK inhibition studies, T cell blasts were incubated with 0.6 mg/ml of the ROCK inhibitor Y27632 (Chemdea) in a complete medium or in a lipid-free medium. After incubation, cells were centrifuged and stained for surface markers for 10 minutes prior to fixation in a 2% PFA solution in PBS. The cells were then stained for intracellular markers and analyzed by FACS.
Actin polymerization assay.
Actin polymerization was analyzed by FACS and by immunofluorescence. For the FACS analysis, the cells were stained for the surface marker in a lipid-free medium and then incubated (or not) with different ligands, including 3 or 6 μg/ml SDF1 (BioLegend), 1 μM S1P, 7.7 μM or 15 μM LPA, and 1 μM U46619 (Santa Cruz Biotechnology Inc.). Stimulation was stopped by fixing cells with a 2% PFA solution in PBS for 10 minutes at room temperature. Cells were then washed once with PBS and permeabilized with PBS with 0.1% saponin and 0.5% BSA for 10 minutes. Cells were then stained for 30 minutes with 1.5 μg/ml phalloidin-FITC (MilliporeSigma) in PBS with 0.1% saponin and 0.5% BSA, in the dark. Stained cells were washed 2 times with PBS with 0.1% saponin and once with PBS. Single-cell analysis of actin was then carried on with a MACSQuant analyzer (Miltenyi Biotec).
For the immunofluorescence staining, cells were loaded on fibronectin-coated (40 μg/ml, Takara/Clontech) coverslides or Lab-TeK chambers (Thermo Fisher) and were allowed to adhere for 15–30 minutes in complete medium or lipid-free medium. Cells stimulated (or not) with ligands were then fixed in 2% PFA in PBS for 10 minutes and washed once in PBS. The PFA solution was then quenched with 50 nM glycine in PBS for 20 minutes and washed once with PBS. Cells were then permeabilized and stained with 0.1% saponin, 0.5% BSA, and 1.5 μg/ml phalloidin-FITC in PBS for 30 minutes in the dark. Stained cells were washed twice in 0.1% saponin with PBS and once in PBS. Nuclei were stained with Vectashield H-1200 mounting medium (Vector). Glass slides were then viewed under the microscope.
AKT Ser473 and S6 Ser235/236 phosphorylation assays.
AKT Ser473 phosphorylation was analyzed on T cell blasts cultured for between 5 and 7 days after the beginning of IL-2 propagation. Cells were stimulated with 6 μg/ml SDF1 in the presence or absence of 20 nM S1P in lipid-free medium for 10 minutes. In experiments (including ROCK inhibition or RhoA activation experiments), cells were pretreated (as in the actin assay) prior to stimulation with ligand. Cells were then fixed with 4% formaldehyde in PBS for 10 minutes. The cells were then permeabilized in ice-cold methanol for 30 minutes and washed twice with 0.5% BSA in PBS. The AKT Ser473-AF647 antibody (clone D9E, catalog 4075S, Cell Signaling Technology) was used to stain for AKT Ser473. The staining was performed in 0.5% BSA in PBS in the dark for 1 hour. The cells were washed once with 0.5% BSA in PBS and then resuspended in PBS prior to analysis on the MACSQuant analyzer (Miltenyi Biotec).
Ex vivo assessment of S6 phosphorylation was performed on total blood using an antibody against phosphorylated S6 Ser235/236 (clone D57.2.2E, catalog 8520S, Cell Signaling Technology) and the PerFix EXPOSE kit, according to the manufacturer’s recommendations (Beckman Coulter). Briefly, 100 μl of freshly drawn blood was incubated in the presence of surface antibody for 10 minutes at 37°C in a water bath. The reaction was stopped using buffer 1, and red blood cells were lysed with buffer 2 for 5 minutes at 37°C. The samples were centrifuged, and the pellets underwent intracellular staining with buffer 3 for 1 hour. The cells were then washed with the dedicated buffer, resuspended, and analyzed with a MACSQuant analyzer (Miltenyi Biotec).
Active RhoA assay.
Active RhoA was measured using the RhoA G-LISA Activation Assay Kit, according to the manufacturer’s recommendations (Cytoskeleton). Briefly, lymphocytes were washed and pelleted prior to 1 minute of lysis with the dedicated buffer. Lysates were clarified by a 1-minute centrifugation at 21,000 g, and supernatants were snap-frozen in liquid nitrogen. Total protein contents were assayed using the Micro BCA Kit (Thermo Fisher), and a minimum of 25 μg/ml protein samples were loaded onto the precoated plates provided with the RhoA G-LISA kit.
Chemotaxis assay.
Boyden chambers (MilliporeSigma) were used to perform chemotaxis experiments. Briefly, 3 × 105 cells were seeded on the upper part of the chamber with the lower part containing 600 μl of RPMI 1640 GlutaMAX medium supplemented with 1% penicillin/streptomycin and various concentrations of SDF1. After 4 hours of incubation at 37°C, cells in the upper and lower chambers were recovered and stained for surface markers. Cells of the 2 compartments were then counted with a MACSQuant analyzer (Miltenyi Biotec). The number of migrating cells was calculated as the ratio of cells in the lower part over the total (cells in the upper plus lower part).
Immunoblot analysis.
For Western blot analysis, 2 to 5 million lymphocytes were lysed with a RIPA buffer containing 50 mM Tris pH7.4, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, and 2 mM EDTA. From 30 to 50 μg protein was separated and stained with several antibodies, including ARHGEF1 (clone E-4, catalog sc-166301, Santa Cruz Biotechnology Inc.), GAPDH (clone 6C5, catalog sc-32233, Santa Cruz Biotechnology Inc.), and RhoA (clone 67B9, catalog 2117S, Cell Signaling Technology).
Viral transductions.
T cell blasts or B-EBV cells were transduced with the retroviral vectors MSCV-IRES-Thy1.1 (empty construction) and MSCV-IRES-Thy1.1 (full-length human ARHGEF1), provided by Jagan Muppidi (UCSF, San Francisco, California, USA) and Jason Cyster (UCSF), or the lentiviral vector psd44-G13WT (full-length human GNA13) provided by Agnese Mariotti (University of Lausanne, Lausanne, Switzerland; Addgene, plasmid no. 46829) according to a previously described protocol (36). Briefly, viruses were introduced into a 96-well plate precoated with 40 μg/ml fibronectin (Takara, Clontech) and centrifuged for 98 minutes at 500 g at 30°C. T cell blasts at day 3 after IL-2 propagation were seeded onto the virus-containing plate and centrifuged for 30 minutes at 14 g at 30°C. The cells were then incubated overnight prior to the addition of complete medium. For GNA13 transduction, cells were puromycin selected for 1 week prior to regular culture. ARHGEF1 transduction efficiency was monitored by the surface expression of Thy1.1 marker (staining with an antibody against Thy1.1 [CD90.1-PE; clone His51, catalog 130-102-636, Miltenyi Biotec]), using FACS. Viral productions were obtained from the lentivector production facility/SFR BioSciences Gerland–Lyon Sud (UMS3444/US8), Lyon, France.
Staining of high-affinity conformation of LFA-1.
For the staining of high-affinity conformation of LFA-1, an equal number of cells were incubated for 5 minutes at 37°C in nonactivating medium (RPMI 1640 supplemented with 10 mM EDTA) or activating medium (RPMI 1640 supplemented with 1 mM magnesium chloride and 100 ng/ml SDF1) before the addition of the antibody recognizing the high-affinity conformation of LFA-1 (CD11α/CD18; clone m24, catalog 363410, BioLegend) and an additional incubation for 2 minutes at 37°C. Afterward, cells were immediately fixed with 2% PFA For 10 minutes at 4°C. Then cells were washed and stained for other surface markers before being analyzed by flow cytometry. Expression analysis of LFA-1 and VLA-4 components was performed with the following fluorescent-conjugated antibodies: CD11a (clone HI111, catalog 301206, BioLegend), CD18 (clone 1B4/CD18, catalog 373408, BioLegend), CD29 (clone TS2/16, catalog 303004, BioLegend), and CD49d (clone 9F10, catalog 304308, BioLegend).
Hematology and tissue immunohistochemistry.
Hematological analyses were performed by the Haematology Department at Necker Children’s Hospital. Myelograms were performed on blood smears stained with May-Grünwald Giemsa reagents, and representative pictures were obtained after acquisition with a Leica microscope coupled to a Sony camera.
Mediastinal lymph nodes were obtained after treatment for mucoepidermoid carcinoma in P2. As a control, a mesenteric lymph node of a patient diagnosed with exudative enteropathy was used. The analyses were performed through the Pathology Department at Necker Childrens’ Hospital. Briefly, 3 μm paraffin-embedded sections were stained with H&E and with antibodies against the following human antigens: CD3 (polyclonal, Agilent, catalog A045201), CD4 (clone 4B12, Microm Microtech France, catalog F/MS-1528-S1), CD5 (clone 4C7, Leica, catalog CD5-4C7-L-CE), CD8 (clone C8/144B, Agilent, catalog M710301), CD20 (clone L26, Agilent, catalog M075501), IgA (polyclonal, Agilent, catalog A026201), IgM (polyclonal, Agilent, catalog A0425), IgD (polyclonal, Agilent, catalog F018901), CD79a (clone JCB117, Agilent, catalog M705001), CD21 (clone 2G9, Leica, catalog CD21-2G9-L-CE), PD1 (clone NAT105, Abcam, catalog AB52587), CD10 (clone 56C6, Leica, catalog CD10-270-L-CE), CD138 (clone MI15, Agilent, catalog M722801), BCL6 (clone PG-B6p, Agilent, catalog M721101), and Ki-67 (clone MIB-1, Agilent, catalog M724001), using a Leica Bond III Automate (Leica Biosystems).
Immunofluorescence microscopy and live cell imaging.
Immunofluorescence images were obtained following acquisition on a Zeiss Axioplan 2 coupled to a QI imaging camera (Roper Scientific) and a Zeiss Observer Z1 inverted microscope coupled to an Orca Flash 4.0 sCMOS camera (Hamamatsu). For all conditions tested, between 5 and 10 fields were acquired. The fluorescence was quantified using ImageJ software (NIH).
For live-cell imaging, cells were seeded on fibronectin-coated IBD chambers and cultured at 37°C in a 5% CO2 atmosphere. Data were acquired with a video-imaging microscope (Eclipse Ti-E; Nikon) at ×20 magnification during a minimum of 5 hours with an image acquisition every 50 seconds. Images were acquired with a QuantEM 512 SC camera (Roper Technologies) and NIS-Elements AR software (version 3.1; Nikon). Object tracking and lengths were respectively evaluated with ICY (37) and ImageJ software in the manual mode. For motility evaluation, a minimum of 13–20 cells for each condition were followed every 50 seconds during a minimum of 2 hours to determine the mean displacement (pixels/frame) of each cell. The maximum tail length (pixels) represents the longest trailing edge measured for a cell during a minimum 2-hour time period.
Statistics.
Data are represented as mean ± SEM. Independent sample measurements and different biological samples are indicated in figure legends or within graphs. Independent experiments of a B-EBV cell line and independent T cell blast cultures originated from blood samples from the same donor were considered as independent samples. Statistical analysis of RhoA activity and migration index was performed on data as indicated in the figure legends using a 1-sample, 2-tailed t test using Graph Prism 6.0 software (GraphPad Software). Differences between populations with normal distribution were probed using an unpaired 2-tailed Student’s t test running in Prism software (GraphPad Software). Comparisons of groups with unequal variances (analysis of mean displacement and maximal tail length) were performed on log2-transformed data using a 2-tailed Welch’s t test using Microsoft Excel software. Comparisons between 2 groups presenting data related to frequencies were performed using unpaired 2-tailed nonparametric Mann-Whitney U test using Graph Prism 6.0 software (GraphPad Software). P < 0.05 was considered significant for all statistical analysis.
Study approval.
The study was approved by the local independent ethic committee (Comité de Protection des Personnes Ile de France II, Paris, France; reference no. CPP: 2015-01-05) and the French Advisory Committee on Data Processing in Medical Research (reference 15.297bis). Written, informed consent was obtained from all subjects prior to participation in the study.
Author contributions
AB and SK designed and analyzed experiments. AB, SL, LC, HL, MCD, and LH performed experiments. SK supervised research. MC, IAS, AF, and AD contributed to data analysis. CP provided centralized immunophenotyping results. JB, VM, and TJM contributed to histological data and image analysis. AT and EM contributed to hematological evaluation and image analysis. MB performed computational analysis, and OA performed WES. AF and EO took care of patients and provided clinical information, and EO provided human samples. AB and SK wrote the manuscript, and all the coauthors revised, edited, and approved the manuscript.
Supplementary Material
Acknowledgments
We wish to thank Meriem Garfa-Traoré and Nicolas Goudin from Imagine Institute’s cell imaging facility and Nathalie Yvart from the Department of Pathology for technical assistance; Nicolas Cagnard from Imagine Institute’s Bioinformatics Facility for advice on statistical analysis; the CRB for generating EBV-transformed lymphoblastoid cell lines; and the clinical research team at the Imagine Institute for their support. For viral production, we thank Gisèle Froment, Didier Nègre, and Caroline Costa from the lentivectors production facility/SFR BioSciences Gerland–Lyon Sud (UMS3444/US8). Philippe Tisserant treated the lung complications in both patients. SK is a Centre National de la Recherche Scientifique staff researcher. The study was funded by INSERM, the Agence National de la Recherche, as part of the Investment for the Future Program (ANR-10-IAHU-01) and by ANR-15-CE15-0020 (ANR-PIKimmun), the Ligue Contre le Cancer–Comité de Paris, the Fondation ARC Pour la Recherche sur le Cancer, and the Centre de Référence Déficits Immunitaires Héréditaires (CEREDIH).
Version 1. 12/06/2018
In-Press Preview
Version 2. 02/04/2019
Electronic publication
Version 3. 03/01/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
License: Copyright 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(3):1047–1060.https://doi.org/10.1172/JCI120572.
See the related Commentary at ARHGEF1 deficiency reveals Gα13-associated GPCRs are critical regulators of human lymphocyte function.
Contributor Information
Amine Bouafia, Email: amine.bouafia@inserm.fr.
Sébastien Lofek, Email: sebastien.lofek@gmail.com.
Julie Bruneau, Email: julie.bruneau@aphp.fr.
Loïc Chentout, Email: loic.chentout@institutimagine.org.
Hicham Lamrini, Email: hlamrini@gmail.com.
Amélie Trinquand, Email: amelie.trinquand@inserm.fr.
Marie-Céline Deau, Email: deaum@igbmc.fr.
Lucie Heurtier, Email: lucie.heurtier@inserm.fr.
Véronique Meignin, Email: veronique.meignin@aphp.fr.
Capucine Picard, Email: capucine.picard@nck.ap-hop-paris.fr.
Elizabeth Macintyre, Email: davimac@wanadoo.fr.
Olivier Alibeu, Email: olivier.alibeu@institutimagine.org.
Marc Bras, Email: marc.bras@institutimagine.org.
Thierry Jo Molina, Email: thierry.molina@nck.aphp.fr.
Marina Cavazzana, Email: m.cavazzana@nck.aphp.fr.
Isabelle André-Schmutz, Email: isabelle.andre-schmutz@inserm.fr.
Anne Durandy, Email: anne.durandy@inserm.fr.
Alain Fischer, Email: alain.fischer@aphp.fr.
Eric Oksenhendler, Email: eric.oksenhendler@aphp.fr.
Sven Kracker, Email: sven.kracker@inserm.fr.
References
- 1.Whitehead IP, Khosravi-Far R, Kirk H, Trigo-Gonzalez G, Der CJ, Kay R. Expression cloning of lsc, a novel oncogene with structural similarities to the Dbl family of guanine nucleotide exchange factors. J Biol Chem. 1996;271(31):18643–18650. doi: 10.1074/jbc.271.31.18643. [DOI] [PubMed] [Google Scholar]
- 2.Aasheim HC, Pedeutour F, Smeland EB. Characterization, expression and chromosomal localization of a human gene homologous to the mouse Lsc oncogene, with strongest expression in hematopoetic tissues. Oncogene. 1997;14(14):1747–1752. doi: 10.1038/sj.onc.1200994. [DOI] [PubMed] [Google Scholar]
- 3.Girkontaite I, et al. Lsc is required for marginal zone B cells, regulation of lymphocyte motility and immune responses. Nat Immunol. 2001;2(9):855–862. doi: 10.1038/ni0901-855. [DOI] [PubMed] [Google Scholar]
- 4.Glaven JA, Whitehead IP, Nomanbhoy T, Kay R, Cerione RA. Lfc and Lsc oncoproteins represent two new guanine nucleotide exchange factors for the Rho GTP-binding protein. J Biol Chem. 1996;271(44):27374–27381. doi: 10.1074/jbc.271.44.27374. [DOI] [PubMed] [Google Scholar]
- 5.Chen Z, Guo L, Hadas J, Gutowski S, Sprang SR, Sternweis PC. Activation of p115-RhoGEF requires direct association of Gα13and the Dbl homology domain. J Biol Chem. 2012;287(30):25490–25500. doi: 10.1074/jbc.M111.333716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubtsov A, Strauch P, Digiacomo A, Hu J, Pelanda R, Torres RM. Lsc regulates marginal-zone B cell migration and adhesion and is required for the IgM T-dependent antibody response. Immunity. 2005;23(5):527–538. doi: 10.1016/j.immuni.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 7.Muppidi JR, et al. Loss of signalling via Gα13 in germinal centre B-cell-derived lymphoma. Nature. 2014;516(7530):254–258. doi: 10.1038/nature13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durandy A, Kracker S, Fischer A. Primary antibody deficiencies. Nat Rev Immunol. 2013;13(7):519–533. doi: 10.1038/nri3466. [DOI] [PubMed] [Google Scholar]
- 9.Jaiswal M, et al. Mechanistic insights into specificity, activity, and regulatory elements of the regulator of G-protein signaling (RGS)-containing Rho-specific guanine nucleotide exchange factors (GEFs) p115, PDZ-RhoGEF (PRG), and leukemia-associated RhoGEF (LARG) J Biol Chem. 2011;286(20):18202–18212. doi: 10.1074/jbc.M111.226431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green JA, et al. The sphingosine 1-phosphate receptor S1P2 maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat Immunol. 2011;12(7):672–680. doi: 10.1038/ni.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harenberg A, Girkontaite I, Giehl K, Fischer KD. The Lsc RhoGEF mediates signaling from thromboxane A2 to actin polymerization and apoptosis in thymocytes. Eur J Immunol. 2005;35(6):1977–1986. doi: 10.1002/eji.200425769. [DOI] [PubMed] [Google Scholar]
- 12.Rieken S, et al. Lysophospholipids control integrin-dependent adhesion in splenic B cells through G(i) and G(12)/G(13) family G-proteins but not through G(q)/G(11) J Biol Chem. 2006;281(48):36985–36992. doi: 10.1074/jbc.M605287200. [DOI] [PubMed] [Google Scholar]
- 13.Tan W, Martin D, Gutkind JS. The Gα13-Rho signaling axis is required for SDF-1-induced migration through CXCR4. J Biol Chem. 2006;281(51):39542–39549. doi: 10.1074/jbc.M609062200. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt G, Sehr P, Wilm M, Selzer J, Mann M, Aktories K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature. 1997;387(6634):725–729. doi: 10.1038/42735. [DOI] [PubMed] [Google Scholar]
- 15.Ricker E, Chowdhury L, Yi W, Pernis AB. The RhoA-ROCK pathway in the regulation of T and B cell responses. F1000Res. 2016;5:2295. doi: 10.12688/f1000research.7522.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dubash AD, Wennerberg K, García-Mata R, Menold MM, Arthur WT, Burridge K. A novel role for Lsc/p115 RhoGEF and LARG in regulating RhoA activity downstream of adhesion to fibronectin. J Cell Sci. 2007;120(pt 22):3989–3998. doi: 10.1242/jcs.003806. [DOI] [PubMed] [Google Scholar]
- 17.Li Z, et al. Regulation of PTEN by Rho small GTPases. Nat Cell Biol. 2005;7(4):399–404. doi: 10.1038/ncb1236. [DOI] [PubMed] [Google Scholar]
- 18.Blaho VA, Hla T. An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res. 2014;55(8):1596–1608. doi: 10.1194/jlr.R046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heurtier L, et al. Mutations in the adaptor-binding domain and associated linker region of p110δ cause activated PI3K-δ syndrome 1 (APDS1) Haematologica. 2017;102(7):e278–e281. doi: 10.3324/haematol.2017.167601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown JP, et al. Arhgef1 is required by T cells for the development of airway hyperreactivity and inflammation. Am J Respir Crit Care Med. 2007;176(1):10–19. doi: 10.1164/rccm.200702-270OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carbone ML, et al. Leukocyte RhoA exchange factor Arhgef1 mediates vascular inflammation and atherosclerosis. J Clin Invest. 2017;127(12):4516–4526. doi: 10.1172/JCI92702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muppidi JR, Lu E, Cyster JG. The G protein-coupled receptor P2RY8 and follicular dendritic cells promote germinal center confinement of B cells, whereas S1PR3 can contribute to their dissemination. J Exp Med. 2015;212(13):2213–2222. doi: 10.1084/jem.20151250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guilluy C, et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med. 2010;16(2):183–190. doi: 10.1038/nm.2079. [DOI] [PubMed] [Google Scholar]
- 24.Lanzi G, et al. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. J Exp Med. 2012;209(1):29–34. doi: 10.1084/jem.20110896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kahr WH, et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat Commun. 2017;8:14816. doi: 10.1038/ncomms14816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobbs K, et al. Inherited DOCK2 deficiency in patients with early-onset invasive infections. N Engl J Med. 2015;372(25):2409–2422. doi: 10.1056/NEJMoa1413462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGhee SA, Chatila TA. DOCK8 immune deficiency as a model for primary cytoskeletal dysfunction. Dis Markers. 2010;29(3-4):151–156. doi: 10.1155/2010/397291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crequer A, et al. Human RHOH deficiency causes T cell defects and susceptibility to EV-HPV infections. J Clin Invest. 2012;122(9):3239–3247. doi: 10.1172/JCI62949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lagresle-Peyrou C, et al. X-linked primary immunodeficiency associated with hemizygous mutations in the moesin (MSN) gene. J Allergy Clin Immunol. 2016;138(6):1681–1689.e8. doi: 10.1016/j.jaci.2016.04.032. [DOI] [PubMed] [Google Scholar]
- 30.Abdollahpour H, et al. The phenotype of human STK4 deficiency. Blood. 2012;119(15):3450–3457. doi: 10.1182/blood-2011-09-378158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nehme NT, et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood. 2012;119(15):3458–3468. doi: 10.1182/blood-2011-09-378364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiow LR, et al. The actin regulator coronin 1A is mutant in a thymic egress-deficient mouse strain and in a patient with severe combined immunodeficiency. Nat Immunol. 2008;9(11):1307–1315. doi: 10.1038/ni.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salzer E, et al. RASGRP1 deficiency causes immunodeficiency with impaired cytoskeletal dynamics. Nat Immunol. 2016;17(12):1352–1360. doi: 10.1038/ni.3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lemoine R, et al. Immune deficiency-related enteropathy-lymphocytopenia-alopecia syndrome results from tetratricopeptide repeat domain 7A deficiency. J Allergy Clin Immunol. 2014;134(6):1354–1364.e6. doi: 10.1016/j.jaci.2014.07.019. [DOI] [PubMed] [Google Scholar]
- 35.Balabanian K, et al. Proper desensitization of CXCR4 is required for lymphocyte development and peripheral compartmentalization in mice. Blood. 2012;119(24):5722–5730. doi: 10.1182/blood-2012-01-403378. [DOI] [PubMed] [Google Scholar]
- 36.Barsov EV. Immortalization of human and rhesus macaque primary antigen-specific T cells by retrovirally transduced telomerase reverse transcriptase. Curr Protoc Immunol. 2011;Chapter 7:Unit 7.21B. doi: 10.1002/0471142735.im0721bs95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Chaumont F, et al. Icy: an open bioimage informatics platform for extended reproducible research. Nat Methods. 2012;9(7):690–696. doi: 10.1038/nmeth.2075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.