

Abstract
Our understanding of the mechanisms underlying Parkinson’s disease, the once archetypical non-genetic neurogenerative disorder, has dramatically increased with the identification of α-synuclein and LRRK2 pathogenic mutations. While α-synuclein protein composes the aggregates that can spread through much of the brain in disease, LRRK2 encodes a multi-domain dual-enzyme distinct from any other protein linked to neurodegeneration. In this review, we discuss emergent datasets from multiple model systems that suggests these unlikely partners do interact in important ways in disease, both within cells that express both LRRK2 and α-synuclein as well as through more indirect pathways that might involve neuroinflammation. Although the link between LRRK2 and disease can be understood in part through LRRK2 kinase activity (phospho-transferase activity), α-synuclein toxicity is multi-layered and plausibly interacts with LRRK2 kinase activity in several ways. We discuss common protein interactors like 14–3-3s that may regulate α-synuclein and LRRK2 in disease. Finally, we examine cellular pathways and outcomes common to both mutant α-synuclein expression and LRRK2 activity and points of intersection. Understanding the interplay between these two unlikely partners in disease may provide new therapeutic avenues for PD.
Keywords: Parkinson, mitochondria, mitophagy, autophagy, neuroinflammation, Prion-like propagation, aggregation, neurodegeneration
Graphical Abstract
LRRK2 and α-synuclein probably play key roles in Parkinson’s disease pathogenesis. In this review, we discuss how these unlikely partners do interact both within neurons that express both LRRK2 and α-synuclein as well as through more indirect pathways that might involve regulation of α-synuclein spreading and neuroinflammation. Understanding the interplay between α-synuclein and LRRK2 may provide new therapeutic avenues for Parkinson’s disease.
Introduction to LRRK2 and α-Syn in PD
Parkinson’s disease (PD) is a neurodegenerative disorder affecting approximately seven million people worldwide (Cacabelos, 2017). Early symptoms of disease often include REM sleep disorder, gastroinsteninal disorders (e.g., constipation), and hyposmia. Later, characteristic movement-related symptoms often include shaking (resting tremor), rigidity, and slowness of movement (bradykinesia). The neuropathological hallmarks of PD are a progressive loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc) and the presence of neuronal aggregates composed mainly of α-synuclein protein (α-syn) (Lewy Bodies, LB) and dystrophic Lewy Neurites (LN) in some surviving neurons (H. Braak & Braak, 2000; Gibb, Scott, & Lees, 1991). Numerous theories exist for the causes of α-syn aggregation and preferential sensitivity of DA neurons, but without treatments that intervene in these processes, pathogenic mechanisms underlying PD remain unclear. Currently, there is no treatment capable of slowing the disease progression and although several therapies such as dopaminergic treatment and deep brain stimulation provide temporary remittance of several movement-related symptoms, the progression of the disease and therefore the aggravation of the neurodegeneration cannot be stopped.
PD is mainly a sporadic neurodegenerative disorder but a proportion of patients have demonstrated genetic origins. Rare mutations in the α-synuclein (SNCA) and Leucine-Rich Repeat Kinase 2 (LRRK2) genes can cause autosomal dominant (AD) PD. Recently, genome-wide association studies have identified common genetic variants in both SNCA and LRRK2 in susceptibility to sporadic PD, further supporting the importance of these two genes in the pathogenesis of PD (Nalls et al., 2014; Satake et al., 2009). The overlap between clinical phenotypes associated with mutations in SNCA or LRRK2 suggest these two proteins/genes concurrently play a role in sporadic and genetic PD. A number of questions are thus raised: Does α-syn and LRRK2 interact synergistically in disease susceptibility? Following, does LRRK2 influence the occurrence and triggering of α-syn pathology? Through which molecular and cellular mechanisms do these two proteins interact in PD, and are they involved in both susceptibility as well as progression? Alternatively, does α-syn play a more permissive role in mutant LRRK2 neurotoxicity?
Beyond improving our understanding of PD pathogenesis, clarifying the interplay between α-syn and LRRK2 may help to determine whether LRRK2 could constitute a relevant therapeutic target to slow down PD progression in patients without rare LRRK2 mutations. Indeed, major research efforts have been conducted in the past decade to design and test novel LRRK2 inhibitors with hopes they will benefit a large proportion of PD patients. Potentially supporting this notion, preliminary results in animal models of PD suggest that targeting LRRK2 could be beneficial in both familial and sporadic PD. Thus, the interaction between α-syn and LRRK2 might be central not only in terms of pathogenesis but also in understanding how to best devise effective therapeutic strategies. Here we briefly review both in vitro and in vivo studies in model systems that may shed light on the relationship between α-syn and LRRK2 in PD.
α-Syn and prion-like propagation
In 1997, Polymeropoulos et al. identified the α-syn A53T missense mutation as the first genetic lesion causative for an aggressive form of familial PD (Polymeropoulos et al., 1997). α-Syn is a presynaptic protein highly abundant in the brain with suspected roles in vesicle trafficking, membrane dynamics, and synaptic maintenance (reviewed in Bendor, Logan, and Edwards 2013). α-Syn has also been shown to localize to mitochondria and to be degraded in-part via chaperone-mediated autophagy (detailed in the paragraphs below on mitochondria and autophagy). The clear majority of α-syn studies focus on its dysfunction in PD pathology because of its capacity to aggregate and form LBs and LNs. Duplications, triplications and rare mutations (A53T (Polymeropoulos et al., 1997); A30P (Krüger et al., 1998); E46K (Zarranz et al., 2004)) in the SNCA gene have been found in several families with dominantly inherited PD. They are associated with early-onset forms of PD with an amplification of the α-syn aggregation process (Chartier-Harlin et al., 2004; Singleton et al., 2003). However, while it is generally accepted that aggregation of α-syn leads to neurotoxicity, the underlying mechanisms are still debated. It is possible that α-syn assemblies (aggregates, oligomers, lewy bodies) trigger toxic mechanisms through a gain of function (e.g. novel detrimental interaction with membranes or proteins) or a toxic loss of function of α-syn as a result of the sequestration of α-syn into aggregates. Indeed, multiple studies have shown a toxic effect of α-syn knock out (Tarasova et al., 2018; Benskey et al., 2018; Cali et al., 2012; Gorbatyuk et al., 2010) while others showed that α-syn KO protected against MPTP-induced neurotixicity (Dauer et al., 2002). α-syn is a member of the synuclein protein family together with bêta and gamma-synuclein in mammals. The α-syn protein is composed of three regions: the first one is an amphipathic region (N-term: residues 1–60) containing apolipoprotein lipid-binding motifs and where PD mutations reside. Secondly, α-syn contains a “non-bêta amyloid” component region (NAC region: residues 61–95) which gives it its potential to form bêta-sheets inherent to protein fibrils (aggregation). The last one is a C-terminal region (residues 96–140) that harbors important post-translational modifications of α-syn including phospho-Ser129. Three post-translational modifications were identified and validated in vivo with phosphorylated residues detected on Ser87, Ser129 and Tyr125. In particular, post-mortem biochemical and immunohistological studies showed that in PD brains α-syn is highly phosphorylated on Ser129 in inclusions. This phosphorylation is also found in pre-LB stages suggesting that it is strongly associated to disease progression (Saito et al., 2003). The pharmacological modulation of this phosphorylation may be a relevant strategy for slowing PD progression, although several studies show α-syn related toxicity is enhanced when the Ser129 residue is mutated to an unphosphorylatable alanine residue (da Silveira et al., 2009; Gorbatyuk et al., 2008; Khodr, Pedapati, Han, & Bohn, 2012). Some kinase interactors have been identified that can modulate Ser129 phosphorylation including polo-like kinases, casein kinases, and G-protein receptor coupled kinase (Inglis et al., 2009; Ishii et al., 2007; Pronin et al., 2000). LRRK2, a serine/threonine protein kinase, has also been evaluated as a protein kinase for α-syn. Results suggest that LRRK2 is unlikely to directly phosphorylate Ser129 (Lin et al., 2009; Herzig et al., 2012). The instrumental role of Ser129 phosphorylation on α-syn is unclear and an increase of phosphor-Ser129 might not be toxic per se. Studies using kinase interactors of α-syn to produce hyper-phosphorylation at Ser129 have shown opposite effects (accelerated neurodegeneration vs mitigated toxicity) in the rat midbrain (Sato et al., 2011; Oueslati et al., 2013). Also, Buck and collaborators showed that phosphorylation of α-syn at Ser129 was not enough to trigger neurodegeneration, further questioning the role of phosphor-Ser129 in the pathogenesis of PD (Buck et al., 2015).
The pathological mechanisms underlying α-syn mediated degeneration of DA neurons has been linked to the very high concentrations of α-syn inherent to A9 DA neurons (that can serve as substrates for α-syn inclusions). SNc DA neurons also have relatively high metabolic demands and lowered oxidative capacity and long extremely branched axonal fibers that lack myelin (Braak and Del Tredici 2004; Post, Lieberman, and Mosharov 2018). Experimental evidence also indicates central roles for mitochondrial damage and associated vulnerabilities, and deregulation of the endolysosomal and autophagic pathways. Some evidence, particularly in vitro, clearly demonstrates that α-syn inclusions can be transferred, through some substrate, from one cell to the next (Busquets, Espargaró, Estelrich, & Sabate, 2015; Domert et al., 2016; Kordower, Chu, Hauser, Freeman, & Olanow, 2008). Kordower and collaborators suggested cell-cell transmission of α-syn in 2008. A post mortem observation of PD brains transplanted with fetal neurons in the striatum showed that, fourteen years after transplantation, the grafted nigral neurons presented LB-like inclusions that stained positively for α-syn and ubiquitin and had a reduced expression of dopamine transporter. Alternatively, the transplanted cells could have intrinsically formed inclusions de novo in the environment of PD-affected nerve terminals in the striatum. Moreover, all healthy grafts of human midbrain transplants do not automatically show signs of α-syn pathology (reviewed in Engelender and Isacson, 2017). Experimentally, cell-to-cell transmission in vivo generally involves over-expression of α-syn and exposures to preformed fibrils of α-syn, brain extracts from α-syn transgenic (tg) mice, and total homogenates from post-mortem PD patients. Many experimental results are supportive of the hypothesis of a prion-like propagation of α-syn. α-Syn linked disease is different from prototypical prion disease in that there is no evidence of disease transmissibility in PD and intrinsic susceptibilities inherent to some sub-populations of neurons appears to outweigh the ability of α-syn to successfully transmit in a continuous way across circuits (Abdelmotilib et al., 2017; Desplats et al., 2009; Luk et al., 2012; Mougenot et al., 2012; Paumier et al., 2015; Peelaerts et al., 2015; Recasens & Dehay, 2014) (Reviewed in Surmeier, Obeso, and Halliday 2017b, 2017a).
However, even though there are many experimental studies supporting this prion-like hypothesis, it is important to note that there is a limited number of clinical evidence directly supporting it, and other experimental evidence goes against it. Indeed, Manfredsson and collaborators failed to show any significant spread of α-syn after they inoculated PFFs directly into the colon of rats and NHPs (Manfredsson et al., 2018). Most of the studies supporting α-syn spreading theory use overexpression models, or brain extracts from individuals presenting synucleinopathies, and among them, not all confirm this spreading hypothesis. This contrasted point of view is very well reviewed by Endelender and Isacson, who present a “Threshold theory for PD” (Engelender and Isacson, 2017).
LRRK2 and the Kinase-Hypothesis of LRRK2-linked PD
In the mid-1990s, several families were identified that transmit autosomal dominant PD phenotypes identical to sporadic PD, a feature very uncommon for genetic forms of PD (Wszolek and Pfeiffer, 1992). In 2002, Funayama and collaborators performed a genome wide linkage analysis showing linkage to a 13.6 cM interval in 12p11.2-q13.1, dubbed the PARK8 locus. While the haplotype was shared by all the patients, it was also present in some unaffected carriers, suggesting the disease penetration to be incomplete. The authors hypothesized other parameters could affect the development of the disease such as environmental factors or other genetics factors (Funayama et al., 2002).
Mutations in LRRK2 are the most common genetic “cause” of both familial and sporadic PD, with causation understood as high-odds ratios (e.g., >20) for susceptibility to disease (Clark et al., 2006; Paisán-Ruíz et al., 2004; Ross et al., 2011; Trabzuni et al., 2013; Trinh et al., 2014; Zimprich et al., 2004). Kachergus and collaborators identified the G2019S mutation in 2005 (Kachergus et al., 2005), known as the most prevalent pathogenic mutation in LRRK2 that accounts for 5–6% of autosomal-dominant familial cases and 1–2% of de novo generic PD cases in some Western populations (Gilks et al., 2005; Healy et al., 2008). The prevalence can be much higher depending on the population: a recent report suggested that as many as 70% of late-onset PD patients seen at a neurology clinic in Morocco were positive for G2019S (Bouhouche et al., 2017). However, in far-east Asian populations, the frequency of pathogenic LRRK2 mutations is extremely low (Tan et al., 2005; Tan et al., 2007; Wu et al., 2012; Zabetian et al., 2009). True to the initial description of LRRK2-linked families, patients harboring the G2019S mutation are clinically indistinguishable from sporadic PD cases, including the presence of typical LBs in the majority of cases (Biskup & West, 2009; Yahalom et al., 2014). The mechanisms underlying LRRK2’s neurotoxicity remain unclear as it is a protein that does not itself form the proteinaceous inclusions typical of genetically linked proteins in neurodegeneration (like tau, APP, TDP43, SOD1, etc.). Rather, the LRRK2 gene encodes a large 2527 AA multidomain protein with pathogenic mutations clustered among the central tri-domain region that forms the catalytic core (Cookson, 2010; Mata, Wedemeyer, Farrer, Taylor, & Gallo, 2006). LRRK2 is part of the ROCO protein family identified first in single-celled organisms and contains the tandem Ras-of-complex proteins (Roc) GTPase and C-terminal of Roc (COR) domains. LRRK2 may homodimerize through its COR domain and may function in a dimeric form as a GAD (G proteins activated by nucleotide-dependent dimerization) in which both ROC domains are brought close together and participate in the catalytic activity of their counterpart within the dimer (Civiero et al., 2012). In addition to its GTPase domain, LRRK2 is composed of a kinase domain, with a characteristic activation loop (P-loop) with a DYG-APE motif altered to DYS-APE with the G2019S mutation. Other domains in LRRK2 include multiple protein–protein interaction regions [ankyrin (ANK), armadillo (ARM), and leucine-rich repeats (LRR) and WD40]. The number and conservation of protein-protein interaction domains suggest LRRK2 functions as a scaffolding protein contributing to the formation of multiprotein complexes that may function in trafficking and signaling.
In vitro evidence and limited in vivo evidence suggest pathogenic LRRK2 mutations, like G2019S, enhance kinase activities associated with autophosphorylation and Rab-substrate phosphorylation (Alessi & Sammler, 2018; West et al., 2005, 2007), and LRRK2-linked neurotoxicity originates from this increased activity (Greggio et al., 2006; Lee et al., 2010; Smith et al., 2006; West et al., 2007). Based on these observations, pharmaceutical companies have developed compounds targeting the kinase activity of LRRK2 in order to block the increased kinase activity associated with LRRK2 mutations. So-called first-generation LRRK2 kinase inhibitors like staurosporine, sunitinib, CZC 54252, TAE684 and LRRK2-IN-1 have a good potential to inhibit the kinase activity of LRRK2 (Deng et al., 2011). However, they are not specific for LRRK2 and inhibit other critical kinases, together with poor drug properties, making them difficult to use for modeling or functional studies. Second generation compounds included more selectivity and better in vivo potential, although the inhibition of LRRK2 in the brain was limited (Saez-Atienzar et al., 2014; Choi et al., 2012; Estrada et al., 2014) (reviewed in Taymans and Greggio 2016; West 2015, 2017).
Early studies demonstrated LRRK2 is a highly phosphorylated protein in the N-terminal part between the ankyrin and the LRR domains (S910; S935; S955; S973) (West et al., 2007). This cluster, more specifically Ser910 and 935, is responsible for 14–3-3 protein binding (Dzamko et al., 2010; R. J. Nichols et al., 2010). The 14–3-3 proteins are a highly conserved family of proteins found throughout the evolutionary scale and are implicated in many cellular functions, notably, they act to promote cell survival through inhibition of many known pro-apoptotic factors including the mitochondrial Bcl-2 family member BAD and the transcription factor Forkhead (Yuan & Yankner, 2000). The 14–3-3 proteins bind to serine/threonine-phosphorylated residues, often functioning as direct regulators of the target proteins to which they bind (Loeffler, Klaver, Coffey, Aasly, & LeWitt, 2017). Seven isoforms are described in mammals. Studies show that 14–3-3 proteins are linked to PD pathology, as 14–3-3 also interacts with α-syn (for more details, see the common interactors paragraph). LRRK2 binding of 14–3-3 leads to a uniform LRRK2 distribution throughout the cytoplasm and may inhibit LRRK2 kinase activity (Lavalley, Slone, Ding, West, & Yacoubian, 2016). Decreased 14–3-3 binding to LRRK2 induced by PD-associated mutations leads to a re-localization of LRRK2 in cytoplasmic pools (Nichols et al., 2010; Rudenko and Cookson 2010). LRRK2 may also be subject to autophosphorylation on a cluster of threonines in the Rab-like ROC GTPase domain (T1343; T1368; T1403; T1404; T1410; T1452; T1491; T1503) and on two isolated serine and threonine residues in the LRR domain (S1292) and in the kinase domain (T2031) (Kamikawaji, Ito, & Iwatsubo, 2009; Sheng et al., 2012; Webber et al., 2011). LRRK2 autophosphorylation appears to act as an enzymatic activator for both the GTPase and the kinase activity. In 2011, Webber and collaborators studied the effect of autophosphorylation on GTPase and on kinase activity in G2019S or in WT context. They showed that autophosphorylation on the T1503 that is localized in the GTPase domain alters both kinase and GTP binding activity, concluding that autophosphorylation in the GTPase domain induces functional and conformational changes that enhance the dimerization and leads to upregulation of kinase activity. In 2012, Sheng and collaborators validated in vivo S1292 as a critical autophosphorylation site (Sheng et al., 2012). Familial PD mutations (N1437H, R1441G, R1441C, G2019S and I2020T) all induced significant increases in phospho-S1292 levels compared to WT-LRRK2 in cellular models, and when the S1292 residue was mutated to a non-phosphorylatable alanine residue, the toxicity associated with LRRK2 kinase activity was ablated. Taken together, these data suggest that LRRK2 kinase activity links pathogenic mutations with neurotoxicity.
Rationale for a meaningful interaction between LRRK2 and α-synuclein in PD
Evidence from humans
Clinical phenotypes
Several LRRK2 mutations from a number of different families in different countries have been described and since they are relatively rare compared to sporadic (i.e., idiopathic or iPD) disease in most populations, comparisons that lack clinic bias are difficult to formulate. However, clinic bias tends to skew genetic forms of disease phenotypically away from sporadic disease and with LRRK2, clinical characteristics of the symptomatic carriers are very similar to one another (Adams et al., 2005; Hasegawa et al., 2009; Huang et al., 2007; Hulihan et al., 2008; Khan et al., 2005; Lin et al., 2008; Nichols et al., 2005). Presently, there is no single clinical phenotype or scale that might predict a LRRK2 mutation carrier from typical late-onset PD. Curiously, no major difference seems to exist between homozygous patients and the ones who are heterozygous, although the number of homozygous LRRK2 mutation carriers yet described is very small, consistent with the frequency of the most common LRRK2 mutation G2019S at less than 0.1% in the Caucasian population (where it is most frequent).
LRRK2 mutations do not cause early-onset disease; the age of onset in susceptible carriers is relatively similar for iPD and LRRK2 mutation carriers (~55–65 years old). Motor symptoms of LRRK2 carriers include resting tremor, rigidity, akinesia, postural instability, and gait difficulty, which are reminiscent of those seen in iPD. Unilateral onset is often seen in LRRK2 mutation carriers as it is in iPD. As expected, LRRK2 carriers with PD are responsive to L-dopa treatment. Further, PET imaging studies of the dopaminergic system indicate alterations in symptomatic LRRK2 carriers that are typical of iPD, especially the reduction in 18F-dopa accumulation in striatal DA projections, and reduced binding of 11C-tetrabenazine to the vesicle dopamine transporter (vMAT) and reduced binding of the 11C-methylphenidate to the dopamine transporter (DAT). The relative preservation of the DA projections in the caudate nucleus as compared to the putamen seen in iPD has been also observed in LRRK2 mutation carriers (reviewed in Biskup and West 2009).
Subtle differences have been described regarding the effect of LRRK2 mutations on the timing and progression of certain symptoms in different populations of carriers. However, a prospective longitudinal follow up of a cohort (n=122) of LRRK2 variant carriers over more than five years indicated that the onset and symptoms were not different from iPD (Oosterveld et al., 2015). However, the progression of the disease after onset might be slightly accelerated in LRRK2 carriers when compared to non-carriers (Oosterveld et al., 2015). Finally, some studies highlighted the possibility of differences in the prevalence and timing of onset of non-motor symptoms (REM sleep disorder, cognitive deficits), especially in PD with dementia (Goldwurm et al., 2006; Sun et al., 2016). In summary, the striking clinical overlap between LRRK2 mutation carriers with PD and PD without LRRK2 mutations suggests common disease mechanisms.
LRRK2 and α-syn in Lewy bodies and Lewy neurites.
Two years after the discovery of LRRK2 as a gene responsible for autosomal dominant forms of PD, Zimprich and collaborators speculated that LRRK2 kinase activity may be responsible for the phosphorylation of α-syn and that this effect may trigger its accumulation and aggregation (Zimprich et al., 2004). The authors found typical LB-dominant pathology, comparable to sporadic PD patients, in LRRK2 mutation carriers. These observations suggested that LRRK2, as a kinase, and α-syn, as a kinase substrate, could physically interact. Later, Giasson and collaborators studied via immunohistochemistry three individuals with the G2019S LRRK2 mutation and concluded LRRK2 protein was not found in LBs or in LNs, in contrast to typical α-syn pathology in these cases. However, studies including larger numbers of LRRK2 mutation carriers revealed that a proportion of cases, up to 1/3 of G2019S carriers of both non-Ashkenazi Jewish and European non-Ashkenazi Jewish descent were negative for Lewy bodies (Kalia et al., 2015). Although the total number of brains evaluated remains low, the number without Lewy bodies is high compared to typical late-onset disease brains evaluated from patients clinically diagnosed with PD.
There are several possibilities to interpret G2019S-LRRK2 carrier brains clinically diagnosed with PD that lack Lewy bodies. First, in the presence of the G2019S LRRK2 mutation, LBs may be more toxic than usual and cells that harbor them may not survive through late stages of disease. Second, the G2019S LRRK2 mutation favors smaller less-phosphorylated α-syn inclusions or abnormal oligomers that are more difficult to detect than typical eosinophilic inclusions, and this drives disease in those patients. Third, the proportion of G2019S-LRRK2 carriers negative for LBs may experience disease onset and progression without the contribution of α-syn. For example, some LRRK2 mutation carriers have been described with Tau tangle pathology. Unfortunately, there is typically insufficient clinical data associated with most tissues available to test whether disease characteristics and progression might be different in LRRK2 carriers with and with α-syn LB burden. A PET tracer that might label α-syn inclusions in life would effectively solve the riddle and such tracers are under active development. In the meantime, better clinical phenotyping within brain donation centers supported by LRRK2 mutation carriers seems vital to better understanding pathology linked to LRRK2.
SNCA genetic variants in modifying PD susceptibility in LRRK2 carriers.
Genome-wide association (GWAS) studies indicate that variants in SNCA and LRRK2, independent from rare pathogenic missense mutations, are among the top genetic risk factors for PD susceptibility (Nalls et al., 2014; Satake et al., 2009; Simón-Sánchez et al., 2009). Studies focused on gene-gene interactions effects of SNCA and LRRK2 found no significant evidence of an interaction between the two genes, although power is still limited due to low individual effect sizes of the linked variants (Biernacka et al., 2011). Botta-Orfila and collaborators genotyped the SNCA polymorphism rs356219 in 84 Spanish LRRK2-associated PD patients carrying the G2019S mutation (Botta-Orfila et al., 2012). This single nucleotide polymorphism (SNP) is known to regulate SNCA expression in idiopathic PD-affected brain regions, blood, and plasma. Results showed that LRRK2 mutation carriers carrying one or two SNCA rs356219-G alleles presented with an earlier PD onset than patients carrying the SNCA rs356219-A homozygote (55.37+/−1.54 vs 64.19+/−2.8 years respectively) (Botta-Orfila et al., 2012). A similar study was performed in iPD patients, and the authors found that this SNP also influenced the age of onset (Cardo et al., 2012). These initial studies suggest that SNCA genetic variants could be a modifier of age of onset but not necessarily disease susceptibility (Botta-Orfila et al., 2012). Thus, these observations indirectly suggest that α-syn may play a permissive role in LRRK2 mutation carriers in disease.
Evidence of α-syn and LRRK2 interaction in animal models
In 2009, Lin et al reported results from triple-transgenic mice with inducible A53T-α-syn and LRRK2 (WT or G2019S) under the CaMKII promoter-controlled tetracycline trans activator (tTA) (CaMKII-tTA), or on a LRRK2 knockout background, to examine the role of LRRK2 in the progression of neuropathological alterations and phenotypes associated with mutant α-syn. This heroic effort first determined the number of neurons that remained in the striatum of A53T/LRRK2WT and A53T/G2019S mice after months of expression; while the difference was not statistically significant in comparisons between WT-LRRK2 or G2019S-LRRK2 expression, a trend towards a decrease in the number of neurons in the A53T/G2019S striatum was reported. More convincingly, the inhibition of LRRK2 expression dramatically ameliorated α-syn-induced neuropathology almost to the level of doxycycline-induced silencing of transgenic mutant α-syn expression. Furthermore the number of neurons at 12 months of age in the striatum of mice was significantly higher in the A53T/LRRK2−/− mice compared to A53T/LRRK2+/+ mice. Moreover, no significant increase of astrocytosis, microgliosis, or somatic accumulation of α-syn was detected in these mice. These experiments highlighted the potential link between WT-LRRK2 expression and α-syn protein in neurodegeneration observed in PD, with a more speculative role for G2019S-LRRK2 in exacerbating α-syn neurotoxicity (Lin et al., 2009).
Further testing the link between G2019S-LRRK2 and α-syn neurotoxicity, in 2012 Herzig and collaborators reported tg mice that expressed human LRRK2 (WT or G2019S) with mice that expressed human α-syn (A53T or WT) under the same neuron-specific promoter (Thy1 promoter) (A53T/G2019S; A53T/WT; WT/G2019S) (Herzig et al., 2012). This strain of A53T-α-syn mice (van der Putten et al., 2000) were known to develop motor coordination deficits in the rotarod test at 40 days of age and severe motor deficits, hypokinesia and weight loss at 6 months of age, although they lack the overt neurodegeneration that occurs in the Lin et al study. The G2019S-LRRK2 tg mice showed no obvious brain pathology at 19 months of age, with no further decline of motor performance in double tg mice (Herzig et al., 2012). In 2017, Xiong and collaborators went back to the CamKII-induction approach to obtain a high expression level of G2019S-LRRK2 in the adult mouse forebrain, and consistent with Lin et al, these animals showed a nominal increase in α-syn aggregation, especially in the cortex (Xiong et al., 2017). They obtained similar results in tg mice with a selective overexpression of G2019S-LRRK2 in the SNc, where they showed a degeneration of DA neurons and the aggregation of endogenous mouse α-syn (Xiong et al., 2018). Potentially consistent, A53T-α-syn transgenic mice directed from the PrP promoter (Lee et al., 2002)(see also: Giasson et al., 2002), when crossed with G2019S-LRRK2 mice expressing mutant LRRK2 under the control of the CMVe-PDGFb promoter, demonstrated mild enhancement of phosphorylated inclusions but did not worsen survival rates or other phenotypes (Ramonet et al., 2011; Daher et al., 2012).
Outside of traditional transgenic mice, the acute induction of α-syn overexpression via viral vector or injection of preformed α-syn fibrils also suggests a role for LRRK2 in α-syn neurotoxicity. BAC-mediated expression of G2019S-LRRK2 in rats causes a very mild motor impairment with no dopaminergic cell loss even at 12 months of age (Lee and Cannon 2015; Walker et al., 2014). However, injection of an AAV coding for α-syn (Daher et al., 2015) in the SNc of these G2019S-LRRK2 rats induces a loss of TH-positive neurons that appears slightly more pronounced as compared to that seen in WT-LRRK2 rats. A larger effect was described in LRRK2 knockout rats that may be protected from viral-vector mediated α-syn-induced dopaminergic neurodegeneration (delivered by an AAV) (Daher et al., 2014).
In summary, the effects of the G2019S LRRK2 mutation on α-syn neurotoxicity and aggregation is mixed, with either mild or no effects reported in rodent studies. In contrast, phenotypes are generally more robust in the amelioration of α-syn phenotypes in LRRK2 knockout rats and mice. For transgenic LRRK2 expression, it is important to note that except in Lin’s study, the co-expression of both proteins (α-syn and LRRK2) in the same anatomical region and in the same neurons is not precisely validated thereby introducing a potential confounder to interpretation. In a recent study in primary hippocampal neurons, G2019S-LRRK2 expression mildly increases α-syn fibril induced inclusions but the effects of LRRK2 inhibition are much larger in reducing α-syn inclusion maturation (L. A. Volpicelli-Daley et al., 2016). Thus it remains possible that, at least in part, G2019S-LRRK2 expression may mediate a cell autonomous (albeit subtle) mechanism in the aggregation of α-syn in neurons. More the convincingly interactions appear from LRRK2 inhibition or knockdown which implies a general effect of LRRK2 interacting with α-syn in PD, outside of pathogenic mutations.
Molecular mechanisms to explain the interaction of LRRK2 and α-syn in PD
An important interaction between LRRK2 and α-syn may plausibly occur within cells that express both proteins at the same time (i.e., cell-autonomous), or through the interaction of cells important in disease that express one protein or the other (i.e., non-cell-autonomous). For example, in neurons, LRRK2 might modulate total α-syn levels (synthesis or degradation levels), levels of a post-translational modification in α-syn, modulate α-syn aggregation through changing interactions with α-syn interacting proteins, or change the subcellular compartments where α-syn normally resides. Within cells that express both LRRK2 and α-syn, understanding their interaction may shed light on how key organelles regulate survival, for example through mitochondria or endolysosome function. Alternatively, LRRK2 and α-syn might be functionally linked through mechanisms involving cell-cell interactions. For example, cells of the innate immune system involved in neuroinflammation that carry high levels of LRRK2 (mutated or with particular polymorphisms) could have increased sensitivity to the presence of aggregated α-syn in the brain. The complexity of the potential functional interaction between a-syn and LRRK2 may also involve different types of glial cells that act on one another in neuroinflammatory processes, namely astrocytes and microglial cells (Liddelow et al., 2017).
Neuron-neuron interaction might also play a role in the apparent spread of abnormal α-syn from one susceptible brain area to the next. Whether the cellular mechanisms dictating α-syn interaction with LRRK2 are cell-autonomous or non-cell-autonomous or some combination, therapeutic targeting of LRRK2, for better or worse, is almost exclusively focused on inhibiting LRRK2 kinase activity. In the second part of this review, we shall emphasize the different directions of research that provide yet imperfect but promising answers.
LRRK2 activity in α-syn expression and aggregation
As the endogenous levels of α-syn phosphorylation are low in the unaggregated state, the phosphorylation state of α-syn, notably on Serine 129, is an excellent readout of the aggregation of α-syn. In recent studies, G2019S-LRRK2 KI mice or G2019S-LRRK2 expressing rats exhibited significantly higher levels of pSer129 α-syn in the striatum compared to WT controls (Longo et al., 2017; L. A. Volpicelli-Daley et al., 2016). LRRK2 could affect the subcellular localization of α-syn, notably its membrane association, promoting the aggregation by increasing the cytosolic pool of α-syn. Through G2019S-LRRK2 upregulation of total levels of α-syn, the net effect may also be an increase in the aggregation-prone cytosolic pool. In SH-SY5Y cells studied by Kondo and collaborators (Kondo, Obitsu, and Teshima 2011), the aggregation capacity of α-syn was evaluated in the presence or absence of G2019S-LRRK2. Cell viability decreases when the cells are co-transfected with α-syn and G2019S-LRRK2 but also with WT-LRRK2 whereas viability remains unchanged in presence of each protein alone. In differentiated cells, α-syn-containing vesicles are not produced when the cells are transfected with α-syn alone. In contrast, the co-transfection of α-syn with G2019S-LRRK2 induces the formation of aggregates and vesicles. Most of the α-syn aggregates co-localize with the mitochondrial outer-membrane marker TOM20 in cells co-transfected with α-syn and G2019S-LRRK2 but not with the WT form of LRRK2. In 2014, Skibinski and collaborators demonstrated that the neurotoxicity caused by G2019S-LRRK2 could be mitigated through knockdown of α-syn (Skibinski, Nakamura, Cookson, & Finkbeiner, 2014). More recently, a study in primary cortical neurons from homozygous G2019S-LRRK2 KI mice shows that expression of G2019S-LRRK2 induced the accumulation of endogenous, detergent-insoluble α-syn, and that this effect can be reversed through pharmacological inhibition of LRRK2 kinase activity (Schapansky et al., 2018). Altogether, these observations indicate that LRRK2 can accelerate aggregation of α-syn, and possibly the localization of the aggregates, but the precise mechanisms have yet to be fully elucidated.
LRRK2 mediation of α-syn cell-to-cell propagation
It is now generally established that α-syn can propagate via the synapse in model systems in vitro, particularly those that use triple-chamber systems. In 2011, Volpicelli-Daley and colleagues observed this cell-to-cell transmission in chambered cultured neurons with bath-application of pre-formed α-syn fibrils (Volpicelli-Daley et al., 2011). Kondo and collaborators also studied the cell-to-cell transmission of α-syn through evaluating exocytosis of α-syn in the media and the internalization in neighboring neuroblastoma cells (Kondo et al., 2011). In this system, G2019S-LRRK2 significantly enhances α-syn release into the conditioned media. Interestingly the phosphorylation state of α-syn is not different in the cells co-transfected compared to cells transfected with α-syn alone, suggesting that LRRK2 can influence the propagation mechanism but not the phosphorylation state of α-syn (Kondo et al., 2011). These preliminary observations warrant further investigations into LRRK2 mediation of cell-to-cell transmission of abnormal α-syn.
Impact of LRRK2 on α-syn-induced neuroinflammation
Evidence of on-going neuroinflammation in affected brain regions in PD first come from analysis of brain tissue for reactive microglia and analyses of pro-inflammatory cytokines like Interferon gamma, (IFN-γ), TNF-α, Interleukin-6 (IL-6), and Interleukin-1β, (IL-1β) (Hunot et al., 1999; Mogi et al., 1994; McGeer, Itagaki, and McGeer 1988; Mogi et al., 1996). In addition, GWAS studies point to polymorphisms in loci with genes related to immunity (e.g. MHC-II genes). Neuroinflammation can be seen as a defense reaction that involves both the adaptative and innate immune systems, occurring in the central nervous system (CNS) after an event that perturbates “normal” homeostasis (reviewed in Gelders et al., 2018; Xanthos and Sandkühler, 2014). It is widely accepted that neuroinflammation occurs in many different acute and chronic neurological disorders. Whether it is only a consequence of neurodegeneration or is also actually a key active player favoring or opposing neurodegeneration is unknown. Presenting extensively the pros and cons of these different hypotheses on the role of neuroinflammation in degenerative diseases is out of the scoop of the present review. Comprehensive overview of this aspect of PD pathogenesis can be found extremely well-reviewed elsewhere (e.g. Skaper et al., 2018). In brief, number of experimental observations in animal models and neuropathological findings in post-mortem tissue from PD patients support the view that neuroinflammation modulates neurodegeneration. For example, in the acute model of PD in animals using the mitochondrial toxin MPTP, signs of neuroinflammation, including activated microglial cells, appear in the substantia nigra pars compacta (SNpc) before actual loss of dopaminergic cells (Liberatore et al., 1999). Similar findings have been recently reported in a model using intrastriatal injection of pre-formed α-syn fibrils (Duffy et al., 2018) and in a model of α-syn overexpression using AAV injection into the SN. In these synuclein related models, the presence of reactive microglia cells and proinflammatory molecules precedes neurodegeneration of dopaminergic neurons. Blockage/inhibition of signaling pathways linked to neuroinflammation reduces the loss of dopaminergic cells induced by MPTP (e.g. Wu et al., 2002; Maatouk et al., 2018). In models of PD using overexpression of α-syn, cellular pathways that trigger activation of microglial cells (such as Toll-like Receptors) favor neurodegeneration (Wong and Krainc, 2017). In line with this, it has been recently showed that the inhibition of the microglia-induced activation of astrocytes using agonists of the Glucagon-like peptide-1 receptor (GLP1R) into their neurotoxic phenotype protect against α-syn toxicity in mouse models (Yun et al., 2018). However, all facets of neuroinflammation are not detrimental and to some extent, there are molecular components and signaling pathways linked to the regulation of immune and glial cells that play a protective role in PD models. For example, the partial ablation of microglial cells increases the vulnerability to MPTP in mice (Yang et al., 2018). The overexpression of α-syn produces changes in activated microglial cells leading not only to the production of pro-inflammatory molecules (e.g. iNOS and IL1-beta) but also anti-inflammatory molecules (e.g. Heme oxygenase-1) (Lastres-Becker et al., 2012).
LRRK2 is expressed not only in neurons in the brain but also in some subsets of immune cells that include myeloid cells (Isolectin B4 positive, CD68 positive) in pathological conditions (Moehle et al., 2012; Daher et al., 2014). In the periphery, LRRK2 is found to be expressed in monocytes with much lower expression in T cells involved in the adaptive immune system, suggesting a role for LRRK2 in the innate immune system (Thévenet, Gobert, van Huijsduijnen, Wiessner, & Sagot, 2011). GWAS studies show LRRK2 variants implicated in the heritability of Crohn’s disease, a type of inflammatory bowel disease (IBD) that may affect any part of the gastrointestinal tract (Barrett, Hansoul, Nicolae, & Cho, 2008), as well as mycobacterium infection (Zhang et al., 2009; Marcinek et al., 2013; Wang et al., 2015). Following, studies by Gardet and Hakami both suggest important roles for LRRK2 in the maturation of myeloid cells that offer host protection from infection (Gardet et al., 2010; Hakimi et al., 2011). This suggests that LRRK2 plays a role in innate immune responses not only in the nervous system but also in the periphery. The exact role of LRRK2 in the inflammatory response has not been characterized but these and other recent studies converge towards the observation that LRRK2 kinase inhibition or LRRK2 knockdown attenuates pro-inflammatory signaling in vitro and in vivo (B. Kim et al., 2012; Ma et al., 2015; Puccini et al., 2015; Russo et al., 2015).
The role of mutated LRRK2 in inflammatory processes has been recently shown. Kozina and colleagues demonstrated that in transgenic mice overexpressing mutant LRRK2, intraperitoneal injection of LPS is able to trigger a long-term inflammation associated with neurodegeneration of DA neurons in the SNpc, which is not the case in mice overexpressing the human wild type LRRK2 or wild type mice (Kozina et al., 2018). This change produced by mutant LRRK2 is not directly linked to activation of innate brain cells but may be linked to a type II interferon immune response in peripheral cells. However, this does not rule out the possible role of LRRK2 in the regulation of brain immune cells, in particular microglia.
Microglial cells are resident innate immune cells and professional phagocytes of the brain, as well as the first line of defense against pathogens. In neurodegenerative disorders there are roles in clearing aggregated proteins and the clearance of dead cells and other debris. Microglia can switch from a resting state (ramified phenotype) into an activated state (ameboid phenotype) through signaling molecules that mediate activation (e.g., MAPK subtypes p38, JNK and ERK). A failure in the appropriate and proportional activation process of microglia can have dramatic consequences. Excessive activation can lead to an abundant secretion of inflammatory mediators and reactive oxygen species that are cytotoxic to neurons. LPS (lipopolysaccharide) treatment, in vitro or in vivo, is known to induce a TLR4-mediated inflammatory response and microglial activation. In primary microglial cells, LPS exposure can induce the switch between the resting phenotype to the activated phenotype with an extension of processes into the environment and eventually a retraction into an amoeboid morphology. In cultured microglial cells isolated from tg mice overexpressing R1441G-LRRK2 or WT littermates, LPS treatment induces an increase in LRRK2 protein expression that is not reflected at the mRNA level, suggesting that LPS-treatment can impact the turn-over of LRRK2 (Gillardon, Schmid, & Draheim, 2012; Moehle et al., 2012). In addition, R1441G-LRRK2 expression increases expression and secretion of proinflammatory cytokines (TNF) compared to WT microglia in culture (Gillardon et al., 2012). LRRK2 inhibitors, LRRK2-IN-1 and sunitinib, were found to attenuate the release and transcriptional induction of TNF and reduce phospho-p38 protein levels. Similarly, knockdown of LRRK2 by a lentivirus coding for an shRNA targeting LRRK2 has the same effect, except for the phosphorylation of p38 that remains unchanged by LRRK2 shRNA. More interestingly, in the presence of LRRK2 inhibitors or LRRK2 shRNA, the LPS treatment fails to induce the switch between the resting phenotype to the activated phenotype (Moehle et al., 2012). The knockdown of LRRK2 in the microglial cell line BV-2 by lentiviral vectors encoding a shRNA significantly reduces the secretion of TNFα and IL-6 with an increase of NO production as compared to control conditions. Treatment with LPS in these knockdown cells induces a decrease in the level of the phosphorylated form of p38 and induces a significant reduction of nuclear factor kappa B (NF-kB) transcriptional activity while JNK and ERK are unaffected (B. Kim et al., 2012). LRRK2 enhances the association of NFAT1 with proteins that seem to block the NFAT transport to the nucleus and thus inhibits the transcription of immune response genes (Liu et al., 2011). Microglia isolated from LRRK2 KO mice have an increased α-syn uptake and clearance when treated with recombinant α-syn, compared to WT mice microglia (Maekawa et al., 2016). In these studies, primary microglia and cell lines in culture adopt a macrophage-like phenotype on a transcriptional and functional level, necessitating studies in vivo with a preserved dynamic innate immune system (Butovsky et al., 2014).
In LRRK2 knockout rats as well as LRRK2 knockout mice, LPS and Tat-induced neuroinflammation (respectively) are dramatically dulled compared to WT rodents (Daher et al., 2014; Puccini et al., 2015). The absence of LRRK2 protects from neurodegeneration induced by an AAV coding for α-syn (Daher et al., 2014). Like LPS, injections of viral vectors coding α-syn are known to induce microglial activation. In this study, the number of CD68-positive cells recruited in response to α-syn expression is reduced in LRRK2 KO rats compared to WT rats. The morphology of IBA1-positive cells shift to the amoeboid (activated) phenotype in WT rats injected with α-syn that was not observed in LRRK2 KO rats also injected with α-syn. In addition, treatment with PF-06447475 attenuates the neuroinflammation in G2019S-LRRK2-BAC Tg rats when compared to untreated animals and to non-Tg rats expressing WT rat LRRK2 after the injection of AAV-α-syn (Daher et al., 2015). Overall it seems likely that LRRK2 modulates the inflammatory response induced by α-syn. It could be imagined that G2019S-LRRK2 could increase the neuroinflammation induced by α-syn, therefore aggravating the deleterious effects of this neuroinflammation. Selective LRRK2 mutant expression or knockdown within cells of the immune system has not yet been described, limiting interpretations of mechanisms.
Many studies have correlated α-syn aggregates with activated microglia, MHC-II expression, and neuroinflammation. Studies in human post-mortem PD brain show that MHC-II detection of microglia performs as well as phosphorylated α-syn in staging brains in PD pathology (Imamura et al., 2003). α-Syn fibrils, perhaps all fibrillized amyloid proteins, are substrates of TLR2 expressed in cells of the innate immune system, and can activate MyD88 pathways similar to TLR4 agonists (e.g., LPS). α-Syn fibrils and aggregates may bind to and activate both TLR2 and TLR4 (Codolo et al., 2013; Daniele et al., 2015; Gustot et al., 2015; C. Kim et al., 2013). The implication is that abnormal α-syn activates an immune response that may be potentiated by mutant LRRK2 expression. Alternatively, LRRK2 activity may initiate an abnormal immune response that causes α-syn to aggregate in neurons, with the immune response further potentiated in perpetuity with α-syn aggregates interacting with toll-like receptors. Future studies can dissect these possibilities.
LRRK2, α-syn, and 14–3-3s
Among all the pathways plausibly affected by α-syn and LRRK2, one possibility is that a common protein interactor could weave together the two proteins in a cell in response to specific stimuli. The 14–3-3 chaperone-like protein family is known to interact robustly with both α-syn and LRRK2, as well as becomes entrapped in LBs in PD (Berg, Holzmann, and Riess 2003; Kawamoto et al., 2002; Nichols et al., 2010). In 2002, Xu and collaborators showed that 14–3-3 proteins interact with α-syn in a 54–83-kD complex (Xu et al., 2002). 14–3-3 sequestration in LBs, plausibly mediated by an interaction with α-syn, may cause loss of 14–3-3 function and lowered anti-apoptotic activity and lead to toxicity and cell death. Moreover, expression of 14–3-3 theta is significantly decreased in tg mice overexpressing human WT-α-syn under the platelet-derived growth factor beta promoter (Yacoubian et al., 2008). In dopaminergic cells, the level of tyrosine hydroxylase can be modulated both with α-syn and 14–3-3 expression. 14–3-3 proteins are known to interact with phosphorylated TH to enhance its activity and to promote dopamine synthesis. α-Syn in turn may bind to unphosphorylated TH to reduce its activity (for more details see Review from Berg, Holzmann, and Riess 2003). This is a compelling example of how 14–3-3 and α-syn binding to each other may manipulate cell phenotypes associated with a rate-limited enzyme (TH) critical for neuronal phenotypes.
As with α-syn and LRRK2, 14–3-3 proteins themselves are subject to phosphorylation. In 2015, Slone and collaborators found that modulation of 14–3-3 phosphorylation reduced their neuroprotective potential (Slone, Lavalley, McFerrin, Wang, & Yacoubian, 2015). 14–3-3 binding to LRRK2 is mediated by the LRRK2 Ser910 and 935 phosphorylation sites (Gloeckner et al., 2010; Nichols et al., 2010; West et al., 2007). Oxidative stress may promote dephosphorylation of LRRK2 causing a loss of 14–3-3 binding to LRRK2 (Mamais et al., 2014). Disruption of 14–3-3 binding to LRRK2 alters LRRK2 localization in the cell and potentially affects LRRK2 kinase activity and membrane-related functions, such as exosome release (Dzamko et al., 2010; Mamais et al., 2014). 14–3-3 binding to LRRK2 could prevent the dephosphorylation of LRRK2 and may stabilize LRRK2 structure. In 2017, Civiero and collaborators showed in vitro that the serine/threonine kinase PAK6 bound and phosphorylated 14–3-3gamma at its Ser59 residue, thus promoting 14–3-3gamma/LRRK2 complex dissociation. In neurons, the activation of this kinase rescues G2019S-LRRK2 neurotoxicity (Civiero et al., 2017). Other recent work concluded that 14–3-3 binding to LRRK2 could control LRRK2 kinase activity (Lavalley et al., 2016). 14–3-3 binding to LRRK2 preventes LRRK2 kinase activity and autophosphorylation, whereas a loss of 14–3-3 expression and binding result in hyper-activated LRRK2 and exacerbate LRRK2 neurotoxicity. In a cell autonomous manner, α-syn-mediated sequestration of 14–3-3 proteins may hyper-activate LRRK2 kinase activity, encourage pro-apoptotic pathways, and reduce levels of dopamine synthesis needed for proper basal ganglia function.
α-syn, LRRK2, and Mitochondria
Mitochondria have been a favorite organelle for study in PD-related research stemming back to post-mortem descriptions of a complex I deficiency in the striatum (Mizuno et al., 1989) and in the SNc (Schapira et al., 1990) of PD patients. Mitochondrial complex I inhibitors (for example rotenone or MPTP) can also induce parkinsonism in exposed people as well as in rodents (Langston and Ballard 1983; Schapira 2008). Mutations in the genes encoding PARKIN, DJ-1 and PINK1 cause autosomal recessive forms of PD, and these proteins are all involved in mitochondrial quality control (reviewed in Dawson, Ko, and Dawson 2010). In rodents, endogenous as well as exogenously administered α-syn (WT or A53T) localizes in-part to mitochondria (Chinta, Mallajosyula, Rane, & Andersen, 2010; Li et al., 2007; Martin et al., 2006; Nakamura et al., 2008; Subramaniam, Vergnes, Franich, Reue, & Chesselet, 2014; Ling Zhang et al., 2008). α-Syn may directly interact with TOM20 (translocase of the outer mitochondrial membrane) and inhibit mitochondrial protein import (Di Maio et al., 2016). Moreover, partial homology to mitochondrial targeting sequence was found in the N-term portion of α-syn. α-Syn binding to mitochondrial membranes appears particularly high in the striatum, in the SNc and in the cortex of PD brains, regions highly affected in PD pathology (Devi, Raghavendran, Prabhu, Avadhani, & Anandatheerthavarada, 2008). α-Syn import to mitochondria may depend on intracellular pH (Cole, DiEuliis, Leo, Mitchell, & Nussbaum, 2008) and on mitochondrial membrane potential and the ATP pool (Devi et al., 2008). In vitro, about 10% of total LRRK2 protein localizes to a mitochondrial enriched protein fraction (West et al., 2005), although a lower percentage is probably associated with the outer mitochondrial membrane without significant levels detected inside mitochondria (Biskup et al., 2006).
Studies in model systems with increased or abnormal α-syn expression, or mutant LRRK2, have demonstrated compromised mitochondrial function such as decreased mitochondrial membrane potential and ATP production and increased oxidative stress (Butler et al., 2012; Mortiboys et al., 2015; Papkovskaia et al., 2012; Parihar et al., 2008; Su and Qi 2013; Wang et al., 2012; Melo et al., 2016; Erustes et al., 2017; Martinez et al., 2018; Martin et al., 2006; Mortiboys et al., 2010; Sarafian et al., 2013). While α-syn null mice are protected against MPTP toxicity, and tg mice overexpressing human α-syn may be more sensitive to MPTP-induced toxicity (Dauer et al., 2002; Klivenyi et al., 2006; Song, Shults, Sisk, Rockenstein, & Masliah, 2004), LRRK2 knockout mice do not appear protected from MPTP (Andres-Mateos et al., 2009). However, mice over-expressing G2019S-LRRK2 appear more sensitive to MPTP (Karuppagounder et al., 2015). Thus, at least through MPTP, both α-syn and LRRK2 appear to have a functional role in mitochondria-dysfunction that can cause neurodegeneration.
While the mechanism of how LRRK2 interacts with mitochondria is speculative at this time, several studies may shed light on α-syn interaction with mitochondria. α-Syn KO mice present with reductions of mitochondria respiration and linked CI/III activity (Ellis et al., 2005), and tg mice expressing α-syn A53T in DA neurons present an age-related mitochondrial CI inhibition (Chinta et al., 2010). Devi and collaborators have identified in 2008 a large decrease in CI activity associated with abnormal α-syn, both in human fetal primary cortical neuronal cultures overexpressing α-syn and in mitochondria isolated from the ST and SN of autopsy brain hemispheres from PD subjects (Devi et al., 2008) and these results have been reproduced in other in vitro models (Loeb, Yakunin, Saada, & Sharon, 2010; Reeve et al., 2015). Mitochondria isolated from Wistar rats exposed to different concentrations of recombinant human α-syn also show a dose-dependent decrease in CI activity (Liu et al., 2009).
Potentially corroborating these results, Tg mice expressing A53T-α-syn display dysmorphic mitochondria (Martin et al., 2006). Double tg mice α-syn/parkin KO have marked alterations of mitochondrial architecture, especially in the SN and cortex (Stichel et al., 2007). Similarly, tg A53T mice show changes in mitochondrial morphology and decreased mitochondrial interconnectivity (Xie & Chung, 2012) and even fragmented mitochondria that appear before DA neuron loss (Chen, Xie, Turkson, & Zhuang, 2015). In vitro, α-syn knock down increases the number of cells presenting elongated mitochondria (Kamp et al., 2010) and overexpression of both WT and A53T-α-syn induced the fragmentation of mitochondria (Butler et al., 2012; Devoto et al., 2017; Guardia-Laguarta et al., 2014; Martinez et al., 2018) that precedes any disturbance in mitochondrial function and cell death (Nakamura et al., 2011). This effect of α-syn on mitochondrial fragmentation was also shown in DA neurons derived from patients with the A53T mutation (Ryan et al., 2018). Moreover, this α-syn-dependent mitochondrial fragmentation requires a direct interaction with the outer mitochondrial membrane in iPS overexpressing α-syn (Devoto et al., 2017). In primary mouse cortical cultures, astrocytes internalize α-syn oligomers causing mitochondrial fragmentation after a 24h exposure (Lindström et al., 2017). In particular, Grassi and collaborators recently identified a conformationally distinct phosphorylated α-syn species (resulting from the incomplete degradation of fibrillar α-syn) that associates with mitochondria to induce mitochondrial dysfunction (Grassi et al., 2018). More precisely, significant reductions of Mnf1 and 2 proteins were observed in tg A53T mice with no changes in Drp1 or OPA1 (Xie and Chung 2012). Drp1 mostly locates in the cytoplasm, but is stimulated after fission stimuli to migrate to the mitochondria. In Drp1 KO mice, the overexpression of WT-α-syn still induces significant fragmentation of mitochondria, even though it is still less important than in the WT mice, implying that α-syn-induced mitochondrial fragmentation might go through an effect on Drp1, but that it would not be the only mechanism (Nakamura et al., 2011). Thus, these findings suggest that the fragmentation of mitochondria caused by α-syn aggregation and expression decreases mitochondrial fusion, rather than an increase in fission.
LRRK2 may interact with Drp1 to enhance Drp1 mitochondrial translocation, and the blockage of Drp1 inhibits mitochondrial fragmentation and dysfunction (Niu, Yu, Wang, & Xu, 2012; Su & Qi, 2013; X. Wang et al., 2012). G2019S-LRRK2 might phosphorylate Drp1 leading to mitochondrial fragmentation (Su & Qi, 2013). Through this pathway, LRRK2 kinase activity could activate Drp1 and enhance its localization to mitochondria, therefore tipping the balance in favor of mitochondrial fission. Supporting this hypothesis, LRRK2 (and not its dead kinase mutant) enhances ROS production, which is known to lead to increased fission (Niu et al., 2012). However, Saez-Atienzar and collaborators observed the opposite effects: in their in vitro study, it was the pharmacological inhibition of LRRK2 that led to a Drp-1-dependent increase in mitochondrial fission (Saez-Atienzar et al., 2014). Similarly, there is no conclusion as to the effect of LRRK2 expression or activity on mitochondrial morphology. In a tg fly model, the expression of G2019S-LRRK2 is associated with mitochondrial deformity (Mortiboys et al., 2015). In vitro, in cell lines, the expression of WT-LRRK2, and even more that of G2019S or R1441C-LRRK2 induces mitochondrial fragmentation (Su & Qi, 2013; X. Wang et al., 2012). Interestingly, this effect seems to be kinase-dependent, which is consistent with the fact that the G2019S mutant has more pronounced effects on mitochondria (Su & Qi, 2013; X. Wang et al., 2012). However, two other studies demonstrated the opposite results in fibroblasts derived from PD patients carrying the G2019S mutation, one observing an increased mitochondrial fragmentation, pointing towards a misbalance in favor of mitochondrial fission (Niu et al., 2012), and the other enhanced mitochondrial elongation and interconnectivity, pointing towards a misbalance in favor of mitochondrial fusion (Mortiboys et al., 2010). Further, the direct effect of LRRK2 on mitochondrial respiration is not clear. In HEK cells, the expression of WT-LRRK2, and to a further degree G2019S, decrease both CI and CIV activities (Su & Qi, 2013). In other model systems, tg flies expressing G2019S, G2385R, or I2020T-LRRK2 are more sensitive to rotenone-induced toxicity (Ng et al., 2009; Venderova et al., 2009), and in C. elegans, WT-LRRK2 but not G2019S-LRRK2 expression protects against rotenone-induced toxicity (Saha et al., 2009).
Taken together, these studies may imply that LRRK2 and α-syn can have a direct effect on the mitochondrial respiratory chain, leading to mitochondrial dysfunction and altered energy production. However, second hypothesis that links LRRK2 and α-syn with the disruption of the equilibrium between mitochondrial fission and fusion also seems plausible. All the data showing an effect of α-syn and LRRK2 on the toxicity of complex I inhibitors point towards a potential effect of α-syn and LRRK2 on mitochondrial respiratory chain, more precisely on CI activity. These complex I defects can lead to oxidative stress, a decrease in ATP production or in mitochondrial respiration (Devi et al., 2008; Loeb et al., 2010; Reeve et al., 2015). All of these events ending up in mitochondrial dysfunction could therefore be a relevant explanation for the mitochondrial deficits induced by α-syn and LRRK2. As mitochondrial homeostasis is a finely regulated process with a precise equilibrium between fission (involving the proteins Drp1 (Dynamin-related protein 1), Fis1 and GDAP1) and fusion (involving the proteins Mfn 1 and 2 and OPA1), mitochondrial fragmentation can reflect an increase in mitochondrial fission or a decrease in mitochondrial fusion. Overall, these results suggest that α-syn and LRRK2 have the potential for a common effect on two major aspects of mitochondrial homeostasis: the energy production and the dynamics of mitochondria. However, there is a tight relationship between the structure and the energy function of the mitochondrial network, and an alteration of one aspect has repercussions on the other. It is therefore difficult to know which effect comes first, but newer model systems with conditional manipulation of LRRK2 and α-syn may shed light onto the relationship between these proteins, mitochondrial respiratory chain function and fusion and fission events.
α-syn, LRRK2 and Autophagy
Proteins can be removed by the ubiquitin-proteasome system (UPS) as well as the autophagy-lysosome pathway (ALP), and both have been extensively evaluated with respect to LRRK2 and α-syn in PD models. α-Syn degradation via the UPS was observed and UPS dysfunction was predicted to lead to α-syn accumulation in early studies (Bennett et al., 1999; McNaught et al., 2002; Tofaris, Layfield, & Spillantini, 2001; Webb, Ravikumar, Atkins, Skepper, & Rubinsztein, 2003). However, other groups failed to show an involvement of the proteasome machinery in this process (Ancolio, Alves Da Costa, Uéda, & Checler, 2000; Rideout, Larsen, Sulzer, & Stefanis, 2001; Rideout & Stefanis, 2002; Vogiatzi, Xilouri, Vekrellis, & Stefanis, 2008) suggesting another pathway to α-syn degradation such as ALP (Petroi et al., 2012). Given the close overlap between LRRK2 and α-syn in other pathways we describe here, LRRK2 function may shed light on this controversy.
Autophagy clears cytoplasmic components through three distinct pathways depending of the way substrates reach the lysosomal lumen and the types of substrates supported: chaperone-mediated autophagy (CMA), macroautophagy, and microautophagy (Ana Maria Cuervo, 2004; Levine & Klionsky, 2004). In contrast, the UPS mainly drives short-lived protein degradation, typically via polyubiquitin chains, whereas autophagy is responsible for long-lived proteins, larger protein aggregates, and cytoplasmic organelles (Zhang et al., 2012). CMA involves chaperone proteins (especially hsc70) which bind target proteins containing a KFERQ peptide motif. These chaperones address the proteins to lysosomes where they interact with the lysosomal receptor, LAMP2a, the rate-limiting step in CMA (A. M. Cuervo & Dice, 1996; Majeski & Fred Dice, 2004; A. Massey, Kiffin, & Cuervo, 2004). At the membrane, LAMP2a multimerizes to allow substrate translocation inside the lysosome with the help of lys-hsc70, an intraluminal chaperone (Agarraberes, Terlecky, and Dice 1997; Cuervo, Dice, and Knecht 1997). Macroautophagy involves the creation of a double membrane structure, named phagophore, engulfing proteins and organelles. Closure completion of the phagophore forms the autophagosome which will fuse with lysosomes to generate the autolysosome, where substrates are degraded. Mitophagy is a subtype of macroautophagy which leads to dysfunctional mitochondria degradation. (Shintani & Klionsky, 2004). The molecular machinery participating in macroautophagy is composed of different type of proteins, including Atg proteins, and Beclin. Microautophagy involves the engulfment and internalization (pinocytosis) of small quantities of cytosol components directly by lysosomes (A. Massey et al., 2004; Müller et al., 2000). In PD brains, autophagic characteristics were shown through immunohistochemistry (Anglade, Vyas, Hirsch, & Agid, 1997) and impairment of autophagy (defective clearance of autophagosomes and lysosomal depletion) was observed in PD patient brains (Dehay et al., 2010). Additionally, LAMP2a and HSC70 protein levels in the SNc of PD patient brains were decreased (Alvarez-Erviti et al., 2010).
The potential degradation of α-syn by CMA is suggested since the protein contains a CMA recognition motif (95-VKKDQ-99) (Cuervo et al., 2004; Dice 1990). First evidence confirming this hypothesis was depicted incubating α-syn with isolated liver lysosomes showing its degradation in a CMA-dependent manner (Cuervo et al., 2004). Evidence of α-syn degradation by CMA is also demonstrated in cell lines and neurons using lysosomal inhibitors which lead to α-syn accumulation (Lee et al., 2004; Paxinou et al., 2001; Webb et al., 2003). In 2004, Cuervo and collaborators also demonstrated that the mutant forms of α-syn (A53T and A30P) can bind to lysosome membranes but can not be engulfed and degraded efficiently by lysosomes. In fact, mutant α-syn interacts more strongly with LAMP2a, blocking CMA (Cuervo et al., 2004). In different cell lines and in primary neuronal cells, mutant α-syn interaction with LAMP2a and impairment of CMA is also demonstrated (Vogiatzi et al., 2008). More recently, decreasing LAMP2a expression through a strategy of shRNA-AAV injections in rat brain results in intracellular accumulation of α-syn and autophagic vacuoles (Xilouri et al., 2016). Additionally, when the recognition motif 95-VDKKQ-99 is mutated, CMA is dysfunctional and compensatory mechanisms occur with the activation of macroautophagy (Vogiatzi et al., 2008; Xilouri, Vogiatzi, Vekrellis, Park, & Stefanis, 2009). In cell culture, LAMP2a silencing blocks CMA but upregulates macroautophagy and sensitizes cells to oxidative stress suggesting also a compensatory mechanism (Massey et al., 2006). α-Syn overexpression inhibits macroautophagy through blockade of autophagosome formation. However, cells expressing α-syn in contact with preformed α-syn aggregates leads to the formation of autophagosomes but their clearance is blocked (Tanik, Schultheiss, Volpicelli-Daley, Brunden, & Lee, 2013). In Atg7 knock out mice, macroautophagy impairment leads to ubiquitinated proteins, α-syn and p62 increased (Ahmed et al., 2012). In another model of Atg7 knock out targeting TH positive cells, accumulation of α-syn in dopaminergic neurons, especially midbrain dopaminergic dendrites which exhibit numerous varicosities containing massive ubiquitinated inclusions, was identified (Friedman et al., 2012). Mechanistically, α-syn compromises autophagy via Rab1a inhibition and causes mislocalization of the autophagy protein Atg9. Atg9 is implicated in the initiation of autophagosome formation (Winslow et al., 2010). Additional experiments are needed in order to decipher the molecular mechanisms implicated in macroautophagy in α-syn toxicity.
While fewer studies have explored LRRK2 function in the UPS and ALP, LRRK2 contains several CMA targeting motifs suggesting its possible degradation by CMA. The role of LRRK2 in CMA has been partially elucidated in a study from Orenstein in 2013. In this study, LRRK2 is degraded by CMA as knock down of LAMP2a increases intracellular levels of LRRK2. Degradation of LRRK2 reaches 50% of normal levels in LAMP2a knock downs whereas G2019S-LRRK2 is limited suggesting impairment of CMA through mutant LRRK2 activity (Orenstein et al., 2013). Degradation of LRRK2 through macroautophagy is supported through experiments in Atg5 KO and Atg7 KO mice showing accumulation of LRRK2 in cells and tissue (Friedman et al., 2012). Moreover, a fraction of LRRK2 is localized to membranes (Berger, Smith, & Lavoie, 2010; Biskup et al., 2006) and more specifically to autophagic vacuoles, amphisomes, and autolysosomes and lysosomes (Alegre-Abarrategui et al., 2009; Mark W. Dodson, Zhang, Jiang, Chen, & Guo, 2012; Higashi et al., 2009; Orenstein et al., 2013). However, contradictory results were obtained on the role of LRRK2 in macroautophagy. On the one hand, several groups observe a positive regulation of autophagy by LRRK2. Saha and collaborators demonstrate that expression of human LRRK2 or dead kinase LRRK2 in C. elegans increases autophagy (Saha et al., 2015). In macrophages and microglial cells, LRRK2 knockdown significantly decreases the autophagic capacity (reduction of LC3 conversion), and LRRK2 inhibitors (GSK2578215A or LRRK2-IN-1) block protein aggregate clearance (Schapansky, Nardozzi, Felizia, & LaVoie, 2014). In 2010, Tong and collaborators showed that LRRK2 KO mice present accumulation of lipofuscin granules and p62 protein and a decrease of LC3-II conversion in renal tubule cells of aged mice (Tong et al., 2010), suggesting slowdown of autophagy in those cells. In flies, knockout of the LRRK2 ortholog dLRRK leads to an impairment of autophagy with enlarged early endosomes, accumulation of autophagosomes which involves a modulation of Rab9 activity (Dodson et al., 2014). On the other hand, knock down of LRRK2 by siRNA produces an induction of autophagy suggesting LRRK2 act as a negative regulator of autophagy (Alegre-Abarrategui et al., 2009). In astrocytes, inhibition of LRRK2 stimulates macroautophagy in an mTor independent manner that requires Beclin (Manzoni et al., 2016). Bravo-San Pedro et collaborators observed an increase of autophagy induction (increase of LC3, LAMP2, Beclin1 and decrease p62) in G2019S-LRRK2 skin fibroblasts from PD patients via a MAPK/ERK dependent pathway resulting in a highest sensitization to cell death (Bravo-San Pedro et al., 2013). As usual, the apparent discrepancies between these different studies may originate from different experimental conditions and cell types used, but as yet there is no clear consensus of LRRK2 effect on ALP.
To go deeper in our understanding of the possible link between α-syn, LRRK2 and ALP, an interesting point is the role of Rab proteins and particularly Rab7. In fact, as mentioned above, Rab7 interacts with LRRK2 and plays a role in autophagosome and lysosome trafficking, positioning, and fusion events. Rab7 promotes clearance of α-syn aggregates (Dinter et al., 2016). Rab7 is also a key regulator of late endosome maturation and autophagosome-lysosome fusion (Jager, 2004). Therefore, it is conceivable that the link between LRRK2 and α-syn in producing autophagy defects would be through Rab-dependent action, for example Rab7, at the membrane of autophagosomes. This modulation could interfere with trafficking of autophagosomes, which need to reach the soma to fuse with lysosomes to have an efficient autophagy.
Conclusion and future directions
A compelling and growing body of evidence points towards a functional interaction between α-syn and LRRK2. Intriguingly, in many studies, larger effects are observed by knocking down LRRK2 expression or activity compared to over-expressing mutated LRRK2, giving hope that LRRK2-targeting therapeutics may offer benefit to all PD patients rather than just those that have LRRK2 mutations. Yet, the underlying mechanisms that dictate LRRK2 and α-syn interaction in PD are not clear. Based on our current understanding of both proteins in disease, cell autonomous and non-cell autonomous pathways are likely relevant in linking the proteins together in pathogenesis and disease progression. When expressed in the same cell, in disease, α-syn and LRRK2 likely interact in response to mitochondria disruption, in autophagy pathways in the endolysosomal system, as well as in normal interactions with common protein partners like 14–3-3 that maintain homeostasis of a number of systems. While α-syn expression in the brain is highly restricted to neurons, LRRK2 is also expressed in the immune system, particularly in cells known to respond to amyloid forms of α-syn, and LRRK2 appears to exacerbate damaging pro-inflammatory responses caused by α-syn. Considering current knowledge, none of the mechanisms we discussed in this review appear to be more prominent than the others. These interactions will all take time and more advanced model systems and experiments to fully resolve. In the meantime, therapeutic targeting of both LRRK2 and α-syn, if our understanding of these proteins in disease is at least partially correct, should provide the best opportunities to intervene in the disease process.
Figure 1: Cellular dysfunction and neurodegeneration through several distinct but non-mutually exclusive mechanisms.
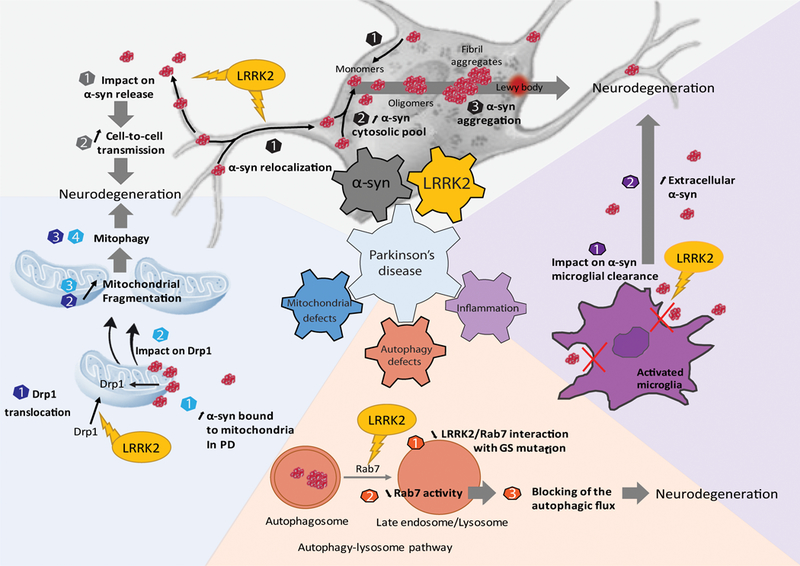
LRRK2 could have an impact on α-syn release (grey numbers) as well as on its relocalization (black numbers) leading to a cell-cell transmission of the pathology and to an increase of the aggregation through an excess of α-syn in the cytosolic pool (grey part). Three mechanisms seem to be impacted: the mitochondrial biology (blue part), the autophagic system (orange part) and the inflammation (violet part). Both LRRK2 and α-syn are known to play a role in mitochondrial biology through Drp1 (blue number) in favor of a mitochondrial fragmentation leading to mitophagy and cell death. Inside the cell, α-syn aggregates are mainly degraded by the autophagic-lysosomal pathway, which is one of the major protein breakdown systems (orange part). LRRK2 was found to decrease the activity of Rab7, which is implicated in the maturation of the autophagosome and thus blocking the degradation of the α-syn aggregates. Outside the cell, α-syn aggregates are captured by microglia to be degraded. One hypothesis is that LRRK2 could alter the machinery of phagocytosis leading to an accumulation of extracellular aggregates.
Acknowledgements:
NC is a recipient of a PhD fellowship from Association France Parkinson (2016). CG is a recipient of the PhD fellowship from the French Ministry of Research. FG is funded by the European Union Horizon 2020 Programme (H2020-MSCA-ITN-2015) under the Marie Skłodowska-Curie Innovative Training Network and Grant Agreement No. 676408. EB is supported by “Fondation de France, Parkinson committee” grants (AAP 2010, Engt 00016819 and AAP 2014, Engt 2014–0052580) and the national “Translational Research Infrastructure for Biotherapies in Neurosciences” (NeurATRIS, “Investissement d’Avenir”, ANR-11-INBS-0011). ABW is supported by NIH grants R01 NS064934, U01 NS097028, and R33 NS097643.
List of Abreviation
- AAV
Adeno-associated virus
- ADPD
Autosomal dominant Parkinson’s disease
- ALP
Autophagy-lysosome pathway
- ANK
Ankyrin
- APP
Amyloid precursor protein
- ARM
Armadillo
- Atg
Autophagy related protein
- ATP
Adenosine Triphosphate
- BAC
Bacterial artificial chromosome
- BAD
Bcl-2-associated death promoter
- Bcl-2
B-cells lymphoma 2
- CaMKII
Calcium/calmodulin-dependent protein kinase II
- CI – CIV
Complex I – Complex IV of mitochondrial respiratory chain
- CMA
Chaperone-mediated autophagy
- CNS
Central nervous system
- COR
C-terminal of Roc
- DA
Dopamine
- DAT
Dopamine transporter
- Drp1
Dynamid related protein 1
- ERK
Extracellular signal-regulated-kinase
- Fis1
Mitochondrial fission protein 1
- GAD
G proteins activated by nucleotide-dependent dimerization
- GDAP1
Ganglioside-induced differentiation-associated protein 1
- GLP1R
Glucagon-like peptide-1 receptor
- GWAS
Genome-wide association
- HEK
Human embryonic kidney (cell line)
- HSC70
Heath shock cognate protein
- IBA1
Ionized calcium-binding adapter molecule 1
- IBD
Inflammatory bowel disease
- IFN-γ
Interferon gamma
- IL-1β
Interleukin-1 beta
- IL-6
Interleukin 6
- iNOS
Inducible nitrite oxide synthase
- iPD
Idiopathic Parkinson’s disease
- iPS
Induced pluripotent stem cells
- JNK
c-Jun N-terminal kinase
- kD
kilo-dalton
- KI
Knock-in
- KO
Knock out
- L-dopa
Levodopa
- LAMP2
Lysosome-associated membrane protein 2
- LB
Lewy bodies
- LC3-II
LC3-phosphatidylethanolamine conjugate
- LN
Lewy neurites
- LPS
Lipopolysaccharides
- LRR
Leucine-rich repeats
- LRRK2
Leucine-rich repeat kinase 2
- MAPK
Mitogen-activated protein kinase
- MHC-II
Major histocompatibility complex II
- Mnf1 – Mnf2
Mitochondrial nucleoid factor 1 – 2
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- mRNA
Messenger RiboNucleic Acid
- mTOR
Mammalian target of rapamycin
- MyD88
Myeloid differentiation primary response 88
- NF-kB
Nuclear factor kappa B
- NFAT
Nuclear factor of activated T-cells
- NHPs
Non-human primates
- NO
Nitrite oxide
- OPA1
mitochondrial dynamin like GTPase
- PARK8
Old name for LRRK2
- PD
Parkinson’s disease
- PET
Positron emission tomography
- PFFs
Pre-formed fibrils
- PrP
Prion protein
- pSer
phosphorylated Serine
- Roc
Ras-of-complex
- Ser
Serine
- SH-SY5Y
Homo sapiens bone marrow neuroblast cell line
- shRNA
Short hairpin RNA
- siRNA
Small interfering RNA
- SNc
Substantia nigra pars compacta
- SNCA
alpha synuclein (gene)
- SNP
Single nucleotide polymorphism
- SOD1
Superoxide dismutase 1
- TDP43
TAR DNA-binding protein 43
- Tg
Transgenic
- TH
thyrosine hydroxylase
- TLR4
Tool-like-receptor 4
- TNF-α
Tumor necrosis factor alpha
- TOM20
Translocase Of Outer Mitochondrial Membrane 20
- tTa
Tetracycline trans activator
- Tyr
Tyrosine
- UPS
Ubiquitin-proteasome system
- vMAT
Vesicle dopamine transporter
- WD40
Beta transducing repeat
- WT
Wild type
- α-syn
α-synuclein
Footnotes
Conflict of interests
The authors of the present review declare to have no conflict of interest.
References
- Abdelmotilib H, Maltbie T, Delic V, Liu Z, Hu X, Fraser KB, Moehle MS, Stoyka L, Anabtawi N, Krendelchtchikova V, Volpicelli-Daley LA & West A (2017) α-Synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic Neurodegeneration. Neurobiol Dis, 105, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JR, Van Netten H, Schulzer M, Mak E, Mckenzie J, Strongosky A, Sossi V, Ruth TJ, Lee CS, Farrer M, Gasser T, Uitti RJ, Calne DB, Wszolek ZK & Stoessl AJ (2005) PET in LRRK2 mutations: Comparison to sporadic Parkinson’s disease and evidence for presymptomatic compensation. Brain, 128, 2777–2785. [DOI] [PubMed] [Google Scholar]
- Agarraberes FA, Terlecky SR, & Dice JF (1997) An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol, 137, 825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed I, Liang Y, Schools S, Dawson VL, Dawson TM, & Savitt JM (2012) Development and Characterization of a New Parkinson’s Disease Model Resulting from Impaired Autophagy. J Neurosci, 32, 16503–16509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alegre-Abarrategui J, Christian H, Lufino MMP, Mutihac R, Venda LL, Ansorge O, & Wade-Martins R (2009) LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet, 18, 4022–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, & Sammler E (2018) LRRK2 kinase in Parkinson’s disease. Science, 360, 36–37. [DOI] [PubMed] [Google Scholar]
- Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, & Schapira AHV (2010) Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol, 67, 1464–1472. [DOI] [PubMed] [Google Scholar]
- Ancolio K, Alves Da Costa C, Uéda K, & Checler F (2000) α-Synuclein and the Parkinson’s disease-related mutant Ala53Thr-α- synuclein do not undergo proteasomal degradation in HEK293 and neuronal cells. Neurosci Lett, 285, 79–82. [DOI] [PubMed] [Google Scholar]
- Andres-Mateos E, Mejias R, Sasaki M, Li X, Lin BM, Biskup S, Zhang L, Banerjee R, Thomas B, Yang L, Liu G, Beal MF, Huso DL, Dawson TM & Dawson VL (2009) Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine). J Neurosci, 29, 15846–15850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglade P, Vyas S, Hirsch EC, & Agid Y (1997) Apoptosis in dopaminergic neurons of the human substantia nigra during normal aging. Histol Histopathol, 12, 603–610. [PubMed] [Google Scholar]
- Barrett J, Hansoul S, Nicolae D, & Cho J (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet, 40, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendor JT, Logan TP, & Edwards RH (2013) The Function of a-Synuclein. Neuron, 79, 1044–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, & Mouradian MM (1999) Degradation of alpha-synuclein by proteasome. J Biol Chem, 274, 33855–33858. [DOI] [PubMed] [Google Scholar]
- Benskey MJ, Sellnow RC, Sandoval IM, Sortwell CE, Lipton JW & Manfredsson FP (2018) Silencing Alpha Synuclein in Mature Nigral Neurons Results in Rapid Neuroinflammation and Subsequent Toxicity. Front Mol Neurosci, 11, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg D, Holzmann C, & Riess O (2003) 14–3-3 Proteins in the Nervous System. Nat Rev Neurosci, 4, 752–762. [DOI] [PubMed] [Google Scholar]
- Berger Z, Smith KA, & Lavoie MJ (2010) Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry, 49, 5511–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernacka JM, Armasu SM, Cunningham JM, Eric Ahlskog J, Chung SJ, & Maraganore DM (2011) Do interactions between SNCA, MAPT, and LRRK2 genes contribute to Parkinson’s disease susceptibility? Parkinsonism Relat Dis, 17, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RLM, Emson PC, Torp R, Ottersen OP, Dawson TM & Dawson VL (2006) Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol, 60, 557–569. [DOI] [PubMed] [Google Scholar]
- Biskup S, & West AB (2009) Zeroing in on LRRK2-Linked Pathogenic Mechanisms in Parkinson’s Disease. Biochim Biophys Acta, 7, 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RLM, Emson PC, Torp R, Ottersen OP, Dawson TM & Dawson VL (2012) Age at onset in LRRK2-associated PD is modified by SNCA variants. J Mol Neurosci, 48, 245–247. [DOI] [PubMed] [Google Scholar]
- Bouhouche A, Tibar H, Haj RBE, El Bayad K, Razine R, Tazrout S, Skalli A, Bouslam N, Elouardi L, Benomar A, Yahyaoui M & Regragui W (2017) LRRK2 G2019S Mutation: Prevalence and Clinical Features in Moroccans with Parkinson’s Disease. Parkinson Dis [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, & Braak E (2000) Pathoanatomy of Parkinson’s disease. J Neurol, 247, II3–II10. [DOI] [PubMed] [Google Scholar]
- Braak H, & Del Tredici K (2004) Poor and protracted myelination as a contributory factor to neurodegenerative disorders. Neurobiol Aging, 25, 19–23. [DOI] [PubMed] [Google Scholar]
- Bravo-San Pedro JM, Niso-Santano M, Gómez-Sánchez R, Pizarro-Estrella E, Aiastui-Pujana A, Gorostidi A, Climent V, López De Maturana R, Sanchez-Pernaute R, López De Munain A, Fuentes JM & González-Polo RA (2013) The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell Mol Life Sci, 70, 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck K, Landeck N, Ulusoy A, Majbour NK, El-Agnaf OM & Kirik D (2015) Ser129 Phosphorylation of Endogenous α-Synuclein Induced by Overexpression of Polo-like Kinases 2 and 3 in Nigral Dopamine Neurons Is Not Detrimental to Their Survival and Function. Neurobiol Dis, 78, 100–114. [DOI] [PubMed] [Google Scholar]
- Busquets MA, Espargaró A, Estelrich J, & Sabate R (2015) Could α -synuclein amyloid-like aggregates trigger a prionic neuronal invasion? Biomed Res Int. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler EK, Voigt A, Lutz AK, Toegel JP, Gerhardt E, Karsten P, Falkenburger B, Reinartz A, Winklhofer KF & Schulz JB (2012) The mitochondrial chaperone protein TRAP1 mitigates alpha-synuclein toxicity. Plos Genet, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP & Weiner HL (2014) Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci, 17, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacabelos R (2017) Parkinson’s disease: From pathogenesis to pharmacogenomics. Int J Mol Scices, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calì T, Ottolini D, Negro A & Brini M (2012) Α-Synuclein Controls Mitochondrial Calcium Homeostasis By Enhancing Endoplasmic Reticulum-Mitochondria Interactions. J Biol Chem, 287, 17914–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardo LF, Coto E, De Mena L, Ribacoba R, Lorenzo-Betancor O, Pastor P, Samaranch Ll., Mata IF, Díaz M, Moris G, Menéndez M, Corao AI & Alvarez V (2012) A search for SNCA 3′ UTR variants identified SNP rs356165 as a determinant of disease risk and onset age in Parkinson’s disease. J Mol Neurosci, 47, 425–430. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M & Destee A (2004) alpha-Synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet, 364, 1167–1169. [DOI] [PubMed] [Google Scholar]
- Chen L, Xie Z, Turkson S, & Zhuang X (2015) Neurobiol Dis A53T Human alpha-Synuclein Overexpression in Transgenic Mice Induces Pervasive Mitochondria Macroautophagy Defects Preceding Dopamine Neuron Degeneration. J Neurosci, 35, 890–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Mallajosyula JK, Rane A, & Andersen JK (2010) Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci Lett, 486, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HG, Zhang J, Deng X, Hatcher JM, Patricelli MP, Zhao Z, Alessi Dario R. & Gray NS (2012) Brain penetrant LRRK2 inhibitor. Acs Med Chem Lett, 3, 658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civiero L, Vancraenenbroeck R, Belluzzi E, Beilina A, Lobbestael E, Reyniers L, Gao F, Micetic I, de Maeyer M, Bubacco L, Baekelandt V, Cookson MR, Greggio E & Taymans JM (2012) Biochemical Characterization of Highly Purified Leucine-Rich Repeat Kinases 1 and 2 Demonstrates Formation of Homodimers. PLoS ONE, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civiero L, Cogo S, Kiekens A, Morganti C, Tessari I, Lobbestael E, Baekelandt V, Taymans J-M, Chartier-Harlin M-C, Franchin C, Arrigoni G, Lewis PA, Piccoli G, Bubacco L, Cookson MR, Pinton P & Greggio E (2017) PAK6 Phosphorylates 14–3-3γ to Regulate Steady State Phosphorylation of LRRK2. Font Mol Neurosci, 10, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark LN, Wang Y, Karlins E, Saito L, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S, Waters C, Ford B, Frucht S, Ottman R & Marder K (2006) Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology, 67, 1786–1791. [DOI] [PubMed] [Google Scholar]
- Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, & de Bernard M (2013) Triggering of Inflammasome by Aggregated α-Synuclein, an Inflammatory Response in Synucleinopathies. PLoS ONE, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole NB, DiEuliis D, Leo P, Mitchell DC, & Nussbaum RL (2008) Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp Cell Res, 314, 2076–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR (2010) The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci, 11, 791–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, & Dice JF (1996) A Receptor for the Selective Uptake and Degradation of Proteins by Lysosomes. Science, 273 501–503. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF, & Knecht E (1997) A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem, 272, 5606–5615. [DOI] [PubMed] [Google Scholar]
- Cuervo AM (2004) Autophagy: In sickness and in health. Trends Cell Biol, 14, 70–7. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, & Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science, 305, 1292–1295. [DOI] [PubMed] [Google Scholar]
- Da Silveira SA, Schneider BL, Cifuentes-Diaz C, Sage D, Abbas-Terki T, Iwatsubo T, Unser M & Aebischer P (2009) Phosphorylation does not prompt, nor prevent, the formation of α-synuclein toxic species in a rat model of Parkinson’s disease. Hum Mol Genet, 18, 872–887. [DOI] [PubMed] [Google Scholar]
- Daher JPL, Pletnikova O, Biskup S, Musso A, Gellhaar S, Galter D, Troncoso JC, Lee MK, Dawson TM, Dawson VL & Moore DJ (2012) Neurodegenerative phenotypes in an A53T α-synuclein transgenic mouse model are independent of LRRK2. Hum Mol Genet, 21, 2420–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher JPL, Volpicelli-Daley LA, Blackburn JP, Moehle MS, & West AB (2014) Abrogation of alpha-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. P Natl A Sci USA, 111, 9289–9294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher JPL, Abdelmotilib HA, Hu X, Volpicelli-Daley LA, Moehle MS, Fraser KB, Needle E, Chen Y, Steyn SJ, Galatsis P, Hirst WD & West AB (2015) Leucine-rich repeat kinase 2 (LRRK2) pharmacological inhibition abates α-synuclein gene-induced neurodegeneration. J Biol Chem, 290, 19433–19444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniele SG, Béraud D, Davenport C, Cheng K, Yin H, & Maguire-Zeiss KA (2015) Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci Signal, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Kholodilov N, Vila M, Trillat A-C, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Jackson-Lewis V, Hersch S, Sulzer D, Przedborski S, Burke R & Hen R (2002) Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. P Natl A Sci USA, 99, 14524–14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Ko HS, & Dawson VL (2010) Genetic animal models of Parkinson’s disease. Neuron, 66, 646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, & Vila M (2010) Pathogenic Lysosomal Depletion in Parkinson’s Disease. J Neurosci, 30, 12535–12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Dzamko N, Prescott A, Davies P, Liu Q, Yang Q, Lee JD, Patricelli MP, Nomanbhoy TK, Alessi DR & Gray NS (2011) Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat Chem Biol, 7, 203–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Lee H-J, Bae E-J, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E & Lee S-J (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. P Natl A Sci, 106, 13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Raghavendran V, Prabhu BM, Avadhani NG, & Anandatheerthavarada HK (2008) Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem, 283, 9089–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devoto VMP, Dimopoulos N, Alloatti M, Pardi MB, Saez TM, Otero MG, Cromberg LE, Marín-Burgin A, Scassa ME, Stokin GB, Schinder AF, Sevlever G & Falzone TL (2017) αsynuclein control of mitochondrial homeostasis in human-derived neurons is disrupted by mutations associated with Parkinson’s disease. Sci Rep, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, Hu X, McCoy J, Chu CT, Burton EA, Hastings TG & Greenamyre JT (2016) α-synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci Transl Med, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinter E, Saridaki T, Nippold M, Plum S, Diederichs L, Komnig D, Fensky L, May C, Marcus K, Voigt A, Schulz JB & Falkenburger BH (2016) Rab7 induces clearance of α-synuclein aggregates. J Neurochem, 758–774. [DOI] [PubMed] [Google Scholar]
- Dodson MW, Zhang T, Jiang C, Chen S, & Guo M (2012) Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum Mol Genet, 21, 1350–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson MW, Leung LK, Lone M, Lizzio MA, & Guo M (2014) Novel ethyl methanesulfonate (EMS)-induced null alleles of the Drosophila homolog of LRRK2 reveal a crucial role in endolysosomal functions and autophagy in vivo. Dis Mol Mech, 7, 1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domert J, Sackmann C, Agholme L, Bergstr�m J, Ingelsson M, Hallbeck M, & Severinsson E (2016) Aggregated alpha-synuclein transfer efficiently between cultured human neuron- like cells and localize to lysosomes. PLoS ONE, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy MF, Collier TJ, Patterson JR, Kemp CJ, Luk KC, Tansey MG, Paumier KL, Kanaan NM, Fischer LD, Polinski NK, Barth OL, Howe JW, Vaikath NN, Majbour NK, El-Agnaf OMA & Sortwell CE (2018) Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J Neuroinflamm, 15, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamko N, Deak M, Hentati F, Reith AD, Prescott AR, Alessi DR, & Nichols RJ (2010) Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser 910 /Ser 935 , disruption of 14–3-3 binding and altered cytoplasmic localization. Biochem J, 430, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis CE, Murphy EJ, Mitchell DC, Golovko MY, Scaglia F, Gwendolyn C, Nussbaum RL & Barcelo GC (2005) Mitochondrial Lipid Abnormality and Electron Transport Chain Impairment in Mice Lacking α -Synuclein Mitochondrial Lipid Abnormality and Electron Transport Chain Impairment in Mice Lacking alpha-Synuclein. Mol Cell Biol, 25, 10190–10201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinter E, Saridaki T, Nippold M, Plum S, Diederichs L, Komnig D, Fensky L, May C, Marcus K, Voigt A, Schulz JB & Falkenburger BH (2017) Overexpression of alpha-synuclein in an astrocyte cell line promotes autophagy inhibition and apoptosis. J Neurosci Research, 96,160–171. [DOI] [PubMed] [Google Scholar]
- Engelender S & Isacson O (2017) The Threshold Theory for Parkinson’s Disease. Trends Neurosci, 40. [DOI] [PubMed] [Google Scholar]
- Estrada AA, Chan BK, Baker-Glenn C, Beresford A, Burdick DJ, Chambers M, Chen H, Dominguez SL, Dotson J, Drummond J, Flagella M, Fuji R, Gill A, Halladay J, Harris SF, Heffron TP, Kleinheinz T, Lee DW, Pichon CEL, Liu X, Lyssikatos JP, Medhurst AD, Moffat JG, Nash K, Scearce-Levie K, Sheng Z, Shore DG, Wong S, Zhang S, Zhang X, Zhu H & Sweeney ZK (2014) Discovery of highly potent, selective, and brain-penetrant aminopyrazole Leucine-rich repeat kinase 2 (LRRK2) small molecule inhibitors. J Med Chemy, 57, 921–936. [DOI] [PubMed] [Google Scholar]
- Fred Dice J (1990) Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci, 15, 305–309. [DOI] [PubMed] [Google Scholar]
- Friedman LG, Lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M, Holstein GR &Yue Z (2012) Disrupted Autophagy Leads to Dopaminergic Axon and Dendrite Degeneration and Promotes Presynaptic Accumulation of -Synuclein and LRRK2 in the Brain. J Neurosci, 32, 7585–7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, & Obata F (2002) A new locus for Parkinson’s Disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol, 51, 296–301. [DOI] [PubMed] [Google Scholar]
- Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD et al. (2010) LRRK2 Is Involved in the IFN-γ Response and Host Response to Pathogens. J Immunol, 185, 5577–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelders G, Baekelandt V & Van Der Perren A (2018) Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J Immunol Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, & Lee VMY (2002) Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron, 34, 521–533. [DOI] [PubMed] [Google Scholar]
- Gibb WRG, Scott T, & Lees AJ (1991) Neuronal inclusions of Parkinson’s disease. Movement Disord, 6, 2–11. [DOI] [PubMed] [Google Scholar]
- Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP, Lynch J, Healy DG, Holton JL, Revesz T & Wood NW (2005) A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet, 365, 415–416. [DOI] [PubMed] [Google Scholar]
- Gillardon F, Schmid R, & Draheim H (2012) Parkinson’s disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience, 208, 41–48. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, Boldt K, Von Zweydorf F, Helm S, Wiesent L, Sarioglu H, & Ueffing M (2010) Phosphopeptide analysis reveals two discrete clusters of phosphorylation in the N-terminus and the Roc domain of the Parkinson-disease associated protein kinase LRRK2. J Proteome Res, 9, 1738–1745. [DOI] [PubMed] [Google Scholar]
- Goldwurm S, Zini M, Di Fonzo A, De Gaspari D, Siri C, Simons EJ, van Doeselaar M, Tesei S, Antonini A, Canesi M, Zecchinelli A, Mariani C, Meucci N, Sacilotto G, Cilia R, Isaias IU, Bonetti A, Sironi F, Ricca S, Oostra BA, Bonifati V & Pezzoli G (2006) LRRK2 G2019S mutation and Parkinson’s disease: A clinical, neuropsychological and neuropsychiatric study in a large Italian sample. Parkinsonism Relat Dis, 12, 410–419. [DOI] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP, Mandel RJ & Muzyczka N (2008) The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. P Natl A Sci USA, 105, 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Nash K, Gorbatyuk M, Lewin AS, Sullivan LF, Mandel RJ, Chen W, Meyers C, Manfredsson FP & Muzyczka N (2010) In Vivo RNAi-Mediated α-Synuclein Silencing Induces Nigrostriatal Degeneration. Mol Ther, 18, 1450–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi D, Howard S, Zhou M, Diaz-Perez N, Urban NT, Guerrero-Given D, Kamasawa N, Volpicelli-Daley LA, LoGrasso P & Lasmézas CI (2018) Identification of a highly neurotoxic α-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease. P Natl A Sci [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K & Cookson MR (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis, 23, 329–341. [DOI] [PubMed] [Google Scholar]
- Guardia-Laguarta C, Area-Gomez E, Rüb C, Liu Y, Magrané J, Becker D, Voos W, Schon E & Przedborski S (2014) α-Synuclein Is Localized to Mitochondria-Associated ER Membranes. J Neurosci, 34, 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustot A, Gallea JI, Sarroukh R, Celej MS, Ruysschaert J-M, & Raussens V (2015). Amyloid fibrils are the molecular trigger of inflammation in Parkinson’s disease. Biochem J, 471, 323–333. [DOI] [PubMed] [Google Scholar]
- Hakimi M, Selvanantham T, Swinton E, Padmore RF, Tong Y, Kabbach G, Venderova K, Girardin SE, Bulman DE, Scherzer CR, Lavoie MJ, Gris D, Park DS, Angel JB, Shen J, Philpott DJ & Schlossmacher MG (2011) Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm, 118, 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Stoessl AJ, Yokoyama T, Kowa H, Wszolek ZK, & Yagishita S (2009) Familial parkinsonism: Study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat Dis, 15, 300–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, Ferreira JJ, Tolosa E, Kay DM, Klein C, Williams DR, Marras C, Lang AE, Wszolek ZK, Berciano J, Schapira AH, Lynch T, Bhatia KP, Gasser T, Lees AJ & Wood NW (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol, 7, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig MC, Bidinosti M, Schweizer T, Hafner T, Stemmelen C, Weiss A, Danner S, Vidotto N et al. (2012) High LRRK2 levels fail to induce or exacerbate neuronal alpha-synucleinopathy in mouse brain. PloS One, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi S, Moore DJ, Yamamoto R, Minegishi M, Sato K, Togo T, Katsuse O, Uchikado H et al. (2009) Abnormal localization of leucine-rich repeat kinase 2 to the endosomal-lysosomal compartment in lewy body disease. J Neuropath Exp Neur, 68, 994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Halliday GM, Vandebona H, Mellick GD, Mastaglia F, Stevens J, Kwok J, Garlepp M, Silburn PA, Horne MK, Kotschet K, Venn A, Rowe DB, Rubio JP & Sue CM (2007) Prevalence and clinical features of common LRRK2 mutations in Australians with Parkinson’s disease. Movement Disord, 22, 982–989. [DOI] [PubMed] [Google Scholar]
- Hulihan MM, Ishihara-Paul L, Kachergus J, Warren L, Amouri R, Elango R, Prinjha RK, Upmanyu R, Kefi M, Zouari M, Sassi SB, Yahmed SB, El Euch-Fayeche G, Matthews PM, Middleton LT, Gibson RA, Hentati F & Farrer MJ (2008) LRRK2 Gly2019Ser penetrance in Arab-Berber patients from Tunisia: a case-control genetic study. Lancet Neurol, 7, 591–594. [DOI] [PubMed] [Google Scholar]
- Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debré P, Agid Y, Dugas B & Hirsch EC (1999) FcepsilonRII/CD23 is expressed in Parkinson’s disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci, 19, 3440–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, & Hashizume Y (2003) Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol, 106, 518–526. [DOI] [PubMed] [Google Scholar]
- Inglis KJ, Chereau D, Brigham EF, Chiou SS, Sch??bel S, Frigon NL, Yu M, Caccavello RJ, Nelson S, Motter R, Wright S, Chian D, Santiago P, Soriano F, Ramos C, Powell K, Goldstein JM, Babcock M, Yednock T, Bard F, Basi GS, Sham H, Chilcote TJ, McConlogue L, Griswold-Prenner I & Anderson JP (2009) Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J Biol Chem, 284, 2598–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Nonaka T, Taniguchi S, Saito T, Arai T, Mann D, Iwatsubo T, Hisanaga S-I, Goedert M & Hasegawa M (2007) Casein kinase 2 is the major enzyme in brain that phosphorylates Ser129 of human alpha-synuclein: Implication for alpha-synucleinopathies. FEBS Lett, 581, 4711–4717. [DOI] [PubMed] [Google Scholar]
- Jager S (2004) Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci, 117, 4837–4848. [DOI] [PubMed] [Google Scholar]
- Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, Gibson JM, Ross OA, Lynch T, Wiley J, Payami H, Nutt J, Maraganore DM, Czyzewski K, Styczynska M, Wszolek ZK, Farrer MJ & Toft M (2005) Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet, 76, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia LV, Lang AE, Hazrati LN, Fujioka S, Wszolek ZK, Dickson DW, Ross OA, Van Deerlin VM, Trojanowski JQ, Hurtig HI, Alcalay RN, Marder KS, Clark LN, Gaig C, Tolosa E, Ruiz-Martínez J, Marti-Masso JF, Ferrer I, López De Munain A, Goldman SM, Schüle B, Langston JW, Aasly JO, Giordana MT, Bonifati V, Puschmann A, Canesi M, Pezzoli G, Maues De Paula A, Hasegawa K, Duyckaerts C, Brice A, Stoessl AJ & Marras C (2015) Clinical correlations with lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol, 72, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamikawaji S, Ito G, & Iwatsubo T (2009) Identification of the autophosphorylation sites of LRRK2. Biochemistry, 48, 10963–10975. [DOI] [PubMed] [Google Scholar]
- Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K, Eimer S, Winklhofer KF & Haass C (2010) Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J, 29, 3571–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppagounder SS, Xiong Y, Lee Y, Lawless MC, Kim D, Nordquist E, Martin I, Ge P Brahmachari S, Jhaldiyal A, Kumar M, Andrabi SA, Dawson TM & Dawson VL (2015) LRRK2 G2019S transgenic mice display increased susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-mediated neurotoxicity. J Chem Neuroanat, 2, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto Y, Akiguchi I, Nakamura S, Honjyo Y, Shibasaki H, & Budka H (2002) 14–3-3 Proteins in Lewy bodies in Parkinson disease and diffuse Lewy body disease brains. J Neuropath Exp Neur, 61, 245–253. [DOI] [PubMed] [Google Scholar]
- Khan NL, Jain S, Lynch JM, Pavese N, Abou-Sleiman P, Holton JL, Healy DG, Gilks WP, Sweeney MG, Ganguly M, Gibbons V, Gandhi S, Vaughan J, Eunson LH, Katzenschlager R, Gayton J, Lennox G, Revesz T, Nicholl D, Bhatia KP, Quinn N, Brooks D, Lees AJ, Davis MB, Piccini P, Singleton AB & Wood NW (2005) Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: Clinical, pathological, olfactory and functional imaging and genetic data. Brain, 128, 2786–2796. [DOI] [PubMed] [Google Scholar]
- Khodr CE, Pedapati J, Han Y, & Bohn MC (2012) Inclusion of a portion of the native SNCA 3′UTR reduces toxicity of human S129A SNCA on striatal-projecting dopamine neurons in rat substantia Nigra. Dev Neurobiol, 72, 906–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Yang MS, Choi D, Kim JH, Kim HS, Seol W, Choi S, Jou I, Kim EY & Joe EH (2012) Impaired inflammatory responses in murine lrrk2-knockdown brain microglia. PLoS ONE, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Lee Sung Joong, Masliah E, Hwang D, Lee HJ & Lee SJ (2013) Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klivenyi P, Siwek D, Gardian G, Yang L, Starkov A, Cleren C, Ferrante RJ, Kowall NW Abeliovich A & Beal MF (2006) Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol Dis, 21, 541–548. [DOI] [PubMed] [Google Scholar]
- Kondo K, Obitsu S, & Teshima R (2011) α-Synuclein aggregation and transmission are enhanced by leucine-rich repeat kinase 2 in human neuroblastoma SH-SY5Y cells. Biol Pharm Bull, 34, 1078–1083. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, & Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med, 14, 504–506. [DOI] [PubMed] [Google Scholar]
- Kozina E, Shankar S, Yun J, Yuchen D, Zhijun M, Haiyan T, Kiran K, Timothy S, Junmin P & Richard JS (2018) Mutant LRRK2 Mediates Peripheral and Central Immune Responses Leading to Neurodegeneration in Vivo. Brain 141, 1753–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schols L & Riess O (1998) AlaSOPro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet, 18, 106–108. [DOI] [PubMed] [Google Scholar]
- Langston JW, & Ballard PA (1983) Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. New Engl J Med, 309, 310. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Ulusoy A, Innamorato NG, Sahin G, Rábano A, Kirik D & Cuadrado A (2012) α-synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum Mol Genet, 21, 3173–3192. [DOI] [PubMed] [Google Scholar]
- Lavalley NJ, Slone SR, Ding H, West AB, & Yacoubian TA (2016) 14–3-3 Proteins regulate mutant LRRK2 kinase activity and neurite shortening. Hum Mol Genet, 25, 109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA & Price DL (2002) Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 Thr mutation causes neurodegenerative disease with -synuclein aggregation in transgenic mice. P Natl A Sci, 99, 8968–8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-J, Khoshaghideh F, Patel S, & Lee S-J (2004) Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci, 24, 1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BD, Shin JH, Vankampen J, Petrucelli L, West AB, Ko HS, Lee YI, Maguire-Zeiss KA, Bowers WJ, Federoff HJ, Dawson VL & Dawson TM (2010) Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat Med, 16, 998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-W, & Cannon JR (2015) LRRK2 mutations and neurotoxicant susceptibility. Exp Biol Med, 240, 752–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, & Klionsky DJ (2004) Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev Cell, 6. [DOI] [PubMed] [Google Scholar]
- Li W-W, Yang R, Guo J-C, Ren H-M, Zha X-L, Cheng J-S, & Cai D-F (2007) Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport, 18, 1543–1546. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM & Przedborski S (1999) Inducible Nitric Oxide Synthase Stimulates Dopaminergic Neurodegeneration in the MPTP Model of Parkinson Disease. Nat Med, 5, 1403–9. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B & Barres BA (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Tzen KY, Yu CY, Tai CH, Farrer MJ, & Wu RM (2008) LRRK2 mutation in familial Parkinson’s disease in a Taiwanese population: Clinical, PET, and functional studies. J Biomed Sci, 15, 661–667. [DOI] [PubMed] [Google Scholar]
- Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z & Cai H (2009) Leucine-Rich Repeat Kinase 2 Regulates the Progression of Neuropathology Induced by Parkinson’s-Disease-Related Mutant α-synuclein. Neuron, 64, 807–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström V, Gustafsson G, Sanders LH, Howlett EH, Sigvardson J, Kasrayan A, Ingelsson M, Bergström J & Erlandsson A (2017) Extensive uptake of α-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol Cell Neurosci, 82, 143–156. [DOI] [PubMed] [Google Scholar]
- Liu G, Zhang C, Yin J, Li X, Cheng F, Li Y, Yang H, Uéda K Chan P & Yu S (2009) α-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci Lett, 454, 187–192. [DOI] [PubMed] [Google Scholar]
- Liu Z, Hamamichi S, Lee BD, Yang D, Ray A, Caldwell GA, Caldwell KA, Dawson TM, Smith WW & Dawson VL (2011) Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Paarkinson’s disease models. Hum Mol Genet, 20, 3933–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb V, Yakunin E, Saada A, & Sharon R (2010) The transgenic overexpression of alph-synuclein and not its related pathology associates with complex I inhibition. J Biol Chem, 285, 7334–7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler DA, Klaver AC, Coffey MP, Aasly JO, & LeWitt PA (2017) Increased Oxidative Stress Markers in Cerebrospinal Fluid from Healthy Subjects with Parkinson’s Disease-Associated LRRK2 Gene Mutations. Front Aging Neurosci, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo F, Mercatelli D, Novello S, Arcuri L, Brugnoli A, Vincenzi F, Russo I, Berti G, Mabrouk OS, Kennedy RT, Shimshek DR, Varani K, Bubacco L, Greggio E & Morari M (2017) Age-dependent dopamine transporter dysfunction and Serine129 phospho-α-synuclein overload in G2019S LRRK2 mice. Acta Neuropathol Communications, 5, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, & Lee VMY (2012) Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science, 338, 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Xu L, Pan X, Sun L, Ding J, Xie C, Koliatsos VE & Cai H (2015) LRRK2 modulates microglial activity through regulation of chemokine (C-X3-C) receptor 1 -mediated signalling pathways. Hum Mol Genet, 25, 3515–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maatouk L, Yi C, Carrillo-de Sauvage M-A, Compagnion A-C, Hunot S, Ezan P, Hirsch EC, Koulakoff A, Pfrieger FW, Tronche F, Leybaert L, Giaume C, Vyas S (2018) Glucocorticoid receptor in astrocytes regulates midbrain dopamine neurodegeneration through connexin hemichannel activity. Cell Death Differ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa T, Sasaoka T, Azuma S, Ichikawa T, Melrose HL, Farrer MJ, & Obata F (2016) Leucine-rich repeat kinase 2 (LRRK2) regulates α-synuclein clearance in microglia. BMC Neurosci, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeski AE, & Fred Dice J (2004) Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol, 36, 2435–44. [DOI] [PubMed] [Google Scholar]
- Mamais A, Chia R, Beilina A, Hauser DN, Hall C, Lewis PA, Cookson MR & Bandopadhyay R (2014) Arsenite stress down-regulates phosphorylation and 14–3-3 binding of leucine-rich repeat kinase 2 (LRRK2), promoting self-association and cellular redistribution. J Biol Chem, 289, 21386–21400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredsson FP, Luk KC, Benskey MJ, Gezer A, Garcia J, Kuhn NC, Sandoval IM, Patterson JR, O’Mara A, Yonkers R & Kordower JH (2018) Induction of Alpha-Synuclein Pathology in the Enteric Nervous System of the Rat and Non-Human Primate Results in Gastrointestinal Dysmotility and Transient CNS Pathology. Neurobiol Dis, 112, 106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni C, Mamais A, Roosen DA, Dihanich S, Soutar MPM, Plun-Favreau H, Bandopadhyay R, Hardy J et al. (2016) MTOR independent regulation of macroautophagy by Leucine Rich Repeat Kinase 2 via Beclin-1. Sci Rep, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinek P, Jha AN, Shinde V, Sundaramoorthy A, Rajkumar R, Suryadevara NC, Neela SK, van Tong H, Balachander V, Valluri VL, Thangaraj K & Velavan TP, (2013) LRRK2 and RIPK2 Variants in the NOD 2-Mediated Signaling Pathway Are Associated with Susceptibility to Mycobacterium leprae in Indian Populations. PLoS ONE, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL & Lee MK (2006) Parkinson’s Disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci, 26, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez JH, Alaimo A, Gorojod RM, Porte Alcon S, Fuentes F, Coluccio Leskow F, & Kotler ML (2018) Drp-1 dependent mitochondrial fragmentation and protective autophagy in dopaminergic SH-SY5Y cells overexpressing alpha-synuclein. Mol Cell Neurosci, 88, 107–117. [DOI] [PubMed] [Google Scholar]
- Massey A, Kiffin R, & Cuervo AM (2004) Pathophysiology of chaperone-mediated autophagy. Int J Biochem Cell B. [DOI] [PubMed] [Google Scholar]
- Massey AC, Kaushik S, Sovak G, Kiffin R, & Cuervo AM (2006). Consequences of the selective blockage of chaperone-mediated autophagy. P Natl A Sci USA, 103, 5805–5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, & Gallo KA (2006) LRRK2 in Parkinson’s disease: protein domains and functional insights. Trends Neurosci, 29, 286–93. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, & McGeer EG (1988) Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol, 76, 550–557. [DOI] [PubMed] [Google Scholar]
- McNaught KSP, Mytilin C, JnoBaptiste R, Yabut J, Shashidharan P, Jenner P, & Olanow CW (2002) Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J Neurochem, 81, 301–306. [DOI] [PubMed] [Google Scholar]
- Melo TQ, van Zomeren KC, Ferrari MFR, Boddeke HWGM, & Copray JCVM (2016) Impairment of mitochondria dynamics by human A53T α-synuclein and rescue by NAP (davunetide) in a cell model for Parkinson’s disease. Exp Brain Res, 235, 731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T & Kagawa Y (1989) Deficiencies in Complex I subunits of the respiratory chain in Parkinson’s disease. Biochem Bioph Res Co, 163, 1450–1455. [DOI] [PubMed] [Google Scholar]
- Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, Cowell RM & West AB (2012) LRRK2 Inhibition Attenuates Microglial Inflammatory Responses. J Neurosci, 32, 1602–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, & Nagatsu T (1994) Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett, 165, 208–210. [DOI] [PubMed] [Google Scholar]
- Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, & Nagatsu T (1996) Interleukin (IL)-1β, IL-2, IL-4, IL-6 and transforming growth factor-α levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci Lett, 211, 13–16. [DOI] [PubMed] [Google Scholar]
- Mortiboys H, Johansen KK, Aasly JO, & Bandmann O (2010) Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology, 75, 2017–2020. [DOI] [PubMed] [Google Scholar]
- Mortiboys H, Furmston R, Bronstad G, Aasly J, Elliott C, & Bandmann O (2015) UDCA exerts beneficial effect on mitochondrial dysfunction in LRRK2 G2019S carriers and in vivo. Neurology, 85, 846–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougenot AL, Nicot S, Bencsik A, Morignat E, Verchère J, Lakhdar L, Legastelois S & Baron T (2012). Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging, 33, 2225–2228. [DOI] [PubMed] [Google Scholar]
- Müller O, Sattler T, Flötenmeyer M, Schwarz H, Plattner H, & Mayer A (2000) Autophagic tubes: Vacuolar invaginations involved in lateral membrane sorting and inverse vesicle budding. J Cell Biol, 151, 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Nemani VM, Wallender EK, Kaehlcke K, Ott M, & Edwards RH (2008) Optical reporters for the conformation of alpha-synuclein reveal a specific interaction with mitochondria. J Neurosci, 28, 12305–12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J, Sesaki H, Cheng Y, Finkbeiner S, Nussbaum RL, Masliah E & Edwards RH (2011) Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein α-synuclein. J Biol Chem, 286, 20710–20726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma J, Schulte J, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, Ikram MA, Ioannidis JPA, Hadjigeorgiou GM, Joshua C, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L, Eriksson N, Foroud T & Singleton AB (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet, 46, 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng C-H, Mok SZS, Koh C, Ouyang X, Fivaz ML, Tan E-K, Dawson VL, Dawson TM, Yu F & Lim K-L (2009) Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci, 29, 11257–11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols WC, Pankratz N, Hernandez D, Paisán-Ruíz C, Jain S, Halter CA, Michaels VE, Reed T, Rudolph A, Shults CW, Singleton A & Foroud T (2005) Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet, 365, 410–412. [DOI] [PubMed] [Google Scholar]
- Nichols RJ, Dzamko N, Morrice NA, Campbell DG, Deak M, Ordureau A, Macartney T, Tong Y, Shen J, Prescott AR & Alessi DR (2010) 14–3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem J, 430, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J, Yu M, Wang C, & Xu Z (2012) Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via dynamin-like protein. J Neurochem, 122, 650–658. [DOI] [PubMed] [Google Scholar]
- Oosterveld LP, Allen JC, Ng EYL, Seah SH, Tay KY, Au WL, Tan EK & Tan LCS (2015) Greater motor progression in patients with Parkinson disease who carry LRRK2 risk variants. Neurology, 85, 1039–1042. [DOI] [PubMed] [Google Scholar]
- Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D & Cuervo AM (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci, 16, 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueslati A, Schneider BL, Aebischer P & Lashuel HA (2013) Polo-like Kinase 2 Regulates Selective Autophagic alpha-Synuclein Clearance and Suppresses Its Toxicity in Vivo. P Natl A Sci USA, 110, E3945–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, Van Der Brug M, De Munain AL, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Peňa AS, De Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW & Singleton AB (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron, 44, 595–600. [DOI] [PubMed] [Google Scholar]
- Papkovskaia TD, Chau KY, Inesta-vaquera F, Papkovsky DB, Healy DG, Nishio K, Staddon J, Duchen MR, Hardy J, Schapira AHV & Cooper JM (2012) G2019s leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum Mol Genet, 21, 4201–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar MS, Parihar A, Fujita M, Hashimoto M, & Ghafourifar P (2008) Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci, 65, 1272–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, Steece-Collier K, Kemp CJ, Celano S, Schulz E, Sandoval IM, Fleming S, Dirr E, Polinski NK, Trojanowski JQ, Lee VM & Sortwell CE (2015) Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis, 82, 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, Trojanowski JQ, Lee VM & Ischiropoulos H (2001) Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci, 21, 8053–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peelaerts W, Bousset L, Van Der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van Den Haute C, Melki R & Baekelandt V (2015) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature, 522, 340–344. [DOI] [PubMed] [Google Scholar]
- Petroi D, Popova B, Taheri-Talesh N, Irniger S, Shahpasandzadeh H, Zweckstetters M, Outeiro TF & Braus GH (2012) Aggregate clearance of alpha-synuclein in Saccharomyces cerevisiae depends more on autophagosome and vacuole function than on the proteasome. J Biol Chem, 287, 27567–27579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI & Nussbaum RL (1997) Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science, 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Post MR, Lieberman OJ, & Mosharov EV (2018) Can interactions between α-synuclein, dopamine and calcium explain selective neurodegeneration in Parkinson’s disease? Font Neurosci, 12, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronin a N., Morris a J., Surguchov a, & Benovic JL (2000) Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem, 275, 26515–26522. [DOI] [PubMed] [Google Scholar]
- Puccini JM, Marker DF, Fitzgerald T, Barbieri J, Kim CS, Miller-Rhodes P, Lu SM, Dewhurst S& Gelbard HA (2015) Leucine-Rich Repeat Kinase 2 Modulates Neuroinflammation and Neurotoxicity in Models of Human Immunodeficiency Virus 1-Associated Neurocognitive Disorders. J Neurosci, 35, 5271–5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing H, Wong W, McGeer EG, & McGeer PL (2009) Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem Bioph Res Co, 387, 149–152. [DOI] [PubMed] [Google Scholar]
- Ramonet D, Daher JPL, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L, Liu Y, Swing DA, Beal MF, Troncoso JC, McCaffery JM, Jenkins NA, Copeland NG, Galter D, Thomas B, Lee MK, Dawson TM, Dawson VL & Moore DJ (2011) Dopaminergic Neuronal loss, Reduced Neurite Complexity and Autophagic Abnormalities in Transgenic Mice Expressing G2019S Mutant LRRK2. PLoS ONE, 6, 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recasens A, & Dehay B (2014) Alpha-synuclein spreading in Parkinson’s disease. Front Neuroanat, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve AK, Ludtmann MHR, Angelova PR, Simcox EM, Horrocks MH, Klenerman D, Gandhi S, Turnbull DM & Abramov AY (2015) Aggregated α-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons. Cell Death Dis, 6, e1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout HJ, Larsen KE, Sulzer D, & Stefanis L (2001) Proteasomal inhibition leads to formation of ubiquitin/alpha-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem, 78, 899–908. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, & Stefanis L (2002) Proteasomal inhibition-induced inclusion formation and death in cortical neurons require transcription and ubiquitination. Mol Cell Neurosci, 21, 223–238. [DOI] [PubMed] [Google Scholar]
- Ross OA, Soto-Ortolaza AI, Heckman MG, Aasly JO, Abahuni N, Annesi G, Bacon JA, Bardien S, Bozi M, Brice A, Brighina L, Van Broeckhoven C, Carr J, Chartier-Harlin MC, Dardiotis E, Dickson DW, Diehl NN, Elbaz A, Ferrarese C, Ferraris A, Fiske B, Gibson JM, Gibson R, Hadjigeorgiou GM, Hattori N, Ioannidis JPA, Jasinska-Myga B, Jeon BS, Kim YJ, Klein C, Kruger R, Kyratzi E, Lesage S, Lin CH, Lynch T, Maraganore DM, Mellick GD, Mutez E, Nilsson C, Opala G, Park SS, Puschmann A, Quattrone A, Sharma M, Silburn PA, Sohn YH, Stefanis L, Tadic V, Theuns J, Tomiyama H, Uitti RJ, Valente EM, van de Loo S, Vassilatis DK, Vilariño-Güell C, White LR, Wirdefeldt K, Wszolek ZK, Wu RM & Farrer MJ (2011) Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: A case-control study. Lancet Neurol, 10, 898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudenko IN, & Cookson MR (2010) 14–3-3 proteins are promising LRRK2 interactors. Biochem J, 430, e5–e6. [DOI] [PubMed] [Google Scholar]
- Russo I, Berti G, Plotegher N, Bernardo G, Filograna R, Bubacco L, & Greggio E (2015) Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF- κB p50 signaling in cultured microglia cells. J Neuroinflamm, 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan T, Bamm VV, Stykel MG, Coackley CL, Humphries KM, Jamieson-Williams R, Ambasudhan R, Mosser DD, Lipton SA, Harauz G & Ryan SD (2018) Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat Commun, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez-Atienzar S, Bonet-Ponce L, Blesa JR, Romero FJ, Murphy MP, Jordan J, & Galindo MF (2014) The LRRK2 inhibitor GSK2578215A induces protective autophagy in SH-SY5Y cells: involvement of Drp-1-mediated mitochondrial fission and mitochondrial-derived ROS signaling. Cell Death Dis, 5, e1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Guillily MD, Ferree A, Lanceta J, Chan D, Ghosh J, Hsu CH, Segal L, Raghavan K, Matsumoto K, Hisamoto N, Kuwahara T, Iwatsubo T, Moore L, Goldstein L, Cookson M & Wolozin B (2009) LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J Neurosci, 29, 9210–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Ash P. E. a., Gowda V, Liu L, Shirihai O, & Wolozin B (2015) Mutations in LRRK2 potentiate age-related impairment of autophagic flux. Mol Neurodegener, 10, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Kawashima A, Ruberu NN, Fujiwara H, Koyama S, Sawabe M, Arai T, Nagura H, Yamanouchi H, Hasegawa M, Iwatsubo T & Murayama S (2003) Accumulation of phosphorylated alpha-synuclein in aging human brain. J Neuropath Exp Neur, 62, 644–654. [DOI] [PubMed] [Google Scholar]
- Sarafian TA, Ryan CM, Souda P, Masliah E, Kar UK, Vinters HV, Mathern GW, Faull KF, Whitelegge JP & Watson JB (2013) Impairment of Mitochondria in Adult Mouse Brain Overexpressing Predominantly Full-Length, N-Terminally Acetylated Human alpha-Synuclein. PLoS ONE, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y & Toda T (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet, 41, 1303–1307. [DOI] [PubMed] [Google Scholar]
- Sato H, Arawaka S, Hara S, Fukushima S, Koga K, Koyama S & Kato T (2011) Authentically Phosphorylated alpha-Synuclein at Ser129 Accelerates Neurodegeneration in a Rat Model of Familial Parkinson’s Disease. J Neurosci, 31, 16884–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapansky J, Nardozzi JD, Felizia F, & LaVoie MJ (2014) Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet, 23, 4201–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapansky J, Khasnavis S, DeAndrade MP, Nardozzi JD, Falkson SR, Boyd JD, Sanderson JB, Bartels T, Melrose HL & LaVoie MJ (2018) Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble α-synuclein in neurons. Neurobiol Dis, 111, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, & Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem, 54, 823–827. [DOI] [PubMed] [Google Scholar]
- Schapira AH (2008). Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol, 7, 97–109. [DOI] [PubMed] [Google Scholar]
- Sheng Z, Zhang S, Bustos D, Kleinheinz T, Le Pichon CE, Dominguez SL, Solanoy HO, Drummond J, Zhang X, Ding X, Cai F, Song Q, Li X, Yue Z, van der Brug MP, Burdick DJ, Gunzner-Toste J, Chen H, Liu X, Estrada AA, Sweeney ZK, Scearce-Levie K, Moffat JG, Kirkpatrick DS & Zhu H (2012) Ser1292 Autophosphorylation Is an Indicator of LRRK2 Kinase Activity and Contributes to the Cellular Effects of PD Mutations. Sci Transl Med, 4, 164ra161-164ra161. [DOI] [PubMed] [Google Scholar]
- Shintani T, & Klionsky DJ (2004) Autophagy in health and disease: A double-edged sword. Science, 306, 990–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez KH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, Van Der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA & Gasser T (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet, 41, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J & Gwinn-Hardy K (2003) α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science, 302, 841. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Facci L, Zusso M, & Giusti P (2018) An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front Cell Neurosci, 12, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G, Nakamura K, Cookson MR, & Finkbeiner S (2014) Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. J Neurosci, 34, 418–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slone SR, Lavalley N, McFerrin M, Wang B, & Yacoubian TA (2015) Increased 14–3-3 phosphorylation observed in Parkinson’s disease reduces neuroprotective potential of 14–3-3 proteins. Neurobiol Dis, 79, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, & Ross CA (2006) Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci, 9, 1231–1233. [DOI] [PubMed] [Google Scholar]
- Song DD, Shults CW, Sisk A, Rockenstein E, & Masliah E (2004) Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol, 186, 158–172. [DOI] [PubMed] [Google Scholar]
- Stichel CC, Zhu XR, Bader V, Linnartz B, Schmidt S, & Lübbert H (2007) Mono- and double-mutant mouse models of Parkinson’s disease display severe mitochondrial damage. Hum Mol Genet, 16, 2377–2393. [DOI] [PubMed] [Google Scholar]
- Su YC, & Qi X (2013) Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum Mol Genet, 22, 4545–4561. [DOI] [PubMed] [Google Scholar]
- Subramaniam SR, Vergnes L, Franich NR, Reue K, & Chesselet MF (2014) Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol Dis, 70, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Wang T, Jiang T, Huang P, Li D, Wang Y, Xiao Q, Liu J & Chen S (2016) Effect of a Leucine-rich Repeat Kinase 2 Variant on Motor and Non-motor Symptoms in Chinese Parkinson’s Disease Patients. Aging Dis, 7, 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Obeso JA, & Halliday GM (2017a) Parkinson’s Disease Is Not Simply a Prion Disorder. J Neurosci, 37, 9799–9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Obeso JA, & Halliday GM (2017b) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci, 18, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan EK, Shen H, Tan LCS, Farrer M, Yew K, Chua E, Jamora RD, Puvan K, Puong KY, Zhao Y, Pavanni R, Wong MC, Yih Y, Skipper L & Liu JJ (2005) The G2019S LRRK2 mutation is uncommon in an Asian cohort of Parkinson’s disease patients. Neurosci Lett, 384, 327–329. [DOI] [PubMed] [Google Scholar]
- Tan EK, Zhao Y, Tan L, Lim HQ, Lee J, Yuen Y, Pavanni R, Wong MC, Fook-Chong S & Liu JJ (2007) Analysis of LRRK2 Gly2385Arg genetic variant in non-Chinese Asians. Movement Disord, 22, 1816–1818. [DOI] [PubMed] [Google Scholar]
- Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR, & Lee VMY (2013) Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem, 288, 15194–15210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasova TV, Lytkina OA, Goloborshcheva VV, Skuratovskaya LN, Antohin AI, Ovchinnikov RK & Kukharsky MS (2018) Genetic Inactivation of Alpha-Synuclein Affects Embryonic Development of Dopaminergic Neurons of the Substantia Nigra, but Not the Ventral Tegmental Area, in Mouse Brain. PeerJ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taymans J-M, & Greggio E (2016) LRRK2 Kinase Inhibition as a Therapeutic Strategy for Parkinson’s Disease, Where Do We Stand? Current Neuropharmacology, 14, 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenet J, Gobert R, van Huijsduijnen RH, Wiessner C, & Sagot YJ (2011) Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS ONE, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Layfield R, & Spillantini MG (2001) α-Synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett, 509, 22–26. [DOI] [PubMed] [Google Scholar]
- Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ, & Shen J (2010) Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of -synuclein, and apoptotic cell death in aged mice. P Natl A Sci, 107, 9879–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabzuni D, Ryten M, Emmett W, Ramasamy A, Lackner KJ, Zeller T, Walker R, Smith C, Lewis PA, Mamais A, de Silva R, Vandrovcova J, Hernandez D, Nalls MA, Sharma M, Garnier S, Lesage S, Simon-Sanchez J, Gasser T, Heutink P, Brice A, Singleton A, Cai H, Schadt E, Wood NW, Bandopadhyay R, Weale ME, Hardy J & Plagnol V (2013) Fine-Mapping, Gene Expression and Splicing Analysis of the Disease Associated LRRK2 Locus. PLoS ONE, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh J, Amouri R, Duda JE, Morley JF, Read M, Donald A, Vilariño-Güell C, Thompson C, Szu Tu C, Gustavsson EK, Ben Sassi S, Hentati E, Zouari M, Farhat E, Nabli F, Hentati F & Farrer MJ (2014) A comparative study of Parkinson’s disease and leucine-rich repeat kinase 2 p.G2019S parkinsonism. Neurobiol Aging, 35, 1125–1131. [DOI] [PubMed] [Google Scholar]
- Van der Putten H, Wiederhold KH, Probst A, Barbieri S, Mistl C, Danner S, Kauffmann S, Hofele K, Spooren W, Ruegg MA, Lin S, Caroni P, Sommer B, Tolnay M & Bilbe G (2000) Neuropathology in mice expressing human α-synuclein. J Neurosci, 20, 6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venderova K, Kabbach G, Abdel-Messih E, Zhang Y, Parks RJ, Imai Y, Gehrke S, Ngsee J, Lavoie MJ, Slack RS, Rao Y, Zhang Z, Lu B, Haque ME & Park DS (2009) Leucine-rich repeat kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson’s disease. Hum Mol Genet, 18, 4390–4404. [DOI] [PubMed] [Google Scholar]
- Vogiatzi T, Xilouri M, Vekrellis K, & Stefanis L (2008) Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem, 283, 23542–23556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ & Lee VMY (2011) Exogenous α-Synuclein Fibrils Induce Lewy Body Pathology Leading to Synaptic Dysfunction and Neuron Death. Neuron, 72, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Abdelmotilib H, Liu Z, Stoyka L, Daher JPL, Milnerwood AJ, Unni VK, Hirst WD, Yue Z, Zhao HT, Fraser K, Kennedy RE & West AB (2016) G2019S-LRRK2 Expression Augments -Synuclein Sequestration into Inclusions in Neurons. J Neurosci, 36, 7415–7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MD, Volta M, Cataldi S, Dinelle K, Beccano-Kelly D, Munsie L, Kornelsen R, Mah C, Chou P, Co K, Khinda J, Mroczek M, Bergeron S, Yu K, Cao LP in., Funk N, Ott T, Galter D, Riess O, Biskup S, Milnerwood AJ, Stoessl AJ, Farrer MJ & Sossi V (2014) Behavioral deficits and striatal DA signaling in LRRK2 p.G2019S transgenic rats: a multimodal investigation including PET neuroimaging. J Parkinson Dis, 4, 483–498. [DOI] [PubMed] [Google Scholar]
- Wang X, Yan MH, Fujioka H, Liu J, Wilson-delfosse A, Chen SG, Perry G, Casadesus G & Zhu X (2012) LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet, 21, 1931–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Xu L, Lv L, Su LY, Fan Y, Zhang DF, Bi R, Yu D, Zhang W, Li XA, Li YY & Yao YG (2015) Association of the LRRK2 genetic polymorphisms with leprosy in Han Chinese from Southwest China. Genes Immun, 16, 112–119. [DOI] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, & Rubinsztein DC (2003) α-synuclein Is Degraded by Both Autophagy and the Proteasome. J Biol Chem, 278, 25009–25013. [DOI] [PubMed] [Google Scholar]
- Webber PJ, Smith AD, Sen S, Renfrow MB, Mobley JA, & West AB (2011) Autophosphorylation in the leucine-rich repeat kinase 2 (LRRK2) GTPase domain modifies kinase and GTP-binding activities. Journal of Molecular Biology, 412, 94–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross C. a, Dawson VL & Dawson TM (2005) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. P Natl A Sci USA, 102, 16842–16847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL & Dawson TM (2007) Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet, 16, 223–232 [DOI] [PubMed] [Google Scholar]
- West AB (2015) Ten years and counting: Moving leucine-rich repeat kinase 2 inhibitors to the clinic. Movement Disord, 30, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB (2017) Achieving neuroprotection with LRRK2 kinase inhibitors in Parkinson disease. Exp Neurol, 298, 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O’Kane CJ & Rubinsztein DC (2010) α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J Cell Biol, 190, 1023–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, & Krainc D (2017) α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat Med, 23, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wszolek ZK & Pfeiffer RF (1992) Genetic considerations in movement disorders. Curr Opin Neurol Neurosurg, 5, 324–330. [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V,Vila M, Tieu K, Teismann P, Vadseth C, Choi D-K, Ischiropoulos H & Przedborski S (2002) Blockade of Microglial Activation Is Neuroprotective in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson Disease. J Neurosci, 22,1763–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YR, Tan LC, Fu X, Chen CM, Au WL, Chen L, Chen YC, Prakash KM, Zheng Y, Lee-Chen GJ, Zhao Y, Zeng JS, Tan EK& Pei Z (2012) LRRK2 A419V is not associated with Parkinson’s disease in different Chinese populations. PLoS ONE, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthos DN, & Sandkühler J (2014) Neurogenic neuroinflammation: Inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci, 15, 43–53. [DOI] [PubMed] [Google Scholar]
- Xie W, & Chung KKK (2012) Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson’s disease. J Neurochem, 122, 404–414. [DOI] [PubMed] [Google Scholar]
- Xilouri M, Vogiatzi T, Vekrellis K, Park D, & Stefanis L (2009) Abberant α-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xilouri M, Brekk OR, Polissidis A, Chrysanthou-Piterou M, Kloukina I, & Stefanis L (2016) Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy, 12, 2230–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Neifert S, Karuppagounder SS, Stankowski JN, Lee BD, Grima JC, Chen G, Ko HS, Lee Y, Swing D, Tessarollo L, Dawson TM & Dawson VL (2017). Overexpression of Parkinson’s Disease-Associated Mutation LRRK2 G2019S in Mouse Forebrain Induces Behavioral Deficits and α-Synuclein Pathology. Eneuro, 4, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Neifert S, Karuppagounder SS, Liu Q, Stankowski JN, Lee BD, Ko Han SeokLee, YunjongGrima JC, Mao X, Jiang H, Kang SU, Swing DA, Iacovitti L, Tessarollo L, Dawson TM & Dawson VL (2018) Robust kinase- and age-dependent dopaminergic and norepinephrine neurodegeneration in LRRK2 G2019S transgenic mice. P Natl A Sci, 115, 201712648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kao S-Y, Lee FJS, Song W, Jin L-W, & Yankner BA (2002) Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med, 8, 600–606. [DOI] [PubMed] [Google Scholar]
- Yacoubian TA, Cantuti-Castelvetri I, Bouzou B, Asteris G, McLean PJ, Hyman BT, & Standaert DG (2008) Transcriptional dysregulation in a transgenic model of Parkinson disease. Neurobiol Dis, 29, 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahalom G, Orlev Y, Cohen OS, Kozlova E, Friedman E, Inzelberg R, & Hassin-Baer S (2014) Motor progression of Parkinson’s disease with the leucine-rich repeat kinase 2 G2019S mutation. Movement Disord, 29, 1057–1060. [DOI] [PubMed] [Google Scholar]
- Yang X, Ren H, Wood K, Li M, Qiu S, Shi F-D, Ma C & Liu Q (2018) Depletion of microglia augments the dopaminergic neurotoxicity of MPTP. FASEB J, 32, 3336–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, & Yankner BA (2000) Apoptosis in the nervous system. Nature, 407, 402–9. [DOI] [PubMed] [Google Scholar]
- Yun SP, Kam T, Panicker N, Kim S, Oh Y, Park J, Kwon S-H, Park YJ, Karuppagounder SS, Park H, Kim S, Oh N, Kim NA, Lee S, Brahmachari S, Mao X, Lee JH, Kumar M, An D, Kang S-U, Lee Y, Lee KC, Na DH, Kim D, Lee SH, Roschke VV, Liddelow SA, Mari Z, Barres BA, Dawson VL & Lee S (2018) Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med, 24, 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabetian CP, Yamamoto M, Lopez AN, Ujike H, Mata IF, Izumi Y, Kaji R, Maruyama H, Morino H, Oda M, Hutter CM, Edwards KL, Schellenberg GD, Tsuang DW, Yearout D, Larson EB & Kawakami H (2009) LRRK2 mutations and risk variants in Japanese patients with Parkinson’s disease. Movement Disord, 24, 1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, Del Ser T, Muñoz DG & De Yebenes JG (2004) The New Mutation, E46K, of α-Synuclein Causes Parkinson and Lewy Body Dementia. Ann Neurol, 55, 164–173. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhang C, Zhu Y, Cai Q, Chan P, Uéda K, Yu S & Yang H (2008) Semi-quantitative analysis of α-synuclein in subcellular pools of rat brain neurons: An immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res, 1244, 40–52. [DOI] [PubMed] [Google Scholar]
- Zhang F-R, Huang W, Chen S-M, Sun L-D, Liu H, Li Y, Cui Y, Yan X-X, Yang H-T, Yang R-D, Chu T-S, Zhang C, Zhang L, Han J-W, Yu G-Q, Quan C, Yu Y-X, Zhang Z, Shi B-Q, Zhang L-H, Cheng H, Wang C-Y, Lin Y, Zheng H-F, Fu X-A, Zuo X-B, Wang Q, Long H, Sun Y-P, Cheng Y-L, Tian H-Q, Zhou F-S, Liu H-X, Lu W-S, He S-M, Du W-L, Shen M, Jin Q-Y, Wang Y, Low H-Q, Erwin T, Yang N-H, Li J-Y, Zhao X, Jiao Y-L, Mao L-G, Yin G, Jiang Z-X, Wang X-D, Yu J-P, Hu Z-H, Gong C-H, Liu Y-Q, Liu R-Y, Wang D-M, Wei D, Liu J-X, Cao W-K, Cao H-Z, Li Y-P, Yan W-G, Wei S-Y, Wang K-J, Hibberd ML, Yang S, Zhang X-J & Liu J-J (2009) Genomewide association study of leprosy. New Engl J Med, 361, 2609–2618. [DOI] [PubMed] [Google Scholar]
- Zhang L, Dong Y, Xu X, & Xu Z (2012) The role of autophagy in Parkinson’s disease. Neural Regen Res, 7, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK & Gasser T (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron, 44, 601–607. [DOI] [PubMed] [Google Scholar]