

Abstract
Deletion and truncation mutations in the X-linked gene CASK are associated with severe intellectual disability (ID), microcephaly and pontine and cerebellar hypoplasia in girls (MICPCH). The molecular origin of CASK-linked MICPCH is presumed to be due to disruption of the CASK-Tbr-1 interaction. This hypothesis, however, has not been directly tested. Missense variants in CASK are typically asymptomatic in girls. We report three severely affected girls with heterozygous CASK missense mutations (M519T (2), G659D (1)) who exhibit ID, microcephaly, and hindbrain hypoplasia. The mutation M519T results in the replacement of an evolutionarily invariant methionine located in the PDZ signaling domain known to be critical for the CASK-neurexin interaction. CASKM519T is incapable of binding to neurexin, suggesting a critically important role for the CASK-neurexin interaction. The mutation G659D is in the SH3 (Src homology 3) domain of CASK, replacing a semi-conserved glycine with aspartate. We demonstrate that the CASKG659D mutation affects the CASK protein in two independent ways: 1) it increases the protein’s propensity to aggregate; and 2) it disrupts the interface between CASK’s PDZ (PSD95, Dlg, ZO-1) and SH3 domains, inhibiting the CASK-neurexin interaction despite residing outside of the domain deemed critical for neurexin interaction. Since heterozygosity of other aggregation-inducing mutations (e.g., CASKW919R) does not produce MICPCH, we suggest that the G659D mutation produces microcephaly by disrupting the CASK-neurexin interaction. Our results suggest that disruption of the CASK-neurexin interaction, not the CASK-Tbr-1 interaction, produces microcephaly and cerebellar hypoplasia. These findings underscore the importance of functional validation for variant classification.
Keywords: microcephaly, CASK, neurexin, X-linked, missense mutation
Introduction
Mutations in the X-linked gene CASK (calcium/calmodulin-activated serine kinase) are associated with many neurodevelopmental disorders. Severe mutations such as deletion mutations are associated with a condition known as mental retardation and microcephaly with pontine and cerebellar hypoplasia (MICPCH) in girls (Burglen et al. 2012; Moog et al. 2011; Najm et al. 2008), and epileptic encephalopathies such as Ohtahara syndrome and infantile spasms in boys (Fallon et al. 2002; Michaud et al. 2014). We have recently used a rodent model to demonstrate that the neurodevelopmental phenotypes associated with CASK mutation indeed represent CASK loss-of-function. Postnatal progressive microcephaly, cerebellar hypoplasia, optic nerve hypoplasia, growth retardation and scoliosis can all be reliably replicated in a rodent model that is heterozygous for CASK deletion (Liang et al. 2017; Srivastava et al. 2016).
The CASK gene encodes a MAGUK (membrane-associated guanylate kinase) protein in which the N-terminus half is comprised of a calcium/calmodulin-dependent kinase (CaMK) domain and two LIN-2,7 (L27) domains and the C-terminus half is comprised of a PDZ (PSD-95, Dlg, ZO-1) domain, a Src homology 3 (SH3) and a guanylate kinase (GuK) domain (LaConte and Mukherjee 2013). Missense mutations throughout the protein have been associated with intellectual disability and autistic traits, especially in boys (Hackett et al. 2010; LaConte et al. 2014). A clear genotype-phenotype correlation has been hard to define in cases of MICPCH (Moog et al. 2013), although mutations in the C-terminus are more often associated with nystagmus (Hackett et al. 2010). Females with missense mutations may either be asymptomatic or have very mild cognitive deficits with no microcephaly or cerebellar hypoplasia, resulting in the inheritance of some CASK mutations (Hackett et al. 2010).
One major challenge in association studies with missense mutations is to establish a causal link. Although a number of bioinformatics algorithms are available to predict the pathogenicity of missense mutations, they rely on differing parameters and often do not produce a consensus with any given missense mutation (LaConte et al. 2014). Previously we reported that a simple phylogenetic analysis, accompanied by transient overexpression of the mutant version of CASK in cell culture, may be a reliable method to identify CASK mutations that affect protein structure (LaConte et al. 2014). Our results indicated that two missense mutations at evolutionarily conserved sites, CASKY728C and CASKW919R, altered structure, whereas three mutations (i.e. CASKR28L, CASKY268H and CASKP396S) at non-conserved sites were not structurally damaging (LaConte et al. 2014). It is important to note that both CASKY728C and CASKW919R are mutations that were maternally inherited, and mothers display negligible cognitive defects and randomly inactivated X-chromosomes (Hackett et al. 2010). These findings suggest that, despite a propensity to aggregate, both CASKY728C and CASKW919R retain some functional properties, and the observed aggregates may not be disordered or entirely non-functional.
The molecular mechanism by which CASK mutations produce microcephaly remains unknown, although it has been presumed to be due to disruption of a CASK-Tbr1 pathway (Hsueh et al. 2000; Moog et al. 2013; Najm et al. 2008). The CASK-Tbr1 interaction and subsequent purported nuclear translocation of CASK has been proposed to be mutually exclusive with the binding of CASK to adhesion molecules like syndecan and neurexin (Hsueh et al. 2000). In the current study, we report a novel mutation, M519T in two subjects, which occurs in a completely conserved site in the PDZ domain (Mukherjee et al. 2010) and is known to be critical for PDZ domain interactions (Li et al. 2014). The biochemical and cell biological analysis of the M519T mutation reported here demonstrates that it specifically disrupts the CASK-neurexin interaction, providing a compelling molecular mechanism for the development of microcephaly. In support of this notion, we describe a third case of a heterozygous missense mutation in CASK (G659D; Clinvar variant ID:180215; dBSNP rs727505397) that is associated with microcephaly and cerebellar hypoplasia, which occurs in a semi-conserved site in the SH3 domain of the CASK protein. We demonstrate that CASKG659D forms aggregates similar to previously described variants such as CASKW919R, but unlike CASKW919R, heterozygous CASKG659D is associated with microcephaly and cerebellar hypoplasia in a girl. Despite being in the SH3 domain, the G659D mutation inhibits the interaction of CASK with neurexin, an interaction classically assumed to be via the PDZ domain. Thus the induction of microcephaly by CASKG659D is more likely due to the disruption of the CASK-neurexin interaction than due to protein aggregation. Our data imply that CASK missense mutations that abolish interactions known to involve CASK’s PDZ domain lead to microcephaly and cerebellar hypoplasia. The findings with CASKG659D may have implications for other multi-domain proteins, in which missense mutations produce functional loss that extends beyond the domain in which the mutations reside. Furthermore, our work demonstrates the importance of functional validation of individual missense variants for precise genotype-phenotype correlation and clinical variant classification.
Results
We report three girls with similar clinical presentations, including intellectual disability, microcephaly, and hindbrain hypoplasia, all carrying a heterozygous missense mutation in the CASK gene.
Subject 1 is a 12-year-old girl, born after an uncomplicated term pregnancy at 2579 grams, and the only child born to non-consanguineous healthy Caucasian parents. After an unremarkable postnatal course, concerns arose at 5 months of age when she was found to have increased muscle tone and nystagmus. An MRI scan of the brain showed, in addition to microcephaly, global hypoplasia of the cerebellum and cerebellar vermis, moderate brain stem hypoplasia, and diminished size of the middle and superior cerebellar peduncles (Figure 1A). Her developmental milestones were delayed across all areas (Figure 1B). She did not roll over until 12 months and did not walk independently until 36 months of age. She started to develop a few single words but never progressed in her language development. A developmental assessment at 7 years of age placed her at a developmental age of approximately 2 years. At the initial genetics evaluation at 9 years of age, she presented essentially non-verbal, with proportionate short stature (height below the 2nd percentile and at the 50th percentile of a 6.5-year-old girl), microcephaly (occipitofrontal head circumference significantly below the 2nd percentile and at the 50th percentile of a 2-year-old girl) (Figure 1B). A few mild and non-specific craniofacial dysmorphic features were noted, including deep-set eyes with long, narrow palpebral fissures, long eyelashes, long and arched eyebrows, and pronounced transverse creases across the central forehead, mild midface hypoplasia, and a wide mouth with mild prognathism and dental misalignment. Except for bilateral prominent thumbs, no other skeletal anomalies were present. Neurological exam revealed nystagmus, mildly increased muscle tone more pronounced in the lower extremities, fine motor discoordination, and mild gait ataxia. Whole exome sequencing (WES) and Sanger confirmation revealed a non-maternally inherited heterozygous variant in exon 16 of the X-linked CASK gene, c.1556T>C (p.M519T), classified as variant of uncertain significance (Supplemental Figure 1). The father was not available for analysis. Analysis of the polymorphic CAG repeat in the androgen receptor locus (Xq13) in indicated random (non-skewed) X Chromosome Inactivation (XCI). This variant was not observed in the NHLBI Exome Sequencing Project (ESP), ExAC cohort, 1000 Genomes Project and not listed in the Database of Single Nucleotide Polymorphisms (dbSNP). The patient’s clinical presentation and characteristic findings on brain MRI in the context of the observed CASKM519T variant were consistent with the diagnosis of X-linked dominant mental retardation and microcephaly with pontine and cerebellar hypoplasia (MICPCH, MIM# 300749). A second case of [c.1556T>C (exon16) P.M519T] CASK mutation (Supplemental Figure 1) is a 34-month girl (Subject 2). She was initially diagnosed with global development delay at 9 months and microcephaly at 11 months. CASK mutation was identified at 17 months. She developed intractable seizures at the age of 22 months old. She has hindbrain hypoplasia and muscle hypotonia. No nystagmus has been noted in the second case although strabismus is observable.
Figure 1. CASKM519T and CASKG659D phenotypic characteristics.
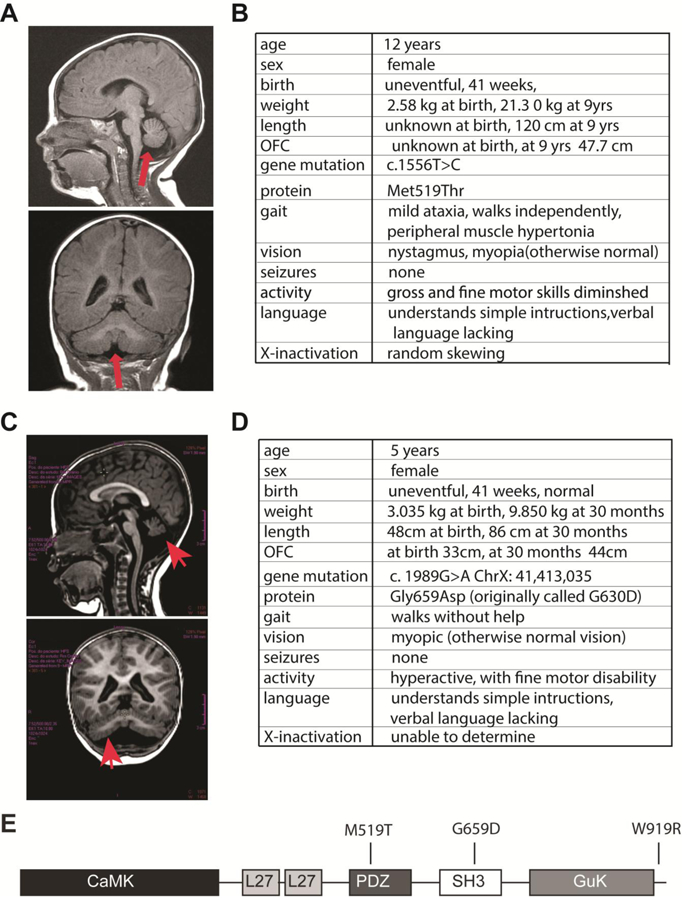
A) Magnetic resonance imaging brain scan of Subject 1 with the heterozygous missense mutation, CASKM519T. Sagittal and coronal views of Subject 1 (T1-weighted images). In addition to microcephaly, global hypoplasia of the cerebellum, including the cerebellar vermis, is noted (arrows). B) Phenotype and genotype of Subject 1. C) Magnetic resonance imaging brain scan of Subject 3 with heterozygous missense mutation, CASKG659D. B) Phenotype and genotype of Subject 3. C) The five major domains of CASK, with the locations of M519T, G659D and W919R mutations indicated in the linear structure. Note: in some CASK conformations, the SH3 and GUK domains are integrated into a single structural entity.
Subject 3 is a 5-year-old girl born after an uneventful pregnancy who displayed progressively apparent secondary microcephaly (within 10th percentile at birth and below 5th percentile by 3 years of age; Figure 1C,D). Magnetic resonance imaging (MRI) showed hypoplasia of the cerebellar hemispheres, cerebellar vermis and part of the brain stem in addition to microcephaly (Figure 1C). On exam, she walked well and was not severely ataxic. There is no history of seizures. She has impaired expressed language and fine motor activity. Her receptive language is also underdeveloped. Ophthalmological evaluation revealed myopia and strabismus (Figure 1D). Whole exome sequencing uncovered a heterozygous variant in the gene CASK c.1989G>A : p.G659D (REF:seq GRCh37) previously reported as ClinVar variant ID:180215; dBSNP rs727505397. This variant was not present in 8000 controls, and Sanger sequencing of the parental DNA suggested that the mutation event was de novo.
Since to our knowledge, these are the first reported cases of microcephaly and cerebellar hypoplasia in a female with a heterozygous missense mutation in CASK, we decided to investigate the molecular impact of these CASK mutations using a combination of in silico, biochemical, and cell biological approaches. The methionine at position 519 in CASK is completely conserved among all known orthologs of CASK. In silico analyses with Polyphen and SIFT both indicated that the CASKM519T variant is pathogenic (possibly damaging in Polyphen and deleterious in SIFT; not shown) (Flanagan et al. 2010). Position 519 is located in the PDZ signaling domain (Mukherjee et al. 2010) and has been shown to be critical for PDZ domain-mediated interactions (Li et al. 2014), strongly suggesting a source for the pathogenesis observed upon mutation. We thus investigated the evolutionarily conserved interaction with neurexin that is mediated by CASK’s PDZ domain (Hata et al. 1996; Mukherjee et al. 2014) in the context of the M519T mutation. CASKM519T was produced in the GFP-CASK cDNA background previously reported (Mukherjee et al. 2008), using site-directed mutagenesis. The interaction of CASK with neurexin can be easily demonstrated using a recruitment assay (LaConte et al. 2016b), because GFP-CASK is a cytosolic protein but when co-expressed with the transmembrane protein neurexin in HEK293T cells, CASK gets recruited to the membrane due to the direct interaction between CASK and neurexin (Figure 2 A&B). This change in CASK’s cellular localization is readily visualized with confocal microscopy (LaConte et al. 2016b). In the recruitment assay, CASKM519T failed to interact with neurexin, indicating that M519T specifically disrupts the CASK-neurexin interaction (Figure 2C&D). A GST pull-down experiment using the neurexin-1β cytosolic tail confirmed the disrupted interaction with CASKM519T but not with native CASK present in HEK (Figure 2E), supporting a straight-forward hypothesis that disruption of CASK-neurexin interaction is sufficient to induce MICPCH.
Figure 2. CASKM519T disrupts CASK-neurexin interaction.
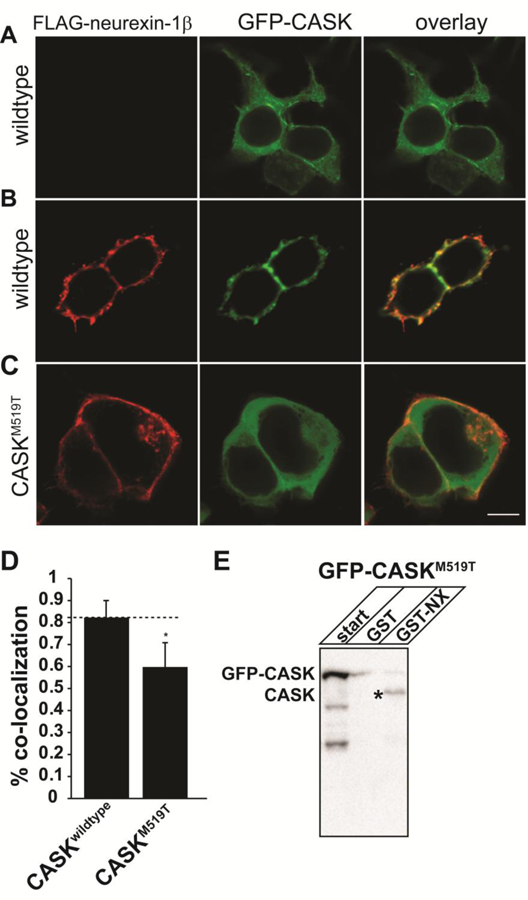
A) Images of HEK293FT cells transfected with GFP-CASK without neurexin (A) and (B,C) GFP-CASK or GFP-CASKM519T plasmid DNA each co-expressed with neurexin-1β-FLAG. After 48 hours, cells were fixed, permeabilized and immunostained for neurexin. Scale bar = 5μm. C) Colocalization analysis performed from 22 different images collected from 3 separate experiments. Results are plotted as mean±SD. * indicates p < 0.05. E) GST pull-down using either GST or GST-neurexin cytosolic tail fusion protein (GST-NX) from HEK293FT cell lysates transfected with GFP-CASKM519T. ‘Start’ indicates the cell lysate. The precipitated proteins were immunoblotted for CASK. * indicates CASK endogenously expressed in HEK293FT cells.
Unlike M519T, the glycine at position 659 that is mutated to aspartate in Subject 3 does not immediately suggest a pathogenic mechanism. It is located within the SH3 domain of CASK (Figure 1E), and an alignment of different CASK orthologs indicates that this glycine is a semi-conserved residue, with only glycine or alanine found at this site (Supplemental Figure 2A). The change of glycine, an aliphatic amino acid, to aspartate, an acidic amino acid, is likely to alter at least the secondary structure in the protein, as predicted by a battery of web-based algorithms that use sequence conservation to assess the pathogenicity of single amino acid changes within a given protein (Supplemental Figure 2B). Algorithms that use existing structural models of proteins to calculate an estimated change in free energy attributable to single amino acid changes were also used to examine the putative impact of the G659D mutation, and in general, G659D was predicted to result in a greater increase in free energy than a previously studied CASK mutation shown to aggregate in cells (LaConte et al. 2014) (Supplemental Figure 2C), suggesting that the mutation at position 659 potentially exerts its pathogenic effect by altering the structure of CASK.
A molecular model of CASK’s SH3-GuK domain (LaConte et al. 2014) demonstrates that replacing the glycine at position 659 with aspartate might lead to the formation of additional, potentially destabilizing contacts (Figure 3A). Global folding of CASKG659D was examined in a GFP-fusion protein aggregation assay previously reported (LaConte et al. 2014). CASK mutations Y728C and W919R, predicted to destabilize CASK’s structure (LaConte et al. 2014), have been shown to produce aggregates of GFP-CASK that are easily visualized by laser scanning confocal microscopy when over-expressed in HEK293FT cells. HEK293FT cells were therefore transiently transfected with GFP-CASKWT, GFP-CASKW919R and GFP-CASKG659D plasmids, and the distribution pattern of these molecules in the cytoplasmic region of the cell was examined (Figure 3B). In contrast to GFP-CASKWT or GFP-CASKM519T, which are uniformly distributed throughout the cytoplasm, GFP-CASKG659D produced aggregates similar to those seen with GFP-CASKW919R (Figure 3B and Supplemental Figure 3). As previously observed with GFP-CASKW919R (LaConte et al. 2014), not all cells transiently expressing GFP-CASKG659D contain visible protein aggregates, suggesting a shift in the equilibrium governing soluble protein structure due to the missense mutation. To estimate aggregation propensity, we quantified the number of cells in which transiently expressed protein (GFP-CASKG659D or GFP-CASKW919R) accumulated as visible aggregates and found that GFP-CASKG659D has a substantially lower propensity to aggregate compared to GFP-CASKW919R (Figure 3C), since only ~17% of transfected cells contain aggregates of GFP-CASKG659D compared to almost 30% of cells transiently expressing GFP-CASKW919R.
Figure 3. Structural impacts of the G659D mutation.
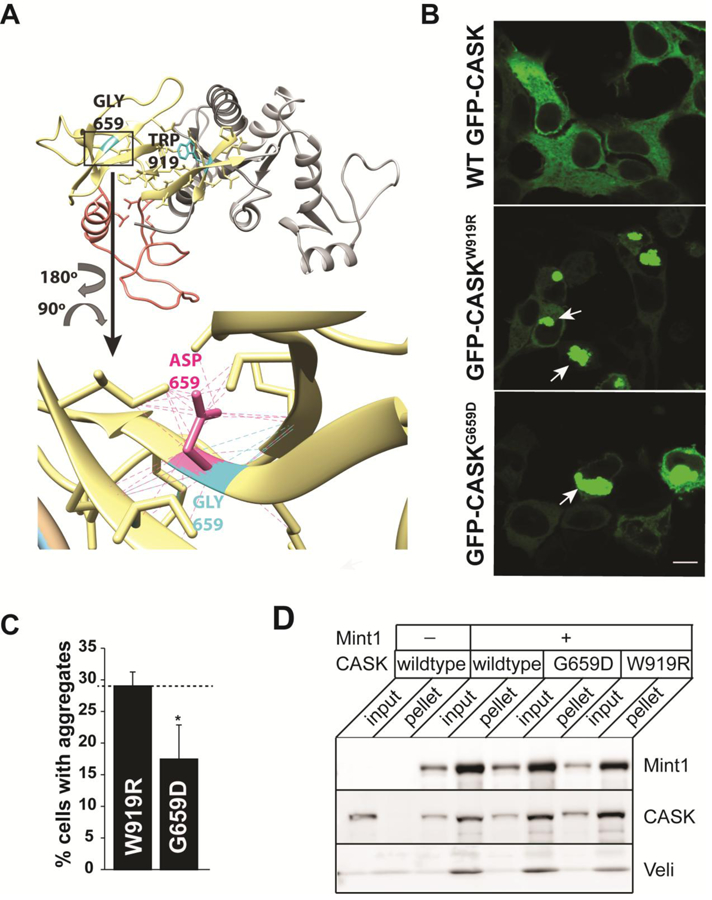
A) Homology model of CASK’s integrated SH3-GUK domain structure (LaConte et al. 2014) showing the location of native residues G659 and W919 (cyan), as well as mutant side chains (G659D, magenta) and associated contacts (dotted lines). SH3 region, yellow. Hinge region, orange. GUK region, gray. B) Representative images of HEK293FT cells expressing GFP-CASK plasmid as indicated. Note aggregation of protein (indicated by arrows) for both GFP-CASKW919R and GFP-CASKG659D. Scale bar = 10μm. C) Quantitation of percentage of cells displaying protein aggregates. The data is represented as mean ± S.E.M., n = 8. * indicates p < 0.05. D) Representative blot showing immunoprecipitation of FLAG-tagged Mint1. HEK293FT cells were transiently transfected with cDNA for GFP-CASK (wild-type and mutants) and FLAG-Mint1 as indicated. FLAG-Mint1 was precipitated from solubilized cells using the M2 beads and blotted for indicated antigens.
Although both CASKW919R and CASKY728C variants exhibit an apparently higher propensity to aggregate than CASKG659D (LaConte et al. 2014), it has been previously reported (Hackett et al. 2010) that neither CASKW919R nor CASKY728C produces microcephaly in females (in other words, the observed inheritance pattern is X-linked recessive), indicating that the G659D mutation likely disrupts a molecular function that remains intact in CASKW919R and CASKY728C, such as an interaction with a known binding partner. Accordingly, we tested the ability of CASKW919R and CASKG659D to participate in an evolutionarily conserved tripartite complex consisting of CASK (lin-2), Veli (lin-7) and Mint1 (X-11α) (Butz et al. 1998; Kaech et al. 1998; Mukherjee et al. 2014). CASKW919R mutation serves as a useful comparison for CASKG659D, since CASKW919R does not produce an obvious phenotype in humans in the heterozygous condition and presumably retains some functionality lost in CASKG659D. GFP-CASKWT, GFP-CASKW919R or GFP-CASKG659D together with FLAG-tagged Mint-1 were overexpressed in HEK293FT cells, which endogenously express Veli. Immunoprecipitation using anti-FLAG M2 beads suggests that both CASKW919R and CASKG659D are able to interact with Mint1 and Veli (Figure 3D).
Microcephalies due to CASK mutation have often been attributed to disruption of a CASK-TBR1-CINAP-dependent pathway (Hsueh et al. 2000; Najm et al. 2008). We therefore next examined the effect of CASK missense mutations on the CASK-TBR1 interaction using an assay previously described by a different group (Deriziotis et al. 2014). This assay depends on translocation of CASK to the nucleus upon co-expression of TBR1. Surprisingly, the distribution of wildtype GFP-CASK did not change when co-expressed with mCherry-TBR1 in HEK293FT cells (Figure 4A). To verify these results in a more native setting, GFP-CASK and mCherry-TBR1 were transiently expressed in primary murine cortical neurons. GFP-CASK and mCherry-Tbr-1 localized to two distinct neuronal compartments, the cytoplasm and the nucleus, respectively (Figure 4B). Finally, because of the surprising nature of these results, untagged CASK and Tbr1 were co-expressed in HEK293FT cells to rule out the possibility that the fluorescently tagged proteins were behaving differently than their untagged counterparts, but no co-localization was observed with the untagged proteins either (Figure 4C). Co-immunoprecipitation experiments from transiently transfected HEK293FT cells demonstrated that GFP-CASK efficiently precipitated another protein, liprin-α3, known to interact with CASK (LaConte et al. 2016b; Wei et al. 2011) but did not co-precipitate Tbr-1 (Figure 4D). Most relevant to the question of how the G659D mutation affects CASK function, no difference in cellular distribution was observed between GFP-CASKG659D and wildtype GFP-CASK upon co-expression of mCherry-TBR1, indicating that in the case of the CASKG659D heterozygous female subject, microcephaly does not arise from disruption of this pathway (Supplemental Figure 4).
Figure 4. CASK do not translocate to nucleus upon Tbr1 co-expression.
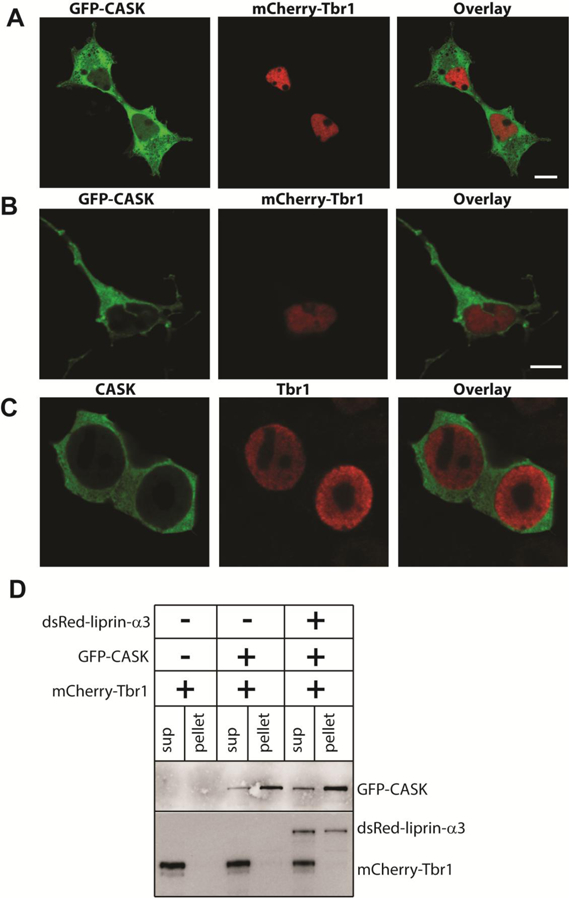
Images of A) HEK293FT cells and B) cortical neurons co-transfected with GFP-CASK and mCherry-Tbr1. C) Images of HEK293FT cells co-transfected with CASK and Tbr1 and immunostained with antibodies for CASK and Tbr1. Scale bars = 5μm. D) Blot showing immunoprecipitation of GFP-CASK using GFP-trap beads. Transfected cDNA is indicated at the top, and antigens for which immunoblotting has been performed is indicated on the right.
Because experimental evidence suggests that, although a subpopulation forms cellular aggregates, a substantial fraction of CASKG659D exists in a monomeric form (Figure 3C), molecular dynamics simulations were employed to further explore the impact of this mutation on the structure of CASK’s SH3-GuK domain. Three 100 ns trajectories were run at 300K for three CASK variants (CASKWT, CASKW919R, CASKG659D). All three structures reached a plateau in a plot of RMSD (root mean square deviation from starting structure backbone) over the time course of the simulations. A root mean square fluctuation (RMSF) calculation was performed at all alpha-carbons for the three CASK variants. The RMSF values were then used to calculate B-factors at each amino acid position (Figure 5A); B-factors are a useful way to assess local changes in flexibility due to each mutation in a molecular dynamics simulation (Rueda et al. 2007). The most dramatic differences in B-factors between the wild-type structures and the two CASK mutants, CASKW919R (data not shown) and CASKG659D, occur not in the region of the specific amino acid mutations but between residues 677 and 727, a region described as the hinge region in CASK (Figure 5).
Figure 5. Molecular dynamics simulations predict increased mobility of CASK’s hinge region and disruption of the SH3 domain.
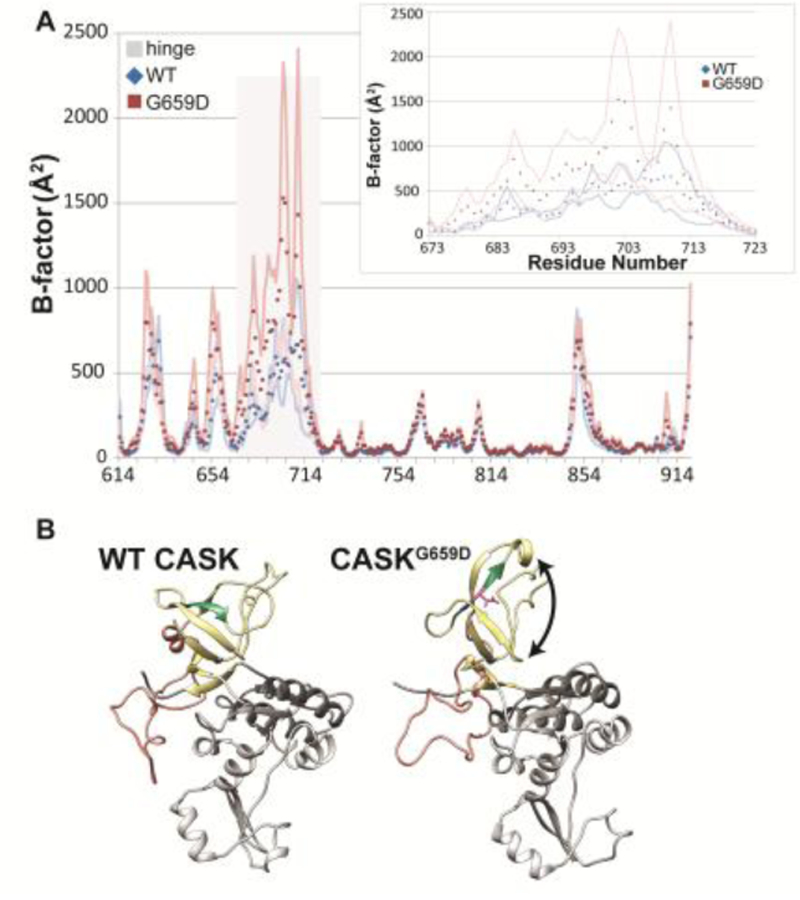
A) B-factors (Å2) calculated from RMS fluctuations of α-carbons in a homology model of CASK’s SH3-GuK (CASK-WT, blue; CASKG659D, red) domain during 100 ns of molecular dynamics simulations at each residue. Points are the average B-factor at a given position, and lines are upper and lower SEM boundaries (n = 3 trajectories). The residues that compose the hinge region are indicated by gray bar. Inset, hinge region. B) Most populated CASKWT (left) and CASKG659D (right) structures during three 100 ns molecular dynamics trajectories based on cluster analysis. SH3 domain, yellow. Hinge region, orange. GUK domain, gray. β strand containing site of G659D mutation, green. Black arrow indicates disrupted β-barrel structure.
A structure cluster analysis of the molecular dynamics trajectories was used to identify structures that were predominantly populated during the course of the molecular dynamics simulations and provides insight into the structural impact of the G659D mutation. As shown in the homology model (McGee et al. 2001) in Figure 5B, CASK’s SH3 domain is composed of a β barrel, with two of the six β strands separated in sequence from the others by both CASK’s hinge (sometimes referred to as the HOOK; shown in orange) domain and GuK domain (shown in gray) (McGee et al. 2001). The average structure that was most populated during the course of the CASKG659D molecular dynamics simulations does not retain this compact β barrel structure; the β strand that contains residue 659 (shown in green, Figure 5B) and the adjacent β strand are no longer parallel to the adjacent strands, essentially unwrapping and forcing the β barrel into a more extended structure (indicated by double-sided arrow in Figure 5B).
Work from other groups supports the idea that in MAGUK proteins, the PDZ, SH3 and GuK domains form a supramodule stabilized by inter-domain interactions predominantly between the PDZ and SH3 domains (Li et al. 2014; Reissner and Missler 2014); (Pan et al. 2011; Zeng et al. 2017). In PALS1, the structure of this module is critical for a high-affinity interaction with a protein binding partner via the PDZ motif, and any disruption of the supramodule weakens this protein-protein interaction. A homology model of CASK was created using the PALS1 PDZ-SH3-GuK supramodule as a template (Figure 6A) to gain a better understanding of how an altered fold of the SH3 domain might impact the PDZ-SH3 interface deemed to be critical for protein-protein interaction with a protein containing a PDZ-binding motif. An examination of the homology model of the CASK supramodule structure (Figure 6A, left) suggests that an unwrapped β barrel (indicated by arrow) would likely occlude the groove that was found to be important for the PALS1-Crumbs tail interaction (Figure 6A, right). The disrupted SH3 β barrel predicted by molecular dynamics simulation in CASKG659D (Figure 5B) suggests that this particular mutation likely destabilizes two putative CASK conformations—the integrated SH3-GuK conformation shown in Figure 5B and a reorganized structure (Figure 6A) purported to favor interactions with proteins containing PDZ binding motifs.
Figure 6. CASKG659D disrupts CASK-neurexin interaction.
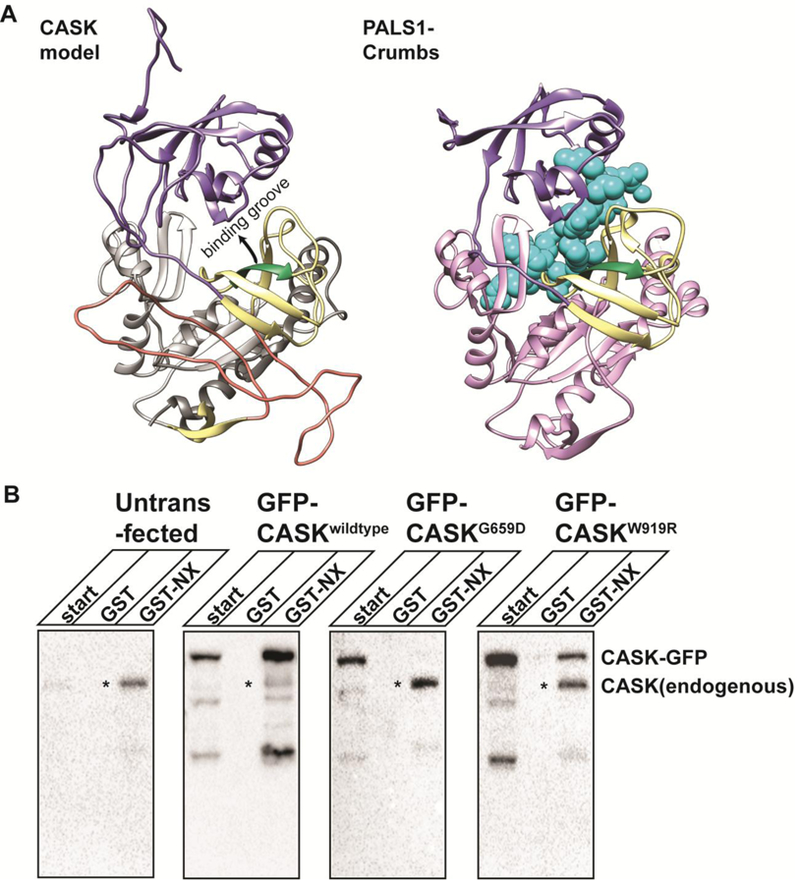
A) Homology model of the CASK PSG supramodule (left), based on PALS1-Crumbs PSG supramodule structure (right; 4wsi.pdb). PDZ domain, purple. SH3 domain, yellow. Hinge region, orange. CASK GuK domain, gray. PALS1 GuK domain, lavender. β strand containing site of G659D mutation, green. Crumbs, cyan. B) GST pull-down using either GST or GST-neurexin cytosolic tail (GST-NX) fusion protein from HEK293FT cell lysates transfected with cDNA indicated at the top. ‘Start’ indicates the cell lysate. The precipitated proteins were immunoblotted for CASK. * indicates CASK endogenously expressed in HEK293FT cells.
Because computational modeling suggests that the G659D mutation might impact CASK’s ability to interact with binding partners that contain a PDZ-binding domain, we next investigated CASKG659D’s ability to interact with neurexin as described above for CASKM519T. Results from the GST pull-down using immobilized neurexin1 cytosolic tail showed that, although neurexin efficiently bound both endogenous CASK from HEK293FT cells and over-expressed GFP-CASK, it failed to pull down GFP-CASKG659D (Figure 6B); GFP-CASKW919R was pulled down by neurexin, but to a lesser extent than wildtype GFP-CASK. Biochemical approaches such as the GST pull-down assay and immunoprecipitation allow protein-protein interactions to be assessed in bulk but not at a single cell level. A bulk analysis can cause problems when variant proteins exhibit aggregates in a subset of cells, as seen with CASKG659D and CASKW919R, because it makes it difficult to determine whether affinity for a given interacting partner is globally reduced or whether the non-aggregated population of the variant protein can interact normally with binding partners. Fortunately, the recruitment assay shown above for CASKM519T (LaConte et al. 2016b) allows for visualization of the interaction between neurexin and CASK at the level of the individual cell. Results from the recruitment assay are shown in Figure 7, in which GFP-CASKW919R and GFP-CASKG659D were co-expressed with FLAG-tagged neurexin and stained with anti-FLAG antibody. Whereas CASKWT (Figure 2B), CASKW919R (Figure 7A), and several other known CASK mutants (R28L, Y728H, P396S, and Y728C; Supplemental Fig 5) interact with neurexin, CASKG659D fails to get recruited to the membrane by neurexin (Figure 7B&C), confirming that the G659D mutation disrupts CASK’s interaction with neurexin. In summary, these data suggest that although the G659D mutation is present in the SH3 domain and may induce aggregation, microcephaly is most likely caused by disruption of a PDZ-mediated interaction.
Figure 7. CASKG659D but not CASKW919R disrupts CASK-neurexin interaction.
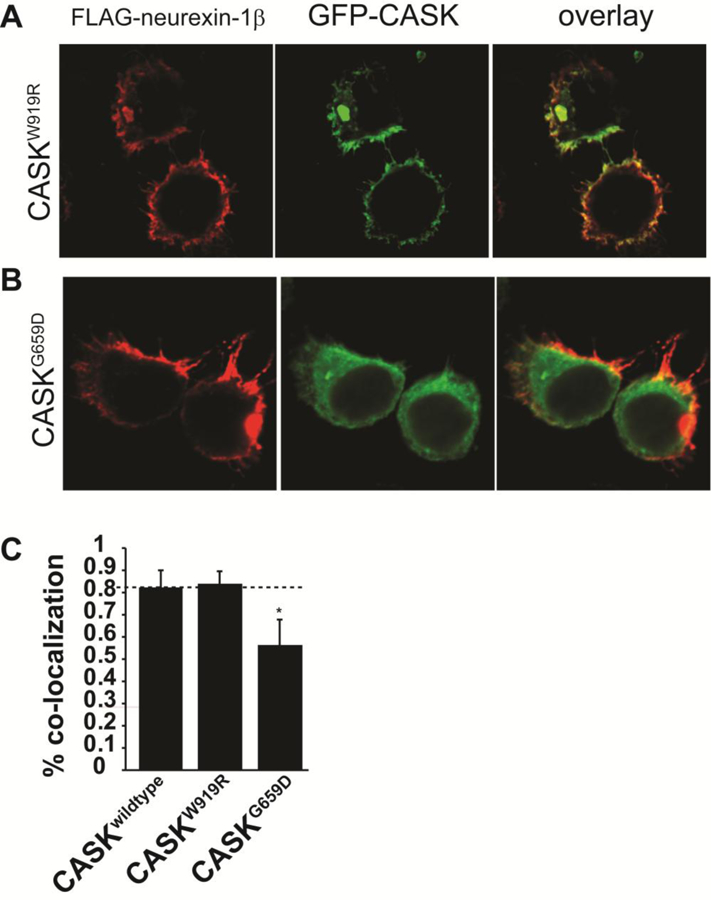
Images of HEK293FT cells transfected with GFP-CASKW919R or GFP-CASKG659D plasmid DNA co-expressed with neurexin-1β-FLAG (A,B). After 48 hours, cells were fixed, permeabilized and immunostained for neurexin. Scale bar = 5μm. C) Co-localization analysis of neurexin and CASK performed from 22 different images collected from 3 separate experiments. Results are plotted as mean±SD. * indicates p < 0.05.
Discussion
The investigation of missense mutations in proteins associated with disorders is of both diagnostic importance and may provide critical insight into the physiological function of a protein. Missense mutations in CASK are frequent in boys with X-linked intellectual disability, but until now, only less severe, if any, phenotypes have been documented in girls with CASK missense mutations, suggesting an X-linked recessive inheritance pattern.
CASK is a multi-domain protein, and mutations throughout the protein are known to produce intellectual disability. In multi-domain proteins, individual domains may interact with each other in ways that are crucial for function at the cellular level. It has been previously shown that CASK evolved as an extremely slow kinase with an inability to use magnesium as a co-factor due to critical residue substitutions (Mukherjee et al. 2010). CASK’s slow enzyme kinetics, however, are partially offset by the fact that CASK phosphorylates its target protein when the target, neurexin, is bound via CASK’s PDZ domain (Mukherjee et al. 2010; Mukherjee et al. 2008).
Here we have reported two CASK heterozygous missense mutations (CASKM519T and CASKG659D) in female subjects with microcephaly and intellectual disability (suggestive of X-linked dominance). The results of the XCI study supports M519T being a loss of function mutation consistent with phenotype. Microcephaly with pontine and cerebellar hypoplasia (MICPCH) is considered an X-linked dominant condition associated with loss of CASK gene function (via nonsense, splice site, deletion or duplication mutation) with only very few reported affected males, which is likely a result of early male lethality (Atasoy et al. 2007). Consequently, random XCI in a female carrying a de novo pathogenic CASK mutation is predicted to result in the more severe phenotype of MICPCH, whereas skewing towards the non-mutated de novo allele is expected to cause the milder X-linked intellectual disability. In contrast, skewing toward the mutated allele is expected to result in decreased fetal viability in a female fetus. The CASKM519T variant provided a straightforward and testable structural hypothesis for the molecular pathology responsible for the severe phenotype because it occurs in CASK’s PDZ domain, and we confirmed that this mutation does indeed disrupt protein-protein interactions mediated by this domain such as that with neurexin (Figure 2). The CASKG659D mutation, present in CASK’s SH3 domain, does not initially offer a similarly straightforward structural mechanism for causing microcephaly in the heterozygous state. The CASKG659D variant produces structural changes and a propensity to aggregate when over-expressed (Figure 3). Molecular dynamics simulations suggest that the CASKG659D mutation leads to increased flexibility of the hinge region of the SH3 domain (Figure 5A). This hinge region has attracted researchers’ attention in structural studies of other MAGUK proteins (PSD-95 and DLG1) (Nix et al. 2000). It has been proposed that MAGUK proteins can participate in a type of conformational change referred to as domain swapping, in which homomultimers form when the intramolecular interactions that maintain the integrated SH3-GuK domain structure of a MAGUK protein dissolve to create a more extended structure, allowing intermolecular interactions between SH3 and GuK domains of neighboring MAGUKs (McGee et al. 2001). The conversion from MAGUK monomer to multimer relies on flexibility in the hinge region. The predicted increase in flexibility of the hinge region of CASKG659D, as demonstrated by the dramatically increased B-factors (Figure 5A), suggests that this mutation may increase the likelihood that CASKG659D exists in an extended conformation which might favor multimer formation and thus account for the observed aggregates (Figure 3B). Determining how a preference for the extended CASK structure impacts both molecular function and multimer formation will require further structural investigation.
Evidence provided here supports the conclusion that neurexin binding is abolished for the CASKG659D variant (Figure 6). A closer examination of the theoretical impact of the G659D mutation on CASK’s structure offers some hypotheses for why this occurs, since this mutation likely disrupts the core SH3 fold (Figure 5B). The crystal structure (Li et al. 2014) of a PDZ-SH3-GuK (PSG) supramodule from another MAGUK protein, PALS1, in complex with its PDZ binding partner, Crumbs, suggests that the PSG supramodule can exist in a conformation in which the PDZ domain and a compactly folded SH3 domain form a groove that interacts with the PDZ domain binding protein, Crumbs (Figure 6A). In the case of CASKG659D, if the core SH3 fold is not compact but instead has a disrupted β barrel structure (Figure 5B), the groove will likely no longer be compatible with binding of the neurexin cytoplasmic tail (Figure 6A) and may in fact be occluded. The importance of the integrity CASK’s PSG supramodule structure for neurexin binding is supported by recent NMR results suggesting that ligand binding (i.e., neurexin) induces coupling between CASK’s PDZ domain and SH3-GuK domain, resulting in much higher affinities than between neurexin and CASK’s isolated PDZ domain. (Zeng et al. 2017).
The CASK-neurexin interaction is evolutionarily conserved and purportedly occurs through the PDZ domain (Hata et al. 1996; LaConte et al. 2014). Experimentally, however, a large portion of CASK, including the SH3-GuK domain, is necessary for this interaction, indicating that neurexin interacts with CASK via multiple domains (Hata et al. 1996; Li et al. 2014; Reissner and Missler 2014). In fact for both neurexin and syndecan, additional secondary sites of interaction have been hypothesized (Daniels et al. 1998). The disruption of CASK-neurexin binding by the G659D mutation, which is in the SH3 domain instead of the PDZ domain traditionally associated with neurexin binding, thus adds to the growing body of evidence that the interaction between CASK and neurexin is not mediated simply by the binding of a few residues at the end of neurexin’s cytoplasmic tail to CASK’s PDZ domain, but rather involves multiple domains and a defined three-dimensional structure for optimal interaction.
Our findings expand the known genotype-phenotype correlations in CASK-related disorders. The M519T and G659D mutations in CASK are noteworthy because they are missense mutation associated with microcephaly and cerebellar hypoplasia in female patients, a phenotype typically seen only in females with deletion mutations. This suggests that both mutations have a profound impact on the function of CASK. We show here that these single amino acid changes disrupt the CASK-neurexin interaction, but the question remains whether this disruption alone is responsible for the severe phenotype observed. Supporting the importance of the CASK-neurexin interaction, we have previously demonstrated that the CASK-neurexin interaction is critical for the stabilization of neurexin via interaction with liprin-α (LaConte et al. 2016b). Mutations in neurexin alter CASK expression (Pak et al. 2015) and are also associated with intellectual disability and microcephaly (Zweier et al. 2009). CASK, however, potentially interacts with multiple proteins via its PDZ, SH3 and GuK domains, and it is reasonable to ask whether the M519T and G659D mutations interrupt multiple protein-binding interactions. For example, CASK’s SH3 domain is also proposed to interact with the polyproline tracts of other proteins such as the alpha subunit of a neuronal calcium channel (Maximov et al. 1999), and the GuK domain of CASK may interact with the transcription factor TBR1 (Hsueh et al. 2000). It is unlikely, however, that the interruption of any of these interactions causes the microcephaly seen here, since the deletion of CASK does not affect the functioning of presynaptic calcium channels (Atasoy et al. 2007), and the disruption of the CASK-TBR1 interaction in mouse does not produce microcephaly (Huang and Hsueh 2017). In fact, nuclear translocation of GFP-CASK with TBR1 was not observed even for wildtype CASK (Figure 4), further weakening the hypothesis that the CASK-TBR-1 interaction is responsible for the microcephaly component of CASK-related phenotypes. The PDZ motif itself may interact with multiple different proteins including SynCAMs (Biederer et al. 2002), syndecan-2 (Hsueh et al. 2000; Hsueh et al. 1998) and Parkin (Fallon et al. 2002). Thus although our data suggest that a PDZ-mediated interaction is critical for CASK function and that in particular, CASK’s interaction with neurexin is disrupted by both mutations described here, we cannot entirely rule out the possibility that other critical interactions via CASK’s PDZ domain contribute to the microcephaly phenotype associated with CASK dysfunction. The precise signaling mechanism that leads to microcephaly in CASK mutation remains to be described.
Eighty percent of known eukaryotic proteins are multi-domain in nature (Apic et al. 2003). Most folding and structural studies have focused on small globular domains, and yet evidence suggests that inter-domain interactions in multi-domain proteins significantly affect stability, folding and unfolding rates (Batey et al. 2008). Here we propose an additional result of inter-domain cooperativity in terms of protein function. Based on our analysis of this pathogenic mutation and previously published results, we suggest that the C-terminus of CASK, encompassing the PDZ-SH3-GuK domains, is critical for high-affinity interaction with target proteins like neurexin, and the N-terminus of CASK, including the CaMK domain and L27 domains, are responsible for phosphorylation of the target protein in a divalent-ion sensitive fashion. Thus, the evolutionarily conserved domain arrangement of CASK may facilitate specific phosphorylation of target proteins when the cytosolic levels of unchelated ATP are high. This study thus also offers insight into how mutations identified in domains throughout CASK, not just the PDZ domain, are associated with microcephaly.
Materials and Methods
Ethical statement
The Virginia Tech Institutional Review Board approved the collection and use of data from subjects. Informed consent was obtained from the families prior to participation.
DNA analysis
Genomic DNA was extracted from peripheral blood using standard methods. Routine diagnostic exome sequencing was performed at a commercial laboratory. Sanger sequencing was used according to standard procedures to confirm the CASK c.1556T>C (p.M519T) mutation and perform segregation analysis. Shodair Genetics Laboratory is a clinical laboratory certified by the Centers for Medicare and Medicaid Services (CMS) through Clinical Laboratory Improvement Amendments (CLIA; certificate number: 27D0652530).
X chromosome inactivation (XCI) studies
Extracted DNA examined at the androgen receptor locus for a pattern of skewed x-inactivation. The CAG trinucleotide repeat within the androgen receptor gene was amplified by PCR, before and after digestion with methylation-sensitive restriction enzyme HpaII, using specific primers labeled with fluorescent dye as previously described (Allen et al. 1992). Peak ratios greater than 80:20 are considered to be indicative of skewed X inactivation. Allele sizes and peak area ratios were determined using a high resolution laser-induced fluorescence capillary electrophoresis system with internal size standard.
Plasmid and Point Mutagenesis
CASK was cloned in the pEGFP-C3 vector and has been previously described (LaConte et al. 2014; Mukherjee et al. 2008). Using the Phusion polymerase (NEB), we generated CASKG659D and CASKM519T mutations by site-directed mutagenesis in the pEGFP CASK background. The CASKW919R mutant plasmid was previously generated (LaConte et al. 2014). pCMV5-CASK (no fluorescent tag) was a kind gift from Tom Südhof. The YFP-Tbr1 plasmid was a kind gift from Dr Pelagia Derizioti (Max Planck Institute for Psycholinguistics). The YFP was exchanged with mCherry for this study, and another Tbr1 plasmid with no fluorescent tag was also generated.
Sequence-based predictions of mutation effects
Phylogenetic analysis was performed using a previously published alignment (Mukherjee et al. 2010). CASK reference sequence NP_003679.2 was used for all analyses. The conservation score for CASKG659D was calculated with ConSurf (Ashkenazy et al. 2010) using the Uniref90 database, the BLOSUM62 matrix, and MUSCLE for multiple sequence alignment (MSA). Clustal Omega v1.1.0 (Goujon et al. 2010), (http://www.ebi.ac.uk/Tools/msa/clustalo/), using default settings, was used to generate a 30-sequence MSA that was analyzed in Jalview (Waterhouse et al. 2009).
Prediction of whether a CASKG659D mutation is pathogenic or non-pathogenic was done using different webserver-based algorithms: i-MutantDDG-Seq 3.0 (Capriotti et al. 2005), ScPred (Dosztanyi et al. 1997), PhD-SNP v. 2.0.6 (Capriotti et al. 2006), PolyPhen-2, v. 2.0.22 (Adzhubei et al. 2010), SIFT, v. 4.0.3 (Kumar et al. 2009), SNAP, v. 1.0.8 (Bromberg and Rost 2007), PON-P (Olatubosun et al. 2012), and PMut (Ferrer-Costa et al. 2004).
Structure-based algorithm predictions of mutation effects
A homology model of CASK’s SH3-GuK domain (LaConte et al. 2014) was used for all algorithms requiring structure data. Changes in free energy (∆∆G) between wild-type and CASKG659D were measured using PoPMuSiC (Dehouck et al. 2009; Dehouck et al. 2011), Eris (Yin et al. 2007), and FoldX 4.0 (Schymkowitz et al. 2005).
Modeling of mutations on a SH3-GuK domain model
The UCSF Chimera software package (Pettersen et al. 2004) was used for molecular visualization, editing, and analysis. Chimera’s Rotamers tool was used to replace the native glycine residue at the position analogous to 659 with an aspartate in the previously published homology model of CASK’s SH3-GuK domain (LaConte et al. 2014). The aspartate rotamer from the Dunbrack backbone-dependent rotamer library with the highest probability was chosen. Chimera’s “Find Clashes/Contacts” tool was used to calculate contacts, defined as pairs of atoms with a separation between their van der Waals radii of less than or equal to 0.4 Å, ignoring intra-residue contacts and contacts of pairs 4 or fewer bonds apart.
Molecular dynamics simulations and analysis
Molecular dynamics simulations were performed using the program GROMACS 5.1.3 (Abraham et al. 2015) on the above-mentioned SH3-GuK domain homology model and corresponding G659D mutant. The AMBER99SB-ILDN force field (Lindorff-Larsen et al. 2010) was used for all simulations. Both structures were solvated with a four-point water model (TIP4P) in an explicit rhombic dodecahedron water box (solute box distance of 1.0 nm) under periodic boundary conditions, with charges neutralized by chloride ions. Starting structures were energy minimized until convergence at Fmax < 1000 kJ/mol/nm. A 200 ps position-restrained NVT equilibration simulation was run for water relaxation at 300K using a modified Berendsen (velocity rescaling) thermostat, followed by a 200 ps NPT equilibration simulation using the Parrinello-Rahman barostat for pressure coupling. After equilibration, an unrestrained 100 ns NPT molecular dynamics simulation was run. Three trajectories initiated with different random seeds were run for each protein structure.
Trajectories were post-processed to correct for periodicity. Analysis of trajectories included calculation of root mean square deviation (RMSD) of the protein backbone from the starting structure at each trajectory time point using the GROMACS rms command and calculation of the radius of gyration at each trajectory time point using the GROMACS gyrate command. The root mean square fluctuation (RMSF) of all alpha carbons from each of the three trajectories was calculated using the rmsf command in GROMACS. B-factors for each residue were calculated from the RMSF values using the equation: B-factor = (8Π2/3) x (RMSF)2. B-factors from all three trajectories were averaged. Cluster analysis of the three concatenated molecular dynamics trajectories (excluding the first 10 ns of each trajectory) for the wild-type and G659D SH3-GuK domains, respectively, was performed using the GROMACS cluster command with a 3Å cutoff employing the gromos algorithm (Daura et al. 1999).
Homology model of CASK’s PDZ-SH3-GuK supramodule
A homology model of CASK’s PDZ-SH3-GuK supramodule structure was created using the Modeller interface within Chimera (Sali and Blundell 1993), with the PALS1 crystal structure as template (4wsi.pdb; (Li et al. 2014)). Loop modeling was done for missing residues. Ten homology models were generated, and the model with the lowest zDOPE was selected.
Cell culture and imaging
Human embryonic kidney (HEK293FT) cells (ATCC) were plated on poly-L-lysine coated coverslips in 24-well plates and maintained in DMEM containing 10% fetal bovine serum. Cells were transfected with 0.5 µg of described plasmid DNA using the calcium phosphate method. Twenty hours post-transfection, cells were washed twice with phosphate buffered saline and fixed using a 4% paraformaldehyde solution. Coverslips were then mounted on microscope slides using Vectashield (Vector Laboratories Inc.) and visualized using confocal laser scanning microscopy (ZEISS Axio Examiner.Z1 LSM 710).
Neuronal culture was done as described previously (Chavan et al. 2015; LaConte et al. 2016a). Briefly, cortices from newborn pups were dissected out in ice-cold Hank’s balanced salt solution. The cortices were digested with 0.25% trypsin for 20 min at 37 °C. Trypsin was inactivated by using DMEM with 10% FBS. Digested pieces of cortices were then triturated using a fire-polished Pasteur pipette and filtered through a 100 μm nylon cell strainer (Corning, cat. no. 431752). The cells were centrifuged and resuspended in complete neuronal media (Lonza PNGM™ SingleQuots, cat. no. CC-4462 and PNBM media) and plated on polylysine-coated coverslips. The neurons were transfected using modified calcium phosphate technique on days in vitro 7 (DIV7) as described in (Chavan et al. 2015; Mukherjee et al. 2008).Cells were fixed with 4% PFA, mounted with Vectashield™ and imaged 24 hours after transfection.
To immunostain for the FLAG tag, the anti-FLAG M2 antibody was used following a protocol described previously (LaConte et al. 2016b). Immunostaining for CASK was done with a CASK antibody from Neuromab (clone number K56A/50). Tbr1 immunostaining was done with Anti-Tbr1 antibody from Millipore (AB2261).
The percentage of transfected cells with visible aggregates was counted using the cell counter plugin of the FIJI distribution of Image J (Schindelin et al. 2012). Total cells and cells containing aggregates were visually identified and tallied from 8 individual images at 20x magnification, and percent of total cells containing aggregates was then calculated from each image for each mutation and averaged. Colocalization analysis was performed using the Colocalization Threshold plugin in FIJI; cells containing visible aggregates were excluded from analysis, and only cells containing both expressed proteins were included. Twenty-two images (63X) from three different sets of experiments were analyzed from each condition, and colocalization coefficients and standard deviations were calculated.
Pull-down assay using anti-FLAG M2 beads
HEK293FT cells maintained in a 6-well plate were transfected with 2 μg GFP-CASK, GFP-CASKG659D, or GFP-CASKW919R plasmid DNA and co-transfected with 5 μg of Mint1-FLAG plasmid. After 48h, cells were harvested and solubilized in PBS containing 1% Triton X-100, 2 mM EDTA and protease inhibitors (aprotinin, leupeptin, pepstatin, PMSF). Lysate was then centrifuged at 15,000 rpm for 15 min at 4°C. Supernatant was incubated on a rocker for 2h at 4°C with washed anti-FLAG M2 Affinity beads (30 μl per reaction; Sigma). Beads were washed three times with TBS (50 mM Tris–HCl, pH 7.4; 150 mM NaCl) and boiled in SDS sample buffer. Protein samples were separated using SDS-PAGE and immunoblotted for specific proteins as described.
Pull-down assay using GST-neurexin cytoplasmic tail (GST-NX) and GST beads
HEK293FT cells maintained in 100mm2 plates were transfected with 10 μg GFP-CASK, GFP-CASKG659D, GFP-CASKM519T, or GFP-CASKW919R plasmid. After 48h, cells were harvested and solubilized in PBS containing 1% Triton X-100, 0.5% sodium deoxycholate, 2 mM EDTA, 1mM EGTA and protease inhibitors (aprotinin, leupeptin, pepstatin, PMSF). Lysate was then centrifuged at 15,000 rpm for 15 min at 4°C. Supernatant was incubated on a rocker for 2h at 4°C with washed GST-NX or GST beads (30 μl per reaction; as described in (LaConte et al. 2016b). Beads were washed three times with the above mentioned buffer and boiled in SDS sample buffer. Protein samples were separated using SDS-PAGE and immunoblotted for specific proteins as described.
Supplementary Material
Acknowledgement:
The current study is supported by R01EY024712 from the NIH’s National Eye Institute to KM. We gratefully acknowledge the participation and cooperation of families of children with CASK mutation in our studies. We thank Prof. Thomas Südhof for providing pCMV5-CASK plasmid, Dr. Pelagia Derizioti for providing the TBR1 plasmid and Dr. Michael Fox for critically reading the manuscript.
Footnotes
Conflict statement: On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2: 19–25. doi: 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nature methods 7: 248–9. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW (1992) Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 51: 1229–39. [PMC free article] [PubMed] [Google Scholar]
- Apic G, Huber W, Teichmann SA (2003) Multi-domain protein families and domain pairs: comparison with known structures and a random model of domain recombination. J Struct Funct Genomics 4: 67–78. [DOI] [PubMed] [Google Scholar]
- Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic acids research 38: W529–33. doi: 10.1093/nar/gkq399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Schoch S, Ho A, Nadasy KA, Liu X, Zhang W, Mukherjee K, Nosyreva ED, Fernandez-Chacon R, Missler M, Kavalali ET, Sudhof TC (2007) Deletion of CASK in mice is lethal and impairs synaptic function. Proc Natl Acad Sci U S A 104: 2525–30. doi: 0611003104[pii] 10.1073/pnas.0611003104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey S, Nickson AA, Clarke J (2008) Studying the folding of multidomain proteins. HFSP J 2: 365–77. doi: 10.2976/1.2991513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Sara Y, Mozhayeva M, Atasoy D, Liu X, Kavalali ET, Sudhof TC (2002) SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science 297: 1525–31. doi: 10.1126/science.1072356 [DOI] [PubMed] [Google Scholar]
- Bromberg Y, Rost B (2007) SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic acids research 35: 3823–35. doi: 10.1093/nar/gkm238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burglen L, Chantot-Bastaraud S, Garel C, Milh M, Touraine R, Zanni G, Petit F, Afenjar A, Goizet C, Barresi S, Coussement A, Ioos C, Lazaro L, Joriot S, Desguerre I, Lacombe D, des Portes V, Bertini E, Siffroi JP, de Villemeur TB, Rodriguez D (2012) Spectrum of pontocerebellar hypoplasia in 13 girls and boys with CASK mutations: confirmation of a recognizable phenotype and first description of a male mosaic patient. Orphanet J Rare Dis 7: 18. doi: 1750-1172-7-18[pii] 10.1186/1750-1172-7-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butz S, Okamoto M, Sudhof TC (1998) A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell 94: 773–82. [DOI] [PubMed] [Google Scholar]
- Capriotti E, Calabrese R, Casadio R (2006) Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 22: 2729–34. doi: 10.1093/bioinformatics/btl423 [DOI] [PubMed] [Google Scholar]
- Capriotti E, Fariselli P, Casadio R (2005) I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic acids research 33: W306–10. doi: 10.1093/nar/gki375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavan V, Willis J, Walker SK, Clark HR, Liu X, Fox MA, Srivastava S, Mukherjee K (2015) Central presynaptic terminals are enriched in ATP but the majority lack mitochondria. PLoS One 10: e0125185. doi: 10.1371/journal.pone.0125185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DL, Cohen AR, Anderson JM, Brunger AT (1998) Crystal structure of the hCASK PDZ domain reveals the structural basis of class II PDZ domain target recognition. Nat Struct Biol 5: 317–25. [DOI] [PubMed] [Google Scholar]
- Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, Mark AE (1999) Peptide folding: When simulation meets experiment. Angewandte Chemie-International Edition 38: 236–240. doi: Doi 10.1002/(Sici)1521–3773(19990115)38:1/2<236::Aid-Anie236>3.3.Co;2-D [Google Scholar]
- Dehouck Y, Grosfils A, Folch B, Gilis D, Bogaerts P, Rooman M (2009) Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics 25: 2537–43. doi: 10.1093/bioinformatics/btp445 [DOI] [PubMed] [Google Scholar]
- Dehouck Y, Kwasigroch JM, Gilis D, Rooman M (2011) PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC bioinformatics 12: 151. doi: 10.1186/1471-2105-12-151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deriziotis P, O’Roak BJ, Graham SA, Estruch SB, Dimitropoulou D, Bernier RA, Gerdts J, Shendure J, Eichler EE, Fisher SE (2014) De novo TBR1 mutations in sporadic autism disrupt protein functions. Nat Commun 5: 4954. doi: 10.1038/ncomms5954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi Z, Fiser A, Simon I (1997) Stabilization centers in proteins: identification, characterization and predictions. Journal of molecular biology 272: 597–612. doi: 10.1006/jmbi.1997.1242 [DOI] [PubMed] [Google Scholar]
- Fallon L, Moreau F, Croft BG, Labib N, Gu WJ, Fon EA (2002) Parkin and CASK/LIN-2 associate via a PDZ-mediated interaction and are co-localized in lipid rafts and postsynaptic densities in brain. The Journal of biological chemistry 277: 486–91. doi: 10.1074/jbc.M109806200 [DOI] [PubMed] [Google Scholar]
- Ferrer-Costa C, Orozco M, de la Cruz X (2004) Sequence-based prediction of pathological mutations. Proteins 57: 811–9. doi: 10.1002/prot.20252 [DOI] [PubMed] [Google Scholar]
- Flanagan SE, Patch AM, Ellard S (2010) Using SIFT and PolyPhen to Predict Loss-of-Function and Gain-of-Function Mutations. Genetic Testing and Molecular Biomarkers 14: 533–537. doi: 10.1089/gtmb.2010.0036 [DOI] [PubMed] [Google Scholar]
- Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic acids research 38: W695–9. doi: 10.1093/nar/gkq313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett A, Tarpey PS, Licata A, Cox J, Whibley A, Boyle J, Rogers C, Grigg J, Partington M, Stevenson RE, Tolmie J, Yates JR, Turner G, Wilson M, Futreal AP, Corbett M, Shaw M, Gecz J, Raymond FL, Stratton MR, Schwartz CE, Abidi FE (2010) CASK mutations are frequent in males and cause X-linked nystagmus and variable XLMR phenotypes. Eur J Hum Genet 18: 544–52. doi: ejhg2009220[pii] 10.1038/ejhg.2009.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata Y, Butz S, Sudhof TC (1996) CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J Neurosci 16: 2488–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh YP, Wang TF, Yang FC, Sheng M (2000) Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature 404: 298–302. doi: 10.1038/35005118 [DOI] [PubMed] [Google Scholar]
- Hsueh YP, Yang FC, Kharazia V, Naisbitt S, Cohen AR, Weinberg RJ, Sheng M (1998) Direct interaction of CASK/LIN-2 and syndecan heparan sulfate proteoglycan and their overlapping distribution in neuronal synapses. J Cell Biol 142: 139–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TN, Hsueh YP (2017) Calcium/calmodulin-dependent serine protein kinase (CASK), a protein implicated in mental retardation and autism-spectrum disorders, interacts with T-Brain-1 (TBR1) to control extinction of associative memory in male mice. Journal of Psychiatry & Neuroscience 42: 37–47. doi: 10.1503/jpn.150359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Whitfield CW, Kim SK (1998) The LIN-2/LIN-7/LIN-10 complex mediates basolateral membrane localization of the C. elegans EGF receptor LET-23 in vulval epithelial cells. Cell 94: 761–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols 4: 1073–81. doi: 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- LaConte L, Mukherjee K (2013) Structural constraints and functional divergences in CASK evolution. Biochem Soc Trans 41: 1017–22. doi: 10.1042/BST20130061 [DOI] [PubMed] [Google Scholar]
- LaConte LE, Chavan V, Liang C, Willis J, Schonhense EM, Schoch S, Mukherjee K (2016a) CASK stabilizes neurexin and links it to liprin-alpha in a neuronal activity-dependent manner. Cellular and molecular life sciences : CMLS 73: 3599–621. doi: 10.1007/s00018-016-2183-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaConte LE, Chavan V, Mukherjee K (2014) Identification and glycerol-induced correction of misfolding mutations in the X-linked mental retardation gene CASK. PLoS One 9: e88276. doi: 10.1371/journal.pone.0088276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaConte LEW, Chavan V, Liang C, Willis J, Schonhense EM, Schoch S, Mukherjee K (2016b) CASK stabilizes neurexin and links it to liprin-alpha in a neuronal activity-dependent manner. Cellular and Molecular Life Sciences 73: 3599–3621. doi: 10.1007/s00018-016-2183-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wei Z, Yan Y, Wan Q, Du Q, Zhang M (2014) Structure of Crumbs tail in complex with the PALS1 PDZ-SH3-GK tandem reveals a highly specific assembly mechanism for the apical Crumbs complex. Proc Natl Acad Sci U S A 111: 17444–9. doi: 10.1073/pnas.1416515111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Kerr A, Qiu Y, Cristofoli F, Van Esch H, Fox MA, Mukherjee K (2017) Optic Nerve Hypoplasia Is a Pervasive Subcortical Pathology of Visual System in Neonates. Invest Ophthalmol Vis Sci 58: 5485–5496. doi: 10.1167/iovs.17-22399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78: 1950–8. doi: 10.1002/prot.22711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Sudhof TC, Bezprozvanny I (1999) Association of neuronal calcium channels with modular adaptor proteins. J Biol Chem 274: 24453–6. [DOI] [PubMed] [Google Scholar]
- McGee AW, Dakoji SR, Olsen O, Bredt DS, Lim WA, Prehoda KE (2001) Structure of the SH3-guanylate kinase module from PSD-95 suggests a mechanism for regulated assembly of MAGUK scaffolding proteins. Mol Cell 8: 1291–301. [DOI] [PubMed] [Google Scholar]
- Michaud JL, Lachance M, Hamdan FF, Carmant L, Lortie A, Diadori P, Major P, Meijer IA, Lemyre E, Cossette P, Mefford HC, Rouleau GA, Rossignol E (2014) The genetic landscape of infantile spasms. Hum Mol Genet 23: 4846–58. doi: 10.1093/hmg/ddu199 [DOI] [PubMed] [Google Scholar]
- Moog U, Kutsche K, Kortum F, Chilian B, Bierhals T, Apeshiotis N, Balg S, Chassaing N, Coubes C, Das S, Engels H, Van Esch H, Grasshoff U, Heise M, Isidor B, Jarvis J, Koehler U, Martin T, Oehl-Jaschkowitz B, Ortibus E, Pilz DT, Prabhakar P, Rappold G, Rau I, Rettenberger G, Schluter G, Scott RH, Shoukier M, Wohlleber E, Zirn B, Dobyns WB, Uyanik G (2011) Phenotypic spectrum associated with CASK loss-of-function mutations. J Med Genet 48: 741–51. doi: jmedgenet-2011-100218[pii] 10.1136/jmedgenet-2011-100218 [DOI] [PubMed] [Google Scholar]
- Moog U, Uyanik G, Kutsche K (2013) CASK-Related Disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K (eds) GeneReviews(R), Seattle (WA: ) [Google Scholar]
- Mukherjee K, Sharma M, Jahn R, Wahl MC, Sudhof TC (2010) Evolution of CASK into a Mg2+-sensitive kinase. Science signaling 3: ra33. doi: 10.1126/scisignal.2000800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee K, Sharma M, Urlaub H, Bourenkov GP, Jahn R, Sudhof TC, Wahl MC (2008) CASK Functions as a Mg2+-independent neurexin kinase. Cell 133: 328–39. doi: 10.1016/j.cell.2008.02.036S0092-8674(08)00287-0[pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee K, Slawson JB, Christmann BL, Griffith LC (2014) Neuron-specific protein interactions of Drosophila CASK-beta are revealed by mass spectrometry. Front Mol Neurosci 7: 58. doi: 10.3389/fnmol.2014.00058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najm J, Horn D, Wimplinger I, Golden JA, Chizhikov VV, Sudi J, Christian SL, Ullmann R, Kuechler A, Haas CA, Flubacher A, Charnas LR, Uyanik G, Frank U, Klopocki E, Dobyns WB, Kutsche K (2008) Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat Genet 40: 1065–7. doi: 10.1038/ng.194 [DOI] [PubMed] [Google Scholar]
- Nix SL, Chishti AH, Anderson JM, Walther Z (2000) hCASK and hDlg associate in epithelia, and their Src homology 3 and guanylate kinase domains participate in both intramolecular and intermolecular interactions. Journal of Biological Chemistry 275: 41192–41200. doi: DOI 10.1074/jbc.M002078200 [DOI] [PubMed] [Google Scholar]
- Olatubosun A, Valiaho J, Harkonen J, Thusberg J, Vihinen M (2012) PON-P: Integrated predictor for pathogenicity of missense variants. Human mutation 33: 1166–74. doi: 10.1002/humu.22102 [DOI] [PubMed] [Google Scholar]
- Pak C, Danko T, Zhang YS, Aoto J, Anderson G, Maxeiner S, Yi F, Wernig M, Sudhof TC (2015) Human Neuropsychiatric Disease Modeling using Conditional Deletion Reveals Synaptic Transmission Defects Caused by Heterozygous Mutations in NRXN1. Cell Stem Cell 17: 316–328. doi: 10.1016/j.stem.2015.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Chen J, Yu J, Yu H, Zhang M (2011) The structure of the PDZ3-SH3-GuK tandem of ZO-1 protein suggests a supramodular organization of the membrane-associated guanylate kinase (MAGUK) family scaffold protein core. J Biol Chem 286: 40069–74. doi: 10.1074/jbc.C111.293084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry 25: 1605–12. doi: 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Reissner C, Missler M (2014) MAGUKs end a tale of promiscuity. Proc Natl Acad Sci U S A 111: 17350–1. doi: 10.1073/pnas.1420387111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda M, Ferrer-Costa C, Meyer T, Perez A, Camps J, Hospital A, Gelpi JL, Orozco M (2007) A consensus view of protein dynamics. Proceedings of the National Academy of Sciences of the United States of America 104: 796–801. doi: 10.1073/pnas.0605534104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. Journal of molecular biology 234: 779–815. doi: 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nature methods 9: 676–82. doi: 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L (2005) The FoldX web server: an online force field. Nucleic acids research 33: W382–8. doi: 10.1093/nar/gki387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, McMillan R, Willis J, Clark H, Chavan V, Liang C, Zhang H, Hulver M, Mukherjee K (2016) X-linked intellectual disability gene CASK regulates postnatal brain growth in a non-cell autonomous manner. Acta Neuropathol Commun 4: 30. doi: 10.1186/s40478-016-0295-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ (2009) Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25: 1189–91. doi: 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z, Zheng S, Spangler SA, Yu C, Hoogenraad CC, Zhang M (2011) Liprin-mediated large signaling complex organization revealed by the liprin-alpha/CASK and liprin-alpha/liprin-beta complex structures. Molecular cell 43: 586–98. doi: 10.1016/j.molcel.2011.07.021 [DOI] [PubMed] [Google Scholar]
- Yin S, Ding F, Dokholyan NV (2007) Eris: an automated estimator of protein stability. Nature methods 4: 466–7. doi: 10.1038/nmeth0607-466 [DOI] [PubMed] [Google Scholar]
- Zeng M, Ye F, Xu J, Zhang M (2017) PDZ Ligand Binding-Induced Conformational Coupling of the PDZ-SH3-GK Tandems in PSD-95 Family MAGUKs. J Mol Biol doi: 10.1016/j.jmb.2017.11.003 [DOI] [PubMed]
- Zweier C, de Jong EK, Zweier M, Orrico A, Ousager LB, Collins AL, Bijlsma EK, Oortveld MA, Ekici AB, Reis A, Schenck A, Rauch A (2009) CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet 85: 655–66. doi: 10.1016/j.ajhg.2009.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.