

Abstract
PU.1 is an ETS-family transcription factor that plays a broad range of roles in hematopoiesis. A direct regulator of myeloid, dendritic-cell, and B cell functional programs, and a well-known antagonist of terminal erythroid cell differentiation, it is also expressed in the earliest stages of T-cell development of each cohort of intrathymic pro-T cells. Its expression in this context appears to give T-cell precursors initial, transient access to myeloid and dendritic cell developmental competence and therefore to represent a source of antagonism or delay of T-cell lineage commitment. However, it has remained uncertain until recently why T-cell development is also intensely dependent upon PU.1. Here, we review recent work that sheds light on the molecular biology of PU.1 action across the genome in pro-T cells and identifies the genes that depend on PU.1 for their correct regulation. This work indicates modes of chromatin engagement, pioneering, and cofactor recruitment (“coregulator theft”) by PU.1 as well as gene network interactions that not only affect specific target genes but also have system-wide regulatory consequences, amplifying the impact of PU.1 beyond its own direct binding targets. The genes directly regulated by PU.1 also suggest a far-reaching transformation of cell biology and signaling potential between the early stages of T-cell development when PU.1 is expressed and when it is silenced. These cell-biological functions can be important to distinguish fetal from adult T-cell development and have the potential to illuminate aspects of thymic function that have so far remained the most mysterious.
Keywords: transcription factor, developmental gene regulation, chromatin, T lymphocyte development, thymus, gene network, cell signaling, hematopoiesis
Introduction
PU.1 Expression in Precursors of T Cells
PU.1, encoded by the Spi1 gene, is an ETS-family transcription factor with multiple roles in hematopoiesis. It is a lineage-specifying transcription factor that positively regulates many genes in the macrophage, granulocyte, dendritic-cell and B-cell lineages. Expressed at highest levels in monocytes/macrophages, at low or moderate levels in B cells, and transiently in early erythroid precursors, its action is also important or indispensable for sustained generation of all known hematopoietic precursors that have lymphoid developmental potentials (1–9). Thus, B, NK, and T cell development are all affected by defects in PU.1 activity, despite partial complementation by the related factor SpiB that is also activated in B-lineage precursors. Much is known about how PU.1 finds and binds to its sites in the DNA, typically (A/G)AGGAAGTG motifs [e.g., (10, 11)], and it is known to be able to bind either as a pioneer factor which displaces nucleosomes to open sites for other factors (12), or as a collaboration-dependent partner in binding complexes, either with activation-dependent factors like NF-κB or with lineage-defining partners like C/EBPα (or β) or IRF4/8 (13–15) [reviewed by (16–18)].
In myeloid, dendritic, and B lineage cells, PU.1 is a major contributor to the positive regulation of genes that establish lineage-specific identity (4, 17, 19). At the same time, PU.1 can work in an all-or-none gene network switch through mutual antagonism with GATA-1 (20–24), which has been much discussed as a possible mechanism for the irreversibility of erythro-myeloid lineage commitment [(25–29); but also see (30, 31)]. Nevertheless, the developmental scope of PU.1 activity is surprisingly broad, and one of its unexpected domains of action is in the early stages of T-cell development, in both the fetal and the postnatal mammalian thymus. To examine what it does in pro-T cells, this review focuses on recent data based on mouse T-cell development, mostly as it occurs in the postnatal thymus or from late fetal progenitors. The final section places these mechanisms in the context of the variants of T-cell development that characterize different ontogenic stages.
Most mature T cells do not express any detectable PU.1 protein or Spi1 transcripts at all, and the T-cell developmental gene network sharply downregulates Spi1 in precursors of αβ T cells before the expression of rearranged Tcrb genes, i.e., before any TCR-dependent steps of T cell development. However, the precursors that give rise to committed T cells express PU.1 at both RNA and protein levels for multiple cell divisions after these cells begin to differentiate in the thymus (32, 33). A summary of early T-cell developmental stages, is shown in Figure 1, with the approximate pattern of PU.1 expression marked. The downregulation of PU.1 occurs during the transition to commitment, between the DN2 (DN = double negative for CD4 and CD8, and Kit+ CD44+ CD25+) and DN3 (DN, and Kitlow CD44low CD25+) stages. This expression timing relative to other developmentally regulated transcription factors is conserved between human and mouse (35, 36), and as in mouse (37), the downregulation of PU.1 is important to avoid malignancy in human T cells: a specifically aggressive class of human T-acute lymphoblastic leukemias results from translocations that promote abnormally sustained and elevated PU.1 expression (38). In the mouse, where lineage commitment has been studied in depth, there is good agreement between the cells' natural loss of access to the dendritic cell and granulocyte programs, on the one hand, and the timing of PU.1 downregulation, on the other hand (33, 39–42). This is part of a general downregulation of stem/progenitor associated regulatory genes (“phase 1 genes”) (34, 43) and a major reorganization of active chromatin and chromatin interactions, genome-wide, that occurs during this transition (44). One important question is what role PU.1 itself may have in controlling the onset of this transformation.
Figure 1.
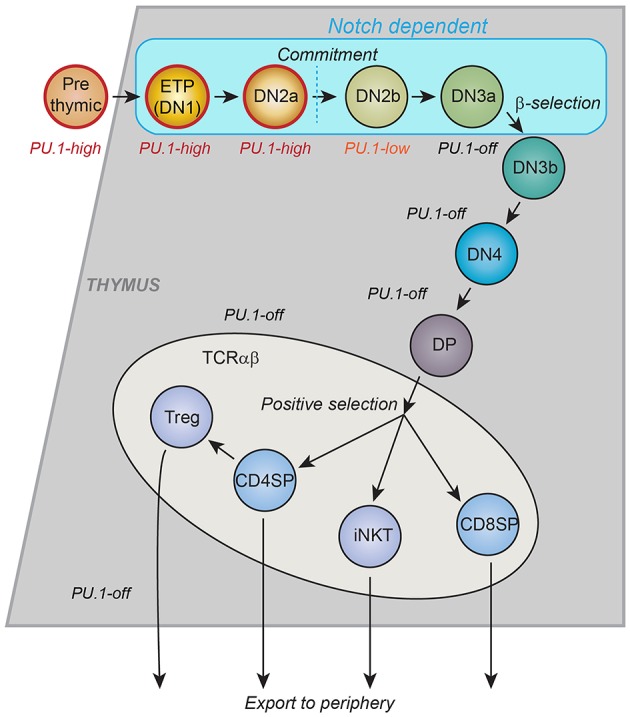
Schematic of T-cell development in the thymus. Major landmarks for T cell developmental stages are CD4 and CD8. CD4− CD8−: “DN”; CD4+ CD8+: “DP,” CD4+ CD8−: “CD4SP”; CD4− CD8+: “CD8SP.” All events described in this review occur within the DN stages, which are divided by other markers. ETP: Kit++ CD44+ CD25−; DN2a: Kit++ CD44+ CD25+; DN2b: Kit+ CD44+ CD25+; DN3a: Kit− CD44− CD25+ CD28−; DN3b: Kit− CD44− CD25+ CD28+; DN4: Kit− CD44− CD25− CD28+. Stages up through DN3a do not depend on T-cell receptor gene rearrangement status and are called “Pro-T cells.” Many cell cycles occur between the ETP stage and commitment, more in post-natal T cell development and fewer in fetal T-cell development. The trends in PU.1 expression, the timing of intrinsic cell commitment to the T-cell lineage, and the stages that depend on Notch signaling from the thymic microenvironment are shown. Gray or blue regions depict thymic cortex. Lighter region depicts thymic medulla, where final maturation of developing T cells takes place. CD4SP: maturing T helper cells. CD8SP: maturing T cytotoxic cells. Treg: thymically derived regulatory T cells. iNKT: Natural Killer T cells with invariant T cell receptors [Schematic adapted from Rothenberg et al. (34)].
PU.1 as an Obstacle to T-Cell Lineage Commitment
The particular interest in PU.1 itself emerged from the hypothesis that it could well be responsible for maintaining the “bridge” to myeloid and dendritic alternative fates before commitment, because of its known roles in many of these alternatives (and in B cells) but not in T cells (45). This hypothesis was supported by the finding that re-expression of PU.1 after T-cell commitment turns on myeloid genes and readily transforms later pro-T cells into dendritic cells, macrophages, or promyelocytic-like cells (46–50). There is a very close relationship between the cells that naturally express PU.1 in the thymus and those that readily exhibit myeloid or dendritic potential in a variety of permissive cell transfer models, in vivo or in vitro. Whereas ETP and DN2 cells can generate myeloid cells if removed from the thymus, pro-T cells that have differentiated past the stage of PU.1 expression in the thymus (i.e., from the DN2 stage to the DN3 stage or later) do not make myeloid cells under these conditions, and this difference between stages up to DN2 and stages from DN3 onward has been a highly consistent observation (33, 40, 41, 51–56). Why, then, do PU.1-expressing early T cell precursors within the thymus almost all go on to produce T cells, not myeloid cells, under normal in vivo conditions (57)? A potential explanation was provided by a key feature of the PU.1 effect: namely, that PU.1 actions are Notch-sensitive. Even artificially high-level PU.1 could only redirect the differentiation of the cells to myeloid or dendritic fates if Notch signaling were reduced (49, 50, 58). In primary fetal-derived pro-T cells and in a DN3-like cell line, the particular genes affected by a given, fixed level of PU.1 in the cells depended strongly on the strength of Notch signaling being induced in the cells at the time (58). Notch ligands are the most important of all the environmental signals that the thymus stroma provides to developing T cells, apparently across all vertebrates (59–61), and Notch activated target genes like Hes1 are expressed throughout the pro-T cell phases (ETP to DN3), until T-cell receptor (TCR) gene rearrangement (62–64) [reviewed in (65)]. Thus, throughout the stages when PU.1 is expressed, the Notch signaling driven in the normal thymus environment could guarantee that PU.1 expression would confer only a potential for differentiation to alternative fates, which the cells would not actually follow unless the thymic environment were disrupted. The silencing of Spi1 expression and permanent loss of PU.1 protein from the cells at a later stage of differentiation would then make their loss of myeloid potential unconditional.
The question raised by such results, however, was why PU.1 should continue to be expressed at all by cells once they entered the thymus. Population dynamic models imply that the stages when PU.1 is expressed occupy a minimum of 7–10 intrathymic cell divisions of pro-T cells (39, 66, 67). If PU.1 was evolutionarily selected to be expressed over such an extended period, it might be playing an important role in pro-T cells, and this could be despite or because of the Notch signaling conditions that were preventing it from diverting the cells to a non-T fate. The earliest stages of T cell development are not well understood, and it until recently it was not obvious what function could be important to the cells at this time a priori, other than proliferation. In the past 5 years, however, a detailed look at the molecular biology of PU.1 action on the genome in pro-T cells has revealed much about the ways that PU.1 works, the complex cell biology of the early precursor states, and previously under-appreciated principles of transcription factor systems operating in development.
Effects of PU.1 Loss on T-cell Development: the Cellular View
A Vital Role for PU.1 in Prethymic T-Cell Progenitors
Disruption of PU.1 has long been known to eliminate or greatly inhibit T-cell development, based on the dramatic phenotypes from the first lines of PU.1-knockout (Spi1 knockout) mice with unconditional, germline mutations (68–71). The question has been how to interpret this severe effect, i.e., whether it is due to loss of a function within the T-cell program itself, or whether it simply reflects a loss of input cells to the pathway. One problem was originally the lethality of the hematopoietic phenotype (death either in late fetal development or immediately after birth), but even when conditional knockouts were developed (2, 72), this remained problematic. All the hematopoietic progenitors that generate either B or T cells appear to originate from PU.1-expressing, PU.1-dependent prethymic cells (2, 27); PU.1 is directly required to maintain the expression of the cytokine receptor Flt3 that is indispensable for progenitors with B and T cell potentials (73). Thus, in postnatal mice, although T cell development is much more severely affected by PU.1 deletion than neutrophil development (2), the effect could still be prethymic. In stark contrast, if Spi1 is conditionally deleted in T-lineage cells only after the cells have passed the DN2 stage, there are very modest effects on T cells as a whole, apparently limited to selective reduction of IL-9-producing T-cells (74), and some loss of restraint on γδ T cells and T follicular helper cell activity (75, 76). Is PU.1 actually needed within the T-cell pathway for T-cell development at all, or is it simply needed to guarantee a supply of prethymic progenitors?
Addressing this question in vivo was handicapped by difficulties in the methods of inducing stage-specific Spi1 deletion. The question about a transient role for PU.1, but one which might have strong effects on viability, makes it important to have high penetrance and high synchrony of deletion as well as fine developmental stage control, both of the deletion and through the analysis afterwards. The widely-used T-cell specific Cre expression constructs that might be appropriate for thymocyte analysis, pLck-Cre and CD4-Cre, actually begin to be expressed too late: pLck-Cre turns on just as PU.1 is turning off, and CD4-Cre is expressed even later, after the rearrangement of the first TCR genes. Constructs like Il7r-Cre or Rag1-Cre, which may have prethymic expession but are also expressed much more strongly during later pro-T cell stages, could make output cell phenotypes difficult to interpret because of uncertainty about when the deletion actually has become complete. Fortunately, pro-T-cell differentiation cultures on OP9-DL1 or OP9-DL4 stroma that constitively present Notch ligands (77, 78) are ideal for examining the stages relevant to PU.1 function, and a variety of efficient retroviral vectors can transduce the cells at these stages with high efficiency to introduce gain or loss of function agents. These systems have proven to be valuable tools not only for verifying the coarse-grained roles of PU.1 in pro-T cells, but also for investigation of their molecular mechanisms.
PU.1 Promotes Proliferation While Slowing Differentiation of Pro-T Cells
PU.1 is indeed important within the T-cell program as well as before thymic entry, as shown by using in vitro differentiation to provide conditions where PU.1 could be removed acutely in a synchronized cohort of precursors and the fates of the cells could be monitored immediately afterwards. In these studies, floxed Spi1 was disrupted in the input cells few days after T-lineage development had begun, using a Cre-encoding retroviral vector (79). The deletion of PU.1 reduced viable cell yield, but a co-transduced Bcl-xL transgene was added with Cre to prevent specific effects on development and proliferation from being masked by cell death. Similar results were obtained independently using Cas9 plus Spi1-specific guide RNA to delete PU.1, and supporting cell viability with a Bcl2 transgene (80). In both experimental setups, PU.1 disruption reduced T-cell precursor proliferation substantially as compared to controls. PU.1-deficient cells underwent fewer cycles per unit time than controls both in ETP stage and in DN2a/2b stages (79), suggesting that even once the cells have begun to express definitive T-lineage markers, they need PU.1 to sustain optimal proliferation. However, of the cells that were generated from PU.1-disrupted precursors, a substantially larger fraction progressed to DN3 stage than in control cells, over the same length of absolute developmental time, suggesting that they were liberated from a differentiation constraint (79, 80).
Thus, endogenous PU.1 does have a functional role within early T-cell development. It slows developmental progression of pro-T cells even as it supports their early proliferation. While this may seem paradoxical, it could fit well with a role to build the size of the pre-selection pool of T-cell precursors before they progress to commitment and then TCR gene rearrangement, so as to maximize TCR gene rearrangement diversity in the population as a whole before selection occurs (66, 81). The effect of PU.1 on proliferation is conditional and dose-dependent, however. While added PU.1 can enhance pro-T cell proliferation in response to cytokine cocktails containing high levels of Stem Cell Factor (Kit ligand) and Flt3 ligand or myeloid growth factors (38, 50), it strongly inhibits the proliferation of pro-T cells under conditions that do not reward the cells for lineage switching (49). Such dose dependent effects are common for transcription factors as for signaling molecules, in part because high concentrations of these factors bind to inappropriate genomic sites, leading to off-target effects. The target genes stimulated by PU.1 include both pro-proliferative and G1-prolonging cell cycle effectors, whereas some important proliferative genes are repressed when PU.1 levels are high (82, 83). Thus, both too much and too little PU.1 can have negative impacts on proliferation of the cells within a similar developmental time window.
The in vitro assays used to define these roles (discussed in more depth in the next section) are powerful because of the easy accessibility of the developing cells during differentiation and because of the ability to follow differentiation of a synchronized cohort of cells in absolute time. As described below, however, the genes most sensitively regulated by PU.1 in developing T cells suggest that this factor may be important to endow cells with additional functions as well, functions that may only contribute to their development specifically in the thymus in vivo.
Defining the PU.1 Regulome in Early T-cell Precursors
Cell Line and Primary-Cell Assay Systems for PU.1 Manipulation
To explain the roles of PU.1 in T cell development, it is crucial to take into account its developmental expression pattern. Its high expression in early-stage pro-T cells followed by downregulation during commitment means that its direct effects have to be correlated with developmental stage. Thus, any inferred role must be validated by developmental stage-dependence of putative target gene expression patterns or of chromatin features that characterize its binding sites. To look more closely at how PU.1 actually regulates specific target genes, acute gain and loss of function experiments are needed. Despite some overlap in occupancy, PU.1 binding sites and PU.1 binding partners are not the same in early T-cell precursors as in myeloid cells or B lineage cells (13, 80, 84, 85), a similar situation to its early role in erythroblasts (86). Therefore, these assay systems need to be based on pro-T cells (Figure 2). Exogenous PU.1 can easily be introduced into developing murine T-cell precursors using retroviral vectors for gain of function studies (46–50, 58, 80, 85). For loss of function, retrovirally transduced Cre can induce acute deletion in pro-T cells from Spi1fl/fl strain mice (79); and in Cas9-transgenic pro-T cells, retrovirally transduced guide RNAs (sgSpi1) can target rapid, biallelic disruption of the Spi1 locus (80, 85). Figure 2 introduces the way the primary-cell and cell-line models can be manipulated to relate experimental gain-of-function and loss-of-function PU.1 experiments to the normal dynamics of endogenous PU.1 expression.
Figure 2.
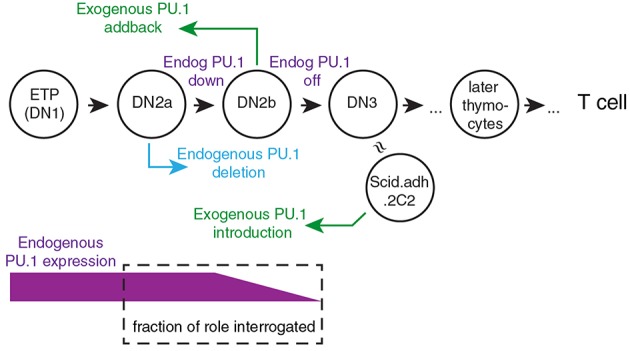
Framework for experimental perturbation studies to define functions of PU.1 in early thymic development. Stages of cells are as in Figure 1 (DN3: primarily DN3a). Exogenous PU.1 is added by retroviral transduction. Note the dependence of the PU.1 functions tested upon the timing of the experimental perturbation. Endogenous PU.1 can be deleted by Cas9 plus single-chain guide RNAs (sgRNA) against Spi1, or by introducing Cre into cells with floxed Spi1 alleles. PU.1 can also be neutralized by adding a dominant negative construct. The DN2a-DN2b interval is accessible to experimental perturbation. The approximate developmental stage represented by the Scid.adh.2C2 cell line (see text) is also shown [Schematic adapted from Ungerbäck et al. (85)].
A very useful model cell line, Scid.adh.2C2, has made it possible to study PU.1 gain of function in a pro-T cell-like context (47). These cells are convenient because they are readily transfectable, retrovirally transducible, and fast-growing, so that cell numbers are not limiting and the developmental baseline is mostly static, all major advantages for genomic comparisons. These cells were a subclone derived from the Scid.adh cell line (87) and are similar to developmentally arrested versions of committed DN3 pro-T cells, lacking any expression of endogenous PU.1 (47). Despite being an immortal cell line, these cells are developmentally transformed by introduction of exogenous PU.1. They respond in an all-or-none way to forced expression of PU.1, coordinately upregulating myeloid- or dendritic-cell associated genes and downregulating T-cell genes in a discrete fraction of the cells that increases with increasing levels of PU.1 (47, 58), resembling responses of primary fetal or postnatal pro-T cells (46–50, 58, 80, 85). The switch-like nature of this response was an important early clue to the regulatory circuit interaction between PU.1 and the Notch signaling pathway (49, 58).
Useful and informative as it is, this system is limited as a way to study the roles of endogenous PU.1 in vivo. The sites occupied by exogenous PU.1 in Scid.adh.2C2 cells overlap highly with the sites occupied by endogenous PU.1 in normal pro-T cells, but the match is by no means complete (85). Even with PU.1 transduction, Scid.adh.2C2 cells do not restore the full chromatin accessibility landscape of ETP and DN2a stage pro-T cells, and despite detectable upregulation of a few other early pro-T cell genes (e.g., Bcl11a and Lyl1), the transduced cells as a whole reactivate little of the program that forms the normal context for endogenous PU.1 activity in pro-T cells (58, 79, 85). Therefore, PU.1 has to be manipulated acutely in dynamically differentiating primary cells.
To focus the introduction of PU.1 into cells at a particular developmental stage, it has proven to be very useful to exploit the powerful in vitro T-cell development systems based on co-culture of primary-cell precursors on OP9-DLL1 (aka OP9-DL1) or OP9-DLL4 (OP9-DL4) stroma with IL-7 and Flt3L (78, 88), or similar systems using other stromal cell lines to express the Notch ligands DLL1 or DLL4. Either fetal-liver-derived precursors or adult bone marrow-derived precursors develop efficiently along the T cell lineage in these systems with strong proliferation through the stages around commitment, allowing the stages to be separated both by flow cytometric phenotypes and by absolute times of differentiation. In these open systems, the cells can be harvested easily at any time point, transduced with vectors, treated with drugs, and/or sorted, and then shifted to the same or a different culture condition for further development. These systems have been indispensable for deeper analysis of the molecular mechanisms that PU.1 uses to regulate development of pro-T cells. However, two issues have to be taken into account in these analyses, both arising from features that amplify the developmental impact of PU.1. These are reviewed in the next sections.
Developmental Challenges: Implications of a Gene Regulatory Network Switch
The ideal conceptual framework of PU.1 gain of function experiments is to start with pro-T cells that have recently turned off their endogenous PU.1 expression and to assess how their developmental state is affected by re-introducing PU.1 expression, comparing the impact of exogenous PU.1 with the pre-commitment gene expression pattern. Ideally in this scenario, restoring PU.1 after commitment should promote some aspect(s) of retrograde differentiation. Both Scid.adh.2C2 and normal DN2b/DN3 pro-T cells make strong responses to forced expression of PU.1, as noted above, and often the response includes downregulation of multiple later T-cell differentiation genes. Does this shed light on PU.1's natural role in earlier T-cell development, or is it simply an inhibitory artifact of overexpression? Clues that the gain-of-function phenotype is linked with a genuine role of PU.1 in earlier T-cell development come from PU.1's (re-)activation of a group of genes that are specifically associated with the early progenitor state, including Bcl11a, Mef2c, Hhex, and Lmo2 (58, 79, 85). Some of these are also upregulated in human T-acute lymphoblastic leukemias with highly expressed PU.1 fusions, as well (38).
However, the power of the response raises caveats about interpretation because of an important systemic feature of the PU.1 role in development. In primary pro-T cells and Scid.adh.2C2 cells, highly overexpressed PU.1 appears to inhibit Notch signaling, as measured by downregulation of Notch target genes and even Notch1 itself. Whether cause or effect, this collapses the balance between Notch signaling and PU.1 activity that is fundamental to channel the natural role of PU.1 in early pro-T cells (see above) (49, 58). The most pronounced effects of PU.1 are thus a nonlinear response to PU.1 dosage mediated through a gene network switch (Figure 3), and this gene network switch underlies the stochastic, switch-like behavior of individual pro-T or Scid.adh.2C2 cells when forced to express high-level PU.1 (47, 58). Importantly, the combination of PU.1 with Notch signaling desensitization pushes the cells out of the T-cell program completely, rather than simply reversing their progression through the T-cell program. Instead of re-acquiring aspects of a progenitor-like state, the cells appear to trans-differentiate to a dendritic-cell or macrophage developmental program (48, 50, 58).
Figure 3.
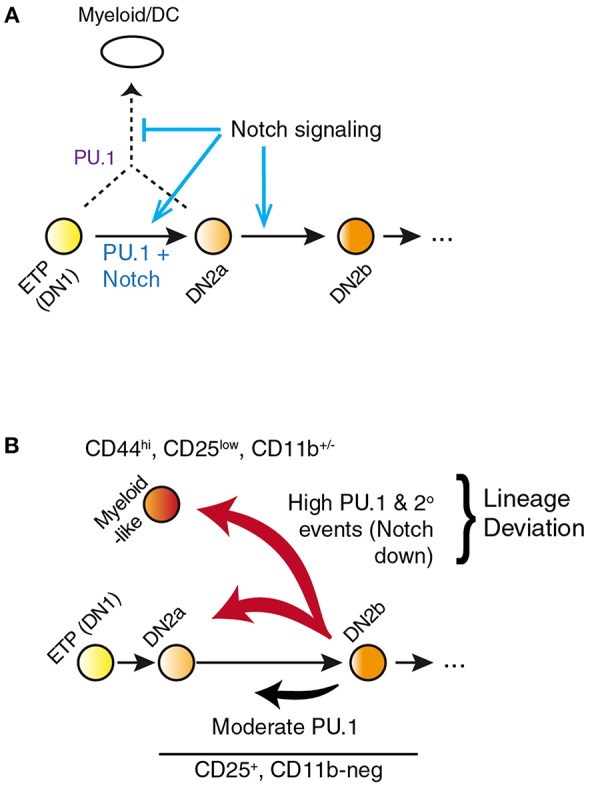
PU.1 and Notch collectively determine T vs. myeloid lineage fates. (A) Notch signaling within the thymus normally constrains PU.1 effects to support early pro-T cell development while blocking alternative fates that PU.1 would otherwise promote. Notch signaling itself does not repress PU.1 expression; however, other transcription factors induced by Notch signaling eventually silence expression of Spi1 during the DN2b stage. (B) Separation of the effects of PU.1 within the T-cell pathway from effects of PU.1 to promote lineage deviation, in PU.1 gain of function experiments. A fraction of cells expressing high levels of PU.1 shift to a myeloid-like state that can be phenotypically distinguished from cells remaining within the T-cell state. This distinction is necessary to relate gain-of-function effects of PU.1 in pro-T cells to effects of loss of endogenous PU.1 in perturbation experiments. Lineage deviation is associated with a broad loss of Notch signal response in the cells, suggesting that the constraint mechanism shown in A has been overwhelmed in these cells. Biochemical mechanisms of these effects remain to be fully defined. Schematic in A, adapted from (58); in B, adapted from (85).
In newly-committed primary pro-T cells forced to express PU.1, the cells crossing this developmental boundary are seen to downregulate the Notch-dependent DN2/DN3 stage marker, CD25 (Il2ra), and often upregulate the myeloid-associated marker CD11b (Mac1; Itgam). The gene expression profiles of cells losing CD25 and upregulating CD11b are radically transformed from the state of newly-committed pro-T cells within 2 days after transduction, with widespread repression of T-lineage-affiliated transcription factor genes and Notch target genes as well as upregulation of multiple Cebp and Irf family transcription factor genes (Figure 4) (85). This response is quite different in gene expression pattern from retrograde differentiation to an ETP- or DN2a-like phenotype. In contrast, cells remaining within the T-cell pathway, continuing to express CD25 and remaining negative for CD11b, show relatively modest and selective changes in gene expression driven by upregulated PU.1, with minimal loss of T-cell regulatory gene expression (79, 85). Details of these transcriptome effects are discussed in a later section, but the point here is that they include qualitative as well as quantitative differences in the gene expression responses. The differences in average Spi1 overexpression levels between cells making these two responses are only on the order of ~2–3 fold (pink, dark red bars in Figure 4), so it is very likely that the additional changes in other regulatory genes contribute strongly to this global shift. Thus, the effect of PU.1 expression per se may be part of the normal T-cell program, but under high-level expression conditions it combines with additional, conditionally induced mechanisms to produce a much broader spectrum of developmental effects that may not only be direct responses to PU.1 itself.
Figure 4.

Profound changes in regulatory gene expression distinguish PU.1-induced lineage deviation from PU.1 effects within the T-cell pathway. Charts show changes in expression of the indicated genes (log2 Fold Change relative to controls) induced by introduction of PU.1 into post-commitment pro-T cells (DN2b-DN3). Panels compare effects on T-lineage regulatory genes (top) and non-T regulatory and signaling genes (bottom) between cells remaining within the T-lineage pathway (light blue bars) and cells undergoing lineage deviation (dark blue bars). Light, dark red bars show corresponding measured levels of exogenous PU.1 in these samples, as log2 fold changes over controls, which have downregulated most of their endogenous PU.1 expression at this stage. Results are from Ungerbäck et al. (85).
Kinetic Challenges: Protein Half-Lives vs. Developmental Progression
Loss of function approaches are indispensable to confirm the roles of endogenous PU.1, especially in view of the potential for indirect effects in gain of function experiments, just described. Here, the challenge has been to find a way to remove or neutralize the endogenous factor quickly enough to see effects robustly, while keeping the controls and the experimental samples at comparable developmental stages. One problem is that the long half-life of PU.1 protein (82) can mask some loss effects at time points <2 days after deletion, while development of the pro-T cells can proceed to new stages if time windows are extended further. There are thus several problems with generating high-quality samples for analysis of transcriptome changes caused by PU.1 loss of function. Cre-dependent deletion of a loxP-flanked Spi1 allele (Spi1fl.fl) is asynchronous, and in an early T-cell population with mixed degrees of Spi1 deletion, cells with inadequate PU.1 levels appear to be at a selective disadvantage, even in vitro. Ironically, because PU.1 protein can persist longer than a cell cycle (82), the very slowdown of cell division caused by deletion of Spi1 (see above) can also interfere with the dilution needed to complete the clearance of the PU.1 protein. As a result of the enrichment of cells with undeleted alleles, and this persistence of pre-existing PU.1 protein even from the cells that have successfully deleted its coding gene, the effects on target gene regulation appear very weak at timepoints up to 2 days after PU.1 deletion, despite the fact that the reduced cell yields from the knockout cells show that PU.1 is biologically important (79). If timepoints are taken too long after deletion, the controls progress to the point when endogenous PU.1 is downregulated, so that any truly PU.1-dependent targets are expressed weakly in the controls, and comparisons with the knockout samples again lose statistical power. A very intriguing new prospect for fast antagonism of PU.1 activity is the discovery of small-molecule inhibitors, some of which are highly potent and specific at blocking PU.1 action in leukemia cells; however, these have not yet been tested for effects on normal T-cell development (89).
A relatively fast way to neutralize PU.1 protein activity directly has been to transduce the cells with a “dominant negative” obligate repressor derivative of PU.1, a fusion protein of the PU.1 DNA binding domain with the repression domain of Drosophila melanogaster Engrailed (PU.1-ENG), to compete for binding against endogenous PU.1 (79) (comparison with wildtype PU.1 shown in Figure 5). The obligate repressor should affect PU.1 positive regulation targets in the opposite direction from wildtype PU.1, and in theory should affect PU.1 negative regulation targets in the same direction, an “algebraic sign” distinction that could be used in principle to dissect indirect effects as well (79). This construct has been useful to reveal quick impacts on expression of positively regulated PU.1 target genes, many of which have been confirmed later by other approaches (85). For example, whereas PU.1 itself can upregulate progenitor-associated genes Bcl11a, Lmo2, Mef2c, and Hhex above their normal levels in DN2a and DN2b primary cells, PU.1-ENG can downregulate them (79). However, PU.1-ENG also has some spurious effects and cannot access closed chromatin sites as well as full-length PU.1 (79, 85).
Figure 5.
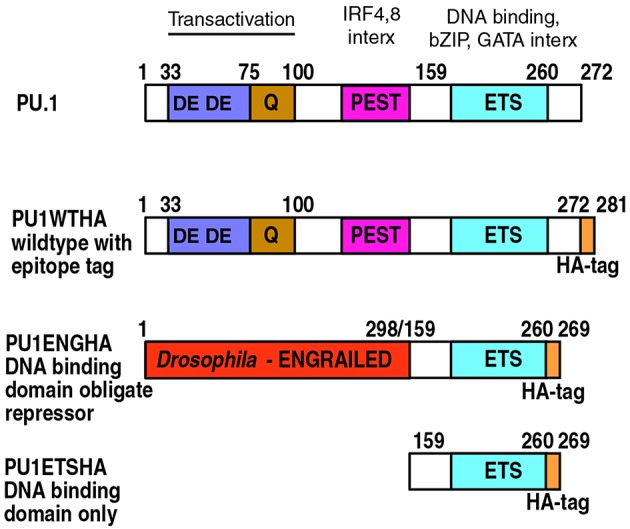
PU.1 structure and derivatives of PU.1 used for functional analysis. Domain boundaries within the amino acid sequence of murine PU.1 are depicted with their associated functions indicated at the top (4, 18, 90, 91). Epitope-tagged wildtype PU.1 (PU1WTHA) and two epitope-tagged, modified constructs are shown (PU1ENGHA, PU1ETSHA); these are used to interrupt endogenous PU.1 activity (79, 82). PU1ENGHA and PU1ETSHA have a full DNA binding domain and efficiently enter open chromatin, but are deficient in entering closed chromatin (85). DE: Acidic residue-rich transactivation domain. Q: Glutamine-rich transactivation domain. PEST: Proline, Glutamate, Serine and Threonine-rich domain, site of IRF4 and IRF8 interaction (interx). Note that in PU.1 this “PEST” domain does not make the protein unstable. ETS: E-twenty-six proto-oncogene homology domain, the DNA binding domain of PU.1. This is also the region that interacts with basic leucine zipper (bZIP) factors such as Jun and C/EBP factors, and GATA family factors.
Cas9-dependent acute deletion of the Spi1 locus can be fast and highly efficient due to the availability of Cas9-transgenic mice (92) and vectors that can be used for high-level, synchronous expression of guide RNAs. However, deletion and clearance of PU.1 protein in this system still require analysis >2 days after introduction of the guide RNAs (80, 85), and the continuing developmental progression of both knockout samples and controls needs to be taken into account in interpreting the results. The strongest evidence for specific physiological PU.1 effects therefore comes from the consensus results from two or more of these perturbation systems. The highest confidence list of potential PU.1 target genes in pro-T cells could be defined as genes that responded reciprocally to gain and loss of PU.1 function within the same DN2a-DN2b developmental interval, and these genes are listed in Table 1. While this list under-represents some PU.1 targets that are only expressed in ETP stage, rigorous definition of the genes that are directly regulated by PU.1 in pro-T cells has made it possible to investigate the range of mechanisms used by the PU.1 protein to exert these transcriptional effects.
Table 1.
High confidence targets of PU.1 regulation in pro-T cells.
| Consensus PU.1-repressed target genes | Consensus PU.1-activated target genes |
|---|---|
| 2900079G21Rik | 3110043O21Rik |
| Ablim1 | 5430427O19Rik |
| Adamts9 | 9930111J21Rik1 |
| Adora2a | 9930111J21Rik2 |
| Arsi | Abcb1b |
| Bcl2 | Acer3 |
| Ccnd3 | Actn1 |
| Cd247 | Actr2 |
| Cd28 | Acy1 |
| Cdc25b | Adam11 |
| Cecr5 | Adam15 |
| Clec2i | Adap1 |
| Clic5 | Adgre1 |
| Csrnp1 | Adrbk2 |
| Cx3cr1 | Alcam |
| Cxcr5 | Alox5ap |
| Cxxc5 | Anks3 |
| Dgka | Antxr2 |
| E2f2 | Ap1s3 |
| Eng | Apbb1ip |
| Epcam | Aqp9 |
| Fam160a2 | Arhgap6 |
| Gbp10 | Arhgef40 |
| Gbp11 | Atp13a2 |
| Gbp4 | Atp6v0a1 |
| Gbp6 | Avpi1 |
| Gimap1 | B3gnt7 |
| Gimap4 | BC035044 |
| Gimap6 | Bcat2 |
| Gimap8 | Bex6 |
| Gimap9 | Bloc1s2 |
| Hdac4 | Bri3bp |
| Hid1 | Btk |
| Hsd11b1 | Cacnb2 |
| Il12rb1 | Ccdc180 |
| Ilvbl | Ccl9 |
| Irak3 | Ccnd1 |
| Itk | Cd180 |
| Jph2 | Cd300a |
| Lmo7 | Cd300lf |
| Lztfl1 | Cd33 |
| Mbp | Cd34 |
| Mir1903 | Cd44 |
| Neil1 | Cdh1 |
| Nipal1 | Clec10a |
| Pik3ip1 | Col9a3 |
| Pitpnc1 | Coro2a |
| Pitpnm2 | Cotl1 |
| Ppm1h | Creg1 |
| Prf1 | Crtac1 |
| Ptprf | Csf2rb2 |
| Rab27a | Csgalnact2 |
| Rdh10 | Ctbp2 |
| Repin1 | Cyp4f18 |
| Selplg | Dnase2a |
| Sh2d5 | Dock5 |
| Sh3bp5 | Dstyk |
| Slc11a2 | Ebi3 |
| Slc12a7 | Entpd1 |
| Slc27a1 | Erlin1 |
| Sox13 | Erp29 |
| Spata6 | Fam101b |
| Spib | Fam217b |
| Spo11 | Fam49a |
| Spry1 | Fcgr2b |
| Ssbp2 | Ffar2 |
| Sstr2 | Fgd2 |
| Tas1r1 | Fgr |
| Tecpr1 | Fh1 |
| Tlr12 | Fig4 |
| Tmc8 | Flnb |
| Tnfsf11 | Gapt |
| Tox2 | Gfod1 |
| Traf3ip2 | Gm16712 |
| Trat1 | Gm16897 |
| Trp53inp1 | Gm2a |
| Tspan13 | Gng10 |
| Tspan32 | Gng2 |
| Utrn | Gns |
| Wnt5b | Gpx1 |
| Gucy1a3 | |
| Gusb | |
| Haao | |
| Hbb-b1 | |
| Hbb-b2 | |
| Hbb-bs | |
| Hbb-bt | |
| Hck | |
| Hfe | |
| Hpse | |
| Hsd17b6 | |
| Idh2 | |
| Il12rb2 | |
| Il13ra1 | |
| Il1r2 | |
| Il5ra | |
| Inpp5j | |
| Irf5 | |
| Irf6 | |
| Itgad | |
| Itgam | |
| Itgax | |
| Jak2 | |
| Kcnk12 | |
| Kcnk6 | |
| Khdc1a | |
| Khdc1c | |
| Klhl18 | |
| Kmo | |
| Krt80 | |
| Lair1 | |
| Lmo1 | |
| Lpcat2 | |
| Lrba | |
| Lrrc25 | |
| Lrrc75a | |
| Lst1 | |
| Ltb4r1 | |
| Ltbr | |
| Lyn | |
| March1 | |
| Matk | |
| Mb21d1 | |
| Mef2c | |
| Megf8 | |
| Met | |
| Mfsd12 | |
| Myo1f | |
| Naaa | |
| Nccrp1 | |
| Ncf1 | |
| Ncf2 | |
| Ndst1 | |
| Ndufb8 | |
| Nedd9 | |
| Neurl3 | |
| Nfam1 | |
| Nlrc4 | |
| Nlrp10 | |
| Nlrp1b | |
| Nod2 | |
| Npl | |
| Nuak2 | |
| Oas1a | |
| Oas2 | |
| Ogfrl1 | |
| P2ry13 | |
| P2ry14 | |
| Padi2 | |
| Pak1 | |
| Pdxk | |
| Phactr2 | |
| Pik3ap1 | |
| Pik3r6 | |
| Piwil2 | |
| Pla2g4a | |
| Plac8 | |
| Pld4 | |
| Plek | |
| Plxnd1 | |
| Pmvk | |
| Pqlc1 | |
| Prex1 | |
| Prkcd | |
| Prtn3 | |
| Ptpn6 | |
| Ptpre | |
| Rab31 | |
| Ralb | |
| Rcn3 | |
| Relt | |
| Rgs18 | |
| Rnf149 | |
| Rogdi | |
| Rufy1 | |
| Samhd1 | |
| Sema3c | |
| Serpina3g | |
| Sh2b2 | |
| Sh3pxd2a | |
| Siglece | |
| Siglecf | |
| Siglecg | |
| Skap2 | |
| Sla | |
| Slc16a7 | |
| Slc35d3 | |
| Slc8a1 | |
| Snx10 | |
| Sorl1 | |
| Spi1 | |
| Stx7 | |
| Susd3 | |
| Svip | |
| Syk | |
| Tbc1d24 | |
| Tbxas1 | |
| Tdrd7 | |
| Tgm1 | |
| Themis2 | |
| Tlr9 | |
| Tmc5 | |
| Tmem51 | |
| Tmprss3 | |
| Tnni2 | |
| Tor3a | |
| Trim55 | |
| Trmt2a | |
| Trpm2 | |
| Tyrobp | |
| Ufsp2 | |
| Unc93b1 | |
| Vamp8 | |
| Vps18 | |
| Wdfy4 | |
| Ywhag | |
| Zc3h12d | |
| Zfp385a | |
| Zfp52 |
This table lists high-confidence PU.1-regulated genes which show reciprocal responses in the loss of function and gain of function perturbations of PU.1. The lists give the three-way intersection of genes affected in loss of function, and (reciprocally) in gain of function within cells remaining CD25+, and in gain of function within cells becoming CD25− CD44+. Consensus PU.1-repressed target genes: genes with expression that goes down in PU.1-transduced DN2b with or without lineage diversion (CD25+ or CD44+ cells), and also increases in normal DN2a cells after deletion of endogenous PU1. Consensus PU.1-activated target genes: genes with expression that increases in PU.1-transduced DN2b with or without lineage diversion, and also goes down in normal DN2a cells after deletion of endogenous PU.1. Data compiled from Ungerbäck et al. (85).
PU.1 Action on the Genome via Direct Binding
PU.1 Protein Is Stable and Active Across the Genome in Early T Cells
Most of the initial hypotheses about PU.1's role in T-cell precursors were based on Spi1 RNA expression patterns and on forced expression of exogenous PU.1 to supra-physiological levels (46, 48–50). With the advent of ChIP-seq data, though, it was confirmed that endogenous, naturally expressed PU.1 is indeed a prominent actor across the genome in T-cell precursors before commitment. PU.1 was found binding to >30,000 genomic sites in these cells at the earliest stages (84), and intracellular protein staining confirmed that some PU.1 expression is still detectable at later stages, in the same individual cells that go through T-cell commitment (marked by activation of the Bcl11b gene) (33, 93). In addition, although the RNA transcript levels are modest in absolute terms, the impact of PU.1 on the cells can be magnified by the high stability of PU.1 protein (82). Although PU.1 occupancy of genomic sites declines as development proceeds, PU.1 occupancy is still detectable through T-lineage commitment at ~5,000 sites before disappearing (84).
PU.1 Binding Site Characteristics
The sites where PU.1 binds are enriched for open chromatin as defined by DNase accessibility or ATAC-seq [assay of transposase-accessible chromatin (94)], and circumstantial evidence suggests that PU.1 is a major factor at those sites that change activity during commitment. PU.1 recognition motifs are the most highly enriched of all defined motifs at sites that start out highly accessible in early pro-T cell stages, when PU.1 is present, and lose accessibility during commitment, i.e., as PU.1 levels decline (44, 85, 95). PU.1 itself is functionally important for the open status of these chromatin sites in the early stages, for many of these sites in fact do not remain as open if the PU.1 is removed acutely from primary pro-T cells by Cas9-mediated deletion (85). This is consistent with PU.1's activity as a site-specific chromatin opening factor in B cells (96, 97), with the ability of PU.1 to eject nucleosomes from sites where it binds in macrophage lines (12), and with its ability to cause rapid increases in ATAC accessibility at the sites it occupies when introduced into Scid.adh.2C2 cells (85). While PU.1 binds at both promoters and non-promoter sites, the evidence from both gain and loss of function studies shows that PU.1 is most associated with chromatin accessibility when it is binding at non-promoter sequences, within introns of genes or in intergenic regions. As described in detail below, such sites, where PU.1 itself is important to maintain chromatin accessibility, are the ones most often linked to genes that are positively regulated in their expression by PU.1 (85). Thus, PU.1 action to keep sites open in chromatin may be an important way that it promotes transcriptional activation in pro-T cells.
Because PU.1 mediates different effects in the rather different regulatory contexts of B, dendritic, myeloid, erythroid progenitors and pro-T cells, an important question is how much of PU.1's binding choice hierarchy is dependent on the prior epigenetic history of cells. PU.1 cannot enter all genomic sites. Notably, PU.1 appears to be excluded from genomic regions that are packaged in Polycomb Repressive Complex 2-modified chromatin, as marked by trimethylation of Histone H3 Lysine 27 (H3K27me3) (84). However, approximately half of the PU.1 occupancy sites in early pro-T cells appear to be relatively “inaccessible” in chromatin by the criterion of ATAC-seq at the stages when PU.1 is seen to be binding there, showing that PU.1 binding can occur without opening the chromatin. These sites in closed chromatin have particularly high-quality matches to the consensus PU.1 binding position weight matrix (85), which several lines of evidence show to be a good indicator of PU.1 binding affinity (11, 85). This suggests that closed chromatin may be less permissive to PU.1 binding than open chromatin, so that only high-affinity site recognition allows binding in closed regions. However, this stringent specificity criterion also shows that these are not “off-target” sites: the tradeoff between site accessibility and the affinity of binding needed for occupancy indicates that PU.1 itself is identifying these sites in closed chromatin to establish its occupancy. By the ability to enter closed chromatin at its own high-affinity sites, and by its functional role in controlling chromatin accessibility at other sites, PU.1 meets the criteria for “pioneer” factor activity in early pro-T cells (98), and the scope of its binding suggests a broad role in genomic architecture of these cells.
Still to be determined are the rules determining when PU.1 binding to closed chromatin results in opening of the closed site. A priori, one could imagine that PU.1 establishes occupancy using its DNA-binding domain and then uses its protein-interaction domains to recruit chromatin modifying complexes (Figure 5, diagram of structures). Many of the known protein-protein interactions between PU.1 and other transcription factors on the DNA are also mediated through parts of the DNA binding domain, consistent with a compartmentalized role of this domain of the protein for binding site choice [reviewed in (18)]. However, PU.1 also contains non-DNA binding domains, acidic and glutamine-rich “transactivation” domains and an IRF4/8 binding domain, that are also clearly implicated in PU.1 function, as selective deletions of these domains greatly reduce PU.1 developmental impacts (24, 47, 99). Recent evidence has pointed to another role of the non-DNA binding domains of PU.1, a function needed for PU.1 to enter closed chromatin. Genome-wide, exogenously introduced full-length PU.1 and the isolated PU.1 binding domain establish quantitatively similar patterns of occupancy at open sites and especially at open promoters all across the genome. However, they show a marked difference in binding between open and closed chromatin, especially at non-promoter sites. Full-length PU.1 binds closed sites nearly as well as open sites, whereas the isolated PU.1 DNA binding domain binds open sites as well as full-length but selectively fails to bind at closed sites (85) (Figure 6). This suggests that an additional process beyond simple DNA sequence recognition is required to establish PU.1 occupancy in closed chromatin, even without an overt change in chromatin accessibility as measured by ATAC-seq. A similar mode of action has already been described for a separate, non-DNA binding domain to enable EBF1 entry into closed chromatin (100). It will be interesting to see if this is a general feature of pioneer transcription factors.
Figure 6.

Summary of PU.1 binding features in pro-T cells. (1) PU.1 binding is preferentially retained at high-affinity sites as pro-T cells progressively reduce their PU.1 levels. This feature indicates that PU.1 primarily uses mass action (concentration × affinity) to determine its genomic site choices in these cells. (2) PU.1 works as a positive regulator in pro-T cells primarily by binding and controlling accessibility of sites distal to the transcriptional start sites, not at promoters. (3) The binding profiles of full-length PU.1 (PU1WT) show a tradeoff between binding site affinity and binding site accessibility in chromatin; however, constructs with the PU.1 DNA binding domain but lacking the transactivation domains (PU1 DBD) are poor at engaging sites in closed chromatin no matter how high their potential affinities.
PU.1 Action via Cofactor Recruitment
Direct Gene Regulation by PU.1: Activation by Distal Enhancer Engagement and Opening
The major problem with connecting PU.1 binding to PU.1 regulatory function is that PU.1 binds to too many genomic sites in ETP and DN2a pro-T cells (84). It shows high fidelity in terms of sequence recognition, but its binding is not confined to functionally responsive target genes. It is found at a large fraction of open, accessible chromatin elements during the stages when it is expressed, and often bound at promoters as well as distal elements. However, only a minority of the genes linked to its binding sites change expression at all across the developmental interval when PU.1 goes from full expression to silence (84). Much of PU.1 binding in pro-T cells thus appears to be either functionally redundant or opportunistic. Identifying PU.1's functional mechanisms of target gene regulation has required a way to link an experimentally inducible change in PU.1 binding at a given site with the rapid, measurable change in expression of the target gene linked to that site. This is considerably easier to do in a gain of function format than in loss of function, as an epitope-tagged exogenous PU.1 construct can be introduced with fast kinetics and its newly established binding tested for association with local gene expression responses (85). Note that the gain-of-function experimental design makes it necessary to use another criterion to screen out genes that are only indirectly affected, as described above. For this reason, in our recent study (85) only cells that remained CD25+ CD11b− (see above) were used for ChIP-seq analysis of exogenous PU.1 binding.
The results showed that PU.1 exerts its main functional regulatory impacts in pro-T cells via non-promoter sites, and especially via sites that are normally developmentally changing in chromatin accessibility (85) (Figure 6). In the aggregate, most of the responses of genes linked directly to PU.1 binding sites were positive; direct repression targets were much rarer. Genes responding to the addition of exogenous PU.1 usually had the exogenous PU.1 binding to distal (intronic or neighboring intergenic) sites, whereas genes that had PU.1 binding only to their promoter regions usually did not change expression at all. In the “blank slate” background of the Scid.adh.2C2 cell line, exogenous PU.1 binding opened chromatin at its non-promoter sites within 2 h, increasing the “activating” H3K27Ac marks at these sites a few hours later, and the linked genes were predominantly upregulated within 8–24 h. The genomic sites that were most highly associated with these responses in primary pro-T cells were developmentally dynamic in chromatin accessibility: normally open in early stages of T-cell development (endogenous PU.1-expressing) but closed once the cells went through commitment (endogenous PU.1-low or negative). Thus, the sites in pro-T cells with the strongest sensitivity to exogenous PU.1 for transcriptional impact were also sites where endogenous PU.1 might be important for maintaining chromatin accessibility.
The Problem of Pro-T Cell Gene Repression by PU.1
The impact of PU.1 on pro-T cell gene expression overall is at odds with the biochemical and genomic evidence for its mode of action in one respect: PU.1 introduced into primary pro-T cells or Scid.adh.2C2 cells causes downregulation of many T-cell genes, especially those associated with Notch signaling and TCR gene rearrangement after commitment. This response is fast, reducing existing transcript pools for many repressed genes even before most positively regulated PU.1 target genes are seen to be turned on (80). However, the local impact of PU.1 binding is strongly biased toward activation of genes linked to the binding sites. While much of the data showing T-lineage affiliated gene downregulation comes from forced PU.1 re-expression or overexpression experiments, and might therefore be a high-dose artifact, it is important to note that the developmental speed-up observed in primary pro-T cells when PU.1 is knocked out also points to a normal PU.1 role as a brake on developmental progression (79). Thus, to account for PU.1's overall role, some explanation for the repressive outcomes is essential.
There is a long history of research on PU.1 as a repressor of genes associated with non-myeloid pathways, especially in the context of PU.1—GATA-1 antagonism in hematopoiesis [reviewed in (4, 7, 25)]. At high levels, PU.1 has been found to block DNA binding by GATA-1 (23), while at lower levels it is reported to antagonize GATA-1-mediated transactivation by forming complexes with it that recruit Rb through the PU.1 acidic transactivation domain (101). It is also reported to act as a repressor by direct recruitment of Dnmt3b (102). In most of these cases, PU.1 is observed to bind directly to the regulatory DNA of its repression targets (86, 103). However, in the pro-T cells, the genes that are repressed when exogenous PU.1 is introduced are not necessarily linked to the sites that the exogenous PU.1 actually binds. In fact, results with the PU.1-ENG obligate repressor construct implied that some kind of indirect effect must be involved: while the obligate repressor downregulated genes that are positive regulatory targets of wildtype PU.1, it actually upregulated many genes that wildtype PU.1 represses, completely inconsistent with a direct repression mechanism (79).
To date, three mechanisms appear to be involved. First, as noted above, high-dose PU.1 can inhibit expression of multiple Notch target genes and Notch1 itself (58, 85). It is possible that the fast downregulation of Notch response genes, including Hes1, Nrarp, Dtx1, Lef1, and Il2ra, by overexpressed PU.1 is due to the loss of positive Notch signaling input rather than to a gene-specific mechanism. This Notch-inhibitory mechanism is not operating in cells that remain within the T-cell pathway, but it becomes prominent in cells that PU.1 causes to transdifferentiate, and would be expected to affect all T-cell genes that use Notch signaling as an obligate positive input, whether or not PU.1 binds them directly. A related scenario in which PU.1 could interfere with a T-lineage specific positive regulatory input might be through repression of GATA-3 by PU.1, by analogy with the cross-inhibition of PU.1 and GATA-1. However, in pro T cells, both PU.1 and GATA-3 are active together and both functionally important throughout the ETP to DN2a stages (79, 104), and there is more evidence for GATA-3 repression of PU.1 than for PU.1 repression of GATA-3 (58, 79, 104, 105). However, GATA-3 function also may become a casualty of PU.1 action when Notch signaling is inhibited (58).
Second, in pro-T cells forced to express PU.1, those that make the lineage jump (i.e. lose CD25, gain CD11b) not only silence Notch1 but also activate myeloid regulatory genes (85). They also begin to express multiple transcription factors of the Egr and IRF families, and in the case of primary cells, they also upregulate C/EBP family factors. These factors probably contribute independently to the repression of pro-T cell genes. Egr2, for example, can collaborate with PU.1 in positive regulation when co-bound with it (106), but has also been implicated as a PU.1-stimulated repressor of the mir17~92 complex (107). Although not required for Notch1 repression in Scid.adh.2C2 cells (58), when activated in primary cells, C/EBPα itself can also repress Notch1 (50). PU.1 does not strongly upregulate transcription factors annotated as repressors, however, in cells remaining within the T-cell program (see below). Therefore, this indirect repressive activity, too, would only be deployed under conditions of lineage shift.
The third mechanism that could play a role in repression within the T-cell program comes from PU.1's own ability to recruit other transcription factors to collaborate with it at PU.1 binding sites. This is a hallmark of pioneering activity in developmental gene regulation (98, 108), but in this case it exposes a particularly intricate post-transcriptional relationship between PU.1 and the factors required for progression of the T-cell program.
System Consequences of Cofactor Recruitment: Repression by Theft
PU.1 is a powerful organizer of the occupancy patterns of other transcription factors genome-wide. PU.1 binding shifts the disposition of other factors in the cell across the genome, even when their own expression levels and total numbers of binding sites remain essentially unchanged (80). The positive regulatory significance of these kinds of shifts is well established; many factors recruit others to collaborate with them in functional complexes at active enhancers [e.g., reviews by (17, 109–111)], and PU.1 is known to establish preferential binding sites for multiple other transcription factors in myeloid cells. However, in this case the positive impact is coupled with a negative regulatory consequence, via action at a distance (Figure 7). For PU.1 in pro-T cells, IRF and C/EBP family partners are mostly not available, but a key positive regulatory partner is Runx1 (previously known as AML1 or CBFα2), which has long been known to interact with PU.1 (and C/EBPα) to form a functional complex at its myeloid positive regulatory target sites (9, 112, 113). In pro-T cells, PU.1 binding sites in open chromatin genome-wide are highly enriched for Runx motifs, raising the possibility that Runx factors assist in the chromatin opening process (85), and proteomic analysis provides support for a strong representation of Runx1 in PU.1-containing complexes formed in the pro-T cell like Scid.adh.2C2 cell line (80). However, Runx1 also has sites at a large fraction of all enhancers active in the cells without PU.1 expression (Figure 7).
Figure 7.

Repression by PU.1 can be caused by redistribution of a limiting co-regulator (co-regulator theft). Schematic depicts the complementary changes in factor binding patterns as well as in transcriptional activity of genes that are normally PU.1-dependent (right) or PU.1-inhibited (left), across the developmental stages when PU.1 goes from high to low (top) and then in an experimental condition when PU.1 is re-introduced into committed cells that have already turned off endogenous PU.1 (bottom). The figure shows that the redistribution of partner factors Runx1 (R) and Satb1 (S) by recruitment to PU.1 (P) binding sites occurs at the expense of sites that these factors would otherwise occupy together with other T-cell factors (T). Broken-line arrows (lower left) indicate that redistribution probably involves the dynamic equilibrium of binding of these factors between genomic sites that are differentially preferred in the presence and absence of PU.1. In at least some cases, the “theft” of the cofactors also results in relative closing of the chromatin at sites from which these cofactors are removed. Schematic modified from Hosokawa et al. (80).
When epitope-tagged PU.1 was introduced into Scid.adh.2C2 cells and the complexes were isolated for proteomic analysis, much enrichment was seen for SWI/SNF complex components as well as some other chromatin modifiers (80). The preponderance of SWI/SNF complex interactions was consistent with the evidence that PU.1 usually acts as an activator. Based on the longstanding literature of PU.1–GATA factor antagonism through protein interaction (20–23), GATA-3 was expected to be present as well, and it was detectably enriched over background in these complexes. However, by far the most highly enriched sequence-specific transcription factor proteins interacting with PU.1 in these cells were Rest and Runx1 (80). Runx1 was of particular interest because of its sequence motif enrichment at PU.1 sites. Although Runx1 can act as a global chromatin accessibility organizer (114), PU.1 itself does not depend on Runx1 for establishing permissive sites for its binding, even in the “blank slate” context of the Scid.adh.2C2 cells (85). However, PU.1 strongly affected the sites where Runx1 bound, resulting in a dramatic shift in Runx1 binding site choices in tests of gain of PU.1 function (80). Supporting the physiological relevance of this mechanism, many of the same genomic sites where Runx1 was shifted by PU.1 in Scid.adh.2C2 cells underwent the reverse changes in Runx1 occupancy in normal primary pro-T cells, as they progressed from PU.1-high to PU.1-low developmental stages.
As expected, PU.1 recruits Runx1 to sites where Runx1 exerts measurable functional collaboration with PU.1, mostly to help in the positive regulation of PU.1 targets (80). However, the aspect of this redistribution that is most notable is that Runx1 is depleted in the process from alternative sites, and the sites that it abandons are themselves highly functional sites. The analysis is somewhat complicated by the fact that many developmentally important genes are linked with multiple Runx1 and/or PU.1 binding sites, only some of which gain or lose Runx1 occupancy. However, focusing on those genes that have Runx1 binding sites but not PU.1 binding sites, the genes that “lose” Runx1 binding when PU.1 is expressed clearly include a large fraction that depend quantitatively on Runx1 for their own expression. These genes show weak downregulation when Runx1 is disrupted by Cas9 and they show stronger downregulation when Runx1 is neutralized by a Runx1 dominant negative construct (80). Thus, the competition for Runx1 protein by PU.1 directly causes coupled positive and negative regulation, to cause a switch-like alteration in genome-wide cell state (Figure 7).
Three features of this mechanism are noteworthy (80). First, PU.1 does not appear to bind, even transiently, at the sites from which Runx1 is lost: Runx1 is competitively redistributed, but is not displaced. Thus, the PU.1 effect differs from “squelching” or other negative regulatory mechanisms where transcription factors are expelled by chromatin closing (115, 116). Second, one might expect that the Runx sites available for redistribution could have been vulnerable to dissociation because they were marginal quality binding sites in the first place; however, motif analysis shows that many of the Runx occupancy sites that are emptied when PU.1 is in the cell are high quality Runx sites in the upper half of the position weight matrix score distribution (80). Considered only as Runx sites, they are likely to be much higher affinity than the ones to which Runx1 moves, to occupy together with PU.1. Thus, the ternary (or higher-order) complexes nucleated by PU.1 are more favored binding sites for Runx1 when PU.1 is present than functionally relevant, high-quality Runx sites elsewhere. Finally, it is clear that this is a system-level mechanism. It is the limited pool of Runx1 operationally available for action across the genome that makes the impact of PU.1 a “zero-sum” outcome. Thus, the regulated level of Runx1 protein contributes to the switch-like impact made by the developmental shift from high-level PU.1 to PU.1 shutoff. However, given the high frequency of Runx factor utilization at multiple lymphoid enhancer sites, this kind of mechanism can propagate local PU.1 impacts to a much broader genomic scale.
The “theft” mechanism of repression by partner factor redistribution is not unique to the PU.1-Runx1 pair. PU.1 has an even stronger effect on binding site choice of Satb1, another transcription factor that is expressed throughout early T-cell development, and GATA-3 also shifts, when PU.1 is added, to occupy sites together with PU.1 (80). Although Satb1 in DN2-DN3 stages appears to have weaker effects on gene expression than Runx1, the PU.1-repressed genes that appear to be responding to Satb1 loss are different from those that are most dependent on Runx1, broadening the full impact of this mechanism (80). A very similar phenomenon has been reported earlier by Jenner and colleagues for the effect of T-bet on GATA-3 in establishing the Th1 cell program (117, 118). Thus, “partner factor theft” can be an integral part of the machinery for program choice operated by lineage-determining transcription factors.
The PU.1 Regulome in Early pro-T cells and Its physiological Roles
PU.1 Target Genes: Gene Network Roles and Developmental Timing
While indirect regulation plays a large role in its developmental impact, the target genes that PU.1 directly regulates are ultimately crucial for understanding what this factor contributes to the T-cell program. PU.1's action as a positive regulator implies that most of its direct target genes should be expressed in a pattern concordant with its own expression. Indeed, PU.1-activated target genes are preferentially expressed in the earliest stages of T-cell development (examples shown in Figure 8). Many of them are expressed also in at least one of the other contexts where PU.1 is active: in myeloid and dendritic lineage cells, in B lineage cells, and particularly also in multipotent progenitor cells (Figure 8). Among the smaller number of genes that appear to be directly repressed by PU.1, most are specific for later stages of T-cell development. These patterns reinforce the case for PU.1's impact in shaping the developmental timecourse of gene expression in pro-T cells.
Figure 8.

PU.1 globally regulates genes involved in multiple signaling and cell biological properties as well as other “phase 1” transcription factors across the stages when PU.1 is expressed. Summary of normal expression patterns of representative groups of PU.1 regulated genes is shown, illustrated using the ImmGen (119) (www.immgen.org) “My Gene Set” browser (http://rstats.immgen.org/MyGeneSet_New/index.html; Microarray V1). Natural levels of expression are shown in different “Stem and Progenitor” cell sets (120) (under light blue bar) and in successive stages of “αβ T cell” development (121) (under purple bar), where the color scale represents z score (warm colors, high expression; cold colors, low expression). Vertical line between “preT_DN2A_Th” and “preT_DN2B_Th” relates these stages to the timing of commitment, when PU.1 levels decline. (A) Genes encoding transcription factors activated by PU.1 in multiple tests (58, 79, 85). (B) Representative genes activated by PU.1 that encode tyrosine protein kinases, cytokine receptors, and G protein coupled receptors, from Table 1 [data from Ungerbäck et al. (85)]. (C) Genes activated by PU.1 that encode additional cell surface molecules, signaling receptors and adhesion molecules, from Table 1. (D) Genes repressed by PU.1, encoding chemokine receptors and G-protein coupled receptors, from Table 1. Functional clusters used in this summary were as defined by DAVID Gene Functional Classification tool (DAVID 6.8) (https://david.ncifcrf.gov/gene2gene.jsp).
Developmentally potent transcription factors often transform a cell's identity by positively or negatively regulating the expression of other transcription factors. As noted above, PU.1 can have this effect on early pro-T cells when it is overexpressed and the cells switch to a non-T cell lineage program. But to what extent does PU.1 control the expression of other transcription factors within the T-cell program? It has become clear that the progression of cells through T-cell commitment involves the ordered downregulation of a substantial set of progenitor-specific transcription factors, called “Phase 1” factors in this context, concomitant with the upregulation of T-lineage affiliated factors (34, 65, 84, 121, 122). PU.1 itself is downregulated at stage when multiple other Phase 1 factors are downregulated, and a key question is whether the withdrawal of positive PU.1 input plays a role in the downregulation of these progenitor factors. With respect to PU.1-mediated repression, some T-cell factors are already upregulated while PU.1 is still highly expressed (GATA-3, TCF-1, and the Notch target Hes1), but others are upregulated only during the period when PU.1 declines (Bcl11b, Ets1, Lef1) and could, in principle, have their expression timing affected by PU.1 negative regulation. To what extent does PU.1 actually control these gene expression patterns under the circumstances of actual pro-T cell development, i.e. with strong Notch signaling that prevents lineage switching?
Data from multiple studies show that PU.1 regulates a subset of developmental control genes but is not alone in its actions. Varied PU.1 gain and loss of function perturbations in the DN2-DN3 stages show that PU.1 does provide positive input into a discrete subset of Phase 1 regulatory genes, with Mef2c, Lmo2, Bcl11a, and often also Hhex responding over a range of different tests (58, 79, 80, 85). Consistent with an evolutionarily conserved program in regulating these genes, the developmental expression patterns of these genes and PU.1 (Spi1) in human pro-T cells (Thy1—Thy4) are similar to their patterns in murine pro-T cells (35). PU.1 is not uniquely responsible for the Phase 1 gene expression pattern, however, for other Phase 1 genes are either unaffected or moderately inhibited by PU.1, as discussed elsewhere (79, 123). Additional factors also probably collaborate with PU.1 to fine-tune the responses of Mef2c and Lmo2, for they are already declining by the end of ETP stage, multiple cell cycles before DN2b stage when PU.1 itself declines (65, 67, 84, 121, 124) (Figure 8A). Thus, PU.1 is likely to be one of several important positive regulators for these genes. PU.1 can indeed have negative regulatory effects on some of the T-cell factors that are upregulated during commitment, but these effects are greatly limited when the analysis is confined to PU.1 activities within the T-cell program. Hes1, Tcf12 (HEB), Ets1, and Lef1 are strongly affected in cells making a lineage switch, but none of these are measurably repressed in cells within the T-cell path. The cell cycle-regulatory locus E2f2, which is also upregulated during commitment, is rare among transcription factor coding genes in that it does appear to be under active repression by PU.1 until the transition to commitment. Thus, within the T-cell pathway, PU.1 has a specific role in promoting maintenance of certain Phase 1 regulatory genes before commitment, but little role in repressing T-cell differentiation regulators directly.
PU.1 as a Choreographer of Thymocyte Cell Biology
An important result from the genome-wide analysis of PU.1 target genes has been recognition of the major gene sets that it does actively control in early pro-T cells. The number of high confidence PU.1 target genes within the T-cell pathway that code for transcription factor genes is low (Figure 8A). In contrast, Gene Ontology and Pathway analyses as well as simple gene lists reveal that PU.1 directly controls major systems of cytokine receptors, chemokine receptors, tyrosine protein kinases, G-protein receptor signaling molecules, and adhesion or cytoskeletal system molecules (Table 2) (85). These directly regulated targets, some of them studied little, if at all, in T-cell development to date, might have a transformative impact on the cell biology of the developing lymphocytes between the stages when PU.1 is present and when it is shut off.
Table 2.
Gene ontology and pathway classifications of genes regulated by PU.1 in pro-T cells.
| (A) Genes upregulated by PU.1 in CD25+ cells with gain of function, downregulated with sgRNA, relative to all genes expressed in cells | ||
|---|---|---|
| GO biological process complete (top 21) | Fold enrichment | Adjusted P-value |
| Peptidyl-tyrosine phosphorylation (GO:0018108) | 7.18 | 2.40E-02 |
| Myeloid leukocyte activation (GO:0002274) | 7.05 | 9.14E-03 |
| Reactive oxygen species metabolic process (GO:0072593) | 6.97 | 3.05E-02 |
| Regulated exocytosis (GO:0045055) | 6.87 | 3.43E-02 |
| Peptidyl-tyrosine modification (GO:0018212) | 6.87 | 3.43E-02 |
| Exocytosis (GO:0006887) | 5.11 | 1.14E-02 |
| Immune response-activating signal transduction (GO:0002757) | 4.99 | 1.46E-02 |
| Inflammatory response (GO:0006954) | 4.76 | 7.00E-06 |
| Immune response-regulating signaling pathway (GO:0002764) | 4.74 | 2.53E-02 |
| Activation of immune response (GO:0002253) | 4.63 | 6.83E-03 |
| Myeloid cell differentiation (GO:0030099) | 4.55 | 3.80E-03 |
| Positive regulation of protein secretion (GO:0050714) | 4.21 | 2.11E-02 |
| Regulation of body fluid levels (GO:0050878) | 4.21 | 4.39E-02 |
| Positive regulation of peptide secretion (GO:0002793) | 4.15 | 1.23E-02 |
| Adaptive immune response (GO:0002250) | 4.1 | 2.92E-02 |
| Positive regulation of defense response (GO:0031349) | 4.07 | 1.59E-02 |
| Regulation of MAP kinase activity (GO:0043405) | 3.95 | 2.32E-02 |
| Defense response to other organism (GO:0098542) | 3.93 | 3.28E-03 |
| Regulation of inflammatory response (GO:0050727) | 3.87 | 2.96E-02 |
| Immune effector process (GO:0002252) | 3.74 | 5.31E-04 |
| Innate immune response (GO:0045087) | 3.73 | 3.02E-04 |
| (B) Genes downregulated by PU.1 in CD25+ cells with gain of function, upregulated with sgRNA, relative to all genes expressed in cells | ||
| GO biological process complete | Fold enrichment | Adjusted P-value |
| Defense response to protozoan (GO:0042832) | 27.62 | 1.72E-02 |
| Response to protozoan (GO:0001562) | 25.11 | 2.57E-02 |
| Cell activation (GO:0001775) | 4.44 | 2.43E-03 |
| Immune response (GO:0006955) | 3.55 | 6.41E-03 |
| Immune system process (GO:0002376) | 2.86 | 2.26E-04 |
| Cellular response to stimulus (GO:0051716) | 1.75 | 4.20E-02 |
| (C) Genes upregulated by PU.1 in CD44+ CD25- cells with gain of function, downregulated with sgRNA, relative to all genes expressed in cells | ||
| GO biological process complete (top 21) | Fold enrichment | Adjusted P-value |
| Regulation of coagulation (GO:0050818) | 8.85 | 5.55E-03 |
| Regulation of blood coagulation (GO:0030193) | 8.58 | 2.49E-02 |
| Regulation of hemostasis (GO:1900046) | 8.36 | 2.98E-02 |
| Positive regulation of inflammatory response (GO:0050729) | 7.17 | 3.76E-05 |
| Myeloid leukocyte activation (GO:0002274) | 6.03 | 1.61E-02 |
| Regulated exocytosis (GO:0045055) | 5.93 | 4.94E-02 |
| Defense response to bacterium (GO:0042742) | 5.16 | 5.18E-03 |
| Positive regulation of stress-activated protein kinase signaling cascade (GO:0070304) | 5.04 | 2.95E-03 |
| Inflammatory response (GO:0006954) | 4.98 | 5.37E-09 |
| Positive regulation of stress-activated MAPK cascade (GO:0032874) | 4.77 | 1.29E-02 |
| Regulation of body fluid levels (GO:0050878) | 4.62 | 1.78E-04 |
| Leukocyte activation involved in immune response (GO:0002366) | 4.53 | 4.90E-02 |
| Positive regulation of defense response (GO:0031349) | 4.51 | 2.71E-05 |
| Positive regulation of MAP kinase activity (GO:0043406) | 4.42 | 6.86E-03 |
| Exocytosis (GO:0006887) | 4.29 | 4.18E-02 |
| Regulation of inflammatory response (GO:0050727) | 4.29 | 6.51E-05 |
| Positive regulation of response to external stimulus (GO:0032103) | 4.09 | 2.38E-03 |
| Regulation of MAP kinase activity (GO:0043405) | 4.01 | 8.19E-04 |
| Activation of immune response (GO:0002253) | 3.85 | 3.77E-02 |
| Positive regulation of protein serine/threonine kinase activity (GO:0071902) | 3.8 | 1.28E-02 |
| Defense response to other organism (GO:0098542) | 3.7 | 8.48E-04 |
| (D) Genes downregulated by PU.1 in CD44+ CD25- cells with gain of function, upregulated with sgRNA, relative to all genes expressed in cells | ||
| GO biological process complete | Fold enrichment | Adjusted P-value |
| T cell activation (GO:0042110) | 4.77 | 3.82E-04 |
| Lymphocyte activation (GO:0046649) | 4.51 | 7.36E-07 |
| Cell-cell adhesion (GO:0098609) | 4.22 | 9.44E-03 |
| Cell activation (GO:0001775) | 4.19 | 1.78E-08 |
| Lymphocyte differentiation (GO:0030098) | 4.18 | 1.07E-02 |
| Leukocyte activation (GO:0045321) | 4.12 | 1.14E-06 |
| Regulation of cell-cell adhesion (GO:0022407) | 3.98 | 6.49E-04 |
| Leukocyte differentiation (GO:0002521) | 3.52 | 2.88E-02 |
| Positive regulation of cell adhesion (GO:0045785) | 3.45 | 2.10E-02 |
| Regulation of defense response (GO:0031347) | 3.11 | 1.15E-02 |
| Regulation of cell adhesion (GO:0030155) | 3.09 | 1.01E-03 |
| Biological adhesion (GO:0022610) | 2.9 | 2.29E-02 |
| Positive regulation of transcription by RNA polymerase II (GO:0045944) | 2.46 | 2.21E-03 |
| Regulation of immune system process (GO:0002682) | 2.35 | 3.85E-03 |
| Immune system process (GO:0002376) | 2.3 | 3.44E-05 |
| Regulation of multicellular organismal process (GO:0051239) | 1.74 | 3.15E-02 |
| Positive regulation of biological process (GO:0048518) | 1.48 | 6.54E-03 |
The table shows PANTHER Overrepresentation Analysis (www.geneontology.org) of categories of genes upregulated or downregulated by PU.1. In each case, responding genes were defined by reciprocal changes in expression in PU.1 gain of function and PU.1 loss of function experiments in the DN2a-DN2b interval as in Table 1. Whereas Table 1 shows the three-way intersection of genes affected in loss of function, in gain of function for cells remaining CD25+, and in gain of function for cells becoming CD25− CD44+, here the effects of the gain of function perturbations were separated to allow comparison of results from cells remaining in the T-cell pathway (CD25+) with results from cells likely deviating toward another fate (CD44+). Database for comparison was all genes expressed in control DN2 cells. Statistical results shown are for a Fisher Test with Bonferroni correction for multiple sample testing. The PANTHER Overrepresentation Test version was released 2018-10-10 using the GO Ontology database released 2018-10-08. For PU.1 activated genes, only the top 21 enriched categories are shown.
Potentially important clues to PU.1 roles are the prominence among positively regulated PU.1 targets of genes encoding specific cytokine receptors not yet studied in T-cell biology (e.g., Pdgfrb); multiple protein tyrosine kinases (Btk, Syk, Hck, Lyn); and G-protein coupled receptors (Ffar2, P2ry13, P2ry14) and G protein signaling mediators (Gng2 and Rgs18); while PU.1 represses other G protein signaling mediators (Gimap and Gbp family members). In addition, PU.1 directly promotes expression of cell surface molecules (CD33, CD34, CD44) used as markers for stages in early T-cell development, but which in vivo work to mediate environmental interactions, and it drives expression of adhesion molecules (integrins and Siglecs) as well as cytoskeletal components such as Coro2a and Myo1f. Representative samples of the expression patterns of such genes are shown in Figures 8B–D. The result is that not only signaling capability but basic properties of adhesion, motility and chemoresponsiveness of the cells can be under PU.1 control in the early stages of T-cell development. While these effects are not seen as direct transcriptional regulation of other transcription factor coding loci, such target genes should have numerous impacts on activation pathways in the cells that induce transcriptional as well as migratory responses to environmental signals.
The PU.1-high stages of thymocyte development are relatively obscure in the context of the whole thymus, yet their accurate regulation is crucial for establishment of immune system homeostasis and avoidance of leukemia (125, 126). These stages span multiple cell cycles in vivo and in vitro (33, 39, 67, 124). However, cells in these stages are hard to visualize in the intact thymus, as only a few cells per day are granted regulated entry into the thymic antechamber (127), then migrate slowly through the cortex, dispersing among a vast excess of more advanced T-cell precursors, as they begin to differentiate toward commitment (128). In postnatal mice, the entry point is thought to be formed by specialized endothelial cells at the cortical/medullary border of the thymus (129). Following an unknown triggering signal, after a variable delay (125), the cells in each cohort then begin to migrate centrifugally toward the outer thymic cortex, and cell surface marker expression patterns imply that it is somewhere midway in the course of this migration that the individual cells undergo lineage commitment [reviewed by (60, 130)]. Because of the extreme rarity of these very immature cells relative to the later-stage thymocytes at any given time, they were almost impossible to study in depth before the development of in vitro culture systems (88), which have continued to be informative to the present. However, the types of genes positively regulated by PU.1 are overwhelmingly in categories likely to be involved in mediating the interaction of the cells with very specific environments. The tests of PU.1 function in these early pro-T cells that have been done so far present the cells with Notch ligands and cytokines, but could be fundamentally lacking in other molecules presented by the normal thymic environment. It will be of great interest to discover which anatomical subdomains of the thymus actually supply the molecules that interact with the potentially important receptors and adhesion molecules that PU.1 enables the cells to express, and what responses they trigger in these earliest T-cell precursors.
PU.1 and the Regulation of Lymphoid Development in Ontogeny
The studies reviewed throughout this paper have characterized the roles of PU.1 in T-cell development in the young postnatal mouse or in late fetal life. Very recent work has now placed these roles of PU.1 into a wider developmental perspective.
The cells used for in vitro differentiation as well as in vivo analysis in the work reviewed above have all been derived from waves of hematopoiesis that begin with definitive hematopoietic stem cells, which first appear in the mouse fetal liver by about day 11.5 of gestation and may be followed by additional stem-cell waves through the end of gestation (day 20) (131). Thus, postnatal thymocytes and any in vitro differentiation cultures seeded with cells from bone marrow or fetal liver from E15 onward are likely to come from true stem cell origins. However, there are earlier hematopoietic progenitors in the embryo that derive from yolk sac, cells with varied developmental potentials but without true stem-cell self-renewal. The first wave of T cell development in the fetus is now thought to arise from these non-stem-cell precursors in the yolk sac (132, 133). It has long been recognized that the earliest fetal thymocytes are different from later waves of developing thymocytes in terms of their abilities to generate particular classes of TCRγδ cells (134) and in terms of their extremely fast differentiation kinetics, both in vivo and in fetal thymic organ culture or stromal coculture systems (104, 133, 135). This is now understood to be intrinsically programmed (136) and due to an altered pathway of differentiation in the first-wave cells, which results in T-cell lineage commitment even before entry into the thymus (137–139).
Remarkable differences have been reported between genetic requirements for T cell development derived from earlier and later waves of prethymic progenitors. For example, the crucial T-lineage transcription factor TCF-1, which plays roles in numerous phases of thymocyte development (95, 140–145), is essential to maintain adult T-cell production but dispensable in fetal and early postnatal T cell development (146). A wave of fetal T cell development can also, apparently, be generated without PU.1 (71). In the case of PU.1, the change in its role occurs within fetal life, and this has now been sharply situated in the transition from precociously committed “first-wave” precursors to precursors that enter the thymus while still multipotent (147). ETPs derived from these precursors naturally express lower levels of PU.1 than adult ETPs, but they are almost unchanged in their ability to generate early fetal T cells when the level of PU.1 is reduced still further (~5 fold) by deletion of the major upstream regulatory element of PU.1 (147). In contrast, the same five-fold diminished level of PU.1 sharply degrades the ability of later fetal hematopoietic stem and progenitor cells to generate T cells at all, in vivo or in vitro, with functional and phenotypic defects evident in the mutants in both multipotent progenitors and newly-entered intrathymic ETPs, as early as in the late fetus. This difference in PU.1 dependence accompanies a subtly different T-cell developmental program. Gene expression differences have been noted between the normal first-wave fetal and adult pro-T cells in the thymus at corresponding stages which indicate that the fetal program drives accelerated development (148, 149). Montecino-Rodriguez et al. point out that these differences conspicuously include reduced initial expression of multiple PU.1-dependent genes in the fetal cells (147). Thus, not only is the first-wave fetal program less dependent on PU.1, but also it may rely on relatively low PU.1 activity for its very distinctiveness. These results therefore support a role for PU.1 in delaying differentiation in order to allow more extended proliferation before commitment, showing how the importance of this role is ontogenically scaled to the needs of the developing organism.
The first-wave precursor cells, also uniquely, enter the thymus by a different route than all subsequent waves. Instead of entering through the blood vessels near the cortical-medullary junction, these early cells migrate through cervical-region mesenchyme to the thymic anlage before it is vascularized. The thymus does not yet have a capsule to present a physical barrier, and the first-wave cells enter directly through the future outer cortex. Thus, they may not use the same interactions with basement membrane, endothelial cells, or chemokine gradients as any future wave of thymic precursors. Not only are these cells intrinsically programmed to cut short the stages supported by PU.1-dependent transcriptional regulators, but also they can dispense with many of the cell biological tools that PU.1 may provide to later-wave successors to navigate the adult or late-fetal thymus.
Conclusions and Future Questions
PU.1 is a broad regulator of the properties of the cells that first enter the thymus, and it helps to determine their proliferation and rate of progression to commitment after they arrive. While dysregulated PU.1 can cause trans-differentiation to myeloid or dendritic-cell fates, endogenous PU.1 normally plays a protracted role within early T-cell development. Its target genes are occasionally repressed but mostly activated by PU.1 binding, and they confer on the cells distinctive stage-specific transcription factor expression patterns as well as a rich array of stage-specific cell biological features that await proper functional analysis. This positive regulatory role is one result of PU.1's strong pattern of binding across the genome, its prominent occupancy of open chromatin sites, and the evidence that it helps to maintain the open chromatin states at bound regulatory sites as long as it is expressed. The number of genes that respond quickly to changes in PU.1 activity may only account for a minority of all the genomic sites where PU.1 is found engaged; at other sites, its role could be structural or redundant with other factors. However, it is clear that PU.1 also affects the activity of certain genes that it does not bind to directly, via creating preferential interaction sites for other factors that can deplete the regulatory elements of those factors' alternative target genes. Through chromatin state placeholding and “coregulator theft” as well as through its own direct transcriptional activities, PU.1 pervades the regulatory state of early T cells as long as it is expressed.
This phase comes to an end when other transcription factors finally accumulate to the point where they can shut PU.1 off. The best current candidates for this silencing activity include GATA-3 (104, 105), TCF-1 or LEF-1 (37), and especially Runx1 (80, 150–152), probably working in a dose-dependent combination, although the mechanism through which they finally achieve the ability to repress PU.1 has not yet been reported. Importantly, the duration of the PU.1 activity phase is regulated to vary among different ontogenic waves of T cell development. It probably extends for over 10 days for the thymocytes in young adult mice (153), where it is crucial for successful T-cell generation (2, 147). In contrast, for many first-wave fetal thymocytes it may last only a day or two, and is mostly or entirely dispensable (71, 147). This indicates that the specific constellation of functions that PU.1 serves in T-cell development is a module within the larger T-cell developmental program that can be deployed optionally to serve a particular role. Perhaps it is more important for scaling the population dynamics of T-cell production as the animal finishes gestation and grows, or for promoting accurate migration through distinct thymic microenvironments, than for making T cell precursors per se.
The pioneering role of PU.1 on the genome raises fascinating questions for future study that connect mechanism with developmental lineage selection. Hematopoietic progenitors express PU.1 before they enter the thymus, but the pattern of its occupancy is not well defined at that stage, so the onset of PU.1's pioneering activity in precursors that will eventually generate T cells is not easy to study. The mechanisms discussed in this review show that it establishes a pre-pattern that can influence the binding of the other transcription factors expressed in the cell throughout multiple cell cycles in the thymus. It is not clear, though, how this particular pre-pattern is set, to be distinguished from PU.1 binding patterns in B cells and myeloid cells (84). The question could be linked with the deeper mystery of the factors involved in designating some multipotent precursors to enter the thymus in the first place, as opposed to remaining in the bone marrow for programming into B cells, natural killer cells, or innate lymphoid cells. So far the innate lymphoid cell developmental program in particular appears to resemble the intrathymic T-cell program in many respects (154–157), enough to raise the question of what makes T-cell precursors wait to activate genes like Tcf7 and Gata3 until they reach the thymus. Is PU.1 part of the answer? The system-wide impact of PU.1 on other factors suggests that in scenarios where PU.1 is absent, the same T-cell transcription factors might initially choose different binding sites. Indeed, pro-T cells that have PU.1 acutely deleted at an early stage do not only differentiate faster along the T lineage; they also tend to shift to a natural killer-like program more readily than controls (79). Thus, activity of PU.1 may be important, also, to block certain alternative differentiation paths for pro-T cells. In the end, is T-cell lineage fidelity itself partly a legacy of PU.1's transient role?
Author Contributions
ER wrote the paper, contributed to ideas in the review, and directed research that led to this review. HH and JU carried out research that led to this review, contributed to ideas in the review, provided some figures and edited the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Rothenberg lab and members of the laboratories of Mikael Sigvardsson, Tomoaki Tanaka, and Barbara Wold for stimulating discussions. The authors' own research on this subject was supported by fellowships from the Swedish Research Council (JU) and the Manpei Suzuki Diabetes Foundation (HH), by grants from the USPHS, R01AI095943 and R01HD076915 (ER), and by the Albert Billings Ruddock Professorship (ER).
References
- 1.Singh H, DeKoter RP, Walsh JC. PU.1, a shared transcriptional regulator of lymphoid and myeloid cell fates. Cold Spring Harbor Symp.Quant. Biol. (1999) 64:13–20. 10.1101/sqb.1999.64.13 [DOI] [PubMed] [Google Scholar]
- 2.Dakic A, Metcalf D, Di Rago L, Mifsud S, Wu L, Nutt SL. PU.1 regulates the commitment of adult hematopoietic progenitors and restricts granulopoiesis. J Exp Med. (2005) 201:1487–502. 10.1084/jem.20050075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iwasaki H, Somoza C, Shigematsu H, Duprez EA, Iwasaki-Arai J, Mizuno SI, et al. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood (2005)106:1590–600. 10.1182/blood-2005-03-0860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman AD. Transcriptional control of granulocyte and monocyte development. Oncogene (2007) 26:6816–28. 10.1038/sj.onc.1210764 [DOI] [PubMed] [Google Scholar]
- 5.Houston IB, Kamath MB, Schweitzer BL, Chlon TM, DeKoter RP. Reduction in PU.1 activity results in a block to B-cell development, abnormal myeloid proliferation, and neonatal lethality. Exp Hematol. (2007) 35:1056–68. 10.1016/j.exphem.2007.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonifer C, Hoogenkamp M, Krysinska H, Tagoh H. How transcription factors program chromatin–lessons from studies of the regulation of myeloid-specific genes. SeminImmunol (2008) 20:257–63. 10.1016/j.smim.2008.05.001 [DOI] [PubMed] [Google Scholar]
- 7.Burda P, Laslo P, Stopka T. The role of PU.1 and GATA-1 transcription factors during normal and leukemogenic hematopoiesis. Leukemia (2010) 24:1249–57. 10.1038/leu.2010.104 [DOI] [PubMed] [Google Scholar]
- 8.Carotta S, Wu L, Nutt SL. Surprising new roles for PU.1 in the adaptive immune response. Immunol Rev. (2010) 238:63–75. 10.1111/j.1600-065X.2010.00955.x [DOI] [PubMed] [Google Scholar]
- 9.Imperato MR, Cauchy P, Obier N, Bonifer C. The RUNX1-PU.1 axis in the control of hematopoiesis. Int J Hematol. (2015) 101:319–29. 10.1007/s12185-015-1762-8 [DOI] [PubMed] [Google Scholar]
- 10.Poon GMK, MacgregorJr RB. A thermodynamic basis of DNA sequence selectivity by the ETS domain of murine PU.1. J Mol Biol. (2004) 335, 113–127. 10.1016/j.jmb.2003.09.046 [DOI] [PubMed] [Google Scholar]
- 11.Pham T-H, Minderjahn J, Schmidl C, Hoffmeister H, Schmidhofer S, Chen W, et al. Mechanisms of in vivo binding site selection of the hematopoietic master transcription factor PU.1. Nucleic Acids Res. (2013) 41:6391–402. 10.1093/nar/gkt355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barozzi I, Simonatto M, Bonifacio S, Yang L, Rohs R, Ghisletti S, et al. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol Cell (2014) 54:844–57. 10.1016/j.molcel.2014.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell (2010) 38:576–89. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heinz S, Romanoski CE, Benner C, Allison KA, Kaikkonen MU, Orozco LD, et al. Effect of natural genetic variation on enhancer selection and function. Nature (2013) 503:487–92. 10.1038/nature12615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, et al. Latent enhancers activated by stimulation in differentiated cells. Cell (2013) 152:157–71. 10.1016/j.cell.2012.12.018 [DOI] [PubMed] [Google Scholar]
- 16.Kanno Y, Levi BZ, Tamura T, Ozato K. Immune cell-specific amplification of interferon signaling by the IRF-4/8-PU.1 complex. J Interferon Cytokine Res. (2005) 25:770–9. 10.1089/jir.2005.25.770 [DOI] [PubMed] [Google Scholar]
- 17.Natoli G, Ghisletti S, Barozzi I. The genomic landscapes of inflammation. Genes Dev (2011) 25:101–6. 10.1101/gad.2018811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Riel B, Rosenbauer F. Epigenetic control of hematopoiesis: the PU.1 chromatin connection. Biol Chem. (2014) 395:1265–74. 10.1515/hsz-2014-0195 [DOI] [PubMed] [Google Scholar]
- 19.Turkistany SA, DeKoter RP. The transcription factor PU.1 is a critical regulator of cellular communication in the immune system. Arch Immunol Ther Exp (Warsz) (2011) 59:431–40. 10.1007/s00005-011-0147-9 [DOI] [PubMed] [Google Scholar]
- 20.Rekhtman N, Radparvar F, Evans T, Skoultchi A. Direct interaction of hematopoietic transcription factors PU.1 and GATA-1: functional antagonism in erythroid cells. Genes Dev. (1999) 13:1398–411. 10.1101/gad.13.11.1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang P, Behre G, Pan J, Iwama A, Wara-aswapati N, Radomska HS, et al. Negative cross-talk between hematopoietic regulators: GATA proteins repress PU.1. Proc Natl Acad Sci USA. (1999) 96:8705–10. 10.1073/pnas.96.15.8705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nerlov C, Querfurth E, Kulessa H, Graf T. GATA-1 interacts with the myeloid PU.1 transcription factor and represses PU.1-dependent transcription. Blood (2000) 95:2543–51. Available online at: www.bloodjournal.org/content/95/8/2543 [PubMed] [Google Scholar]
- 23.Zhang P, Zhang X, Iwama A, Yu C, Smith KA, Mueller BU, et al. PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood (2000) 96:2641–48. Available online at: www.bloodjournal.org/content/96/8/2641 [PubMed] [Google Scholar]
- 24.Stopka T, Amanatullah DF, Papetti M, Skoultchi AI. PU.1 inhibits the erythroid program by binding to GATA-1 on DNA and creating a repressive chromatin structure. EMBO J. (2005) 24:3712–23. 10.1038/sj.emboj.7600834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laiosa CV, Stadtfeld M, Graf T. Determinants of lymphoid-myeloid lineage diversification. Annu Rev Immunol. (2006) 24:705–38. 10.1146/annurev.immunol.24.021605.090742 [DOI] [PubMed] [Google Scholar]
- 26.Swiers G, Patient R, Loose M. Genetic regulatory networks programming hematopoietic stem cells and erythroid lineage specification. Dev Biol. (2006) 294:525–40. 10.1016/j.ydbio.2006.02.051 [DOI] [PubMed] [Google Scholar]
- 27.Arinobu Y, Mizuno S, Chong Y, Shigematsu H, Iino T, Iwasaki H, et al. Reciprocal activation of GATA-1 and PU.1 marks initial specification of hematopoietic stem cells into myeloerythroid and myelolymphoid lineages. Cell Stem Cell (2007) 1:416–27. 10.1016/j.stem.2007.07.004 [DOI] [PubMed] [Google Scholar]
- 28.Huang S, Guo YP, May G, Enver T. Bifurcation dynamics in lineage-commitment in bipotent progenitor cells. Dev Biol (2007) 305:695–713. 10.1016/j.ydbio.2007.02.036 [DOI] [PubMed] [Google Scholar]
- 29.Chickarmane V, Enver T, Peterson C. Computational modeling of the hematopoietic erythroid-myeloid switch reveals insights into cooperativity, priming, and irreversibility. PLoS Comput Biol. (2009) 5:e1000268. 10.1371/journal.pcbi.1000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walsh JC, DeKoter RP, Lee HJ, Smith ED, Lancki DW, Gurish MF, et al. Cooperative and antagonistic interplay between PU.1 and GATA-2 in the specification of myeloid cell fates. Immunity (2002) 17:665–76. 10.1016/S1074-7613(02)00452-1 [DOI] [PubMed] [Google Scholar]
- 31.Hoppe PS, Schwarzfischer M, Loeffler D, Kokkaliaris KD, Hilsenbeck O, Moritz N, et al. Early myeloid lineage choice is not initiated by random PU.1 to GATA1 protein ratios. Nature (2016) 535:299–302. 10.1038/nature18320 [DOI] [PubMed] [Google Scholar]
- 32.Tydell CC, David-Fung ES, Moore JE, Rowen L, Taghon T, Rothenberg EV. Molecular dissection of prethymic progenitor entry into the T lymphocyte developmental pathway. J Immunol. (2007) 179:421–38. 10.4049/jimmunol.179.1.421 [DOI] [PubMed] [Google Scholar]
- 33.Yui MA, Feng N, Rothenberg EV. Fine-scale staging of T cell lineage commitment in adult mouse thymus. J Immunol. (2010) 185:284–93. 10.4049/jimmunol.1000679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rothenberg EV, Ungerback J, Champhekar A. Forging T-Lymphocyte identity: intersecting networks of transcriptional control. Adv Immunol. (2016) 129:109–74. 10.1016/bs.ai.2015.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casero D, Sandoval S, Seet CS, Scholes J, Zhu Y, Ha VL, et al. Long non-coding RNA profiling of human lymphoid progenitor cells reveals transcriptional divergence of B cell and T cell lineages. Nat Immunol. (2015) 16:1282–91. 10.1038/ni.3299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ha VL, Luong A, Li F, Casero D, Malvar J, Kim YM, et al. The T-ALL related gene BCL11B regulates the initial stages of human T-cell differentiation. Leukemia (2017) 31:2503–14. 10.1038/leu.2017.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosenbauer F, Owens BM, Yu L, Tumang JR, Steidl U, Kutok JL, et al. Lymphoid cell growth and transformation are suppressed by a key regulatory element of the gene encoding PU.1. Nat Genet. (2006) 38:27–37. 10.1038/ng1679 [DOI] [PubMed] [Google Scholar]
- 38.Seki M, Kimura S, Isobe T, Yoshida K, Ueno H, Nakajima-Takagi Y, et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat Genet (2017) 49:1274–81. 10.1038/ng.3900 [DOI] [PubMed] [Google Scholar]
- 39.Masuda K, Kakugawa K, Nakayama T, Minato M, Katsura Y, Kawamoto H. T cell lineage determination precedes the initiation of TCRβ rearrangement. J Immunol. (2007) 179:3699–706. 10.4049/jimmunol.179.6.3699 [DOI] [PubMed] [Google Scholar]
- 40.Bell JJ, Bhandoola A. The earliest thymic progenitors for T cells possess myeloid lineage potential. Nature (2008) 452:764–7. 10.1038/nature06840 [DOI] [PubMed] [Google Scholar]
- 41.Wada H, Masuda K, Satoh R, Kakugawa K, Ikawa T, Katsura Y, et al. Adult T-cell progenitors retain myeloid potential. Nature (2008) 452:768–72. 10.1038/nature06839 [DOI] [PubMed] [Google Scholar]
- 42.De Obaldia ME, Bell JJ, Bhandoola A. Early T-cell progenitors are the major granulocyte precursors in the adult mouse thymus. Blood (2013) 121:64–71. 10.1182/blood-2012-08-451773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rothenberg EV, Zhang J, Li L. Multilayered specification of the T-cell lineage fate. Immunol Rev. (2010) 238:150–68. 10.1111/j.1600-065X.2010.00964.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu G, Cui K, Fang D, Hirose S, Wang X, Wangsa D, et al. Transformation of accessible chromatin and 3D nucleome underlies lineage commitment of early T cells. Immunity (2018) 48:227–242 e228. 10.1016/j.immuni.2018.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson MK, Hernandez-Hoyos G, Diamond RA, Rothenberg EV. Precise developmental regulation of Ets family transcription factors during specification and commitment to the T cell lineage. Development (1999) 126:3131–48. [DOI] [PubMed] [Google Scholar]
- 46.Anderson MK, Weiss AH, Hernandez-Hoyos G, Dionne CJ, Rothenberg EV. Constitutive expression of PU.1 in fetal hematopoietic progenitors blocks T cell development at the pro-T cell stage. Immunity (2002) 16:285–96. 10.1016/S1074-7613(02)00277-7 [DOI] [PubMed] [Google Scholar]
- 47.Dionne CJ, Tse KY, Weiss AH, Franco CB, Wiest DL, Anderson MK, et al. Subversion of T lineage commitment by PU.1 in a clonal cell line system. Dev Biol. (2005) 280:448–66. 10.1016/j.ydbio.2005.01.027 [DOI] [PubMed] [Google Scholar]
- 48.Lefebvre JM, Haks MC, Carleton MO, Rhodes M, Sinnathamby G, Simon MC, et al. Enforced expression of Spi-B reverses T lineage commitment and blocks β-selection. J Immunol. (2005) 174:6184–94. 10.4049/jimmunol.174.10.6184 [DOI] [PubMed] [Google Scholar]
- 49.Franco CB, Scripture-Adams DD, Proekt I, Taghon T, Weiss AH, Yui MA, et al. Notch/Delta signaling constrains reengineering of pro-T cells by PU.1. Proc Natl Acad Sci USA. (2006) 103:11993–8. 10.1073/pnas.0601188103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laiosa CV, Stadtfeld M, Xie H, de Andres-Aguayo L, Graf T. Reprogramming of committed T cell progenitors to macrophages and dendritic cells by C/EBPα and PU.1 transcription factors. Immunity (2006) 25:731–44. 10.1016/j.immuni.2006.09.011 [DOI] [PubMed] [Google Scholar]
- 51.Wu L, Li CL, Shortman K. Thymic dendritic cell precursors: relationship to the T lymphocyte lineage and phenotype of the dendritic cell progeny. J Exp Med. (1996) 184:903–11. 10.1084/jem.184.3.903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee CK, Kim JK, Kim Y, Lee MK, Kim K, Kang JK, et al. Generation of macrophages from early T progenitors in vitro. JImmunol. (2001) 166:5964–9. 10.4049/jimmunol.166.10.5964 [DOI] [PubMed] [Google Scholar]
- 53.King AG, Kondo M, Scherer DC, Weissman IL. Lineage infidelity in myeloid cells with TCR gene rearrangement: a latent developmental potential of proT cells revealed by ectopic cytokine receptor signaling. Proc Natl Acad Sci USA. (2002) 99:4508–13. 10.1073/pnas.072087899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balciunaite G, Ceredig R, Rolink AG. The earliest subpopulation of mouse thymocytes contains potent T, significant macrophage, and natural killer cell but no B-lymphocyte potential. Blood (2005) 105:1930–6. 10.1182/blood-2004-08-3087 [DOI] [PubMed] [Google Scholar]
- 55.Ikawa T, Hirose S, Masuda K, Kakugawa K, Satoh R, Shibano-Satoh A, et al. An essential developmental checkpoint for production of the T cell lineage. Science (2010) 329:93–6. 10.1126/science.1188995 [DOI] [PubMed] [Google Scholar]
- 56.Luc S, Luis TC, Boukarabila H, Macaulay IC, Buza-Vidas N, Bouriez-Jones T, et al. The earliest thymic T cell progenitors sustain B cell and myeloid lineage potential. Nat Immunol. (2012) 13:412–9. 10.1038/ni.2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schlenner SM, Madan V, Busch K, Tietz A, Laufle C, Costa C, et al. Fate mapping reveals separate origins of T cells and myeloid lineages in the thymus. Immunity (2010) 32:426–36. 10.1016/j.immuni.2010.03.005 [DOI] [PubMed] [Google Scholar]
- 58.Del Real MM, Rothenberg EV. Architecture of a lymphomyeloid developmental switch controlled by PU.1, Notch and Gata3. Development (2013) 140:1207–19. 10.1242/dev.088559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Radtke F, Wilson A, Mancini SJ, MacDonald HR. Notch regulation of lymphocyte development and function. Nat Immunol. (2004) 5:247–53. 10.1038/ni1045 [DOI] [PubMed] [Google Scholar]
- 60.Petrie HT, Zuniga-Pflucker JC. Zoned out: functional mapping of stromal signaling microenvironments in the thymus. Annu Rev Immunol. (2007) 25:649–79. 10.1146/annurev.immunol.23.021704.115715 [DOI] [PubMed] [Google Scholar]
- 61.Bajoghli B, Aghaallaei N, Hess I, Rode I, Netuschil N, Tay BH, et al. Evolution of genetic networks underlying the emergence of thymopoiesis in vertebrates. Cell (2009) 138:186–97. 10.1016/j.cell.2009.04.017 [DOI] [PubMed] [Google Scholar]
- 62.Koch U, Lacombe TA, Holland D, Bowman JL, Cohen BL, Egan SE, et al. Subversion of the T/B lineage decision in the thymus by lunatic fringe- mediated inhibition of Notch-1. Immunity (2001) 15:225–36. 10.1016/S1074-7613(01)00189-3 [DOI] [PubMed] [Google Scholar]
- 63.Yui MA, Rothenberg EV. Deranged early T cell development in immunodeficient strains of nonobese diabetic mice. J Immunol. (2004) 173:5381–91. 10.4049/jimmunol.173.9.5381 [DOI] [PubMed] [Google Scholar]
- 64.Geimer Le Lay AS, Oravecz A, Mastio J, Jung C, Marchal P, Ebel C, et al. The tumor suppressor Ikaros shapes the repertoire of Notch target genes in T cells. Sci Signal (2014) 7:ra28. 10.1126/scisignal.2004545 [DOI] [PubMed] [Google Scholar]
- 65.Yui MA, Rothenberg EV. Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol. (2014) 14:529–45. 10.1038/nri3702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu M, Tayu R, Ikawa T, Masuda K, Matsumoto I, Mugishima H, et al. The earliest thymic progenitors in adults are restricted to T, NK, and dendritic cell lineage and have a potential to form more diverse TCRβ chains than fetal progenitors. J Immunol. (2005) 175:5848–56. 10.4049/jimmunol.175.9.5848 [DOI] [PubMed] [Google Scholar]
- 67.Manesso E, Chickarmane V, Kueh HY, Rothenberg EV, Peterson C. Computational modelling of T-cell formation kinetics: output regulated by initial proliferation-linked deferral of developmental competence. J R Soc Interface (2013) 10:20120774. 10.1098/rsif.2012.0774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science (1994) 265:1573–7. 10.1126/science.8079170 [DOI] [PubMed] [Google Scholar]
- 69.McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, et al. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. (1996) 15:5647–58. 10.1002/j.1460-2075.1996.tb00949.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scott EW, Fisher RC, Olson MC, Kehrli EW, Simon MC, Singh H. PU.1 functions in a cell-autonomous manner to control the differentiation of multipotential lymphoid-myeloid progenitors. Immunity (1997) 6:437–47. 10.1016/S1074-7613(00)80287-3 [DOI] [PubMed] [Google Scholar]
- 71.Spain LM, Guerriero A, Kunjibettu S, Scott EW. T cell development in PU.1-deficient mice. J. Immunol. (1999) 163:2681–87. [PubMed] [Google Scholar]
- 72.Polli M, Dakic A, Light A, Wu L, Tarlinton DM, Nutt SL. The development of functional B lymphocytes in conditional PU.1 knockout mice. Blood (2005) 106:2083–90. 10.1182/blood-2005-01-0283 [DOI] [PubMed] [Google Scholar]
- 73.Carotta S, Dakic A, D'Amico A, Pang SH, Greig KT, Nutt SL, et al. The transcription factor PU.1 controls dendritic cell development and Flt3 cytokine receptor expression in a dose-dependent manner. Immunity (2010) 32:628–41. 10.1016/j.immuni.2010.05.005 [DOI] [PubMed] [Google Scholar]
- 74.Chang HC, Sehra S, Goswami R, Yao W, Yu Q, Stritesky GL, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat. Immunol. (2010) 11:527–34. 10.1038/ni.1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jabeen R, Chang HC, Goswami R, Nutt SL, Kaplan MH. The transcription factor PU.1 regulates γδ T cell homeostasis. PLoS ONE (2011) 6:e22189. 10.1371/journal.pone.0022189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Awe O, Hufford MM, Wu H, Pham D, Chang HC, Jabeen R, et al. PU.1 Expression in T follicular helper cells limits CD40L-dependent germinal center B Cell development. J Immunol. (2015) 195:3705–15. 10.4049/jimmunol.1500780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schmitt TM, Zuniga-Pflucker JC. T-cell development, doing it in a dish. Immunol Rev. (2006) 209:95–102. 10.1111/j.0105-2896.2006.00353.x [DOI] [PubMed] [Google Scholar]
- 78.Mohtashami M, Shah DK, Nakase H, Kianizad K, Petrie HT, Zúñiga-Pflücker JC. Direct comparison of Dll1- and Dll4-mediated Notch activation levels shows differential lymphomyeloid lineage commitment outcomes. J Immunol. (2010) 185:867–76. 10.4049/jimmunol.1000782 [DOI] [PubMed] [Google Scholar]
- 79.Champhekar A, Damle SS, Freedman G, Carotta S, Nutt SL, Rothenberg EV. Regulation of early T-lineage gene expression and developmental progression by the progenitor cell transcription factor PU.1. Genes Dev. (2015) 29:832–48. 10.1101/gad.259879.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hosokawa H, Ungerbäck J, Wang X, Matsumoto M, Nakayama KI, Cohen SM, et al. Transcription factor PU.1 represses and activates gene expression in early T cells by redirecting partner transcription factor binding. Immunity (2018) 48:1119–34.e1117. 10.1016/j.immuni.2018.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kawamoto H, Ohmura K, Fujimoto S, Lu M, Ikawa T, Katsura Y. Extensive proliferation of T cell lineage-restricted progenitors in the thymus: an essential process for clonal expression of diverse T cell receptor β chains. Eur J Immunol. (2003) 33:606–15. 10.1002/eji.200323461 [DOI] [PubMed] [Google Scholar]
- 82.Kueh HY, Champhekar A, Nutt SL, Elowitz MB, Rothenberg EV. Positive feedback between PU.1 and the cell cycle controls myeloid differentiation. Science (2013) 341:670–3. 10.1126/science.1240831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Staber PB, Zhang P, Ye M, Welner RS, Nombela-Arrieta C, Bach C, et al. Sustained PU.1 levels balance cell-cycle regulators to prevent exhaustion of adult hematopoietic stem cells. Mol Cell (2013) 49:934–46. 10.1016/j.molcel.2013.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang JA, Mortazavi A, Williams BA, Wold BJ, Rothenberg EV. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell (2012) 149:467–82. 10.1016/j.cell.2012.01.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ungerbäck J, Hosokawa H, Wang X, Strid T, Williams BA, Sigvardsson M, et al. Pioneering, chromatin remodeling, and epigenetic constraint in early T-cell gene regulation by SPI1 (PU.1). Genome Res (2018) 28:1508–19. 10.1101/gr.231423.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wontakal SN, Guo X, Will B, Shi M, Raha D, Mahajan MC, et al. A large gene network in immature erythroid cells is controlled by the myeloid and B cell transcriptional regulator PU.1. PLoS Genet (2011) 7:e1001392. 10.1371/journal.pgen.1001392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carleton M, Haks MC, Smeele SA, Jones A, Belkowski SM, Berger MA, et al. Early growth response transcription factors are required for development of CD4−CD8− thymocytes to the CD4+CD8+ stage. J Immunol. (2002) 168:1649–58. 10.4049/jimmunol.168.4.1649 [DOI] [PubMed] [Google Scholar]
- 88.Schmitt TM, Zúñiga-Pflücker JC. Induction of T cell development from hematopoietic progenitor cells by Delta-like-1 in vitro. Immunity (2002) 17:749–56. 10.1016/S1074-7613(02)00474-0 [DOI] [PubMed] [Google Scholar]
- 89.Antony-Debré I, Paul A, Leite J, Mitchell K, Kim HM, Carvajal LA, et al. Pharmacological inhibition of the transcription factor PU.1 in leukemia. J Clin Invest. (2017) 127:4297–313. 10.1172/JCI92504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fisher RC, Scott EW. Role of PU.1 in hematopoiesis. Stem Cells (1998) 16:25–37. 10.1002/stem.160025 [DOI] [PubMed] [Google Scholar]
- 91.Marecki S, Fenton MJ. PU.1/Interferon regulatory factor interactions: mechanisms of transcriptional regulation. Cell Biochem.Biophys. (2000) 33:127–48. 10.1385/CBB:33:2:127 [DOI] [PubMed] [Google Scholar]
- 92.Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell (2014) 159:440–55. 10.1016/j.cell.2014.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rothenberg EV, Kueh HY, Yui MA, Zhang JA. Hematopoiesis and T-cell specification as a model developmental system. Immunol Rev. (2016) 271:72–97. 10.1111/imr.12417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods (2013) 10:1213–8. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Johnson JL, Georgakilas G, Petrovic J, Kurachi M, Cai S, Harly C, et al. Lineage-determining transcription factor TCF-1 initiates the epigenetic identity of T cells. Immunity (2018) 48:243–257 e210. 10.1016/j.immuni.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nikolajczyk BS, Sanchez JA, Sen R. ETS protein-dependent accessibility changes at the immunoglobulin æ heavy chain enhancer. Immunity (1999) 11:11–20. 10.1016/S1074-7613(00)80077-1 [DOI] [PubMed] [Google Scholar]
- 97.Marecki S, McCarthy KM, Nikolajczyk BS. PU.1 as a chromatin accessibility factor for immunoglobulin genes. Mol Immunol. (2004) 40:723–31. 10.1016/j.molimm.2003.08.007 [DOI] [PubMed] [Google Scholar]
- 98.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. (2011) 25:2227–41. 10.1101/gad.176826.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.DeKoter RP, Singh H. Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science (2000) 288:1439–41. 10.1126/science.288.5470.1439 [DOI] [PubMed] [Google Scholar]
- 100.Boller S, Ramamoorthy S, Akbas D, Nechanitzky R, Burger L, Murr R, et al. Pioneering Activity of the C-Terminal Domain of EBF1 Shapes the Chromatin Landscape for B Cell Programming. Immunity (2016) 44:527–41. 10.1016/j.immuni.2016.02.021 [DOI] [PubMed] [Google Scholar]
- 101.Rekhtman N, Choe KS, Matushansky I, Murray S, Stopka T, Skoultchi AI. PU.1 and pRB interact and cooperate to repress GATA-1 and block erythroid differentiation. Mol Cell Biol. (2003) 23:7460–74. 10.1128/MCB.23.21.7460-7474.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.de la Rica L, Rodriguez-Ubreva J, Garcia M, Islam AB, Urquiza JM, Hernando H, et al. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. (2013) 14:R99. 10.1186/gb-2013-14-9-r99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ridinger-Saison M, Boeva V, Rimmele P, Kulakovskiy I, Gallais I, Levavasseur B, et al. Spi-1/PU.1 activates transcription through clustered DNA occupancy in erythroleukemia. Nucleic Acids Res. (2012) 40:8927–41. 10.1093/nar/gks659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Scripture-Adams DD, Damle SS, Li L, Elihu KJ, Qin S, Arias AM, et al. GATA-3 dose-dependent checkpoints in early T cell commitment. J Immunol. (2014) 193:3470–91. 10.4049/jimmunol.1301663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Taghon T, Yui MA, Rothenberg EV. Mast cell lineage diversion of T lineage precursors by the essential T cell transcription factor GATA-3. Nat Immunol. (2007) 8:845–55. 10.1038/ni1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Krysinska H, Hoogenkamp M, Ingram R, Wilson N, Tagoh H, Laslo P, et al. A two-step, PU.1 dependent, mechanism for developmentally regulated chromatin remodelling and transcription of the c-fms gene. Mol. Cell Biol. (2007) 27:878–87. 10.1128/MCB.01915-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pospisil V, Vargova K, Kokavec J, Rybarova J, Savvulidi F, Jonasova A, et al. Epigenetic silencing of the oncogenic miR-17-92 cluster during PU.1-directed macrophage differentiation. EMBO J. (2011) 30:4450–64. 10.1038/emboj.2011.317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Soufi A, Donahue G, Zaret KS. Facilitators and impediments of the pluripotency reprogramming factors' initial engagement with the genome. Cell (2012) 151:994–1004. 10.1016/j.cell.2012.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Spitz F, Furlong EEM. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet (2012) 13:613–26. 10.1038/nrg3207 [DOI] [PubMed] [Google Scholar]
- 110.Vahedi G, Kanno Y, Sartorelli V, O'Shea JJ. Transcription factors and CD4 T cells seeking identity: masters, minions, setters and spikers. Immunology (2013) 139:294–8. 10.1111/imm.12113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Glass CK, Natoli G. Molecular control of activation and priming in macrophages. Nat Immunol (2016) 17:26–33. 10.1038/ni.3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Petrovick MS, Hiebert SW, Friedman AD, Hetherington CJ, Tenen DG, Zhang DE. Multiple functional domains of AML1: PU.1 and C/EBPα synergize with different regions of AML1. Mol. Cell. Biol. (1998) 18:3915–25. 10.1128/MCB.18.7.3915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Leddin M, Perrod C, Hoogenkamp M, Ghani S, Assi S, Heinz S, et al. Two distinct auto-regulatory loops operate at the PU.1 locus in B cells and myeloid cells. Blood (2011) 117:2827–38. 10.1182/blood-2010-08-302976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lichtinger M, Ingram R, Hannah R, Muller D, Clarke D, Assi SA, et al. RUNX1 reshapes the epigenetic landscape at the onset of haematopoiesis. EMBO J. (2012) 31:4318–33. 10.1038/emboj.2012.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Guertin MJ, Zhang X, Coonrod SA, Hager GL. Transient estrogen receptor binding and p300 redistribution support a squelching mechanism for estradiol-repressed genes. Mol Endocrinol. (2014) 28:1522–33. 10.1210/me.2014-1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, et al. Cooperative binding of transcription factors orchestrates reprogramming. Cell (2017) 168:442–59. 10.1016/j.cell.2016.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kanhere A, Hertweck A, Bhatia U, Gokmen MR, Perucha E, Jackson I, et al. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat Commun. (2012) 3:1268. 10.1038/ncomms2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Evans CM, Jenner RG. Transcription factor interplay in T helper cell differentiation. Brief Funct Genomics (2013) 12:499–511. 10.1093/bfgp/elt025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Heng TSP, Painter MW, Consortium TIGP. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. (2008) 9:1091–4. 10.1038/ni1008-1091 [DOI] [PubMed] [Google Scholar]
- 120.Gazit R, Garrison BS, Rao TN, Shay T, Costello J, Ericson J, et al. Transcriptome analysis identifies regulators of hematopoietic stem and progenitor cells. Stem Cell Reports (2013) 1:266–80. 10.1016/j.stemcr.2013.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mingueneau M, Kreslavsky T, Gray D, Heng T, Cruse R, Ericson J, et al. The transcriptional landscape of αβ T cell differentiation. Nat Immunol. (2013) 14:619–32. 10.1038/ni.2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.David-Fung ES, Butler R, Buzi G, Yui MA, Diamond RA, Anderson MK, et al. Transcription factor expression dynamics of early T-lymphocyte specification and commitment. Dev Biol. (2009) 325:444–67. 10.1016/j.ydbio.2008.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Longabaugh WJR, Zeng W, Zhang JA, Hosokawa H, Jansen CS, Li L, et al. Bcl11b and combinatorial resolution of cell fate in the T-cell gene regulatory network. Proc Natl Acad Sci USA. (2017) 114:5800–7. 10.1073/pnas.1610617114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kueh HY, Yui MA, Ng KKH, Pease SS, Zhang JA, Damle SS, et al. Asynchronous combinatorial action of four regulatory factors activates Bcl11b for T cell commitment. Nat Immunol. (2016) 17:956–65. 10.1038/ni.3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Goldschneider I. Cyclical mobilization and gated importation of thymocyte progenitors in the adult mouse: evidence for a thymus-bone marrow feedback loop. Immunol Rev. (2006) 209:58–75. 10.1111/j.0105-2896.2006.00354.x [DOI] [PubMed] [Google Scholar]
- 126.Martins VC, Busch K, Juraeva D, Blum C, Ludwig C, Rasche V, et al. Cell competition is a tumour suppressor mechanism in the thymus. Nature (2014) 509:465–70. 10.1038/nature13317 [DOI] [PubMed] [Google Scholar]
- 127.Zietara N, Lyszkiewicz M, Puchalka J, Witzlau K, Reinhardt A, Forster R, et al. Multicongenic fate mapping quantification of dynamics of thymus colonization. J Exp Med. (2015) 212:1589–601. 10.1084/jem.20142143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lind EF, Prockop SE, Porritt HE, Petrie HT. Mapping precursor movement through the postnatal thymus reveals specific microenvironments supporting defined stages of early lymphoid development. J Exp Med. (2001) 194:127–34. 10.1084/jem.194.2.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Buono M, Facchini R, Matsuoka S, Thongjuea S, Waithe D, Luis TC, et al. A dynamic niche provides Kit ligand in a stage-specific manner to the earliest thymocyte progenitors. Nat Cell Biol. (2016) 18:157–67. 10.1038/ncb3299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Love PE, Bhandoola A. Signal integration and crosstalk during thymocyte migration and emigration. Nat Rev Immunol. (2011) 11:469–77. 10.1038/nri2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dzierzak E, Speck NA. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat Immunol. (2008) 9:129–36. 10.1038/ni1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yoshimoto M, Porayette P, Glosson NL, Conway SJ, Carlesso N, Cardoso AA, et al. Autonomous murine T-cell progenitor production in the extra-embryonic yolk sac before HSC emergence. Blood (2012) 119:5706–14. 10.1182/blood-2011-12-397489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ramond C, Berthault C, Burlen-Defranoux O, de Sousa AP, Guy-Grand D, Vieira P, et al. Two waves of distinct hematopoietic progenitor cells colonize the fetal thymus. Nat Immunol. (2014) 15:27–35. 10.1038/ni.2782 [DOI] [PubMed] [Google Scholar]
- 134.Ikuta K, Kina T, MacNeil I, Uchida N, Peault B, Chien YH, et al. A developmental switch in thymic lymphocyte maturation potential occurs at the level of hematopoietic stem cells. Cell (1990) 62:863–74. 10.1016/0092-8674(90)90262-D [DOI] [PubMed] [Google Scholar]
- 135.Kawamoto H, Ohmura K, Hattori N, Katsura Y. Hemopoietic progenitors in the murine fetal liver capable of rapidly generating T cells. J Immunol. (1997) 158:3118–24. [PubMed] [Google Scholar]
- 136.Yuan J, Nguyen CK, Liu X, Kanellopoulou C, Muljo SA. Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis. Science (2012) 335:1195–200. 10.1126/science.1216557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Harman BC, Jenkinson WE, Parnell SM, Rossi SW, Jenkinson EJ, Anderson G. T/B lineage choice occurs prior to intrathymic Notch signalling. Blood (2005) 106:886–92. 10.1182/blood-2004-12-4881 [DOI] [PubMed] [Google Scholar]
- 138.Masuda K, Kubagawa H, Ikawa T, Chen CC, Kakugawa K, Hattori M, et al. Prethymic T-cell development defined by the expression of paired immunoglobulin-like receptors. EMBO J. (2005) 24:4052–60. 10.1038/sj.emboj.7600878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Berthault C, Ramond C, Burlen-Defranoux O, Soubigou G, Chea S, Golub R, et al. Asynchronous lineage priming determines commitment to T cell and B cell lineages in fetal liver. Nat Immunol. (2017) 18:1139–49. 10.1038/ni.3820 [DOI] [PubMed] [Google Scholar]
- 140.Okamura RM, Sigvardsson M, Galceran J, Verbeek S, Clevers H, Grosschedl R. Redundant regulation of T cell differentiation and TCRβ gene expression by the transcription factors LEF-1 and TCF-1. Immunity (1998) 8:11–20. 10.1016/S1074-7613(00)80454-9 [DOI] [PubMed] [Google Scholar]
- 141.Germar K, Dose M, Konstantinou T, Zhang J, Wang H, Lobry C, et al. T-cell factor 1 is a gatekeeper for T-cell specification in response to Notch signaling. Proc Natl Acad Sci USA. (2011) 108:20060–5. 10.1073/pnas.1110230108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang R, Xie H, Huang Z, Ma J, Fang X, Ding Y, et al. T cell factor 1 regulates thymocyte survival via a RORγt-dependent pathway. J Immunol. (2011) 187:5964–73. 10.4049/jimmunol.1101205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Weber BN, Chi AW, Chavez A, Yashiro-Ohtani Y, Yang Q, Shestova O, et al. A critical role for TCF-1 in T-lineage specification and differentiation. Nature (2011) 476:63–8. 10.1038/nature10279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Steinke FC, Yu S, Zhou X, He B, Yang W, Zhou B, et al. TCF-1 and LEF-1 act upstream of Th-POK to promote the CD4+ T cell fate and interact with Runx3 to silence Cd4 in CD8+ T cells. Nat Immunol. (2014) 15:646–56. 10.1038/ni.2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Barra MM, Richards DM, Hansson J, Hofer AC, Delacher M, Hettinger J, et al. Transcription factor 7 limits regulatory T cell generation in the thymus. J Immunol. (2015) 195:3058–70. 10.4049/jimmunol.1500821 [DOI] [PubMed] [Google Scholar]
- 146.Schilham MW, Wilson A, Moerer P, Benaissa-Trouw BJ, Cumano A, Clevers HC. Critical involvement of Tcf-1 in expansion of thymocytes. J Immunol. (1998) 161:3984–91. [PubMed] [Google Scholar]
- 147.Montecino-Rodriguez E, Casero D, Fice M, Le J, Dorshkind K. Differential expression of PU.1 and Key T lineage transcription factors distinguishes fetal and adult T Cell development. J Immunol. (2018) 200:2046–56. 10.4049/jimmunol.1701336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.David-Fung ES, Yui MA, Morales M, Wang H, Taghon T, Diamond RA, et al. Progression of regulatory gene expression states in fetal and adult pro-T-cell development. Immunol Rev. (2006) 209:212–36. 10.1111/j.0105-2896.2006.00355.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Belyaev NN, Biro J, Athanasakis D, Fernandez-Reyes D, Potocnik AJ. Global transcriptional analysis of primitive thymocytes reveals accelerated dynamics of T cell specification in fetal stages. Immunogenetics (2012) 64:591–604. 10.1007/s00251-012-0620-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Huang G, Zhang P, Hirai H, Elf S, Yan X, Chen Z, et al. PU.1 is a major downstream target of AML1 (RUNX1) in adult mouse hematopoiesis. Nat Genet. (2008) 40:51–60. 10.1038/ng.2007.7 [DOI] [PubMed] [Google Scholar]
- 151.Hoogenkamp M, Lichtinger M, Krysinska H, Lancrin C, Clarke D, Williamson A, et al. Early chromatin unfolding by RUNX1: a molecular explanation for differential requirements during specification versus maintenance of the hematopoietic gene expression program. Blood (2009) 114:299–309. 10.1182/blood-2008-11-191890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zarnegar MA, Chen J, Rothenberg EV. Cell type-specific activation and repression of PU.1 by a complex of discrete, functionally specialized cis-regulatory elements. Mol Cell Biol. (2010) 30:4922–39. 10.1128/MCB.00354-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Porritt HE, Gordon K, Petrie HT. Kinetics of steady-state differentiation and mapping of intrathymic-signaling environments by stem cell transplantation in nonirradiated mice. J Exp Med. (2003) 198:957–62. 10.1084/jem.20030837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Shih HY, Sciume G, Mikami Y, Guo L, Sun HW, Brooks SR, et al. Developmental acquisition of regulomes underlies innate lymphoid cell functionality. Cell (2016) 165:1120–33. 10.1016/j.cell.2016.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yu Y, Tsang JC, Wang C, Clare S, Wang J, Chen X, et al. Single-cell RNA-seq identifies a PD-1hi ILC progenitor and defines its development pathway. Nature (2016) 539:102–6. 10.1038/nature20105 [DOI] [PubMed] [Google Scholar]
- 156.Cherrier DE, Serafini N, Di Santo JP. Innate lymphoid cell development: a T cell perspective. Immunity (2018) 48:1091–103. 10.1016/j.immuni.2018.05.010 [DOI] [PubMed] [Google Scholar]
- 157.Harly C, Cam M, Kaye J, Bhandoola A. Development and differentiation of early innate lymphoid progenitors. J Exp Med. (2018) 215:249–62. 10.1084/jem.20170832 [DOI] [PMC free article] [PubMed] [Google Scholar]