

Abstract
Erwinia amylovora is the etiological agent of fire blight, a devastating disease which is a global threat to commercial apple and pear production. The Erwinia genus includes a wide range of different species belonging to plant pathogens, epiphytes and even opportunistic human pathogens. The aim of the present study is to understand, within the Erwinia genus, the genetic differences between phytopathogenic strains and those strains not reported to be phytopathogenic. The genes related to the hydroxamate siderophores iron uptake have been considered due to their potential druggability. In E. amylovora siderophore-mediated iron acquisition plays a relevant role in the progression of Fire blight. Here we analyzed the taxonomic relations within Erwinia genus and the relevance of the genes related to the siderophore-mediated iron uptake pathway. The results of this study highlight the presence of a well-defined sub-group of Rosaceae infecting species taxonomically and genetically related with a high number of conserved core genes. The analysis of the complete ferrioxamine transport system has led to the identification of two genes exclusively present in the Rosaceae infecting strains.
Introduction
Fire blight is one of the major threat to Rosaceae with a potentially disastrous economic impact on apple and pear production1. With favorable environmental conditions, an outbreak can cause the loss of entire annual harvests. The etiological agent of the disease is Erwinia amylovora, one of the top ten known plant pathogens2. The Erwinia genus comprises species that are plant pathogens, non-pathogen, epiphytes, and even opportunistic human pathogens3–5. E. amylovora can infect a wide range of hosts, comprehensive of apple, pear, hawthorn, cotoneaster, rubus, etc6. Earlier comparative genomic studies, focused on virulence genes, suggest that E. amylovora host specificity and pathogenicity is driven by the absence or presence of certain genes7.
In living organisms, iron is a fundamental element, present as a cofactor in proteins and enzymes (e.g., iron-sulfur clusters, heme groups, etc.). A broad spectrum of biological relevant reactions are catalyzed by the reversible Fe(II)/Fe(III) redox pair8. The restriction on the availability of iron led to the development of highly selective systems for its acquisition, either directly (e.g. through transferrin, heme or hemoproteins) or indirectly, through hemophores or siderophores. In particular, siderophores are small molecules (usually < 1 kDa) that complex ferric iron and are among the strongest Fe(III) chelators9. Iron uptake is one of the main molecular pathways involved in the fire blight disease progression10,11. Not all Erwinia species are able to synthetize their own siderophores, suggesting that iron uptake is a potential niche adaptation factor12. E. amylovora mutants, with defective siderophore biosynthesis and uptake, showed an important reduction in their growth on apple flowers compared to the wild-type strain13. In E. amylovora, siderophore mediated iron uptake is dependent on desferrioxamines (DFOs)14: molecules consisting of alternating diamine and dicarboxylic acid building blocks linked by amide bonds. The major product of DFOs biosynthesis in E. amylovora is nocardamine (desferrioxamine E, DFO-E). Three proteins, namely DfoJ, DfoA and DfoC, are responsible for the biosynthesis of DFO-E starting from lysine15,16, these proteins are encoded by a single gene cluster, the dfoJAC operon. Together with the DFOs biosynthetic pathway proteins, the bacterium expresses a specific membrane receptor, called FoxR, necessary for the transport of ferrioxamine complexes across the outer membrane17,18. Transport across the bacterium inner membrane, possibly depends on the periplasmic binding protein-dependent ABC transporter complex FhuABCD, as reported for other hydroxamate siderophores of enteric bacteria e.g. in Escherichia coli19–21 and Salmonella enterica22. Ferrisiderophore complexes are very stable, and the mechanism of iron release can occur with three different mechanisms: hydrolysis of the siderophore, proton-assisted dissociation of the complex, and reduction of the metal center23. The lack of specific hydrolases in E. amylovora and the incompatibility of the cytoplasmic pH with the proton-assisted dissociation suggests that the iron is released after its reduction (ferrioxiamine has a lower affinity for Fe(II) than for Fe(III)), as in E. coli24.
The aim of the present study is to understand the genetic differences within Erwinia spp. between the species reported to be phytopathogenic and the one never reported to be correlated to plant diseases (here considered as non-phytopathogenic). A special focus is dedicated at the genes related to the hydroxamate sidephores iron uptake. We used a comparative genomic approach and selected a specific dataset of Erwinia spp. covering 11 genomes from phytophatogenic species, and 8 genomes from non-pathogenic. The genome selection has been limited to the available ones completed and correctly annotated. Average Nucleotide Identity (ANI), phylogenetic inference based on conserved marker genes, pangenomic analysis and molecular diversity analysis have been performed to get insights about the relevance of siderophore mediated iron acquisition in the evolution of pathogens’ hosts selectivity and virulence.
Results
In order to work on a balanced dataset of the Erwinia genus, 11 plant pathogens and 8 non-pathogens genomes were selected among the sequenced genomes in Erwinia (Table 1). The genome selection has been limited to the ones suitable for the analysis (see Methods section). Seven of the 19 genomes examined have been reported to be Rosaceae infecting pathogens, belonging to 4 different species: E. amylovora, E. pyrifoliae, Erwinia sp Ejp617 and E. piriflorinigrans.
Table 1.
List of the genomes used for this study.
| Strain | Accession number | Habitat/host | Plant pathogenicity |
|---|---|---|---|
| E. amylovora CFBP1430*,• | GCA_000091565.1 | Crataegus (hawthorn) | Pathogen of Rosaceae26 |
| E. amylovora ATCC49946• | GCA_000027205.1 | Malus sp. (apple tree) | Pathogen of Rosaceae26 |
| E. amylovora E-2• | GCA_002803865.1 | Malus sp. (apple tree) | Pathogen of Rosaceae26,27 |
| E. pyrifoliae Ep1/96• | GCA_000027265.1 | Pyrus pyrofolia (asian pear tree/nashi) | Pathogen of Pyrus pyrifolia30 |
| E. pyrifoliae DSM-12163• | GCA_000026985.1 | Pyrus pyrifolia (asian pear tree/nashi) | Pathogen of Pyrus pyrifolia28 |
| Erwinia sp. Ejp617• | GCA_000165815.1 | Pyrus pyrifolia (asian pear tree/nashi) | Pathogen of Pyrus pyrifolia29 |
| E. piriflorinigrans CFBP-5888• | GCA_001050515.1 | Pyrus communis (pear tree) | Pathogen of Pyrus communis25 |
| E. tasmaniensis ET1/99 | GCA_000026185.1 | Malus sp. (apple tree) | Non-pathogen30,47 |
| E. billingiae OSU19-1 | GCF_001269445.1 | Pyrus communis (pear tree) | Non-pathogen48 |
| E. billingiae Eb661 | GCA_000196615.1 | Malus sp. (apple tree) | Non-pathogen47 |
| E. toletana DAPP-PG-7351 | GCA_000336255.1 | Olea sp. (olive tree) | aAssociated to the pathogen of Olea sp.49 |
| E. Oleae DAPP-PG531 | GCA_000770305.1 | Olea europaea (olive tree) | Non-pathogen50,51 |
| E. tracheiphila BuffGH | GCA_000975275.1 | Cucurbita pepo ssp. Texana (squash plant) | Pathogen of Cucurbitaceae52 |
| E. tracheiphila PSU-1 | GCA_000404125.1 | Cucurbita pepo ssp. Texana (squash plant) | Pathogen of Cucurbitaceae52 |
| E. mallotivora BT-MARDI | GCA_000590885.1 | Carica sp. (papaya tree) | Pathogen of Carica sp.53 |
| E. persicina NBRC-102418 | GCA_001571305.1 | Piezodorus guildinii (guts of redbanded stink bug) and Leguminosae (legume plants) | Pathogen of Leguminosae54,55 |
| E. iniecta B149 | GCA_001267545.1 | Diuraphis noxia (wheat aphid) | Non-pathogen56 |
| E. iniecta B120 | GCA_001267535.1 | Diuraphis noxia (wheat aphid) | Non-pathogen56 |
| E. gerundensis EM595 | GCA_001517405.1 | Pyrus communis (pear tree) | Non-pathogen57 |
•These strains are Rosaceae infecting pathogen.
*E. amylovora CFBP1430 is the reference genome where all the DNA gene sequences were extracted.
aFound on olive knots caused by Pseudomonas savastanoi pv. savastanoi. The presence of E. toletana is correlated with the virulence of the disease suggesting a possible interactions with P. savastanoi pv. savastanoi.
Average Nucleotides Identity and Phylogenetic analysis
The ANI values and the phylogenetic analysis consistently highlighted a clear division between the Rosaceae infecting pathogens (RIP) and the other species (Non Rosaceae infecting pathogens, NRIP). The genomes of the RIP group show higher similarity, with pairwise ANI values always higher than the ones from the other species and a large portion of genome aligned (above 6Mbp). Other species instead, had lower pairwise ANI values, also when the alignment lengths was comparable to the ones in the pathogenic species (Fig. 1a). Furthermore, the PhyloPhAn tree (Fig. 1b) reports a shorter phylogenetic distance between RIP species compared to the NRIP species. The tree topology is slightly different from the one in Fig. 1a, however the large basal split between RIP and NRIP is conserved. Erwinia tasmaniensis is an exception: in fact, although it has never been reported to be a pathogen, it shows a high genome similarity with the RIP group.
Figure 1.

Relations within the selected Erwinia species. (a) Heatmap showing ANI analysis of the genomes: on the left the ANI values, on the right the length of the alignment. (b) PhyloPhlAn analysis: the phylogenetic tree was built on a dataset consisting of the concatenation of 400 universally conserved proteins. The phylogenetic distance is represented by the branch length.
Core and accessory genome in Erwinia
Clusters of orthologous proteins were created using Anvi’o, a binary matrix including the presence of genes in the different genomes was exported to perfom a pangenomic study. The core genome shared between RIP and NRIP is plotted in the Fig. 2a. A total of 1551 genes (58.9% of the core genes) are shared between the two groups (RIP and NRIP, hard-core genes), the number of soft-core genes (i.e. core within one of the two groups, but not in the other), is 1034 (39.3%) and 47 (1.8%) in RIP and NRIP, respectively. The multivariate analysis plot (Fig. 2b) displays patterns that are coherent with the findings of our phylogenetic analysis. All the RIP and E. tasmaniensis are concentrated in a closer space forming a narrow cluster in the center of the plot. The other species are scattered in both directions along the NMDS1 and NMDS2 axes within the plot without forming any distinguishable cluster.
Figure 2.

Pangenome analysis. (a) Venn diagram of the core genes shared within the two groups in Erwinia (RIP vs NRIP) (b) Non-metric MultiDimensional Scaling (NMDS): the plot is computed from the presence/absence matrix of all protein clusters in the genomes.
Presence/absence of genes related to hydroxamate siderophores iron uptake and polymorphism
Table 2 summarizes the presence/absence of coding sequences (CDS) for genes related to hydroxamate siderophores iron uptake in the 19 Erwinia genomes taken into account. As shown in Table 2, 9 genes have been considered as marker. All the marker CDS have been found in the RIP and in E. tasmaniensis; in NRIP species the number of the CDS (found with a complete coding sequence) of interest ranged from 2 to 4 (Table 2). The most prevalent gene is fhuB (coding part of the hydroxamate import system), found in 15 out of 19 strains, followed by dfoC, dfoA and foxR (synthesis and transport of ferrioxamine). The least present are fhuD (periplasmic binding protein) and sidE (siderophores utilization protein), both found only in RIP and E. tasmaniensis. The combination of the PhyloPhlAn analysis and the CDS presence is represented in Fig. 3. A monophyletic clade constituted by all the RIP and E. tasmaniensis contains all the CDS screened. Moreover, the CDS for fhuD (in orange) and sidE (in red) can be found only in this clade. The CDS for fhuB (in pink) is almost ubiquitous but it is absent in E. gerundensis, in one of the E. iniecta (E. iniecta B149) and in both E. billingiae.
Table 2.
Distribution of the marker genes in the genomes analyzed. The presence of a CDS for the marker is represented as a black dot.
| Organisms | fhuB | dfoC | dfoA | foxR | fhuA | dfoJ | fhuC | fhuD | sidE | Total markers |
|---|---|---|---|---|---|---|---|---|---|---|
| E. amylovora CFBP1430• | • | • | • | • | • | • | • | • | • | 9 |
| E. amylovora ATCC49946• | • | • | • | • | • | • | • | • | • | 9 |
| E. amylovora E-2• | • | • | • | • | • | • | • | • | • | 9 |
| E. piriflorinigrans CFBP-5888• | • | • | • | • | • | • | • | • | • | 9 |
| E. pyrifoliae DSM-12163• | • | • | • | • | • | • | • | • | • | 9 |
| E. pyrifoliae Ep1/96• | • | • | • | • | • | • | • | • | • | 9 |
| Erwinia.sp Ejp617• | • | • | • | • | • | • | • | • | • | 9 |
| E. tasmaniensis Et1/99 | • | • | • | • | • | • | • | • | • | 9 |
| E. billingiae Eb661 | • | • | • | • | 4 | |||||
| E. billingiae OSU19-1 | • | • | • | • | 4 | |||||
| E. oleae DAPP-PG531 | • | • | • | • | 4 | |||||
| E. iniecta B120 | • | • | • | 3 | ||||||
| E. iniecta B149 | • | • | 2 | |||||||
| E. gerundensis EM595 | • | • | 2 | |||||||
| E. mallotivora BT-MARDI | • | • | 2 | |||||||
| E. persicina NBRC-102418 | • | • | 2 | |||||||
| E. toletana DAPP-PG-7351 | • | • | 2 | |||||||
| E. tracheiphila BuffGH | • | • | 2 | |||||||
| E. tracheiphila PSU-1 | • | • | 2 | |||||||
| Total genomes | 15 | 14 | 13 | 13 | 12 | 9 | 9 | 8 | 8 |
•These strains are Rosaceae infecting pathogen.
Figure 3.
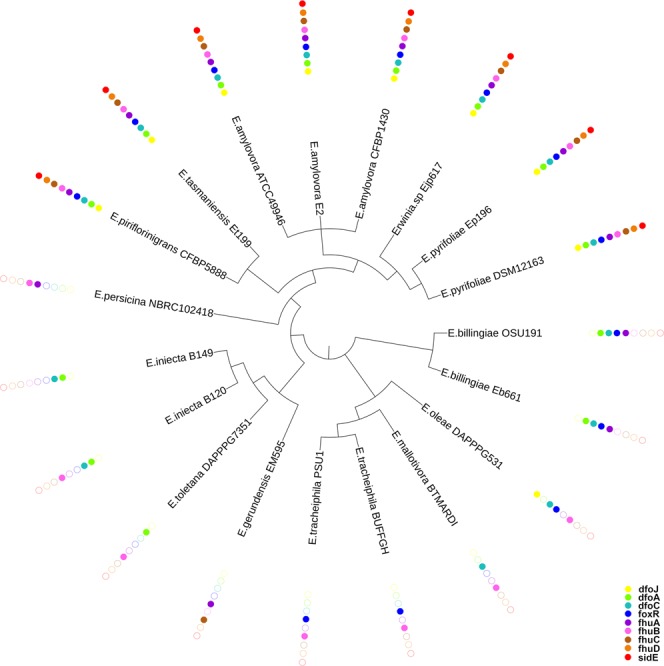
Representation of the genes presence according to the tree generated from PhyloPhlAn analysis (branch length are not shown in this figure to better visualize the topology of the tree). The dots represent the presence of a complete CDS for each gene.
dfoA
A complete CDS for the gene dfoA was found in all RIP, in E. tasmaniensis, both strain of E. iniecta, E. toletana and one of the two E. billingiae strains (EB661). The other E. billingiae strain (OSU 191) missed an in-frame start codon. From none of the remaining species was possible to retrieve the homologous gene neither through blast, nor through the annotated genbank file. In RIP, the gene is conserved, with a Pi value below 0.1, whereas in NRIP there is a highly variable region around 400 bp. Despite the lower conserved of dfoA gene in NRIP, there are two noticeable conserved regions around 200 bp and 1100 bp (Fig. 4a).
Figure 4.

DNA polymorphism analysis of the genes involved in the siderophore mediated iron uptake within the two populations (RIP vs NRIP). RIP in grey, NRIP in orange.
dfoC
A complete CDS for dfoC was found in all the RIP, in E. tasmaniensis, in both E. billingiae and E. iniecta, in E. olea and E. mallotivora. It was not possible to retrieve the CDS from the annotated file. A remarkable feature of this alignment was a large (12 nt), in-frame insertion found in the two E. iniecta strains and E. toletana. In RIP, the gene is very conserved and in particular the region around 400 bp and the last 500 nucleotides. In the NRIP, the gene is generally less conserved than in RIP, with conserved 3 areas (regions around 400 bp, 1300 bp and 2000 bp, Fig. 4b). The variability pattern in this gene show a peak of variation at 1200 bp in NRIP that was not present in RIP.
dfoJ
A complete CDS was found using blast for all E. amylovora, the two E. pyrifoliae, Erwinia sp. Ejp617, E. pyriflorinigrans and E. tasmaniensis. It should be noted that from the genbank file, a feature annotated as dfoJ was present only in one out of three E. amylovora genomes (CFBP1430) and one E. pyrifoliae (DSM12163). A sequence with high identity with the dfoJ gene was also found in E. oleae, however, this gene was lacking an in-frame stop codon, even after manual inspection of the 5′ downstream region. The dfoJ gene was found only in RIP, and it display a relatively low value of Pi all along the sequence compared to the other genes (Fig. 4c).
fhuA
This gene was annotated only in two out of three E. amylovora strains (ATCC49946 and CFBP1430), and in the two E. pyrifoliae. However, a complete coding sequence was found in all E. amylovora and E. pyrifoliae, and in Erwinia sp., E. tasmaniensis, E. persicina, E. piriflorinigrans, both E. billingiae and E. gerundensis. The latter featured a TGA (opal) stop codon, whereas all other species have a TAA (ochre). As for the previous genes, the polymorphism is higher in NRIP than in RIP (Fig. 4d). This gene had a peak of variation in RIP in correspondence of a conserved region of NRIP, at 400 bp.
fhuB
Similar to fhuA, this gene was annotated in the same genomes, but found through blast in 16 out of 19 genomes (leaving out only the two E. billingiae and E. gerundensis). For E. iniecta B149, however, it was not possible to retrieve the whole species due to the fragmentation of the assembly (the 5′ end was cut by a stretch of “N”). Therefore, for the subsequent analysis, fhuB sequences for this genome was removed. The differences in the degree of polymorphism between the two populations (RIP vs NRIP) is evident (Fig. 4e), with maximum in the regions around 900 bp and 1500 bp.
fhuC
This was detected in all rosaceae infecting strains, in E. tasmaniensis and E. gerundensis (that once more had a TGA stop codon, and the most diverging sequence), although it was annotated only in the same four strains as fhuA and fhuB. The gene is conserved in RIP, with slightly higher polymorphism in the last 150 nucleotides (Fig. 4f).
fhuD
This was detected in all Rosaceae-infecting strains and in E. tasmaniensis, although it was annotated only in the same four strains as fhuA and fhuB. The gene is conserved in RIP (Fig. 4g).
foxR
This gene was detected in all RIP plus the two E. billingiae, the two E. tracheiphila, E. oleae, E. tasmaniensis. It was annotated in the genbank files of the two E. pyrifoliae and E. amylovora CFBP1430. The gene polymorphism is higher in NRIP than in RIP (Fig. 4h). In RIP, the most conserved regions are around 400 bp, 750 bp and 1300 bp, while in NRIP the most conserved regions are at 300 bp and 1600 bp. Once again, a peak of nucleotide variation in NRIP was detected in correspondence of a conserved region in RIP, at 1300 bp.
sidE
This gene was detected in all Rosaceae-infecting strains, plus E.tasmaniensis. In RIP, the gene is overall conserved, with a small peak at 700 bp (Fig. 4i).
Discussion
The Erwinia genus includes phytopathogens affecting Rosaceae relevant in fruit production (e.g., Malus sp. and Pyrus sp.). In E. amylovora, the ferrioxamine biosynthesis and transport has been reported to be involved in the progress of the Fire blight disease10–12,15,16 which makes it a worth studying target pathway. In this work, a specific dataset of Erwinia spp. has been analyzed comparing at first the whole genome, then a group of conserved proteins, and at last focusing on specific siderophore genes. The ANI (Fig. 1a), the phylogenetic analysis (Fig. 1b), the pangenome and multivariate analysis based on the presence/absence of all genes (Fig. 2b) are coherent. The whole analysis highlights that a defined group of species is present in the heterogeneous Erwinia genus. All the species in this group, with the exception of E. tasmaniensis25, have been reported to be Rosaceae infecting pathogens25–29. Our results support that the lack of only few specific genes (involved in the biosynthesis and regulation of harpins, effectors and amylovoran) hints for a possible non-pathogenicity of E. tasmaniensis ET1/99 as suggested by Borruso et al.7 and others previously30–32. Erwinia spp. Ejp617 in all analysis clusters very close to the group of E. pyrifoliae although not as ingroup. According to our results Erwinia spp. Ejp617 isolated in Japan28 could be assigned within the species E. pyrifoliae as has been done in other reports28,33. All phylogenetic analysis (Figs 1 and 2b) suggest a different evolutionary rate within the RIP. These difference can be appreciated by looking at the branch length in Fig. 1b: the distance between E. amylovora, E. pyrifoliae, Erwinia sp Ejp617, E. piriflorinigrans and E. tasmaniensis is shorter than any distances among other species. The number of soft-core genes in the two groups is very different (Fig. 2a), suggesting a high level of host-adaptation required in the RIP for infecting the plant. The genomic similarity of RIP is revealed by the selected marker genes. The high level of host-adaptation is reflected in the iron uptake system, where the Pi value/polymorphism for all the genes from NRIP species is higher than in RIP (Fig. 4). Moreover, there are some contrasting nucleotide positions (e.g. 1200 bp in dfoC and 400 bp in fhuA) where there is a maximum of Pi value for one population and minimum for the other one. These regions could play a key role in the enzyme function and/or be part of the active site. Complete CDS for ferroxiamine siderophore biosynthesis (dfoJAC), outer membrane receptors (fhuA and foxR), periplasmic binding protein (fhuD), ABC cassette type receptor components (fhuB and fhuC) and siderophore utilization protein (sidE) are always present in all the Rosaceae infecting Erwinia (plus E. tasmaniensis). The role of ferrioxamine synthesis and foxR receptor in the disease has been previously reported13,14,17,18 and accordingly to our results, the complete transport system is relevant as well. Moreover, while some genes of the biosynthesis and the receptors are shared between RIP and NRIP, the genes coding for the periplasmic binding protein (fhuD) and the siderophore utilization protein (sidE) are strictly conserved in the RIP (Fig. 3). According to the PhyloPhlAn analysis, the epiphytic bacteria E. billingiae is the farthest Erwinia in respect of the RIP. However, it shares both the outer membrane receptors for hydroxamate siderophore and 2 out of 3 enzyme required for the ferrioxamine biosynthesis. E. billingiae tends to invade plants necrotic tissue34,35. E. billingiae could withdraw the intermediates and the ferrioxamine from the RIP, slowing down its growth, while scavenging the iron available in the plant tissues necrotized by pathogens. This behavior suggests a possible symbiosis/antagonism with the RIP36,37 even in the synthesis of siderophores. The absence of the almost ubiquitous gene fhuB from E. billingiae, E. gerundensis, and E. iniecta B149 could be explained with two hypotheses: i) it is possible that the gene, present in the ancestor, has been lost by the two derived species; or ii) a fragmented assembly may result in a missing gene. The latter is the most probable for E. iniecta B149, because the assembly consists of 121 contigs, whereas E. gerudensis consists of 3 contigs. However, in both E. billingiae and in E. gerundensis, the gene was most likely lost.
Methods
In order to have a balanced dataset of the Erwinia genus (11 plant pathogens vs 8 non-pathogens) genomes to be screened were selected among the sequenced genomes in Erwinia according to the list in Borruso et al.7, both fasta and genbank format were downloaded from genbank on the 14th of February 2018. To avoid biases due to the much larger number of sequenced E. amylovora genomes we selected the three genomes with the highest completion score, namely E. amylovora strain CFBP1430, strain 49946 and strain E-2. Eventually, 19 genomes, spanning 13 species, were selected. Seven genomes (in four species), belonged to species known to infect Rosaceae, the remaining 12, spanned 9 species in which this phenotype is not reported (Table 1). Siderophore mediated iron uptake genes of Erwinia spp. were selected for their relevance: dfoA (ordered locus name EAMY_3239), dfoC (EAMY_3240), dfoJ (EAMY_3238), fhuA (annotated as 3 fragments: EAMY_2775, EAMY_2776 and EAMY_2777), fhuB (EAMY_2772), fhuC (EAMY_2774), fhuD (EAMY_2773), foxR (EAMY_3241), sidE (EAMY_3562). The gene sequences from E. amylovora CFBP1430 have been used as reference. A first genome-wide comparison was done using the Average Nucleotide Identity (ANI), using the software PYANI38, and the output was elaborated using the software DiMHepy (https://github.com/lucaTriboli/DiMHepy). A phylogenetic tree was built on the concatenation of 400 conserved proteins using PhyloPhlAn39, and the tree was drawn and annotated (e.g. presence absence of genes was reported on each leaf of the tree) using IToL40. To infer the number of shared protein clusters, genomes were imported in Anvi’o41, the pipeline followed the standard pangenomic workflow with all default parameters. A binary matrix containing all protein clusters of the different genomes was exported and analyzed via Non Multivariate Analysis (NMDS) based on Euclidean distance. Venn diagram was generated to display the number of core genes. Briefly, we considered as core genome the pool of genes present in all genomes of RIP and NRIP separately. Coding sequences (CDS) for each of the 9 genes were searched through BLASTn42 using as query the CDS of E. amylovora CFBP1430. When partial or no CDS were detected, alignments were manually curated by retrieval of the homologous flanking sequences from the fasta files. DNA polymorphism for each selected gene has been analyzed using the sliding window method with the software DnaSP v643 and expressed as Nucleotide diversity (Pi)44,45. The parameters for the analysis have been established in function of the gene length. The window length and the step size values have been settled as 10% and 2.5% respectively (rounded to integer). The Pi value has been calculated for RIP and also for NRIP where the gene was present in a comparable number of species. These outputs have been plotted using Excel. All other graphics, where not explicitly stated, were generated with R46.
Conclusion
The results of this study highlight the presence of a defined sub-group of Rosaceae infecting species taxonomically and genetically related, with a high number of conserved core genes. The importance of the siderophores uptake genes has been extended to the complete transport system of ferrioxamine, with the identification of two genes present exclusively in the strains infecting the Rosaceae. These genes, namely fhuD and sidE, code for two proteins that require further studies and are possible new targets for development of novel control measures against RIP. Our results raise the interest towards E. tasmaniensis for a better understanding of the transition between non-pathogenicity and pathogenicity. Moreover, our data confirm the classification of Erwinia spp. Ejp617 as E. pyrifoliae.
Acknowledgements
This research was funded by the Faculty of Science and Technology, Free University of Bolzano, project: Genomic Selection in Erwinia amylovora, GenSelEa, grant number 1576 and by the Free University of Bolzano, project: Bioinformatics analysis of Erwinia amylovora strains genome sequences, BioinfEa, grant number 1627. This work was supported by the Open Access Publishing Fund of the Free University of Bozen-Bolzano. IP was supported by a predoctoral fellowship from the Free University of Bolzano.
Author Contributions
Conceptualization, I.P. and A.E.; Methodology, L.B., L.T. and A.E.; Formal analysis, I.P., L.B., R.C., L.T. and A.E., Project Administration and Funding acquisition, S.B.; Writing-Original Draft Preparation, all the authors.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Alfonso Esposito, Email: alfonso.esposito@unitn.it.
Stefano Benini, Email: stefano.benini@unibz.it.
References
- 1.Vanneste, J. L. In Fire blight: the disease and its causative agent, Erwinia amylovora (ed. Vanneste, J. L.) (CABI Publishing, New York, NY 10016, 2000).
- 2.Mansfield J, et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant. Pathol. 2012;13:614–629. doi: 10.1111/j.1364-3703.2012.00804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Hara CM, Steigerwalt AG, Hill BC, Miller JM, Brenner DJ. First Report of a Human Isolate of Erwinia persicinus. Journal of Clinical Microbiology. 1998;36:248–250. doi: 10.1128/jcm.36.1.248-250.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prod’homme M, et al. Cutaneous infection and bactaeremia caused by Erwinia billingiae: a case report. New Microbes New Infect. 2017;19:134–136. doi: 10.1016/j.nmni.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shin SY, Song JH, Ko KS. First Report of Human Infection Due to Erwinia tasmaniensis-Like Organism. International Journal of Infectious Diseases. 2008;12:e329–e330. doi: 10.1016/j.ijid.2008.05.881. [DOI] [Google Scholar]
- 6.Mann RA, et al. Comparative analysis of the Hrp pathogenicity island of Rubus- and Spiraeoideae-infecting Erwinia amylovora strains identifies the IT region as a remnant of an integrative conjugative element. Gene. 2012;504:6–12. doi: 10.1016/j.gene.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Borruso L, Salomone-Stagni M, Polsinelli I, Schmitt AO, Benini S. Conservation of Erwinia amylovora pathogenicity-relevant genes among Erwinia genomes. Arch. Microbiol. 2017;199:1335–1344. doi: 10.1007/s00203-017-1409-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miethke M, Marahiel MA. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 2007;71:413–451. doi: 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li K, Chen W, Bruner SD. Microbial siderophore-based iron assimilation and therapeutic applications. Biometals. 2016;29:377–388. doi: 10.1007/s10534-016-9935-3. [DOI] [PubMed] [Google Scholar]
- 10.Vrancken K, Holtappels M, Schoofs H, Deckers T, Valcke R. Pathogenicity and infection strategies of the fire blight pathogen Erwinia amylovora in Rosaceae: state of the art. Microbiology. 2013;159:823–832. doi: 10.1099/mic.0.064881-0. [DOI] [PubMed] [Google Scholar]
- 11.Piqué N, Miñana-Galbis D, Merino S, Tomás J. Virulence Factors of Erwinia amylovora: A Review. International Journal of Molecular Sciences. 2015;16:12836–12854. doi: 10.3390/ijms160612836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smits TH, Rezzonico F, Duffy B. Evolutionary insights from Erwinia amylovora genomics. J. Biotechnol. 2011;155:34–39. doi: 10.1016/j.jbiotec.2010.10.075. [DOI] [PubMed] [Google Scholar]
- 13.Dellagi A, Brisset MN, Paulin JP. & Expert, D. Dual role of desferrioxamine in Erwinia amylovora pathogenicity. Mol. Plant Microbe Interact. 1998;11:734–742. doi: 10.1094/MPMI.1998.11.8.734. [DOI] [PubMed] [Google Scholar]
- 14.Expert, D., Dellagi, A. & Kachadourian, R. In Fire Blight: The Disease and its Causative Agent, Erwinia amylovora (ed. Vanneste, J. L.) 179–195 (Blackwell Science Ltd, Wallingford, UK, 2000).
- 15.Smits TH, Duffy B. Genomics of iron acquisition in the plant pathogen Erwinia amylovora: insights in the biosynthetic pathway of the siderophore desferrioxamine E. Arch. Microbiol. 2011;193:693–699. doi: 10.1007/s00203-011-0739-0. [DOI] [PubMed] [Google Scholar]
- 16.Salomone-Stagni M, et al. A complete structural characterization of the desferrioxamine E biosynthetic pathway from the fire blight pathogen Erwinia amylovora. Journal of Structural Biology. 2018;202:236–249. doi: 10.1016/j.jsb.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Kachadourian R, et al. Desferrioxamine-dependent iron transport in Erwinia amylovora CFBP1430: cloning of the gene encoding the ferrioxamine receptor FoxR. Biometals. 1996;9:143–150. doi: 10.1007/BF00144619. [DOI] [PubMed] [Google Scholar]
- 18.Dellagi A, Reis D, Vian B. & Expert, D. Expression of the ferrioxamine receptor gene of Erwinia amylovora CFBP1430 during pathogenesis. Mol. Plant Microbe Interact. 1999;12:463–466. doi: 10.1094/MPMI.1999.12.5.463. [DOI] [PubMed] [Google Scholar]
- 19.Burkhardt R, Braun V. Nucleotide sequence of the fhuC and fhuD genes involved in iron (III) hydroxamate transport: domains in FhuC homologous to ATP-binding proteins. Mol. Gen. Genet. 1987;209:49–55. doi: 10.1007/BF00329835. [DOI] [PubMed] [Google Scholar]
- 20.Groeger W, Köstert W. Transmembrane topology of the two FhuB domains representing the hydrophobic components of bacterial ABC transporters involved in the uptake of siderophores, haem and vitamin B. Microbiology. 1998;144:2759–2769. doi: 10.1099/00221287-144-10-2759. [DOI] [PubMed] [Google Scholar]
- 21.Clarke TE, Braun V, Winkelmann G, Tari LW, Vogel HJ. X-ray crystallographic structures of the Escherichia coli periplasmic protein FhuD bound to hydroxamate-type siderophores and the antibiotic albomycin. J. Biol. Chem. 2002;277:13966–13972. doi: 10.1074/jbc.M109385200. [DOI] [PubMed] [Google Scholar]
- 22.Kingsley RA, et al. Ferrioxamine-mediated Iron(III) utilization by Salmonella enterica. Appl. Environ. Microbiol. 1999;65:1610–1618. doi: 10.1128/aem.65.4.1610-1618.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schalk IJ, Guillon L. Fate of ferrisiderophores after import across bacterial outer membranes: different iron release strategies are observed in the cytoplasm or periplasm depending on the siderophore pathways. Amino Acids. 2013;44:1267–1277. doi: 10.1007/s00726-013-1468-2. [DOI] [PubMed] [Google Scholar]
- 24.Matzanke BF, Anemuller S, Schunemann V, Trautwein AX, Hantke K. FhuF, part of a siderophorereductase system. Biochemistry. 2004;43:1386–1392. doi: 10.1021/bi0357661. [DOI] [PubMed] [Google Scholar]
- 25.López MM, et al. Erwinia piriflorinigrans sp. nov., a novel pathogen that causes necrosis of pear blossoms. Int. J. Syst. Evol. Microbiol. 2011;61:561–567. doi: 10.1099/ijs.0.020479-0. [DOI] [PubMed] [Google Scholar]
- 26.Mann RA, et al. Comparative Genomics of 12 Strains of Erwinia amylovora Identifies a Pan-Genome with a Large Conserved Core. PLoS One. 2013;8:e55644. doi: 10.1371/journal.pone.0055644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lagonenko AL, Komardina VS, Nikolaichik YA, Evtushenkov AN. First Report of Erwinia amylovora Fire Blight in Belarus. J. Phytopathol. 2008;156:638–640. doi: 10.1111/j.1439-0434.2008.01420.x. [DOI] [Google Scholar]
- 28.Geider K, Auling G, Jakovljevic V, Völksch B. A polyphasic approach assigns the pathogenic Erwinia strains from diseased pear trees in Japan to Erwinia pyrifoliae. Lett. Appl. Microbiol. 2009;48:324–330. doi: 10.1111/j.1472-765X.2008.02535.x. [DOI] [PubMed] [Google Scholar]
- 29.Park DH, et al. Complete genome sequence of Japanese Erwinia strain ejp617, a bacterial shoot blight pathogen of pear. J. Bacteriol. 2011;193:586–587. doi: 10.1128/JB.01246-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kube, M. et al. Genome comparison of the epiphytic bacteria Erwinia billingiae and E. tasmaniensis with the pear pathogen E. pyrifoliae. BMC Genomics11 (2010). [DOI] [PMC free article] [PubMed]
- 31.Malnoy M, et al. Fire blight: applied genomic insights of the pathogen and host. Annu. Rev. Phytopathol. 2012;50:475–494. doi: 10.1146/annurev-phyto-081211-172931. [DOI] [PubMed] [Google Scholar]
- 32.Zhao Y, Qi M. Genes (Basel) 2011. Comparative Genomics of Erwinia amylovora and Related Erwinia Species-What do We Learn? pp. 627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Llop P. Genetic islands in pome fruit pathogenic and non-pathogenic Erwinia species and related plasmids. Front. Microbiol. 2015;6:874. doi: 10.3389/fmicb.2015.00874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eve B, Baker LA. Characteristics of Erwinia‐like Organisms found in Plant Material. J. Appl. Bacteriol. 1963;26:58–65. doi: 10.1111/j.1365-2672.1963.tb01155.x. [DOI] [Google Scholar]
- 35.Mergaert J, Hauben L, Cnockaert MC, Swings J. Reclassification of non-pigmented Erwinia herbicola strains from trees as Erwinia billingiae sp. nov. Int. J. Syst. Bacteriol. 1999;49(Pt 2):377–383. doi: 10.1099/00207713-49-2-377. [DOI] [PubMed] [Google Scholar]
- 36.Palacio-Bielsa A, Roselló M, Llop P, López MM. Erwinia spp. from pome fruit trees: similarities and differences among pathogenic and non-pathogenic species. Trees. 2012;26:13–29. doi: 10.1007/s00468-011-0644-9. [DOI] [Google Scholar]
- 37.Jakovljevic V, Jock S, Du Z, Geider K. Hypersensitive response and acyl-homoserine lactone production of the fire blight antagonists Erwinia tasmaniensis and Erwinia billingiae. Microb. Biotechnol. 2008;1:416–424. doi: 10.1111/j.1751-7915.2008.00043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pritchard L, Glover RH, Humphris S, Elphinstone JG, Toth IK. Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods. 2016;8:12–24. doi: 10.1039/C5AY02550H. [DOI] [Google Scholar]
- 39.Segata N, Bornigen D, Morgan XC, Huttenhower C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 2013;4:2304. doi: 10.1038/ncomms3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eren AM, et al. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ. 2015;3:e1319. doi: 10.7717/peerj.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 43.Rozas J, et al. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017;34:3299–3302. doi: 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
- 44.Nei, M. In Molecular Evolutionary Genetics (1987).
- 45.Nei M, Miller JC. A simple method for estimating average number of nucleotide substitutions within and between populations from restriction data. Genetics. 1990;125:873–879. doi: 10.1093/genetics/125.4.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria., http://www.R-project.org/ (2013).
- 47.Kube M, et al. The genome of Erwinia tasmaniensis strain Et1/99, a non-pathogenic bacterium in the genus Erwinia. Environ. Microbiol. 2008;10:2211–2222. doi: 10.1111/j.1462-2920.2008.01639.x. [DOI] [PubMed] [Google Scholar]
- 48.Klein, J. M. et al. Draft Genome Sequence of Erwinia billingiae OSU19-1, Isolated from a Pear Tree Canker. Genome Announc3, 10.1128/genomeA.01119-15 (2015). [DOI] [PMC free article] [PubMed]
- 49.Passos da Silva, D. et al. Draft Genome Sequence of Erwinia toletana, a Bacterium Associated with Olive Knots Caused by Pseudomonas savastanoi pv. savastanoi. Genome Announc1, 10.1128/genomeA.00205-13 (2013). [DOI] [PMC free article] [PubMed]
- 50.Moretti, C. et al. Draft Genome Sequence of Erwinia oleae, a Bacterium Associated with Olive Knots Caused by Pseudomonas savastanoi pv. savastanoi. Genome Announc2, 10.1128/genomeA.01308-14 (2014). [DOI] [PMC free article] [PubMed]
- 51.Moretti C, et al. Erwinia oleae sp. nov., isolated from olive knots caused by Pseudomonas savastanoi pv. savastanoi. Int. J. Syst. Evol. Microbiol. 2011;61:2745–2752. doi: 10.1099/ijs.0.026336-0. [DOI] [PubMed] [Google Scholar]
- 52.Shapiro LR, et al. Horizontal Gene Acquisitions, Mobile Element Proliferation, and Genome Decay in the Host-Restricted Plant Pathogen Erwinia Tracheiphila. Genome Biol. Evol. 2016;8:649–664. doi: 10.1093/gbe/evw016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Redzuan, R. A. et al. Draft Genome Sequence of Erwinia mallotivora BT-MARDI, Causative Agent of Papaya Dieback Disease. Genome Announc2, 10.1128/genomeA.00375-14 (2014). [DOI] [PMC free article] [PubMed]
- 54.Zhang Z, Nan Z. Erwinia persicina, a possible new necrosis and wilt threat to forage or grain legumes production. Eur. J. Plant Pathol. 2014;139:349–358. doi: 10.1007/s10658-014-0390-0. [DOI] [Google Scholar]
- 55.González A, Tello J, Rodicio M. Erwinia persicina causing chlorosis and necrotic spots in leaves and tendrils of Pisum sativum in southeastern Spain. Plant Dis. 2007;91:460–460. doi: 10.1094/PDIS-91-4-0460A. [DOI] [PubMed] [Google Scholar]
- 56.Campillo T, et al. Erwinia iniecta sp. nov., isolated from Russian wheat aphid (Diuraphis noxia) Int. J. Syst. Evol. Microbiol. 2015;65:3625–3633. doi: 10.1099/ijsem.0.000466. [DOI] [PubMed] [Google Scholar]
- 57.Rezzonico F, et al. Erwinia gerundensis sp. nov., a cosmopolitan epiphyte originally isolated from pome fruit trees. Int. J. Syst. Evol. Microbiol. 2016;66:1583–1592. doi: 10.1099/ijsem.0.000920. [DOI] [PubMed] [Google Scholar]