

Abstract
Taxus (yew) is both the most species-rich and taxonomically difficult genus in Taxaceae. To date, no study has elucidated the complexities of the plastid genome (plastome) or examined the possibility of whole plastomes as super-barcodes across yew species worldwide. In this study, we sequenced plastomes from two to three individuals for each of the 16 recognized yew species (including three potential cryptics) and Pseudotaxus chienii. Our comparative analyses uncovered several gene loss events that independently occurred in yews, resulting in a lower plastid gene number than other Taxaceous genera. In Pseudotaxus and Taxus, we found two isomeric arrangements that differ by the orientation of a 35 kb fragment flanked by “trnQ-IRs”. These two arrangements exist in different ratios within each sampled individual, and intraspecific shifts in major isomeric arrangements are first reported here in Taxus. Moreover, we demonstrate that entire plastomes can be used to successfully discriminate all Taxus species with 100% support, suggesting that they are useful as super-barcodes for species identification. We also propose that accD and rrn16-rrn23 are promising special barcodes to discriminate yew species. Our newly developed Taxus plastomic sequences provide a resource for super-barcodes and conservation genetics of several endangered yews and serve as comprehensive data to improve models of plastome complexity in Taxaceae as a whole and authenticate Taxus species.
Introduction
The plastid genomes (plastomes) of photosynthetic land plants are generally characterized by two unequal single-copy regions separated by a pair of canonical inverted repeats (IRs)1. However, coniferous plastomes lack the canonical IRs and show extensive rearrangements2. Recent comparative plastomics studies have revealed several lineage-specific and actively recombining IRs in conifers. For instance, Pinaceae-specific IRs are recombination substrates associated with the formation of distinct plastomic architectures3,4. In cupressophytes, lineage-specific IRs are able to mediate inversions at a subtle level, ultimately resulting in the existence of major and minor isomeric plastomes that differ based on how a particular region is oriented5–9. Shifts in major isomeric plastomes were observed at interspecific levels in a few Cupressaceous lineages6,9, but such shifts were absent in populations of Calocedrus macrolepis9. Nonetheless, the above observations were mainly based on Cupressaceae. Little is known about the shift in major isomeric plastomes at the intra- and inter-specific level in other cupressophyte families such as Taxaceae.
Plastomic sequences are excellent resources for resolving the tree of life10 and delimiting the species entity11. Plastids are predominantly inherited uniparentally12 and they behave as a single non-recombining locus, which provides a strong signal of phylogenetic history13. Plastid loci have been utilized widely as DNA barcodes for discriminating plant species14. A combination of two plastid loci (matK and rbcL) were suggested as the core barcode to discriminate species of land plants15; however, it generally could not distinguish closely related species or recently evolved species in most groups due to the lack of adequate variation among taxa16. Further specific barcodes could help improve this discriminatory power at the species level17. Therefore, the quest for improved barcodes with universal usage in plants is ongoing18. Although concerns were raised about the possibility of plastid introgression and hybridization19–21, many researchers advocated for the approach that uses whole plastomes as super-barcodes22–24. For example, the super-barcode approach was shown to successfully distinguish closely related species such as Theobroma spp. (Malvaceae)25, Araucaria spp. (Aruacariaceae)26, and Echinacea (Asteraceae)27, especially for taxonomically complex groups, e.g., Camellia spp. (Theaceae)28, Epimedium spp. (Berberidaceae)29, and Fritillaria spp. (Liliacae)30. The increased availability of plastome sequences and reduced cost of next generation sequencing (NGS) technology have recently sparked an interest in the versatility of plastomes. An approach combining the best use of single-locus barcodes and super-barcodes for efficient plant identification was suggested for selected groups of taxa, including specific barcodes that could distinguish closely related plants at the species and population levels17.
Taxaceae includes six genera (Amentotaxus, Austrotaxus, Cephalotaxus, Pseudotaxus, Taxus, and Torreya) and about 30 species of evergreen trees or shrubs, distributed mainly in the Northern Hemisphere31,32. This cupressophyte family likely diverged from its closest sister, Cupressaceae, during the Early Triassic32,33. Taxus (yews), the largest and most widespread genus in Taxaceae34, is famous for its high content of the anticancer compound taxol, a chemotherapeutic drug used in breast and lung cancer treatment35. However, yews have a complex and controversial taxonomic history due to their high degree of morphological similarity between species36–39. For example, Spjut37 recognized 24 species in Taxus, with 16 species and seven varieties in China, whereas Farjon31 only admitted 10 species in the genus, including only five in China. Recently, based on a global scale genetic and distribution analysis, Liu et al.40 approved a total of 15 Taxus species/lineages including the ten recognized by Farjon31, two by Spjut37, one by Möller et al.39 and two cryptics by Liu et al.40,41.
To date, reported plastomes are limited to few Taxus species, and super-barcodes have not been used to elucidate plastomes for Taxus species on a large scale. To this end, we sequenced the complete plastomes from all 16 recognized Taxus species (including three potential cryptics) and the sole species of Pseudotaxus, P. chienii (Cheng) Cheng, sampling three individuals per species except for the Huangshan type of Taxus and P. chienii because wild populations of them were unavailabile. Incorporating the previously elucidated plastomes of other Taxaceous genera, this study aims to address the following questions: 1) Do plastomic characteristics—in terms of genome size, gene content, nucleotide compositions, and structure—vary across the Taxaceae? 2) Are isomeric plastomes common in Taxus? If yes, do their relative abundances vary among species and/or populations? 3) Are whole plastome sequences suitable super-barcodes for discriminating yew species? If not, are there any special plastid genes/intergenic spacers that are promising barcoding loci for identifying yew species?
Results
The Plastome size and gene content vary across Taxaceae
Plastomes were sequenced from 49 samples of all 16 recognized species of Taxus and Pseudotaxus chienii, with two to three individuals sampled per species. These 49 newly sequenced plastomes were assembled into circular molecules (Fig. 1), with an average sequencing coverage of 41 to 2,716 times (Table S1). They are deposited in GenBank under the accession numbers MH390441 to MH390489. The Pseudotaxus and Taxus plastomes are 129,874–130,505 bp and 127,335–129,752 bp long, respectively. They are shorter than previously reported plastomes in other Taxaceous genera (Table 1). The plastomic GC content across Taxaceae ranges from 34.6 to 35.9%, with Taxus being the lowest. A pair of short inverted repeats with a trnQ-UUG in each copy (hereafter called trnQ-IRs based on Guo et al.6) is also common in Taxaceae, with Taxus and Amentotaxus having the shortest and longest trnQ-IRs, respectively (Table 1).
Figure 1.

Circular maps of Pseudotaxus and Taxus plastomes. Colored boxes outside and inside the circle are genes in counterclockwise and clockwise transcribed directions, respectively. Locally co-linear blocks (LCBs 1, 2, and 3) between Pseuodtaxus and Taxus are indicated by the inner colored arrows with their relative orientations. Regions of trnQ-IRs are denoted by purple arrows along LCBs. Scale bars 10 kb in length are given with psbA as the starting point.
Table 1.
Comparison of plastomic characteristics across Taxaceae.
| Genus | Taxus | Pseudotaxus | Torreya | Amentotaxus | Cephalotaxus |
|---|---|---|---|---|---|
| Plastome size (bp) | 127,335‒129,752 | 129,874‒130,505 | 136,949‒137,075 | 136,430‒136,657 | 134,337‒136,196 |
| GC content (%) | 34.6‒34.8 | 35.2 | 35.4‒35.5 | 35.8‒35.9 | 35.1‒35.3 |
| trnQ-IR length (bp) | 216‒267 | 552 | 298 | 534‒564 | 544‒548 |
| No. of genesa | 114 | 116 | 121 | 120 | 116 |
| Variation in gene content b | |||||
| rps16 | − | − | + | + | + |
| trnA-UGC | − | + | + | + | + |
| trnV-GAC | Ψ | + | + | + | + |
| trnS-GGA | − | − | + | + | + |
| trnG-UCC | − | − | + | + | + |
| trnI-GAU | − | − | + | + | + |
| trnV-UAC | + | + | + | + | − |
| trnI-CAU | +, + | +, + | +, + | +, + | + |
| trnT-UGU | + | + | + | + | Ψ |
| trnN-GUU | + | + | +, + | +, Ψ | + |
aShared genes: accD, ccsA, cemA, clpP, infA, matK, rbcL, atpA, B, E, F, H, and I; chlB, L, and N; ndhA, B, C, D, E, F, G, H, I, J, and K; petA, B, D, G, L, and N; psaA, B, C, I, J, M; psbA, B, C, D, E, F, H, I, J, K, L, M, N, T, and Z; rpl2, 14, 16, 20, 23, 32, 33, and 36; rpoA, B, C1, and C2; rps2, 3, 4, 7, 8, 11, 12, 14, 15, 18, and 19; ycf1, 2, 3, and 4; rrn4.5, 5, 16, and 23; trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnH-GUG, trnI-CAU, trnK-UUU, trnL-CAA, trnL-UAA, trnL-UAG, trnM-CAU, trnN-GUU, trnP-GGG, trnP-UGG, trnQ-UUG, trnQ-UUG, trnR-ACG, trnR-UCU, trnS-GCU, trnS-UGA, trnT-GGU, trnW-CCA, and trnY-GUA.
b“−” absent; “+” present; “Ψ” pseudo; “+, +” duplicate; “+, Ψ” duplicate but one of them is pseudo.
The gene content varies from 114 to 121 genes per plastome, with the smallest and largest sets of genes being in Taxus and Torreya, respectively (Table 1). In total, 82 protein-coding genes, 4 ribosomal RNAs, and 25 transfer RNAs are shared across Taxaceae. Variation in gene content includes 1) pseudogenization of trnV-GAC in and loss of trnA-UGC from Taxus; 2) losses of trnS-GGA, trnG-UCC, trnI-GAU, and rps16 from both Pseudotaxus and Taxus; 3) pseudogenization of trnT-UGU in Cephalotaxus, and losses of trnV-UAC, trnV-GAC, and a trnI-CAU copy from Cephalotaxus; and 4) duplication of trnN-GUU in Torreya, but one of the two trnN-GUU copies has become pseudogenized in Amentotaxus. Because Amentotaxus is phylogenetically close to Torreya42, duplication of trnN-GUU might predate the divergence of these two genera.
In addition to loss/duplication of genes, a specific extension of clpP was found in Taxus. ClpP encodes the caseinolytic protease, which contains 339‒537 and 224‒245 amino acids in Taxus and other Taxaceous genera, respectively. Therefore, there is great variation in the length of clpP at both intra- and inter-genus levels. As shown in the clpP alignment (Fig. 1S), a block of Glu (E)-rich insertions separates Taxus from other Taxaceous genera. Whether this Glu-rich insertion has implications in the fundamental function of clpP remains to be answered.
Shifting major isomeric plastome arrangements at intraspecific levels
Three locally co-linear blocks (designated LCBs 1, 2, and 3) between Pseudotaxus and Taxus were identified in the same orientation with four exceptions (Fig. 1). The LCB 2 fragments of approximately 35 kb are inverted in four individuals—T. brevifolia 02, T. globosa 01, and T. floridana 02 and 05 (here named “arrangement B” following Guo et al.6)—and are not in the remaining 45 samples (designated “arrangement A”). Notably, these data also indicate intraspecific variation in the LCB 2 orientation in three taxa—T. brecifolia, T. globosa, and T. floridana. These LCB2 fragments are exclusively flanked by trnQ-IRs, regardless of the relative orientations (Fig. 1). Previously, trnQ-IRs were proposed to facilitate homologous recombination that generates isomeric plastome arrangements in Cephalotaxus5 and Cupressoideae6,9. Accordingly, if trnQ-IRs are active recombinant agents in Pseudotaxus and Taxus, we would expect arrangements A and B to coexist in each sample.
Figure 2A shows that specific regions typifying isomeric arrangements A and B were detected by four primer pairs. For T. brevifolia 02, T. floridana 06, and T. globosa 01, specific amplicons of arrangement A were observed when 20 to 35 PCR cycles were used, while arrangement B appeared only when ≥30 PCR cycles were used (Fig. 2B). Conversely, the amplicons of arrangement B appeared earlier than those of arrangement A in T. chinensis 06, T. florinii 01, and T. phytonii 03. Similar PCR assays were performed for the remaining samples (Fig. S2). Collectively, our PCR results suggest that major and minor isomeric arrangements exist in both Pseudotaxus and Taxus.
Figure 2.

Verification of isomeric plastome arrangements. (A) Primer pairs designed to amplify specific regions of isomeric arrangements. (B) PCR results generated from 20, 25, 30, and 35 cycles of reactions to determine the major and minor isomeric arrangements in six exemplified Taxus accessions. The gel images were cropped from the full-length gels shown in Fig. S3. (C) Counts of Illumina pair-end reads that support isomeric arrangements. Stacked horizontal bars indicate the relative frequency between arrangements A and B.
The Illumina paired-end reads provide the other line of evidence that supports the coexistence of arrangements A and B in both Pseudotaxus and Taxus. For examples, 1,266 reads that spanned trnQ-IRs were gathered from T. brevifolia 02, of which 1,245 (98.3%) and 21 (1.7%) supported arrangements A and B, respectively (Fig. 2C); this is in agreement with the PCR result that suggests arrangement A being overwhelmingly abundant (Fig. 2B). Overall, coexistence of arrangements A and B was detected in 47 of the 49 samples, and the relative frequency of major arrangements was estimated to be 86.2% to 98.4% (Figs 2C; S2). None of the detected reads supported arrangement A in P. chienii 03 and T. brevifolia 03, possibly because 1) for the former, most reads (insertion size approximately 500 bp) were too short to include its entire trnQ-IR (552 bp), and 2) for the latter, there were not enough reads to detect the minor arrangement A, as its sequencing depth is the lowest among the examined samples (Table S1).
Taken together, data from PCRs and pair-end reads are consistent in supporting our plastome assemblies as well as the major isomeric arrangement B. As a consequence, intraspecific variation in plastomic organizations observed in T. brevifolia, T. floridana, and T. globosa (Fig. 1) suggests that shifts in major isomeric arrangements have occurred among populations.
Whole plastomes as super-barcodes for discriminating yew species
As mentioned above, Pseudotaxus and Taxus share three LCBs. Alignments and concatenation of these three LCBs yielded a matrix containing 146,099 characters. An ML tree (Fig. 3) was inferred based on this plastomic matrix using Pseudotaxus as an outgroup. Of note, the four New World yews T. brevifolia, T. globosa, T. floridana, and T. canadensis did not form a monophyletic clade. Instead, T. brevifolia was placed as the earliest diverged yew, and T. canadensis was inferred to be more closely related to Old World yews than to other New World ones (Fig. 3). All Old World yews except T. canadensis were grouped into a clade separate from New World yews. Nonetheless, the conspecific accessions of all species, including those from the three potential cryptic types, were grouped into respective monophyletic clades each with 100% support, thereby suggesting that whole plastomes are effective super-barcodes for identifying yew species. The newly discovered species (Huangshan type) was well supported, which was close to T. chinensis and T. florinii (Fig. 3).
Figure 3.

An ML tree constructed from whole plastome sequences with Pseudotaxus as the outgroup. Values (%) along branches were estimated from 1,000 bootstrap replicates. Inferred relationships of conspecific accessions are shown in grey boxes. The tree was condensed under a 50% majority rule.
Single genic loci as promising special barcodes for discriminating yew species
After excluding loci smaller than 400 bp, 73 syntenic loci, including 45 protein-coding genes and 28 intergenic spacers, were determined to assess their discriminatory power for yew species (Table S2). They are highly variable in length, with two extremes: the longest loci (ycf1 ~6.7 kb long and ycf2 ~7.3 kb) exceed the shortest locus rps11 (~0.4 kb) by over 16-fold. For each locus, pairwise intra- and inter-specific K2P distances were estimated from 46 and 1,035 comparisons, respectively. Among the 73 single loci, the average intra- and inter-specific distances are positively correlated (Fig. 4), suggesting that intraspecific polymorphisms contributed to interspecific divergence increases in Taxus plastomes. In terms of both intra- and inter-specific distances, clpP (~1.2 kb) is a standout due to its Glu (E)-rich insertion. Two intergenic loci, rrn16-rrn23 (~2 kb) and ycf1-chlN (~0.9 kb), also exhibit a great degree of intraspecific variation, implying that they may be useful in population genetics studies. We noted that accD (~2.2 kb) shows a conspicuous discrepancy between intra- and inter-specific K2P distances, with the former being smaller than the latter by approximately 298 times (Fig. 4). A discrete distribution between intraspecific variation and interspecific divergence (termed barcoding gap) is crucial for species discrimination (Hebert et al. 2004)43, so maximum intra- and minimum inter-specific distances were compared across all examined loci. Nonetheless, only three loci accD, ycf1, and rpoB (~3.3 kb) show no overlap (Table S2); this scarcity (3/76 = 3.95%) is attributed to the minimum interspecific distance of many loci being as low as 0%.
Figure 4.
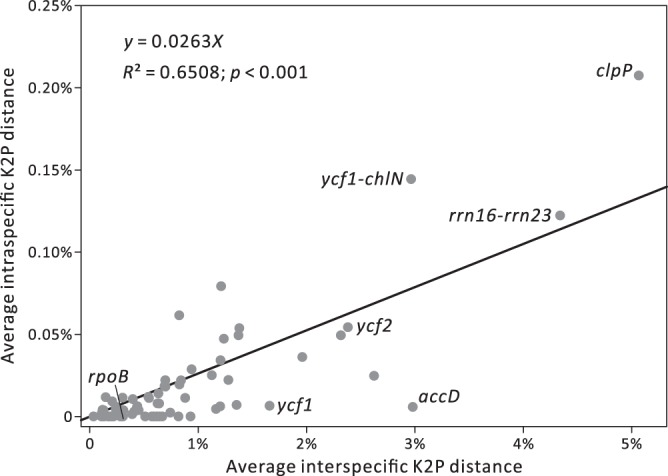
Positive correlation between intra- and inter-specific K2P distances among the 73 examined single genic and intergenic loci.
We applied the NJ tree method to evaluate the discriminatory rate for yew species using 73 single loci. Formation of a monophyletic clade of conspecific samples was treated as successful discrimination when the corresponding BS values were larger than 50%. A fully discriminatory rate was yielded in the trees inferred from accD, ycf1, ycf2, and rrn16-rrn23, but only two of them were diagnosed with a barcoding gap (Table S2). Although a barcoding gap existed in rpoB, the tree inferred from this gene did not discriminate species 100% successfully (Table S2). In contrast, all conspecific accessions formed monophyletic clades with 100% support in the tree inferred from a non-barcoding gap gene ycf2 (Table S2).
Discussion
New sequencing technologies are cost-effective and give data of previously unimaginable mass and quality. They have facilitated the sequencing of plastomes from numerous species26,44–47. In this study, 49 complete plastomes were obtained from P. chienii of the monotypic genus Pseudotaxus and 16 species of Taxus, including samples of three individuals of almost all species. Using P. chienii as the outgroup, our dataset, in terms of taxon sampling, is by far the most comprehensive among comparative plastomics across the genus Taxus. Based on these samples, we estimated the plastomic architecture variation at both intra- and inter-specific levels, examined the power of entire plastomes for discriminating species, and evaluated useful single loci as special DNA barcodes.
It is well accepted that the canonical IRs in plastomes are able to trigger intramolecular recombination to generate equal amounts of isomeric plastomes, one of which differs from the other by the relative orientation of its small single copy region48,49. Despite lacking the canonical IR, cupressophytes have evolved a diverse set of lineage-specific IRs capable of mediating inversions to form isomeric plastomes. For instance, isomeric plastomes associated with trnQ-IRs have been discovered in Cephalotaxus5 and Cupressaceae6,9. In Sciadopitys (Sciadopityaceae), the presence of trnQ-containing tandem repeats has led to the speculation that trnQ-IRs resulted from multistep rearrangements after a tandem duplication50. In addition, Sciadopitys contains specific IRs (called rpoC2-IRs) that are proven to be re-combinable7. The trnN-IRs that were proposed to be responsible for formation of isomeric plastomes8 are common in Podocarpaceae51. All three Araucariaceae genera possess rrn5-IRs, though their recombinant activity has not been assessed51. The diverse set of IRs ubiquitously associates with the presence of isomeric plastomes, which suggests convergent evolution of isomeric plastomes among cupressophyte families.
Our PCR and read mapping analyses show that the isomeric arrangements are not present in equal percentages. Instead, the major arrangements strikingly exceed the minor ones in their relative ratios (Figs 2; S2). This feature suggests that trnQ-IRs mediate recombination at low frequency in both Pseudotaxus and Taxus. In Taxaceae, trnQ-IR lengths are between 216 and 564 bp (Table 1). These lengths of repeats occasionally mediate recombination in mitochondria52. However, our data reveal that major isomeric arrangements have shifted among conspecifics in T. brevifolia, T. floridana, and T. globosa. Intraspecific shifts in major isomeric arrangements might also occur in T. chinensis because an earlier reported plastomes (arrangement A)53,54 and our newly assemblies (arrangement B: Figs 1 and 2) are oriented differently. Guo et al.6 proposed that major isomeric arrangements have shifted multiple times during the diversification of cupressophytes. This proposition is further extended by our findings that major and minor plastomic arrangements could shift at intraspecific levels. In mitochondria, selective amplification was thought to account for alternation of major isomers55. Nevertheless, it remains unclear whether accumulated mutations that benefit amplification would enable a minor isomer to become a major one in plastids. Unfortunately, the Illumina reads used in this study are too short to extensively quantitate mutations between isomers. The PacBio long-read sequencing technology that was recently used to distinguish heteroplastomic DNAs within individuals56 opens a new avenue to deepen our understanding of isomeric plastome evolution in future.
To date, using the entire plastome sequence as a super-barcode has been demonstrated to be useful for discriminating species in diverse lineages, such as rice22, cacao25, Araucaria26, and Stipa57, especially in some taxonomically complex groups29,30. Our ML tree inferred from the entire plastome sequence shows that all conspecific samples were resolved as monophyletic with robust support (Fig. 3), therefore the super-barcoding approach is validated for discriminating Taxus species. It has been proposed that the super-barcoding approach circumvents the issues of gene deletion, locus choice, and low PCR recovery rate often encountered in studies using conventional barcodes26,45,58. Despite sharing the same set of plastid genes (Table 1), the sampled Taxus species differ in their plastome organizations (i.e., arrangement A or B), which hampers the performance of whole plastome alignments. Identification of LCBs before conducting alignments is a prerequisite for using the super-barcoding approach in cupressophyte lineages whose plastomes are highly rearranged51,59. Collectively, the plastome as super-barcode showed a great promise for distinguishing closely related species in Taxus.
Lineage-specific barcodes are also thought to enhance the resolution of species discrimination because they might provide more sufficient information within a particular group than traditional barcodes17. Indeed, the core barcode matK + rbcL suggested by the CBOL Plant Working Group (2009) only distinguished 63% of our sampled Taxus species (Table S2). Although trnL-trnF showed high discriminatory rates for yew species, it did not discern two New World yews (i.e., T. globosa and T. floridana) based on the Tree-based method41. The combination of trnL-trnF and nrITS could effectively discriminate between all the yew species41. In this study, accD, ycf1, ycf2, and rrn16-rrn23 are shown to successfully discriminate all species, with the former two containing distinct barcoding gaps. rpoB contains a barcoding gap, but did not yield a 100% species resolution. In contrast, all conspecific accessions formed monophyletic clades with a 100% support from both non-barcoding loci: ycf2 and rrn16-rrn23. Collectively, our results imply that the existence of a clear barcoding gap is not prerequisite to discriminate species 100% successfully. Considering the length of the loci, we therefore suggest accD and rrn16-rrn23 can be effective special barcoding loci for discriminating Taxus species. Exploration of potential barcodes/mutational hotspots is highly dependent on the estimated sequence divergence60–63. In Taxus, the locus clpP (Fig. 4) exhibits the highest degree of both intra- and inter-specific sequence divergences, indicating its potential for population genetics studies. However, clpP only achieved a discriminatory rate of 81.25% in our practical analysis (Table S2). As a result, we suggest that future researchers perform practical analyses in order to accurately evaluate the discriminatory power of selected loci in barcoding studies.
Conclusion
Continuous advances in sequencing technologies make obtaining complete plastomes from major lineages across a genus more feasible. A total of 49 plastomes from Pseudotaxus chienii and 16 Taxus species were elucidated and compared in the present study. Our PCR and read mapping results together support the existence of trnQ-IR mediated isomeric plastomes in both Pseudotaxus and Taxus. We provide evidence, for the first time, that major isomeric arrangements have shifted among populations. We successfully used entire plastome sequences to distinguish all Taxus species, including three potential cryptic types, supporting that the plastome sequences themselves in Taxus species are effective super-barcodes for species identification and discovery. Moreover, four single loci—accD, ycf1, ycf2, and rrn16-rrn23—are capable of achieving 100% discriminatory rates; of these, accD and rrn16-rrn23, which have never been used before in discrimination of yews, are modest in length, and we therefore suggest that they can be used as special DNA barcodes for yews. Further studies should design primers and examine the PCR recovery rate for these four loci with a more comprehensive set of samples as our previous study40. In conclusion, our newly developed genetic resources of Taxus plastomes and barcoding candidates may aid in conservation and authentication of endangered Taxus species.
Methods
Plant materials, DNA extraction and sequencing
As Taxus is a taxonomically difficult genus and its interspecific classification remains controversial. This study adopts Farjon’s classification31 and follows our recent study40. A total of 16 Taxus species worldwide were sampled and identified on the basis of the morphological and molecular evidence described in our previous studies38–41,64,65, including 13 species (T. brevifolia Nutt., T. globosa Schltdl., T. floridana Nutt., T. canadensis Marshall, T. cuspidata Siebold & Zucc., T. baccata L., T. contorta Griff., T. chinensis (Pilg.) Rehd., T. mairei (Lemée & Lév.) S.Y. Hu, T. wallichiana Zucc., T. calcicola L.M. Gao & Mich. Möller, T. florinii Spjut, T. phytonii Spjut) and three potential cryptic species, of which two (Emei and Qingling types) have been previously described39,41,64 and one (i.e., Huangshan type) is newly discovered (it was formally treated as T. chinensis, described from high elevation mountains in eastern China).
To assess genetic variation at the intraspecific level, two to three individuals per species were sampled from different populations for each Taxus species except T. canadensis and Qinling type. In addition, two individuals of Pseudotaxus chienii were also sampled and used as the outgroup. The specimens and vouchers (Table S1) of these sampled taxa are deposited in the herbarium of Kunming Institute of Botany, Chinese Academy of Science (KUN), Yunnan, China.
Total genomic DNA was extracted from fresh or silica-gel dried leaves using a modified CTAB method66, in which 4% CTAB was used with incorporation of 0.1% DL-dithiothreitol (DTT). After it was quantified using Qubit 2.0 (Invitrogen, Carlsbad, CA, USA), the extracted DNA was sheared into approximately 500 bp fragments for library construction using standard protocols (NEBNext® Ultra II™DNA Library Prep Kit for Illumina®). All samples were sequenced on an Illumina HiSeq X Ten platform in CloudHealth Company (Shanghai, China) to generate approximately 5‒70 million paired-end 150 bp reads (Table S1).
Plastome assembly and annotation
We used the GetOrganelle pipeline67 to de novo assemble plastomes. In this pipeline, plastomic reads were extracted from total genomic reads and were subsequently assembled using SPAdes version 3.1068. Plastid genes were annotated using Geneious 11.0.369 with the published plastome of T. mairei70 as the reference. Transfer RNAs (tRNAs) were confirmed by their specific structure predicted by tRNAscan-SE 2.071. Plastomes were visualized using Circos 0.6772.
Locally co-linear block (LCB) identification and sequence alignment
The locally co-linear blocks (LCBs) between Pseudotaxus and Taxus plastomes were identified using progressMavus73 with the default options and psbA as the initial point. Sequences of LCBs or loci were aligned using MAFFT 7.074. The parameter set was algorithm = auto, scoring matrix = 200PAM/k = 2, gap open penalty = 1.53, and offset value = 0.123.
Tree construction and pairwise distance calculation
The alignments of LCBs were concatenated in DAMBE 5.075. We used jModelTest276 to assess the best models for tree construction under the corrected Akaike Information Criterion (AICc). Maximum likelihood trees inferred from this concatenated alignment were analyzed under the GTRGAMMAI model with 1,000 rapid bootstrap searches in RAxML 8.277. For each single locus or combined multiple loci, estimates of pairwise distances and neighbor-joining (NJ) trees were carried out in MEGA 778 under the Kimura 2-parameter method. The bootstrap supports for NJ trees were computed with 1,000 replicates. All yielded tree topologies were condensed under a 50% majority rule in MEGA 7.
Verification of isomeric arrangements
To verify isomeric arrangements, we adopted two approaches. First, semi-quantitative PCRs involved use of specific primers to yield amplicons unique to the isomeric arrangements (see Fig. 2A). PCR reactions were performed on a GeneAmp PCR System 9700 thermal cycler (PerkinElmer, Foster City, CA, USA). The 20 µL PCR mixture contained 1 µL total DNA (20 ng/µL), 0.5 µL each of the forward and reverse primers (10 µM), 10 µL Tiangen 2 × Taq PCR MasterMix (Tiangen Biotech, Beijing), and 8 µL ddH2O. After an initial denaturation at 94 °C for 5 min, PCR reactions were conducted for 20, 25, 30 and 35 cycles, respectively. Each cycle included 94 °C for 30 s, annealing at 56 °C for 30 s, and elongation at 72 °C for 1 min. The PCR procedure ended up with a final incubation at 72 °C for 7 min. PCR gel images were taken using a G:BOX gel doc system (SYNGENE, USA) under the exposure time of 1s500ms/1s800ms, followed by “color invert” in PhotoImpact 10 (https://www.paintshoppro.com/). Second, we mapped the Illumina paired-end reads onto the regions specific to each of the isomeric arrangements using Geneious with the default setting. Paired-end reads that spanned the entire trnQ-IR were counted if the sequence identity was >90%. The mapping scenario was viewed and checked in Geneious.
Supplementary information
Acknowledgements
We thank Dr. Michael Möller from the Royal Botanic Garden Edinburgh, UK; Prof. Kevin S. Burgess from Columbus State University, USA; Prof. Marc W Cadotte from the University of Toronto, Canada; Prof. Marcos Soto-Hernandez from Postgraduate College, Mexico; Dr. Chun-Neng Wang from National Taiwan University; and our colleagues Prof. Zhi-Yong Zhang, Drs. Jie Cai, and Zeng-Yuan Wu from mainland China for providing samples. We are grateful to Jun-Bo Yang, Xiao-Jian Qu, Jian-Jun Jin, Han-Tao Qing, Jing Yang, Zhi-Rong Zhang, and Ji-Xiong Yang from GBOWS for assisting with the laboratory work and data analysis. This study was supported by the Large-scale Scientific Facilities of the Chinese Academy of Sciences (Grant No: 2017-LSFGBOWS-01), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB31010000), the National Natural Science Foundation of China (41571059 and 31370252), the Interdisciplinary Research Project of Kunming Institute of Botany (KIB2017003), and research grants from the Ministry of Science and Technology, Taiwan (106-2311-B-001-005) to SMC. Laboratory work was performed at the Laboratory of Molecular Biology at the Germplasm Bank of Wild Species, Kunming Institute of Botany, Chinese Academy of Sciences.
Author Contributions
L.M.G., S.M.C., and D.Z.L. conceived of and designed the research; J.L. and L.M.G. collected the samples; C.S.W., C.N.F., and Y.W.C. performed data analysis; L.N.Y., C.N.F., and Z.Q.M. carried out the molecular lab work; C.S.W., C.N.F., and L.M.G. wrote the first draft of the manuscript with critical input from S.M.C., and all authors contributed to manuscript revision.
Data Accessibility
All DNA sequences have been deposited in GenBank with accession numbers MH390441 to MH390489 (Table S1).
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Chao-Nan Fu and Chung-Shien Wu contributed equally.
Contributor Information
Shu-Miaw Chaw, Email: smchaw@sinica.edu.tw.
Lian-Ming Gao, Email: gaolm@mail.kib.ac.cn.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-019-39161-x.
References
- 1.Wicke S, Schneeweiss GM, dePamphilis CW, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 2011;76:273–297. doi: 10.1007/s11103-011-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaw, S. M., Wu, C. S. & Sudianto, E. Evolution of gymnosperm plastid genomes in Advances in Botanical ResearchVol. 85 (ed. Shu, M. C. & Robert, K. J.) 195–222 (Academic Press, 2018).
- 3.Tsumura Y, Suyama Y, Yoshimura K. Chloroplast DNA inversion polymorphism in populations of Abies and Tsuga. Mol. Biol. Evol. 2000;17:1302–1312. doi: 10.1093/oxfordjournals.molbev.a026414. [DOI] [PubMed] [Google Scholar]
- 4.Wu CS, Wang YN, Hsu CY, Lin CP, Chaw SM. Loss of different inverted repeat copies from the chloroplast genomes of pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol. Evol. 2011;3:1284–1295. doi: 10.1093/gbe/evr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yi X, Gao L, Wang B, Su YJ, Wang T. The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): Evolutionary comparison of Cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol. Evol. 2013;5:688–698. doi: 10.1093/gbe/evt042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo W, et al. Predominant and substoichiometric isomers of the plastid genome coexist within juniperus plants and have shifted multiple times during cupressophyte evolution. Genome Biol. Evol. 2014;6:580–590. doi: 10.1093/gbe/evu046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu CY, Wu CS, Chaw SM. Birth of four chimeric plastid gene clusters in Japanese umbrella pine. Genome Biol. Evol. 2016;8:1776–1784. doi: 10.1093/gbe/evw109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vieira LdN, et al. The plastome sequence of the endemic Amazonian conifer, Retrophyllum piresii (Silba) C.N.Page, reveals different recombination events and plastome isoforms. Tree Genet. Genom. 2016;12:10. doi: 10.1007/s11295-016-0968-0. [DOI] [Google Scholar]
- 9.Qu XJ, Wu CS, Chaw SM, Yi TS. Insights into the existence of isomeric plastomes in Cupressoideae (Cupressaceae) Genome Biol. Evol. 2017;9:1110–1119. doi: 10.1093/gbe/evx071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gitzendanner, M. A., Soltis, P. S., Yi, T. S., Li, D. Z. & Soltis, D. E. Plastome phylogenetics: 30 years of inferences into plant evolution in Advances in Botanical ResearchVol. 85 (ed. Shu, M. C. & Robert, K. J.) 293–313 (Academic Press, 2018).
- 11.Dodsworth S. Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 2015;20:525–527. doi: 10.1016/j.tplants.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 12.Birky CW. The inheritance of genes in mitochondria and chloroplasts: Laws, mechanisms, and models. Annu. Rev. Genet. 2001;35:125–148. doi: 10.1146/annurev.genet.35.102401.090231. [DOI] [PubMed] [Google Scholar]
- 13.Petit, R. J. & Vendramin, G. G. Plant phylogeography based on organelle genes: an introduction in Phylogeography of Southern European Refugia: Evolutionary perspectives on the origins and conservation of European biodiversity (ed. Steven, W. & Nuno, F.) 23–97 (Springer Netherlands, 2007).
- 14.Hollingsworth PM, Graham SW, Little DP. Choosing and using a plant DNA barcode. PLoS One. 2011;6:e19254. doi: 10.1371/journal.pone.0019254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.CBOL Plant Working Group. A. DNA barcode for land plants. Proc. Natl. Acad. Sci. USA. 2009;106:12794–12797. doi: 10.1073/pnas.0905845106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li DZ, et al. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. USA. 2011;108:19641–19646. doi: 10.1073/pnas.1104551108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, et al. Plant DNA barcoding: from gene to genome. Biol. Rev. 2015;90:157–166. doi: 10.1111/brv.12104. [DOI] [PubMed] [Google Scholar]
- 18.Hollingsworth, P. M., Li, D. Z., van der Bank, M. & Twyford, A. D. Telling plant species apart withDNA: from barcodes to genomes. Philos. T. R. Soc. B. 371, 10.1098/rstb.2015.0338 (2016). [DOI] [PMC free article] [PubMed]
- 19.Percy DM, et al. Understanding the spectacular failure of DNA barcoding in willows (Salix): Does this result from a trans‐specific selective sweep? Mol. Ecol. 2014;23:4737–4756. doi: 10.1111/mec.12837. [DOI] [PubMed] [Google Scholar]
- 20.Yan LJ, et al. DNA barcoding of Rhododendron (Ericaceae), the largest Chinese plant genus in biodiversity hotspots of the Himalaya–Hengduan Mountains. Mol. Ecol. Resour. 2015;15:932–944. doi: 10.1111/1755-0998.12353. [DOI] [PubMed] [Google Scholar]
- 21.Sullivan AR, Schiffthaler B, Thompson SL, Street NR, Wang XR. Interspecific plastome recombination reflects ancient reticulate evolution in Picea (Pinaceae) Mol. Biol. Evol. 2017;34:1689–1701. doi: 10.1093/molbev/msx111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nock CJ, et al. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 2011;9:328–333. doi: 10.1111/j.1467-7652.2010.00558.x. [DOI] [PubMed] [Google Scholar]
- 23.Kane NC, Cronk Q. Botany without borders: barcoding in focus. Mol. Ecol. 2008;17:5175–5176. doi: 10.1111/j.1365-294X.2008.03972.x. [DOI] [PubMed] [Google Scholar]
- 24.Yang JB, Tang M, Li HT, Zhang ZR, Li DZ. Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 2013;13:84. doi: 10.1186/1471-2148-13-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kane N, et al. Ultra‐barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am. J. Bot. 2012;99:320–329. doi: 10.3732/ajb.1100570. [DOI] [PubMed] [Google Scholar]
- 26.Ruhsam M, et al. Does complete plastid genome sequencing improve species discrimination and phylogenetic resolution in Araucaria? Mol. Ecol. Resour. 2015;15:1067–1078. doi: 10.1111/1755-0998.12375. [DOI] [PubMed] [Google Scholar]
- 27.Zhang N, et al. An analysis of Echinacea chloroplast genomes: Implications for future botanical identification. Sci. Rep. 2017;7:216. doi: 10.1038/s41598-017-00321-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang JB, Yang SX, Li HT, Yang J, Li DZ. Comparative chloroplast genomes of Camellia Species. PLoS One. 2013;8:e73053. doi: 10.1371/journal.pone.0073053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang, Y. et al. The complete chloroplast genome sequences of five Epimedium Species: Lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 710.3389/fpls.2016.00306 (2016). [DOI] [PMC free article] [PubMed]
- 30.Bi Y, et al. Chloroplast genomic resources for phylogeny and DNA barcoding: a case study on Fritillaria. Sci. Rep. 2018;8:1184. doi: 10.1038/s41598-018-19591-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farjon, A. A Handbook of the World’s Conifers (2 vols.). Vol. 1 (Brill, 2010).
- 32.Christenhusz M, et al. A new classification and linear sequence of extant gymnosperms. Phytotaxa. 2011;19:55–70. doi: 10.11646/phytotaxa.19.1.3. [DOI] [Google Scholar]
- 33.Mao K, et al. Distribution of living Cupressaceae reflects the breakup of Pangea. Proc. Natl. Acad. Sci. USA. 2012;109:7793–7798. doi: 10.1073/pnas.1114319109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu, L. G., Li, N. & Mill, R. R. Taxaceae in Flora of China (ed. Wu, Z. Y. & Peter, R. H.) 89–96 (Science Press, 1999).
- 35.Kingston DGI, Newman DJ. Taxoids: cancer-fighting compounds from nature. Curr. Opin. Drug Discov. Devel. 2007;10:130–144. [PubMed] [Google Scholar]
- 36.Möller M, et al. Morphometric analysis of the Taxus wallichiana complex (Taxaceae) based on herbarium material. Bot. J. Linn. Soc. 2007;155:307–335. doi: 10.1111/j.1095-8339.2007.00697.x. [DOI] [Google Scholar]
- 37.Spjut RW. Taxonomy and nomenclature of taxus (taxaceae) J. Bot. Res. Inst. Texas. 2007;1:203–289. [Google Scholar]
- 38.Shah A, et al. Delimitation of Taxus fuana Nan Li & R.R. Mill (Taxaceae) based on morphological and molecular data. Taxon. 2008;57:211–222. doi: 10.2307/25065961. [DOI] [Google Scholar]
- 39.Möller M, et al. A multidisciplinary approach reveals hidden taxonomic diversity in the morphologically challenging Taxus wallichiana complex. Taxon. 2013;62:1161–1177. doi: 10.12705/626.9. [DOI] [Google Scholar]
- 40.Liu J, et al. Integrating a comprehensive DNA barcode reference library with the global map of yews (Taxus L.) for species identification. Mol. Ecol. Resour. 2018;18:1115–1131. doi: 10.1111/1755-0998.12903. [DOI] [PubMed] [Google Scholar]
- 41.Liu J, Möller M, Gao LM, Zhang DQ, Zhu LD. DNA barcoding for the discrimination of Eurasian yews (Taxus L., Taxaceae) and the discovery of cryptic species. Mol. Ecol. Resour. 2011;11:89–100. doi: 10.1111/j.1755-0998.2010.02907.x. [DOI] [PubMed] [Google Scholar]
- 42.Chaw SM, Sung HM, Long H, Zharkikh A, Lie WH. The phylogenetic positions of the conifer genera Amentotaxus, Phyllocladus, and Nageia inferred from 18s rRNA sequences. J. Mol. Evol. 1995;41:224–230. doi: 10.1007/bf00170676. [DOI] [PubMed] [Google Scholar]
- 43.Hebert PD, Stoeckle MY, Zemlak TS, Francis CM, Lie WH. Identification of birds through DNA barcodes. PLoS Biol. 2004;2:e312. doi: 10.1371/journal.pbio.0020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parks M, Cronn R, Liston A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009;7:84. doi: 10.1186/1741-7007-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curci PL, Paola DD, Sonnante G. Development of chloroplast genomic resources for. Cynara. Mol. Ecol. Resour. 2016;16:562–573. doi: 10.1111/1755-0998.12457. [DOI] [PubMed] [Google Scholar]
- 46.Chen Z, et al. Molecular evolution of the plastid genome during diversification of the cotton genus. Mol. Phylogen. Evol. 2017;112:268–276. doi: 10.1016/j.ympev.2017.04.014. [DOI] [PubMed] [Google Scholar]
- 47.Du YP, et al. Complete chloroplast genome sequences of Lilium: insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 2017;7:5751. doi: 10.1038/s41598-017-06210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palmer JD, Chloroplast DNA. exists in two orientations. Nature. 1983;301:92. doi: 10.1038/301092a0. [DOI] [Google Scholar]
- 49.Walker JF, Jansen RK, Zanis MJ, Emery NC. Sources of inversion variation in the small single copy (SSC) region of chloroplast genomes. Am. J. Bot. 2015;102:1751–1752. doi: 10.3732/ajb.1500299. [DOI] [PubMed] [Google Scholar]
- 50.Li J, et al. Evolution of short inverted repeat in cupressophytes, transfer of accD to nucleus in Sciadopitys verticillata and phylogenetic position of Sciadopityaceae. Sci. Rep. 2016;6:20934. doi: 10.1038/srep20934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu CS, Chaw SM. Large-scale comparative analysis reveals the mechanisms driving plastomic compaction, reduction, and inversions in conifers II (Cupressophytes) Genome Biol. Evol. 2016;8:3740–3750. doi: 10.1093/gbe/evw278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alverson AJ, Zhuo S, Rice DW, Sloan DB, Palmer JD. The mitochondrial genome of the legume vigna radiata and the analysis of recombination across short mitochondrial repeats. PLoS One. 2011;6:e16404. doi: 10.1371/journal.pone.0016404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y, et al. The complete chloroplast genome sequence of Taxus chinensis var. mairei (Taxaceae): loss of an inverted repeat region and comparative analysis with related species. Gene. 2014;540:201–209. doi: 10.1016/j.gene.2014.02.037. [DOI] [PubMed] [Google Scholar]
- 54.Jia XM, Liu XP. Characterization of the complete chloroplast genome of the Chinese yew Taxus chinensis (Taxaceae), an endangered and medicinally important tree species in China. Conserv. Genet. Resour. 2017;9:197–199. doi: 10.1007/s12686-016-0649-1. [DOI] [Google Scholar]
- 55.Woloszynska M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes—though this be madness, yet there’s method in’t. J. Exp. Bot. 2010;61:657–671. doi: 10.1093/jxb/erp361. [DOI] [PubMed] [Google Scholar]
- 56.Ruhlman TA, Zhang J, Blazier JC, Sabir JSM, Jansen RK. Recombination‐dependent replication and gene conversion homogenize repeat sequences and diversify plastid genome structure. Am. J. Bot. 2017;104:559–572. doi: 10.3732/ajb.1600453. [DOI] [PubMed] [Google Scholar]
- 57.Krawczyk K, Nobis M, Myszczyński K, Klichowska E, Sawicki J. Plastid super-barcodes as a tool for species discrimination in feather grasses (Poaceae: Stipa) Sci. Rep. 2018;8:1924. doi: 10.1038/s41598-018-20399-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang CY, Grünheit N, Ahmadinejad N, Timmis JN, Martin W. Mutational decay and age of chloroplast and mitochondrial genomes transferred recently to angiosperm nuclear chromosomes. Plant Physiol. 2005;138:1723–1733. doi: 10.1104/pp.105.060327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu CS, Chaw SM. Highly rearranged and size‐variable chloroplast genomes in conifers II clade (cupressophytes): evolution towards shorter intergenic spacers. Plant Biotechnol. J. 2014;12:344–353. doi: 10.1111/pbi.12141. [DOI] [PubMed] [Google Scholar]
- 60.Korotkova N, Nauheimer L, Ter-Voskanyan H, Allgaier M, Borsch T. Variability among the most rapidly evolving plastid genomic regions is lineage-specific: Implications of pairwise genome comparisons in pyrus (rosaceae) and other angiosperms for marker choice. PLoS One. 2014;9:e112998. doi: 10.1371/journal.pone.0112998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Niu, Z. et al. The complete plastome sequences of four orchid species: Insights into the evolution of the orchidaceae and the utility of plastomic mutational hotspots. Front. Plant Sci. 8, 10.3389/fpls.2017.00715 (2017). [DOI] [PMC free article] [PubMed]
- 62.Fu CN, et al. Comparative analyses of plastid genomes from fourteen Cornales species: inferences for phylogenetic relationships and genome evolution. BMC Genomics. 2017;18:956. doi: 10.1186/s12864-017-4319-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song Y, et al. Chloroplast genomic resource of paris for species discrimination. Sci. Rep. 2017;7:3427. doi: 10.1038/s41598-017-02083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gao LM, et al. High variation and strong phylogeographic pattern among cpDNA haplotypes in Taxus wallichiana (Taxaceae) in China and North Vietnam. Mol. Ecol. 2007;16:4684–4698. doi: 10.1111/j.1365-294X.2007.03537.x. [DOI] [PubMed] [Google Scholar]
- 65.Poudel RC, et al. Using morphological, molecular and climatic data to delimitate yews along the hindu Kush-Himalaya and adjacent regions. PLoS One. 2012;7:e46873. doi: 10.1371/journal.pone.0046873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bul. 1987;19:11–15. [Google Scholar]
- 67.Jin, J. J. et al. GetOrganelle: a simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. bioRxiv, 10.1101/256479 (2018).
- 68.Bankevich A, et al. Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kearse M, et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hsu CY, Wu CS, Chaw SM. Ancient nuclear plastid DNA in the yew family (Taxaceae) Genome Biol. Evol. 2014;6:2111–2121. doi: 10.1093/gbe/evu165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lowe TM, Chan PP. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44:W54–W57. doi: 10.1093/nar/gkw413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krzywinski M, et al. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Darling AE, Mau B, Perna NT. ProgressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xia XDAMBE5. A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013;30:1720–1728. doi: 10.1093/molbev/mst064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and high-performance computing. Nat. Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All DNA sequences have been deposited in GenBank with accession numbers MH390441 to MH390489 (Table S1).