

Key Points
Infusion of heat-inactivated S aureus triggers inflammation, coagulopathy, complement activation, organ failure, and death in primates.
Inhibition of FXI activation by FXIIa decreased inflammation and prevented organ failure and death in baboons challenged with S aureus.
Abstract
Staphylococcus aureus infections can produce systemic bacteremia and inflammation in humans, which may progress to severe sepsis or septic shock, even with appropriate antibiotic treatment. Sepsis may be associated with disseminated intravascular coagulation and consumptive coagulopathy. In some types of mouse infection models, the plasma coagulation protein factor XI (FXI) contributes to the pathogenesis of sepsis. We hypothesize that FXI also contributes to the pathogenesis of sepsis in primates, and that pharmacological interference with FXI will alter the outcome of Staphylococcus aureus–induced lethality in a baboon model. Pretreatment of baboons with the anti-FXI antibody 3G3, a humanized variant of the murine monoclonal 14E11 that blocks FXI activation by FXIIa, substantially reduced the activation of coagulation, as reflected by clotting times and plasma complexes of coagulation proteases (FXIIa, FXIa, FIXa, FXa, FVIIa, and thrombin) with serpins (antithrombin or C1 inhibitor) following infusion of heat-inactivated S aureus. 3G3 treatment reduced fibrinogen and platelet consumption, fibrin deposition in tissues, neutrophil activation and accumulation in tissues, cytokine production, kininogen cleavage, cell death, and complement activation. Overall, 3G3 infusion protected the structure and function of multiple vital organs, including lung, heart, liver, and kidney. All treated animals reached the end point survival (7 days), whereas all nontreated animals developed terminal organ failure within 28 hours. We conclude that FXI plays a role in the pathogenesis of S aureus–induced disseminated intravascular coagulation and lethality in baboons. The results provide proof of concept for future therapeutic interventions that may prevent sepsis-induced organ failure and save lives in certain forms of sepsis.
Visual Abstract

Introduction
The contact proteins factor XI (FXI), factor XII (FXII), high-molecular-weight kininogen (HK), and plasma prekallikrein (PK) serve as a central node connecting coagulation to inflammation, and have long been thought to contribute to the pathologic host response in sepsis.1,2 Although FXI deficiency has been linked to infections in Holstein cattle,3 studies with rodents, dogs, and humans with various contact factor deficiencies (including FXI deficiency) have not identified a defect in innate immunity, suggesting the contact pathway is dispensable in terms of host defense. Furthermore, FXII, HK, and PK appear dispensable for hemostasis. Indeed, the only clinically relevant phenotype associated with a contact factor deficiency is the bleeding tendency in some individuals lacking FXI (hemophilia C), a condition considerably milder than the disorders hemophilia A or B.4,5 Thus, although contact activation contributes to a number of proinflammatory and procoagulant effects,6 its absence does not significantly compromise host defense or hemostasis. Inhibiting the contact system, therefore, may safely attenuate systemic inflammation triggered by certain pathogens, and may improve outcomes in septic patients.
Previous work with a baboon model of Escherichia coli sepsis showed that partial inhibition of FXII activity reduced hypotension and inflammation, but without a significant effect on survival.1,7 Our group subsequently showed that the murine monoclonal anti-FXI antibody 14E11, which specifically interferes with activation of FXI by activated FXII (FXIIa), significantly improved outcomes in a murine model of abdominal sepsis, with decreasing markers of inflammation and improving overall survival.8,9 We reported similar findings with FXI-deficient mice9,10 and with 14E11 in a murine model of listeriosis.11 In contrast, we found activated protein C (APC) to be detrimental in the abdominal sepsis model.8 Interestingly, FXI deficiency or treatment with 14E11 was ineffective in mice infected with Yersinia enterocolitica, suggesting the benefits of FXI inhibition may be pathogen specific.12 These findings could be relevant to understanding incongruous preclinical work, discordant outcomes in clinical trials of APC,13,14 and other anticoagulants in sepsis with disseminated intravascular coagulation (DIC),15 in which patients were not stratified based on type of infection
Previous studies on the biochemistry and pathophysiology of FXI in mice raise questions regarding the suitability of this species for modeling human disease. Unlike humans, cattle, and dogs with FXI deficiency, FXI-deficient mice do not seem to have a hemostatic defect.16,17 Furthermore, the majority of FXI in mice is associated with blood vessel walls rather than circulating in plasma, a situation that does not appear to be the case in primates.18 Several studies indicate that FXI-deficient mice are unusually susceptible to inflammation in the lungs after pulmonary injury,19-21 which does not have an obvious correlate in humans.22,23 This is consistent with the substantial body of evidence indicating significant differences in inflammatory processes in mice and humans.24 These observations point to the need for studies in primates to address the role of the contact system in general, and FXI in particular, in sepsis.
The detrimental host response to sepsis often persists after the infectious agent is killed. Thus, exposure to certain bacterial components alone is sufficient to produce a severe systemic inflammatory response,25,26 modeling the commonly encountered clinical scenario in which patients develop sepsis despite use of bactericidal antibiotics. Staphylococcus aureus bacteremia is a prevalent cause of human sepsis and systemic inflammatory response syndrome worldwide, with mortality rates of >30% at 30 days. We used a primate model that closely mimics human responses27 to investigate whether interference with FXI activation would affect outcomes after infusion of a lethal dose of heat-inactivated S aureus. The humanized antibody 3G3 (AB023) used in this study is derived from 14E11, with which it shares functional characteristics.27 The data show that 3G3 reduces procoagulant and inflammatory responses to S aureus and provides a survival benefit.
Material and methods
Preparation of a humanized function-blocking anti-FXI antibody
Recombinant 3G3 was produced by grafting the 14E11 complementarity determining regions into the heavy and light chains of an S241P-stabilized human IgG4 κ antibody as described elsewhere.28
Baboon model of S aureus sepsis
The study was approved by the Interfaculty Animal Ethics committee of the University of the Free State, Bloemfontein, South Africa, and the Institutional Care and Use Committee of Oklahoma Medical Research Foundation. Healthy Papio ursinus baboons (8-20.2 kg body weight) with a leukocyte count less than 13 000/µL and hemoglobin >10 g/dL were randomly distributed between the control and treatment groups.
Bacterial culture and heat inactivation
S aureus subspecies aureus Rosenbach (ATCC 12598) was purchased from American Type Culture Collection (Manassas, VA). To control the standard dosing of bacteria and avoid potentially confounding effects by live organisms,29 exponential-phase cultured S aureus was extensively washed with saline solution and counted, then heated for 1 hour at 70°C. Aliquots were stored at −80°C until use. For uniformity, animals were challenged with bacteria from a single preparation.
The experimental design included 2 arms: a control group (n = 3) and a treated group (n = 4). Animals in both groups were challenged with 3 × 1010 heat-inactivated S aureus (a lethal dose),30 given by IV infusion over 2 hours. The untreated control group received only the bacterial infusion, whereas the treated group received a bolus of 3G3 (1 mg/kg) 30 minutes before the bacterial infusion was started. The time point at which the bacterial infusion began was designated as T0. Eight hours after the start of the bacterial infusion (T+8), the animals were returned to the recovery cage and observed until they exhibited signs of unrecoverable organ failure and septic shock, at which time they were humanely euthanized. Surviving animals were euthanized on day 7. Details on critical care monitoring during and postsepsis challenge are detailed in the supplemental Methods. Hematologic parameters (including differential white blood cell counts, red blood cell (RBC) counts, hematocrit, hemoglobin, and platelet counts), coagulation parameters (activated partial thromboplastin time [aPTT], prothrombin time [PT], and fibrinogen levels), and organ function tests including plasma lactate, blood glucose, blood urea nitrogen, creatinine, alanine aminotransferase, aspartate transaminase, alkaline phosphatase, amylase, potassium, and phosphate ions levels were monitored for all animals. At the time of euthanasia, tissue samples were collected from select organs and processed for microscopy.
Biochemical tests
Blood glucose was measured using a Contour Next blood glucose meter (Bayer HealthCare LLC, Mishawaka, IN). Blood lactate was measured by Lactate Scout (EKF Diagnostics GmbH, Barleben, Germany). Serum alanine aminotransferase, aspartate aminotransferase, amylase, creatinine, blood urea nitrogen, potassium, phosphate, and lactate dehydrogenase levels were measured using standard clinical tests. Myeloperoxidase (MPO) activity in plasma was quantified by using a FluoroMPO myeloperoxidase detection kit (Cell Technology, Fremont, CA).
ELISA assays
Total FXI was measured using a matched-paired antibody set from Affinity Biologicals (Ancaster, ON, Canada). Complexes of activated factors with antithrombin (Kallikrein-AT, FXIIa-AT, FXIa-AT, FIXa-AT, FVIIa-AT, FXa-AT, TAT) or C1 inhibitor (Kallikrein-C1 INH, FXIIa-C1 INH) were measured via custom sandwich enzyme-linked immunosorbent assays (ELISAs), as detailed in the supplemental Methods.
Plasma kininogen (all forms), plasminogen activator inhibitor 1 (PAI-1) and tissue-type plasminogen activator (t-PA) were quantified using DuoSet ELISA kits (R&D Systems, Minneapolis, MN). For plasmin-antiplasmin complexes, we used affinity purified goat anti-human plasminogen (2 µg/mL, Affinity Biologicals) as capture and horseradish peroxidase–conjugated goat anti-human α2-antiplasmin (1 µg/mL, Affinity Biologicals) as detection antibodies. D-dimer ELISA was similarly done, using mouse monoclonal anti-human D-dimer (clone DD1, 1 µg/mL, Novus Biologicals, Littleton, CO) as capture and horseradish peroxidase–conjugated sheep anti-human fibrinogen as detection antibody (2 µg/mL, Affinity Biologicals). Plasma cytokines were quantified using MILLIPLEX MAP Non-Human Primate Cytokine Magnetic Bead Panel (EMD Millipore, Billerica, MA). C3b and C5b-9 were measured as previously described.31 Nucleosome was quantified using a Cell Death Detection ELISA PLUS kit (Roche Diagnostics GmbH, Mannheim, Germany). Nucleohistones preparation (Worthington Biochemical Corporation, Lakewood, NJ) was used as a standard.
Western blot analysis of kininogen cleavage
Samples of baboon plasma (1 μL) were size fractionated on nonreducing 10% polyacrylamide sodium dodecyl sulfate gels. After transfer to nitrocellulose membranes, blots were developed with a monoclonal murine immunoglobulin-G (IgG) raised against human HK (6H1). The secondary antibody was donkey anti-mouse IgG labeled with IRDye 680LT (Abcam). Blots were imaged on an Azure Biosystems c600 system, using excitation λ 680 nm and emission λ 700 nm.
Microscopy
Formalin-fixed, paraffin-embedded sections were used for hematoxylin and eosin staining to reveal the general tissue morphology. The preparations were examined by a veterinary pathologist who was blinded to the experimental condition.31 Double immunofluorescence confocal microscopy on blood smears was performed to detect tissue factor (TF) on leukocytes using mouse monoclonals anti-TF (clone HTF1, ThermoFisher) and anti CD14 (clone 23G4, ATCC HB11637). Paraformaldehyde-fixed cryosections31 were immunostained to detect deposition of fibrinogen/fibrin, platelet aggregates, neutrophils, monocyte/macrophages, citrullinated histones, and complement terminal complex (C5b-9) using the following antibodies: rabbit anti-human fibrinogen IgG, mouse monoclonal anti-CD61 (GPIIIa; clone Y2/51), mouse monoclonal anti-CD68 (clone PG-M1), all from Agilent Technologies (Santa Clara, CA); rabbit anti-human neutrophil elastase IgG (MilliporeSigma, Burlington, MA); rabbit anti citrullinated histone H4 (citrulline 3; EMD Millipore); and mouse anti-C5b-9 neoepitope (clone aE11; Enzo Life Sciences Inc, Farmingdale, NY).
Statistical analysis
Prism (GraphPad Software 7.0d) was used for statistical analysis. Data are given as mean ± standard error of the mean. Comparisons between 2 groups were performed using a 2-tailed Student t test. Results were considered significant at P < .05 (*P < .05, **P < .01, ***P < .001). The log-rank Mantel-Cox test was used for comparison of survival curves.
Results
Effect of 3G3 on survival after S aureus infusion into baboons
Using our established model of S aureus–induced lethality in baboons, we tested whether administration of 3G3 antibody affects survival. Baboons were pretreated with 1 mg/kg 3G3 before IV infusion of a lethal dose of heat-inactivated S aureus. All 4 3G3-treated baboons reached the 7-day survival end point, whereas all identically challenged nontreated animals were terminally ill between 10 and 28 hours postchallenge (Mantel-Cox test P = .0101; Figure 1). Subsequent studies were designed to investigate the mechanisms by which 3G3 provided a survival benefit by studying the effects of 3G3 pretreatment on markers of coagulation, fibrinolysis, inflammation, and complement activation during the first 24 hours for untreated animals, 7 days for treated animals, and by assessments of organ function and damage.
Figure 1.

Pretreatment with the FXI A2 domain binding antibody, 3G3, prevented the death of baboons challenged with a lethal IV injection of heat-inactivated S aureus. Survival plot of animals challenged with an LD of heat-inactivated S aureus and pretreated with the anti-FXI A2 domain antibody 3G3 (LDSA + 3G3; n = 4) vs control animals’ LDSA in the absence of 3G3 pretreatment (n = 3). Survival distribution of the 2 groups was determined using a log-rank Mantel-Cox test; the result is significant at P = .01. LD, lethal dose; LDSA, lethal dose of S aureus.
Effect of 3G3 on the coagulopathic response to S aureus challenge
As shown in Figure 2, IV infusion of heat-inactivated S aureus induced rapid increases in the aPTT and PT, suggesting induction of a consumptive coagulopathy. Increases in activated coagulation protease species, including members of the contact phase system, were observed within the first 2 to 4 hours after bacterial infusion, as reflected by increases in plasma levels of protease-serpin complexes (FXIIa-AT, FXIIa-C1INH, Kal-AT, Kal-C1INH, FXIa-AT, FIXa-AT, FVIIa-AT, FXa-AT, and T-AT; Figure 2C-K). Pretreatment with 3G3 dramatically blunted the activation of coagulation induced by S aureus (Figure 2). Interestingly, this included blunting of coagulation through the extrinsic pathway (FVIIa), which is not affected by 3G3 in vitro. This may be due to significant reduction by 3G3 of S aureus–induced TF expression on circulating blood cells (Figure 2L; supplemental Figure 1).
Figure 2.
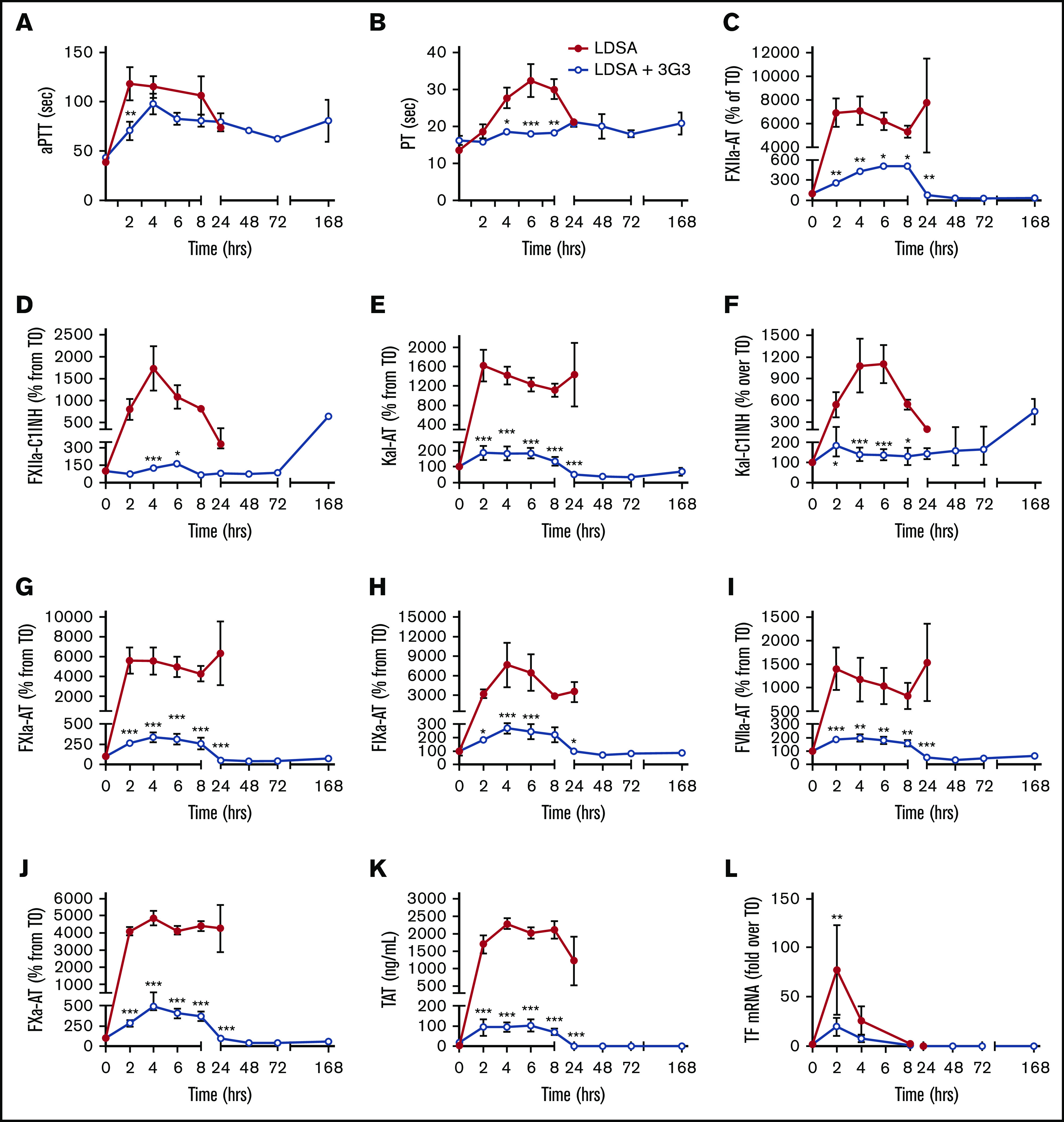
Pretreatment of baboons with 3G3 antibody reduced coagulation activation and prevented coagulopathy following a lethal dose of heat-inactivated S aureus injection. Time course dynamics of clotting times aPTT (A) and PT (B), and plasma hemostatic biomarkers FXIIa-AT (C), FXIIa-C1 inhibitor complexes (D), kal-AT complexes (E), kal-C1 inhibitor complexes (F), FXIa-AT complexes (G), FIXa-AT complexes (H), FVIIa-AT (I), FXa-AT (J), TAT complexes (K), and TF mRNA (L) in leukocytes. Data are represented as mean ± standard error of the mean (SEM). Same time points are compared between LDSA and LDSA plus 3G3-pretreated (LDSA + 3G3) animals using 2-tailed Student t test. *P < .05, **P < .01, ***P < .001. AT, antithrombin; kal, kallikrein; mRNA, messenger RNA; TAT, thrombin-antithrombin.
Consistent with activation of the contact and coagulation systems, S aureus–treated baboons showed early consumption of plasma FXI (supplemental Figure 2A), fibrinogen (Figure 3A), and kininogens (supplemental Figure 3A), with clear evidence for cleavage of HK, a marker of bradykinin generation (Figure 3B).32 The increased clotting times and decreased platelet counts (Figure 4B) indicate rapid development of a consumptive coagulopathy. Examination of vital organs including the lung (Figure 3B) and kidney (Figure 3C) after euthanasia revealed marked accumulation of fibrin and platelet-rich microthrombi. Also, consistent with development of a consumptive coagulopathy, plasma markers of fibrinolysis including t-PA, PAI-1, plasmin (as measured by plasmin-antiplasmin complexes), and D-dimer increased in response to S aureus infusion (Figure 3B-F).
Figure 3.
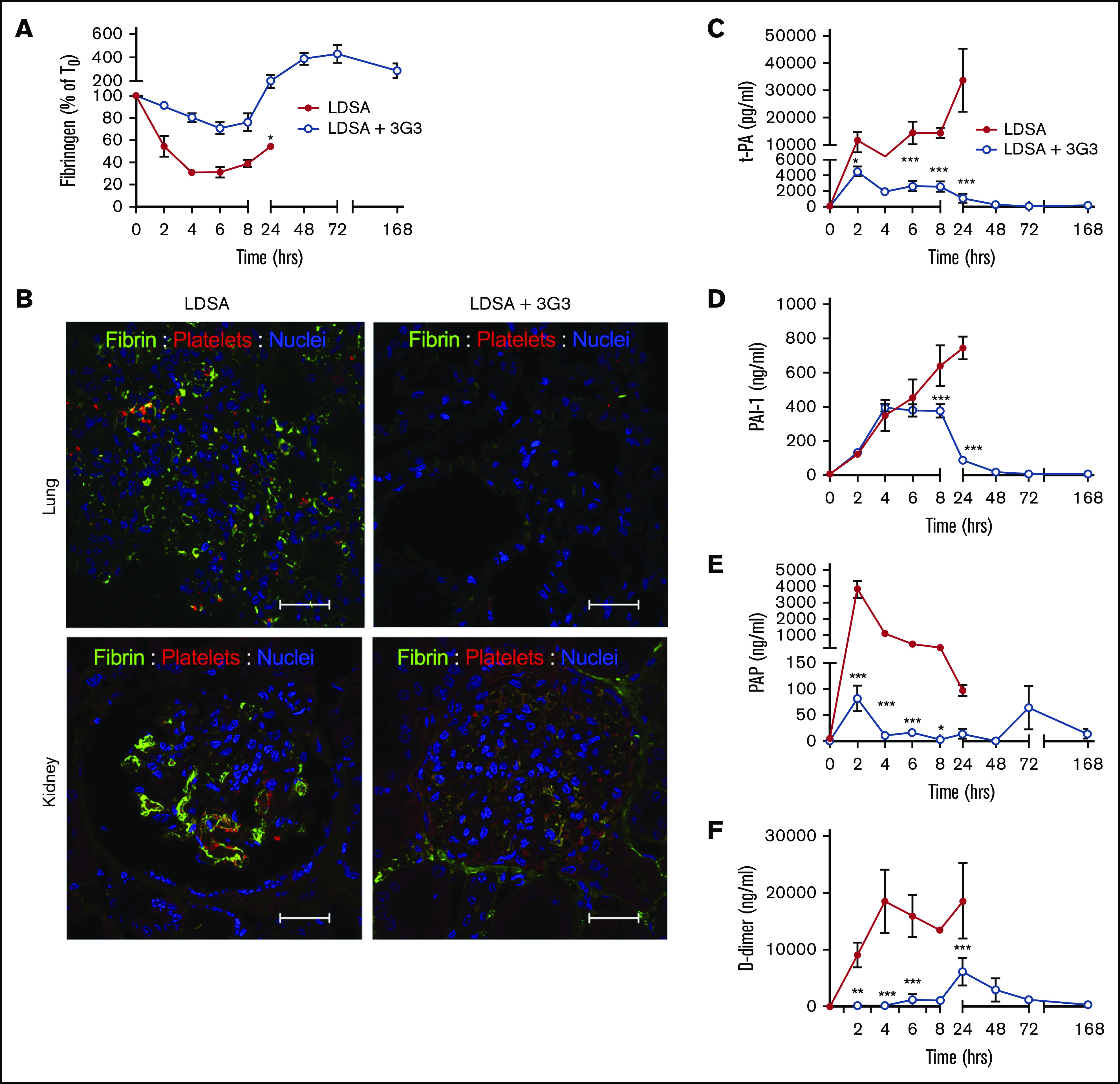
Pretreatment with 3G3 antibody prevented fibrinogen consumption, tissue fibrin deposition, and fibrinolysis activation following a lethal dose of heat-inactivated S aureus injection in baboons. (A) Time course changes of fibrinogen levels. (B) Double immunostaining for fibrin (green) and platelet marker CD41 (red) in the lung and kidney of baboons challenged with LDSA (left) or with anti FXI antibody treatment (LDSA + 3G3; right). Nuclear counterstaining is shown in blue. Confocal images were captured on a Nikon Eclipse TE2000-U inverted microscope equipped with a Nikon C1 scanning head. Images were acquired and processed using EZ-C1 software (v 3.80; Nikon, Melville NY). Scale bars, 100 µm. (C-F) Time course changes of plasma markers of fibrinolysis: t-PA (C), PAI-1 (D), plasmin-antiplasmin complexes (E), and D-dimer (F). *P < .05, **P < .01, ***P < .001.
Figure 4.
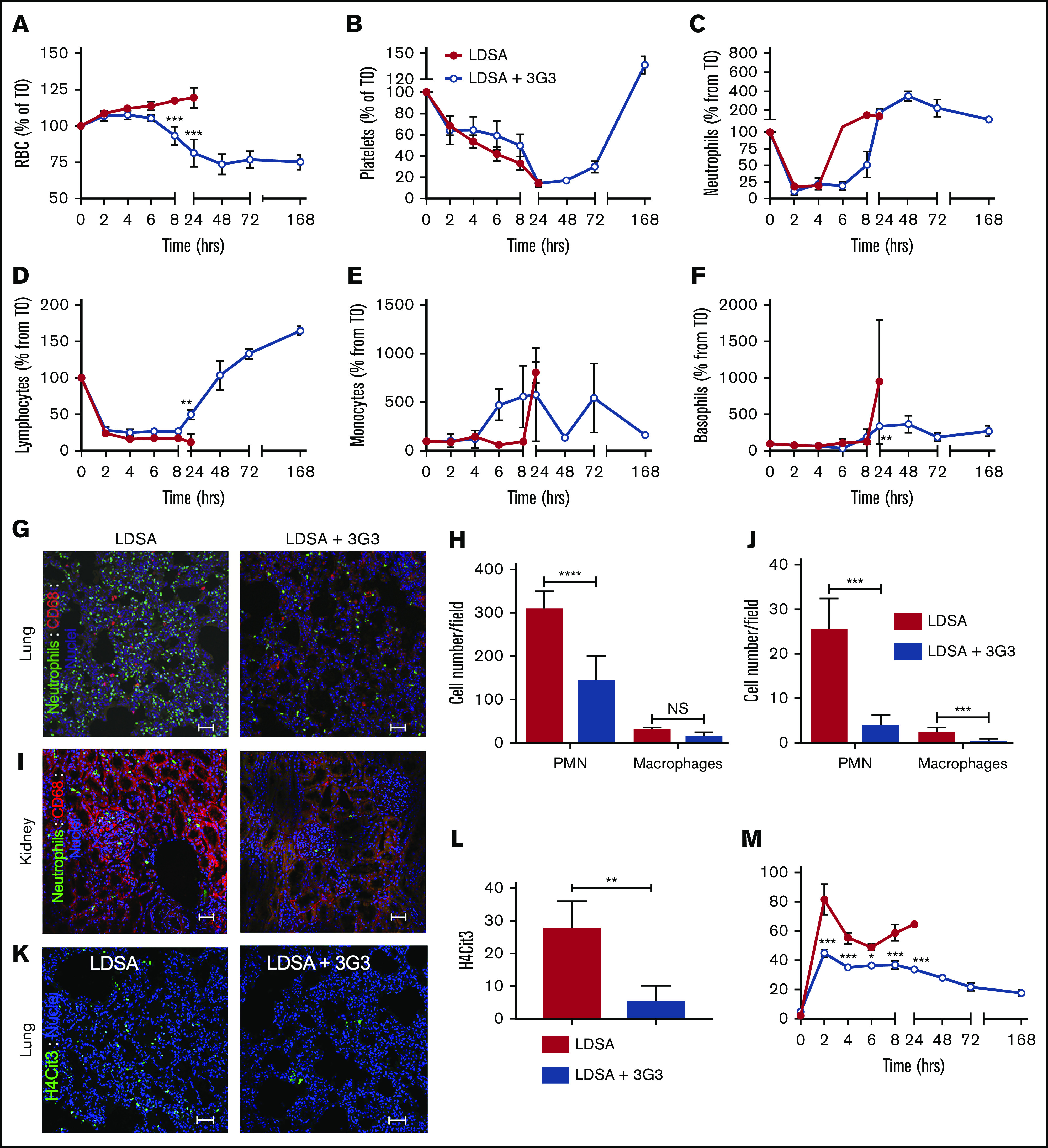
Effect of pretreatment with 3G3 antibody on blood cells, neutrophil activation, and tissue accumulation following a lethal dose of heat-inactivated S aureus injection in baboons. Time course changes of RBCs (A), platelets (B), neutrophils (C), lymphocytes (D), monocytes (E), and basophils (F) in baboons challenged with LDSA or with pretreatment with 3G3 (LDSA + 3G3). (G) Double immunofluorescence staining of elastase (neutrophils, green) and CD68 (monocytes/macrophages, red) in the lung (top) and kidney (bottom) of baboons challenged with S aureus, without (LDSA; left) or with pretreatment with 3G3 antibodies (LDSA + 3G3; right). Confocal images were captured on a Nikon Eclipse TE2000-U inverted microscope equipped with a Nikon C1 scanning head. Images were acquired and processed using EZ-C1 software (v 3.80; Nikon). Scale bars, 100 µm. (H-J) Quantitation of neutrophils and macrophages in 5 microscopic fields is shown (H, lung; J, kidney). (K) Immunostaining of H4cit3 (green) in the lung of baboons challenged with S aureus without (LDSA; left) or with 3G3 pretreatment (LDSA+3G3; right). (L) Quantitation of H4cit3+ cells in 5 microscopic fields. Images were acquired and processed as described previously. Scale bars, 100 µm. (M) Time course changes of MPO (marker of neutrophil degranulation) enzymatic activity in plasma. (A-F,M) Data are presented as mean ± SEM. Same time point data are compared between untreated and 3G3-pretreated animals using 2-tailed Student t test: *P < .05, **P < .01, **P < .001. (H,J) Data are presented as mean ± SEM. The groups are compared by 2-tailed Student t test: ***P < .001, ****P < .0001. H4cit3, citrullinated histone H4; NS, not significant.
The procoagulant and profibrinolytic effects were significantly smaller in 3G3-treated animals. After a dip between 24 and 48 hours postbacteria infusion, total FXI protein rebounded in the 3G3-treated group, reaching levels at 72 to 168 hours that are two- to threefold greater than at T0. Almost all circulating FXI was bound to 3G3 (supplemental Figure 3B-C). We calculated that infusion of 1 mg/kg 3G3 (25 µg/mL;167 nM theoretical peak plasma concentration) provided an approximate fourfold excess of 3G3 over circulating FXI levels during the 7-day course of the experiment (supplemental Figure 3D). Thus, similar to another inhibitory anti-FXI antibody developed by us,33 3G3 mediates its effect through sustained presence in the circulation and its interaction with FXI, not by clearance of the zymogen from the circulation.
Pretreatment of baboons with 3G3 reduced fibrinogen and HK consumption and reduced markers of fibrin formation and fibrinolysis, as well as HK cleavage. There was no evidence of microthrombi in the lungs and kidneys of 3G3-treated animals when evaluated after euthanasia at day 7. These findings strongly indicate that FXI, and perhaps contact activation, contribute to consumptive coagulopathy/DIC in this model.
Effect of 3G3 on the blood cell and microenvironment response to S aureus
Fluid loss from capillary leakage was detected by increases in RBC count, hematocrit, and hemoglobin, reflecting hemoconcentration in untreated baboons (Figure 4; supplemental Figure 4). Infusion of S aureus induced a drop in circulating platelet and lymphocyte count, as well as an initial drop in neutrophil count, with circulating neutrophil, monocyte, and basophil levels subsequently increasing before the 24-hour postinfusion time point (Figure 4A-F). When blood cell content was examined in the lung and kidney, it was evident that infusion of S aureus induced massive accumulation and activation of neutrophils, as assessed by immunofluorescence staining for neutrophil elastase and citrullinated histone H4, a marker of neutrophil extracellular traps (Figure 4G-L). Pretreatment of baboons with 3G3 prevented the rise in RBC and hematocrit, without visible hemolysis, while allowing for recovery of circulating platelets and leukocytes. There was no residual evidence of accumulation or activation of neutrophils to form neutrophil extracellular traps in the kidneys and lungs when assessed at day 7 in surviving 3G3-treated animals. Similarly, the ability of S aureus to induce release of myeloperoxidase from neutrophils into the plasma was inhibited by 3G3 treatment (Figure 4M).
Effect of 3G3 on organ failure in response to S aureus
S aureus infusion induced organ dysfunction and, consequentially, multiple organ failure in untreated baboons. An early sign was hyperdynamic shock, characterized by rapid heart rate and drop in blood pressure, likely from low vascular resistance in untreated baboons (supplemental Figure 5A-B). These animals developed acute respiratory distress, indicated by increased respiration rate (supplemental Figure 5C), a drop in blood oxygen saturation, and increased body temperature (supplemental Figure 5D-E). Baboons pretreated with 3G3 exhibited less respiratory problems, with lower respiration rates after 3 to 8 hours (supplemental Figure 5C), better peripheral perfusion and oxygenation as shown by pulse oximetry (supplemental Figure 5D), and reduced fever after 6 hours (supplemental Figure 5E).
S aureus induced liver and pancreas damage in untreated animals as indicated by elevated plasma transaminases alanine aminotransferase, aspartate aminotransferase, and pancreatic amylase (supplemental Figure 6A-C). Hyperglycemia was observed at 2 hours, followed by hypoglycemia at 4 to 8 hours (supplemental Figure 6D). Moreover, S aureus induced kidney dysfunction as measured by elevated plasma creatinine and blood nitrogen urea and increases in potassium and phosphate ions (supplemental Figure 6E-H). Pretreatment with 3G3 prevented liver and pancreas damage, hypoglycemia, and uremia (supplemental Figure 6).
Markers of cell death including circulating nucleosomes (Figure 5A) and lactate dehydrogenase (Figure 5B) increased over the 24 hours following infusion of S aureus; pretreatment with 3G3 delayed the increase and reduced their levels, suggesting that inhibition of FXI protected against S aureus–induced cytolysis. FXI inhibition also decreased complement activation induced by S aureus, as shown by significantly lower amounts of circulating C3b (Figure 5C) and C5b-9 soluble terminal complement complex (Figure 5D) detected in 3G3-treated animals compared with the nontreated cohort. Immunofluorescence staining of kidney cryosections for C5b-9 showed less staining in the treated vs nontreated animals challenged with S aureus (Figure 5E).
Figure 5.

Pretreatment with 3G3 antibody prevented tissue damage and complement activation following a lethal dose of heat-inactivated S aureus injection in baboons. Time course changes of plasma levels of cell death markers, nucleosomes (A) and lactate dehydrogenase (B), and of complement activation markers C3b (C) and soluble C5b-9 (D). Data are presented as mean ± SEM. Same time point data are compared between untreated (LDSA) and 3G3-pretreated (LDSA + 3G3) animals using 2-tailed Student t test: *P < .05, **P < .01, ***P < .001. (E) Immunofluorescence staining of C5b-9 terminal complement complex (green) in the kidney of S aureus–challenged animals without (LDSA; left) or with 3G3 pretreatment (LDSA + 3G3; right). Note stronger C5b-9 staining in the T and G of nontreated vs treated baboons. Confocal images were captured on a Nikon Eclipse TE2000-U inverted microscope equipped with a Nikon C1 scanning head. Images were acquired and processed using EZ-C1 software (v 3.80; Nikon). Scale bars, 100 µm. G, glomeruli; T, tubule.
Markers of the cytokine storm associated with sepsis were observed in baboons following infusion of S aureus, including elevated plasma levels of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-8, monocyte chemoattractant protein 1, granulocyte-macrophage colony-stimulating factor, and IL-1β. Consistent with our previous observations in which FXI-deficient mice were protected from the cytokine response during sepsis, pretreatment with 3G3 reduced the extent and onset of cytokine production with the exception of the rapid transient increase in TNF-α observed at the 2-hour time point (Figure 6).
Figure 6.

Pretreatment with 3G3 antibody prevented the increase in the levels of many proinflammatory cytokines, except for TNF, following a lethal dose of heat-inactivated S aureus injection in baboons. TNF-α (A), IL-6 (B), IL-8 (C), monocyte chemoattractant protein 1 (MCP1) (D), granulocyte-macrophage colony-stimulating factor (E), and IL-1β (F). Data are presented as mean ± SEM. Same time point data are compared between LD challenge (LDSA) and challenge plus 3G3 treatment (LDSA + 3G3) groups using 2-tailed Student t test: *P < .05, **P < .01, ***P < .001.
Histopathologic analysis confirmed that infusion of heat-inactivated S aureus induced organ damage in multiple vital organs (Figure 7). At 24 hours postinfusion, lungs showed marked congestion with leukocyte infiltration in the interalveolar wall and intravascular leukocyte aggregation with almost complete occlusion observed in some medium-size vessels. Similarly, increased granulocyte accumulation was observed within the perisinusoidal space of the liver accompanied by mild vacuolation of the hepatocytes. The kidneys showed extensive tubular necrosis and apoptosis, characterized by pyknotic nuclei and karyolysis, both indicators of acute renal failure. The spleen displayed marked congestion and granulocyte infiltration within the red pulp and lymphocyte apoptosis in the central follicular areas of the white pulp. Pretreatment with 3G3 preserved organ integrity, with all organs showing essentially normal appearance when examined at day 7 (Figure 7).
Figure 7.

Histopathology of vital organs of euthanized baboons following LD of heat-inactivated S aureus challenge with or without 3G3 pretreatment. Lung (A), liver (B), kidney (C), and spleen (D) were collected at the time of necropsy, embedded in paraffin, and stained with hematoxylin and eosin. Images were acquired on an upright Nikon Eclipse E800M microscope equipped with an Omax high-speed digital camera using the Omax ToupView software. For each organ and experimental condition, representative images showing both low and high magnification insets are shown. Scale bars, 100 µm. av, lung alveoli; F, spleen follicle; G, kidney glomeruli; S, liver sinusoidal capillaries; T, renal tubuli. Black arrows, neutrophils infiltration in the subsinusoidal space; green arrows, tubular necrosis; red arrows, lymphoid apoptosis. *Intravascular leukocyte aggregation.
Discussion
Despite advances in mechanical support and medical management, mortality remains significant in sepsis-associated systemic inflammation, DIC, consumptive coagulopathy, and organ failure, with rates approaching 30%.34 Organ failure remains the primary driver of morbidity, initiated by direct bacterial invasion into organs, systemic exposure to pathogen-derived molecules, cell death from pathogen exposure or proliferation, hypotension provoking diminished tissue perfusion,25 microvascular thrombosis, and systemic inflammation provoking endothelial barrier dysfunction.35 Severe sepsis is often associated with DIC, which can lead to a consumptive coagulopathy with depletion of platelets and fibrinogen, and enhanced fibrino(geno)lysis. These events result in a significant risk for both bleeding and thrombosis, which renders use of traditional anticoagulants unsafe.36 The contact system interacts with several other host defense systems,2 including pathways for bradykinin/angiotensin generation, coagulation, fibrinolysis, and complement that have all been implicated in the pathogenesis of sepsis. This suggests that selective targeting and inhibition of contact activation could potentially limit the activity of some or all of these sepsis-associated processes, blunting the pathologic host response that leads to inflammation and DIC after exposure to certain pathogens.
Currently available systemic anticoagulants target the common pathway of thrombin generation. Agents such as heparin37 have been used in septic patients in an attempt to interrupt the processes that lead to DIC. This approach works in some cases; however, it increases the risk of bleeding and is generally avoided. FXIa is a plasma protease that serves a more limited role in hemostasis.38 During hemostasis, it is thought that thrombin-mediated activation of FXI is required in some situations to control trauma-induced bleeding in certain vascular beds.4 FXI can also be activated during contact activation by FXIIa; however, this reaction does not appear to be required for hemostasis. Therefore, targeting FXI activation by FXIIa, without inhibiting FXI activation by thrombin or the ability of FXIa to activate FIX, may not compromise hemostasis to the same degree as heparin and other anticoagulants.39 Previously, we described a murine monoclonal antibody, 14E11, which exhibits this type of inhibition.27 We used a humanized antibody (3G3) that has the following characteristics in common with 14E1127: (1) binds to the A2 domain of FXI and inhibits FXI activation by FXIIa; (2) reduces the ability of FXIa to activate FXII; (3) interferes with FXI autoactivation in the presence of polyanions; (4) does not affect FXI activation by thrombin or the ability of FXIa to activate FIX; and (5) prolongs plasma aPTT by about twofold when used at and above saturating concentrations. The findings presented in this report indicate that 3G3 attenuates several detrimental processes that develop during sepsis, including DIC and thrombus formation.
FXI deficiency is beneficial in select murine models of sepsis, including Listeria monocytogenes11 and polymicrobial8 sepsis, and some bacterial components such as polyP40 and peptidoglycan41 induce FXI activation in vivo. Activation of FXI during infection has mechanistically been shown to induce DIC as a consequence of thrombin generation, fibrinogen consumption, and platelet activation. The mechanism by which FXI is activated during sepsis is not established, but hypothetically could involve thrombin-mediated feedback activation42 (as in hemostasis), FXIIa (contact activation), and/or unknown mechanisms. Our observation that 3G3 abrogates activation of coagulation proteases “downstream” of FXIa (FIX, FXI, prothrombin), while dampening fibrinogen consumption and microthrombi formation, argues for a role for contact activation (FXIIa) in the process. However, counterintuitively, this anti-FXI antibody also blunted activation of the contact system proteases FXII and PK, 2 enzymes upstream of FXI in classic coagulation models. This suggests that FXI may be affecting the intrinsic coagulation and contact activation systems in a bidirectional manner.10,43 3G3 also suppressed activation of the fibrinolytic system, blunted activation of complement proteins C3 and C5, and prevented increases in the inflammatory cytokines IL-6, IL-8, and029983IL-1β. These observations lend credence to the concept that FXIa contributes to several processes in sepsis, and not just promotion of thrombin generation through activation of FIX. Previously, we reported evidence that FXIa, like its homolog kallikrein, activates FXII.10 Because direct or indirect links between the intrinsic/contact activation and inflammation,44,45 complement,46 or fibrinolysis47 have been documented, reduced activation of both FXI and FXII could explain the effect of 3G3 on fibrinolysis, complement activation, and kinin formation in our sepsis model, all of which are influenced by FXIIa and contact activation.
Leukocyte activation and trafficking are hallmarks of inflammation. We observed a marked increase in leukocyte (predominantly neutrophil) migration at sites of inflammation in the lung and kidney following exposure to S aureus. This was paralleled by increases in markers of leukocyte activation and granule secretion in the circulation and formation of neutrophil extracellular traps in the tissue space, as measured by MPO48and citrullinated H4, respectively. We previously showed that inhibition of complement C531 or administration of APC49 attenuates MPO release in response to a lethal dose of E coli in baboons. Discrepant responses have been reported in mouse models, with FXI deficiency impairing leukocyte infiltration in a model of polymicrobial sepsis,9,11 while increasing neutrophil influx in response to a pneumonia challenge.21 We have shown that activated FXI inhibits neutrophil migration and chemotaxis in a purified system in vitro,50 whereas neutrophil-derived FXIIa promotes neutrophil activation and NETosis in sterile inflammation. In a mouse model of Yersinia pestis sepsis, the ability of neutrophils to combat plague is dependent on fibrin generation,51 demonstrating a link between fibrin formation and the innate immune response against this particular pathogen. FXI had no demonstrable role with this organism. Similarly, in mouse models of lethal Y enterocolitica12 or postinfluenza streptococcal infection, FXI was dispensable regarding survival or antibacterial host defense. Although FXI deficiency was detrimental in a murine pneumonia model,20 this effect was not observed in FXI-deficient humans.23 These differences highlight distinct host responses to inflammation in mice vs humans and emphasize the need for models that more closely reflect human responses to bacteria.48
Our study points to a potential pathogenic role of FXI that contributes to the early development of an ultimately lethal systemic inflammatory response syndrome following systemic exposure to inactivated S aureus bacteria that may be independent of its role in FIX activation. We have shown that infusion of comparable amounts of live or heat-inactivated S aureus in baboons can induce inflammation and coagulopathic responses52 to a similar extent, and can be equally lethal.30 This is consistent with the premise that killing pathogens with antibiotics will not necessarily prevent development of inflammation, DIC, and septic shock. We propose that our data warrant further exploration in preclinical and clinical studies of early combined treatment of various septic conditions with a combination of antibiotics and 3G3. Moreover, the role of contact activation in the development of sepsis should be further investigated experimentally by therapeutic inhibition of other contact factors, including FXIIa.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Stephanie Reitsma and Annachiara Mitrugno (Oregon Health and Science University) for helpful discussions, and Jan Roodt and Seb Lamprecht (University of the Free State) for technical support with animal experimentation.
This work was supported by grants from the National Institutes of Health, National Institute of General Medical Sciences (GM116184 [O.J.T.M. and F.L.] and GM121601 and P30GM114731 [F.L.]); National Heart, Lung, and Blood Institute (HL101972 [O.J.T.M.], HL128016 and HL106919 [A.G., C.U.L., and E.I.T.], and HL140025 [D.G.]); National Institute of Allergy and Infectious Diseases (U19AI062629 [F.L.] and AI088937 [A.G., C.U.L., and E.I.T.]); and National Institute of Neurological Disorders and Stroke (NS077600) (A.G., C.U.L., and E.I.T.).
Footnotes
Presented in abstract form at the meeting of the International Society on Thrombosis and Hemostasis (ISTH2017), Berlin, Germany, 8-13 July 2017.
Authorship
Contribution: R.S., R.S.K., W.J.V.R., and F.L. performed animal experiments; R.S., R.S.K., H.C., G.R., A.S., and C.L. performed assays; R.S., R.S.K., C.L., H.C., and F.L. analyzed data; A.G., E.I.T., and C.U.L. conceived, developed, prepared, and provided the AB023 (3G3) antibody; C.P., J.J.S., C.L., R.S., and R.S.K. contributed to writing of various sections; A.G., D.G., C.P., O.J.T.M., and F.L. designed the study; F.L. and O.J.T.M. supervised the study and wrote the manuscript; A.G. and D.G. provided critical revision of the manuscript; and all authors read and approved the manuscript.
Conflict-of-interest disclosure: A.G., E.I.T., C.U.L., D.G., Aronora, and Oregon Health Sciences University may have a financial interest in the results of this study. The remaining authors declare no competing financial interests.
Correspondence: Florea Lupu, Cardiovascular Biology Research Program, Oklahoma Medical Research Foundation, 825 NE 13th St, Oklahoma City, OK 73104; e-mail: florea-lupu@omrf.org.
References
- 1.Pixley RA, De La Cadena R, Page JD, et al. The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia. In vivo use of a monoclonal anti-factor XII antibody to block contact activation in baboons. J Clin Invest. 1993;91(1):61-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Y. The plasma contact system as a modulator of innate immunity. Curr Opin Hematol. 2018;25(5):389-394. [DOI] [PubMed] [Google Scholar]
- 3.Marron BM, Robinson JL, Gentry PA, Beever JE. Identification of a mutation associated with factor XI deficiency in Holstein cattle. Anim Genet. 2004;35(6):454-456. [DOI] [PubMed] [Google Scholar]
- 4.Wheeler AP, Gailani D. Why factor XI deficiency is a clinical concern. Expert Rev Hematol. 2016;9(7):629-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asakai R, Chung DW, Davie EW, Seligsohn U. Factor XI deficiency in Ashkenazi Jews in Israel. N Engl J Med. 1991;325(3):153-158. [DOI] [PubMed] [Google Scholar]
- 6.Gailani D, Bane CE, Gruber A. Factor XI and contact activation as targets for antithrombotic therapy. J Thromb Haemost. 2015;13(8):1383-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jansen PM, Pixley RA, Brouwer M, et al. Inhibition of factor XII in septic baboons attenuates the activation of complement and fibrinolytic systems and reduces the release of interleukin-6 and neutrophil elastase. Blood. 1996;87(6):2337-2344. [PubMed] [Google Scholar]
- 8.Tucker EI, Verbout NG, Leung PY, et al. Inhibition of factor XI activation attenuates inflammation and coagulopathy while improving the survival of mouse polymicrobial sepsis. Blood. 2012;119(20):4762-4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tucker EI, Gailani D, Hurst S, Cheng Q, Hanson SR, Gruber A. Survival advantage of coagulation factor XI-deficient mice during peritoneal sepsis. J Infect Dis. 2008;198(2):271-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bane CE Jr, Ivanov I, Matafonov A, et al. Factor XI deficiency alters the cytokine response and activation of contact proteases during polymicrobial sepsis in mice. PLoS One. 2016;11(4):e0152968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo D, Szaba FM, Kummer LW, et al. Factor XI-deficient mice display reduced inflammation, coagulopathy, and bacterial growth during listeriosis. Infect Immun. 2012;80(1):91-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo D, Szaba FM, Kummer LW, et al. Protective roles for fibrin, tissue factor, plasminogen activator inhibitor-1, and thrombin activatable fibrinolysis inhibitor, but not factor XI, during defense against the gram-negative bacterium Yersinia enterocolitica. J Immunol. 2011;187(4):1866-1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ranieri VM, Thompson BT, Barie PS, et al. ; PROWESS-SHOCK Study Group. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012;366(22):2055-2064. [DOI] [PubMed] [Google Scholar]
- 14.Bernard GR, Vincent JL, Laterre PF, et al. ; Recombinant human protein C Worldwide Evaluation in Severe Sepsis (PROWESS) study group. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344(10):699-709. [DOI] [PubMed] [Google Scholar]
- 15.Okamoto K, Tamura T, Sawatsubashi Y. Sepsis and disseminated intravascular coagulation. J Intensive Care. 2016;4(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohammed BM, Cheng Q, Matafonov A, Monroe DM, Meijers JCM, Gailani D. Factor XI promotes hemostasis in factor IX-deficient mice. J Thromb Haemost. 2018;16(10):2044-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Cheng Q, Xu L, et al. Effects of factor IX or factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost. 2005;3(4):695-702. [DOI] [PubMed] [Google Scholar]
- 18.Mohammed BM, Cheng Q, Matafonov A, et al. A non-circulating pool of factor XI [abstract]. Res Pract Thromb Haemost. 2017;1(suppl 1):100. Abstract OC 43.1. [Google Scholar]
- 19.Cheng Q, Zhao Y, Lawson WE, et al. The effects of intrinsic pathway protease deficiencies on plasminogen-deficient mice. Blood. 2005;106(9):3055-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stroo I, Yang J, de Boer JD, et al. Factor XI deficiency enhances the pulmonary allergic response to house dust mite in mice independent of factor XII. Am J Physiol Lung Cell Mol Physiol. 2017;312(2):L163-L171. [DOI] [PubMed] [Google Scholar]
- 21.Stroo I, Zeerleder S, Ding C, et al. Coagulation factor XI improves host defence during murine pneumonia-derived sepsis independent of factor XII activation. Thromb Haemost. 2017;117(8):1601-1614. [DOI] [PubMed] [Google Scholar]
- 22.Gailani D, Mohammed BM, Cheng Q. Factor XI and pulmonary infections. Haemophilia. 2018;24(4):519-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salomon O, Preis M, Abu Shtaya A, Kotler A, Stein N, Saliba W. Factor XI deficiency is not associated with an increased risk of pneumonia and pneumonia-related mortality. Haemophilia. 2018;24(4):634-640. [DOI] [PubMed] [Google Scholar]
- 24.Seok J, Warren HS, Cuenca AG, et al. ; Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110(9):3507-3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369(9):840-851. [DOI] [PubMed] [Google Scholar]
- 26.Ramachandran G. Gram-positive and gram-negative bacterial toxins in sepsis: a brief review. Virulence. 2014;5(1):213-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng Q, Tucker EI, Pine MS, et al. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116(19):3981-3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lorentz CU, Verbout NG, Wallisch M, et al. The contact activation inhibitor and factor XI antibody, AB023, produces safe, dose-dependent anticoagulation in a phase 1 first-in-human trial. Arterioscler Thromb Vasc Biol. 2019;ATVBAHA118312328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meliton AY, Meng F, Tian Y, et al. Oxidized phospholipids protect against lung injury and endothelial barrier dysfunction caused by heat-inactivated Staphylococcus aureus. Am J Physiol Lung Cell Mol Physiol. 2015;308(6):L550-L562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hinshaw LB, Emerson TE Jr, Taylor FB Jr, et al. Lethal Staphylococcus aureus-induced shock in primates: prevention of death with anti-TNF antibody. J Trauma. 1992;33(4):568-573. [PubMed] [Google Scholar]
- 31.Keshari RS, Silasi R, Popescu NI, et al. Inhibition of complement C5 protects against organ failure and reduces mortality in a baboon model of Escherichia coli sepsis. Proc Natl Acad Sci USA. 2017;114(31):E6390-E6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suffritti C, Zanichelli A, Maggioni L, Bonanni E, Cugno M, Cicardi M. High-molecular-weight kininogen cleavage correlates with disease states in the bradykinin-mediated angioedema due to hereditary C1-inhibitor deficiency. Clin Exp Allergy. 2014;44(12):1503-1514. [DOI] [PubMed] [Google Scholar]
- 33.Tucker EI, Marzec UM, White TC, et al. Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood. 2009;113(4):936-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar G, Kumar N, Taneja A, et al. ; Milwaukee Initiative in Critical Care Outcomes Research (MICCOR) Group of Investigators. Nationwide trends of severe sepsis in the 21st century (2000-2007). Chest. 2011;140(5):1223-1231. [DOI] [PubMed] [Google Scholar]
- 35.Goldenberg NM, Steinberg BE, Slutsky AS, Lee WL. Broken barriers: a new take on sepsis pathogenesis. Sci Transl Med. 2011;3(88):88ps25. [DOI] [PubMed] [Google Scholar]
- 36.Levi M, Scully M. How I treat disseminated intravascular coagulation. Blood. 2018;131(8):845-854. [DOI] [PubMed] [Google Scholar]
- 37.Zarychanski R, Abou-Setta AM, Kanji S, et al. ; Canadian Critical Care Trials Group. The efficacy and safety of heparin in patients with sepsis: a systematic review and metaanalysis. Crit Care Med. 2015;43(3):511-518. [DOI] [PubMed] [Google Scholar]
- 38.Gailani D, Gruber A. Factor XI as a therapeutic target. Arterioscler Thromb Vasc Biol. 2016;36(7):1316-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simmons J, Pittet J-F. The coagulopathy of acute sepsis. Curr Opin Anaesthesiol. 2015;28(2):227-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi SH, Smith SA, Morrissey JH. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood. 2011;118(26):6963-6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Popescu NI, Silasi R, Keshari RS, et al. Peptidoglycan induces disseminated intravascular coagulation in baboons through activation of both coagulation pathways. Blood. 2018;132(8):849-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gailani D, Broze GJ Jr. Factor XI activation in a revised model of blood coagulation. Science. 1991;253(5022):909-912. [DOI] [PubMed] [Google Scholar]
- 43.Griffin JH. Role of surface in surface-dependent activation of Hageman factor (blood coagulation factor XII). Proc Natl Acad Sci USA. 1978;75(4):1998-2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bender L, Weidmann H, Rose-John S, Renné T, Long AT. Factor XII-driven inflammatory reactions with implications for anaphylaxis. Front Immunol. 2017;8:1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Didiasova M, Wujak L, Schaefer L, Wygrecka M. Factor XII in coagulation, inflammation and beyond. Cell Signal. 2018;51:257-265. [DOI] [PubMed] [Google Scholar]
- 46.Ghebrehiwet B, Kaplan AP, Joseph K, Peerschke EI. The complement and contact activation systems: partnership in pathogenesis beyond angioedema. Immunol Rev. 2016;274(1):281-289. [DOI] [PubMed] [Google Scholar]
- 47.de Maat S, Bjorkqvist J, Suffritti C, et al. Plasmin is a natural trigger for bradykinin production in patients with hereditary angioedema with factor XII mutations. J Allergy Clin Immunol. 2016;138(5):1414-1423. [DOI] [PubMed] [Google Scholar]
- 48.Taylor FB Jr, Kinasewitz GT, Lupu F. Pathophysiology, staging and therapy of severe sepsis in baboon models. J Cell Mol Med. 2012;16(4):672-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Healy LD, Puy C, Fernández JA, et al. Activated protein C inhibits neutrophil extracellular trap formation in vitro and activation in vivo. J Biol Chem. 2017;292(21):8616-8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Itakura A, Verbout NG, Phillips KG, et al. Activated factor XI inhibits chemotaxis of polymorphonuclear leukocytes. J Leukoc Biol. 2011;90(5):923-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luo D, Lin JS, Parent MA, et al. Fibrin facilitates both innate and T cell-mediated defense against Yersinia pestis. J Immunol. 2013;190(8):4149-4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Boer JP, Creasey AA, Chang A, et al. Alpha-2-macroglobulin functions as an inhibitor of fibrinolytic, clotting, and neutrophilic proteinases in sepsis: studies using a baboon model. Infect Immun. 1993;61(12):5035-5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.