

Abstract
Recent insight into the mechanisms of induction of tissue-resident memory (TRM) CD8+ T cells enables the development of novel vaccine strategies against sexually-transmitted infections. In order to maximize both systemic and genital intraepithelial CD8+ T cells against vaccine antigens, we assessed combinations of intramuscular and intravaginal routes in heterologous prime-boost immunization regimens with unrelated viral vectors. Only intramuscular prime followed by intravaginal boost induced concomitant strong systemic and intraepithelial genital-resident CD8+ T cell responses. Intravaginal boost with vectors expressing vaccine antigens was far superior to intravaginal instillation of CXCR3 chemokines receptor ligands or toll-like receptor 3, 7 and 9 agonists to recruit and increase the pool cervicovaginal CD8+ TRM. Transient antigen presentation increased trafficking of cognate and bystander circulating activated, but not naïve, CD8+ T cells into the genital tract and induced in situ proliferation and differentiation of cognate CD8+ TRM. Secondary genital CD8+ TRM were induced in the absence of CD4+ T cell help and shared a similar T cell receptor repertoire with systemic CD8+ T cells. This prime-pull-amplify approach elicited systemic and genital CD8+ T cell responses against high-risk HPV type 16 E7 oncoprotein and conferred CD8-mediated protection to a vaccinia virus genital challenge. These results underscore the importance of the delivery route of non-replicating vectors in prime-boost immunization to shape the tissue distribution of CD8+ T cell responses. In this context, the importance of local antigen presentation to elicit genital CD8+ TRM provides a rationale to develop novel vaccines against sexually-transmitted infections and to treat HPV-neoplasia.
Introduction
Sexually-transmitted infections are a significant cause of mortality and morbidity worldwide(1). Few vaccines against sexually-transmitted infections have been licensed and rely on the action of neutralizing antibodies(2, 3). However, the development of a next generation of vaccines against sexually transmitted infections might require the induction cell-mediated immunity(4, 5). Vaccine-induced long-lived plasma cells can exert their function “remotely” because of the soluble nature of the antibodies they produce. In contrast, T cells require direct interaction with target cells presenting cognate peptides in the groove of MHC molecules expressed at the cell surface. Therefore, vaccines able to elicit long-lived T cell responses in the epithelial portal of entry of sexually-transmitted pathogens might confer immediate protection and enhance vaccine efficacy(6).
Tissue-resident memory CD8+ T cells(CD8+ TRM) are excluded from the blood circulation and reside in non-lymphoid tissues in contrast to conventional effector and central memory CD8+ T cells(7). CD8+ TRM play a key role in immunity against viral pathogens at epithelial surfaces where they can exert immediate effector T cell functions and induce a broad innate antiviral response in surrounding tissue(8-11). Marker of TRM varies with tissue localization(12), however the expression of the activation marker CD69 defines most skin CD8 TRM which exhibit a unique transcriptional program compared to other memory populations(13). Also, to a large extent, the expression of the integrin CD103 defines intraepithelial CD8+ TRM and is involved in the long-term retention of CD8+ TRM in pluristratified epithelia(14, 15).
Recent insight into the mechanisms of induction of CD8+ TRM provide a strong rationale to evaluate novel vaccine strategies against infectious diseases. Topical delivery of host- and pathogen-derived immunomodulatory molecules (e.g. chemokines and Toll-like receptor ligands) have been shown to promote the recruitment of circulating effector and memory T cells into non-lymphoid tissues(16, 17). Such approaches referred to as “prime-and-pull” were shown to confer protection against mucosal or cutaneous viral infections as well as carcinoma(16, 18–21). However, these approaches have not been compared with topical vaccination involving local expression of antigen. Yet a recent study demonstrated that cognate antigen expression is critical to the establishment of a pool of long-lived virus-specific CD8+ TRM upon resolution of skin viral infection(22, 23).
Prime-boost immunization with viral vectors typically requires a heterologous vector vaccination series to overcome the anti-vector neutralizing antibodies induced by the priming dose and to focus the immune response from the boost toward vaccine antigens(24). We have previously shown that adenoviral vectors administered systemically (e.g. intramuscularly) elicit low CD8+ TRM responses in the female reproductive tract despite high systemic memory CD8+ T cell responses. In contrast, non-replicating human papillomavirus (HPV) pseudovirus (PsV) given intravaginally (Ivag) were poor at priming CD8+ T cell responses in the circulation but remarkably recalled high numbers of intraepithelial CD8+ TRM cells to the cervicovaginal mucosa(25). More recently, we have shown that Ivag immunization with adenoviral type 26 and 35 expressing a fusion of high-risk HPV type 16 E6 and E7 oncoproteins preferentially induced genital CD8+ TRM(26) and that heterologous IM/Ivag immunization with these adenovirus (Ad) vectors further induced of both systemic CD8+ and genital mucosal CD8+ TRM responses(26).
The goals of this study were to maximize both circulating and genital intraepithelial CD8+ T cells by vaccination, to characterize the responses and to provide mechanistic insights into their induction. We herein have assessed in mice a combination of intramuscular (IM) and intravaginal routes using Ad vectors and HPV PsV, respectively. We evaluated the consequences of the order of route usage on induction of cervicovaginal and systemic CD8+ T cell responses. We compared local antigen expression to local inflammation (e.g. tissue damage or immunomodulatory molecules) in the induction of cervicovaginal CD8+ TRM. A series of experiments to evaluate the phenotype, functionality and mechanism of induction of the intraepithelial CD8+ TRM generated by our optimized vaccination protocol was then performed. Ex vivo presentation assays and T-cell receptor (TCR) transgenic adoptive transfer experiments were conducted to assess the contribution of recruitment and in situ proliferation to the amplification of CD8+ TRM after Ivag booster immunization. We further evaluated the role of CD4+ T cell help in the induction of secondary cervicovaginal CD8+ TRM and compared the TCR-β repertoire between genital and systemic CD8+ T cells. A genital vaccinia virus challenge model was used to evaluate protection from viral infection by our optimized protocol. Finally, we generated HPV PsV expressing a fusion of E6 and E7 oncoproteins derived from HPV16 as a prototype HPV therapeutic vaccine and evaluated the cytokine production by E7-specific cervicovaginal CD8+ TRM.
Material and Methods
Plasmids, viral vectors and vaccinia virus
Target plasmids expressing model antigen derived from respiratory syncytial virus (RSV) M and M2-1 proteins (MM2), firefly luciferase, alkaline phosphatase, chicken egg ovalbumin (OVA) or a fusion protein derived from mutated HPV16 E6 and E7 oncoproteins(27), and packaging plasmids expressing HPV L1 and L2 capsid proteins are described on the Laboratory of Cellular Oncology website (http://home.ccr.cancer.gov/Lco/plasmids.asp).
Non-replicating HPV PsV were generated as previously described(25). Briefly, 293TT cells were co-transfected with both a target plasmid encoding the gene of interest and a packaging plasmid encoding the L1 and L2 capsid proteins. Cell lysates containing HPV PsV were incubated overnight at 37°C and purified by ultracentrifugation on an Optiprep gradient, fractions containing packaged DNA were pooled. Infectious titer expressed in infectious units (IU) was determined on 293TT cells. Ad5 vector encoding the MM2 fusion protein of RSV was generated and produced by GenVec, infectious titer was determined by plaque assay and expressed as plaque forming units (PFU). Vaccinia virus (WR strain) expressing the minimal RSV epitope M282-90 (VV-M2) was propagated in Hela S3 and titer expressed in PFU was determined on BSC-1 cells as previously described(25).
Mice, immunization and vaccinia virus genital challenge
Mice were housed and bred under specific pathogen-free conditions at the animal care facilities of the National Cancer Institute. Female BALB/c, C57BL/6, C57BL/6 congenic strain carrying CD45.1, OT-I and Pmel-1 were purchased from the Jackson Laboratory(28, 29). OT-I and Pmel-1 carrying CD45.1 were obtained by crossing with C57BL/6.CD45.1. All mice were immunized between 8 and 12 weeks of age. The NCI Animal Care and Use Committee approved all animal protocols used in this study.
All mice were injected subcutaneously prior to immunization with 3mg medroxyprogesterone acetate. Five hours prior to Ivag immunization, mice were treated Ivag with 50μl of 4% nonoxynol-9 (Spectrum Chemicals) gel to expose the cervicovaginal basement membrane. Mice were immunized with 5×107, 1×108 or 4×108 IU HPV diluted in 2% carboxymethyl cellulose (CMC) gel instilled in the vaginal vault. In some experiments, mice were treated Ivag with 2% CMC containing CXCL9/CXCL10 (Preprotech), M282-90 peptide (Genscript), Polyinosinic-polycytidylic acid (Poly I:C) and CpG oligodeoxynucleotide (Invivogen), or IFN-γ (Preprotech). For IM immunization, 5×106 or 5×107 plaque-forming units (PFU) adenoviral vectors were diluted in 50μl of PBS and injected in the quadriceps muscle.
For genital vaccinia challenge, 1×107 PFU of recombinant VV-M2 was diluted in 10μl of 2% CMC and instilled in the vaginal vault. Cervicovaginal tissue homogenates were obtained 3 days after challenge and vaccinia titer was determined by plaque assay.
In vitro HPV neutralization assay
In vitro HPV neutralization has been described previously(30). Briefly, serial dilutions of serum samples from individual mice were mixed with PsV expressing a secreted alkaline phosphatase and added to 293TT cells. Neutralizing titers were calculated using Prism software and expressed as the 50% effective concentration (EC50).
Preparation of cell suspensions
Cervicovaginal mucosa, spleen and lymph nodes (LN) were collected and finely minced using dissection scissors. The minced tissues were placed in RPMI containing 2% fetal bovine serum (SIGMA), 0.5mg/ml Collagenase A or D (Roche) and 0.1mg/ml DNase1 (Roche). All samples were placed at 37°C in a shaker at 250rpm and incubated for 1hr (cervicovaginal mucosa) and 15min (LN and spleen) and filtered through 70μm mesh. To remove erythrocytes, single cell suspensions and EDTA-treated blood were incubated in an Ammonium-Chloride-Potassium buffer (Life Technologies).
Antibody-mediated T cell depletion
Mice were depleted of CD4+ T cells or CD8+ T cells by i.p. injection of 200μg CD4 (GK1.5) or 100μg CD8 (YTS 169.4) antibodies (BioXcell) on days −3/−1/2 and 4 with respect to day 0 of immunization or VV-M2 challenge. Control mice were injected with isotype control antibodies.
Tetramer staining and phenotyping of antigen-specific CD8+ T cells
FACS staining was performed in 96-well plates. After FcR-blocking with CD16/32 antibody (24G2, BioXcell) cells were incubated for 30min at 4°C with H-2Kd/M282-90 or H-2Db/E749-57 tetramers (NIH Tetramer facility) with the following antibodies: CD8-Pacific Orange (5H10, Life Technologies), CD3-Pacific Blue (17A2, BioLegend), CD4-APC/Cy7 (RM4-5, BioLegend), CD127-PE (SB/199, BioLegend), CD62L-FITC (MEL-14, BioLegend), CD103-PERCP/Cy5.5 (1E7, BioLegend) and CD69-PE/Cy7 (H1.2F3, BioLegend) or CXCR3-PE/Cy7 (CXCR3-173, BioLegend).
In vitro peptide stimulation and intracellular cytokine staining
Single cell suspensions were incubated for 5 hours at 37°C in RPMI medium only or supplemented E749-57 peptide (5μg/ml; GenScript). Cells were washed and surface-stained with CD8-PE (53-6.7), CD3-PE/Cy7 (145-2C11) and CD4-APC/Cy7 (RM4-5) antibodies (Biolegend). Dead cells were stained with Live/Dead yellow dye (Life Technologies) and intracellular staining was performed for 30min at 4C with IFN-γ-FITC (XMG1.2), TNF-α-APC (MP6-XT22) and IL-2-PERCP/Cy5.5 (JES6-1A12) antibodies (Biolegend). Data acquisition was performed on a FACS Canto II flow cytometer (BD Biosciences) and analyzed using Flowjo v10 (TreeStar).
Preparation of naïve and in vitro activated TCR transgenic CD8+ T cells
Naïve OT-I CD8+ T cells (>95% purity) were obtained from LN cell suspensions by depletion of CD19+, CD4+, CD16/32+, and GR1+ cells using corresponding purified antibodies (BioXcell) and sheep anti-rat IgG magnetic beads (Dynabeads) following manufacturer’s instruction. Activated OT-I and Pmel-1 TCR transgenic CD8+ T cells were expanded in vitro from spleen cell suspensions. Spleen cells were incubated for three days at 37°C in RPMI medium containing recombinant human IL-2 (100IU/ml, TECIN) and OVA254-262 (20ng/ml, Genscript) or gp10025-33 peptide (100ng/ml, Genscript). Cells were washed and incubated for two more days in RPMI medium containing IL-2 (100IU/ml). OT-I and Pmel-1 in vitro expansion typically yielded >95% purity. Activated TCR transgenic CD8+ T cells were labeled with CellTrace Violet (Life Technologies) and transferred (5×106 cells) i.v. into naïve C57BL/6 mice.
For in vivo proliferation assay, cells were stained with APC-conjugated H-2Kb/OVA257-264 tetramers (NIH Tetramer facility) and 24G2 antibody for 20min at 4°C. Then stained with CD8-PE (53-6.7), CD45.1-PE.Cy7 (A20), CD4-APC/Cy7 (RM4-5), CD103-FITC (1E7) antibodies (Biolegend) for 20min at 4°C.
Immunofluorescence staining and microscopy.
Immunofluorescence analysis of cervicovaginal tissue was performed following the procedure previously described(25). Briefly, ethanol-fixed six-micrometer thick tissue cryosections were stained with purified rabbit polyclonal Ki-67 (Bethyl Laboratory) or Armenian hamster CD103 (2E7, Biolegend) antibodies, followed by a second-step incubation with donkey anti-rabbit IgG-Alexa Fluor 594 or anti-Armenian hamster IgG-Alexa Fluor 594 (BioLegend), respectively, and CD8-Alexa Fluor 488 (53-6.7, Biolegend). Confocal images were acquired on a Zeiss LSM 780 Confocal system at the Confocal Microscopy Core Facility, Center for Cancer Research, National Cancer Institute, NIH.
Statistical analysis
Mann-Whitney U test was used to compare the mean between groups in experiments with two independent groups. For experiments with more than two groups, one-way or two-way analysis of variance (ANOVA) and post-hoc analysis.
Results
Intramuscular priming followed by intravaginal booster immunization maximizes systemic and genital tract memory CD8+ T cell responses
The acquisition of a mucosal-homing phenotype in the draining lymph nodes (dLN) has been proposed as a mechanism of compartmentalization of mucosal immune responses(31). In this line, intravaginal prime-boost immunization (Ivag/Ivag) with HPV PsV preferentially induces intraepithelial cervicovaginal CD8+ T cell responses but poor systemic responses. In contrast, intramuscular prime-boost (IM/IM) Ad vector preferentially induces systemic CD8+ T cell responses but poor cervicovaginal responses(25). Here, we sought to determine in a heterologous prime-boost regimen that combines IM and Ivag routes using distinct vectors, which sequence of immunization Ivag/IM (mucosal priming) or IM/Ivag (mucosal boost) would result in maximal systemic and cervicovaginal CD8+ T cell responses. For Ivag/Ivag prime-boost immunization, we used HPV PsVs (types 16 and 45) which are equally efficient at transducing basal cervicovaginal keratinocytes. For IM and Ivag combined heterologous prime-boost immunization, we used Ad5 vectors and HPV16, respectively. All experimental vaccines expressed the model antigen MM2 derived from RSV. IM (Ad5) priming followed by Ivag (HPV16) booster immunization (IM/Ivag) elicited higher percentage and 10-fold higher numbers of cervicovaginal M2-specific CD8+ T cells than Ivag (HPV16)/Ivag (HPV45) or Ivag (HPV16)/IM (Ad5) (Fig. 1A, 1B and Supplemental Fig. 1A). This suggests that IM-primed systemic CD8+ T cell responses are redirected to the cervicovaginal mucosa upon Ivag mucosal booster immunization. All groups immunized at least once by the IM route showed a higher frequency and number of spleen M2-specific CD8+ T cells compared to Ivag/Ivag (Fig. 1A, 1B and Supplemental Fig. 1A). In the genital dLN, IM/Ivag induced higher number and frequency of M2-specific CD8+ T cells compared to Ivag/IM and IM/IM suggesting that IM primed systemic CD8+ T cell responses are recalled in the genital dLN (Fig. 1A, 1B and Supplemental Fig. 1A). Of all groups, the IM/IM sequence induced the highest spleen M2-specific CD8+ T cell responses albeit the lowest number of cervicovaginal M2-specific CD8+ T cell (Fig. 1 and Supplemental Fig. 1).
FIGURE 1.
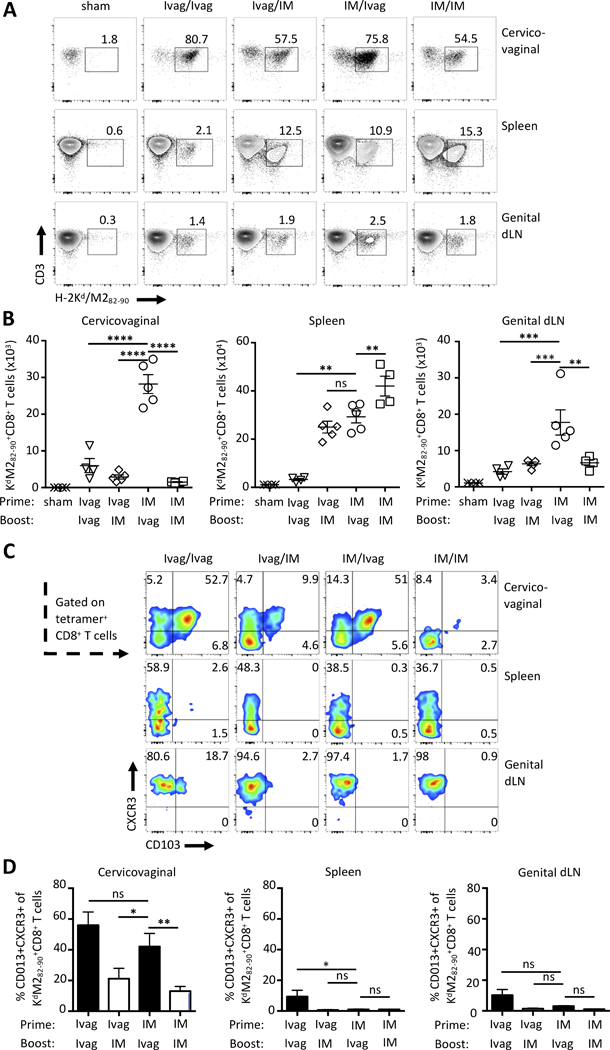
The order of route of immunization shapes the tissue localization of CD8+ T cell responses. BALB/c mice were prime-boost immunized one month apart via Ivag or IM routes with HPV16 (5×107 IU), and Ad5 (5×107 PFU) vectors expressing the RSV fusion protein MM2, respectively. For Ivag/Ivag immunization, mice were primed Ivag with HPV45 (5×107 IU) and boosted Ivag with HPV16 (5×107 IU). Two weeks after the final immunization, cell suspensions from cervicovaginal tissues, spleen and genital dLN were analyzed by FACS for the presence of H-2Kd/M282–90 tetramer+CD8+ T cells and expression of CD103 and CXCR3. (A) Representative FACS plot of H-2Kd/M282–90 tetramer staining of CD8+ T cells. (B) Symbol represent individual mice, bars represent mean total numbers +/- SEM of M2-specific CD8+ T cells. (C) Representative FACS plot of expression of CD103 and CXCR3 by M2-specific CD8+ T cells in the indicated tissues. (D) Histogram bars represent mean percentage +/- SEM of CXCR3+CD103+ M2-specific CD8+ T cells. Data are representative of three independent experiments (n=4–5 mice per group). P values (*≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001) were determined by one-way ANOVA with post-hoc Tukey analysis.
We further characterized M2-specific CD8+ T cell responses in secondary lymphoid organs and in the cervicovaginal mucosa. Specifically, we assessed the expression of the integrin CD103, a marker of intraepithelial lymphocytes, and CXCR3, the receptor of CXCL9 and 10, involved in lymphocyte migration to inflamed tissues(32). CD103+CXCR3+ M2-specific CD8+ T cells were greatly enriched in the cervicovaginal mucosa compared to spleen and genital dLN (Fig. 1C, 1D). In the genital mucosa, IM/Ivag induced 2-fold higher frequency of CD103+CXCR3+ M2-specific CD8+ T cells compared to Ivag/IM and IM/IM but similar to Ivag/Ivag (Fig. 1C, 1D). Interestingly in the genital dLN, most M2-specific CD8+ T cells expressed CXCR3, but not CD103. In contrast, in the spleen, a smaller fraction of M2-specific CD8+ T cells expressed CXCR3 (Fig. 1C). Central memory CD62L+CD127+ CD8+ T cells were enriched in the genital dLN compared to the spleen and absent in the genital mucosa (Supplemental Fig. 1B). Of note, after IM/Ivag immunization, CD8+ T cells in the genital tract mucosa expressed a higher level of CD127+ compared to other groups which suggests an increased responsiveness to the homeostatic cytokine IL-7 (Supplemental Fig. 1B). These results indicate that IM-primed systemic CD8+ T cell responses can further differentiate into cervicovaginal CD103+CD8+ TRM after an Ivag booster immunization.
Transient antigen expression by cervicovaginal keratinocytes redirect systemic memory CD8+ T cells to the cervicovaginal mucosa
Trafficking of circulating CD8+ T cells to inflamed epithelia has been demonstrated and the role of the CXCR3-CXCL9/10 axis in recruitment has been noted in the context of skin viral infection(33). HPV PsV Ivag transduction requires exposure of the cervicovaginal basement membrane with the spermicide nonoxynol-9 (N-9), which causes transient inflammation(21, 34). We assessed whether local antigen expression and/or local inflammation caused by the Ivag procedure contributed to the induction of cervicovaginal CD8+ TRM. Mice were primed IM with Ad5-MM2 and one month later boosted with Ad5-MM2 IM or N-9/HPV16-MM2 Ivag, or treated Ivag with N-9 only or N-9/HPV16-control vector (HPV-ctrl), or sham-treated. M2-specific CD8+ T cell responses were assessed one month after the boost. HPV16-MM2 Ivag booster immunization induced 20-fold and 2-fold higher numbers of cervicovaginal and blood M2-specific CD8+ T cells, respectively, compared to treatments with N-9 alone or HPV16-control vector, respectively (Fig. 2A, 2B). HPV16-MM2 Ivag booster immunization induced a 3- to 5-fold increase in the percentage of CXCR3+CD103+ M2-specific CD8+ T cells in the cervicovaginal mucosa compared to Ivag treatment with N-9 alone or HPV16-ctrl vector, respectively. Notably, we did not detect CXCR3+CD103+ M2-specific CD8+ T cells in blood (Fig. 2C).
FIGURE 2.
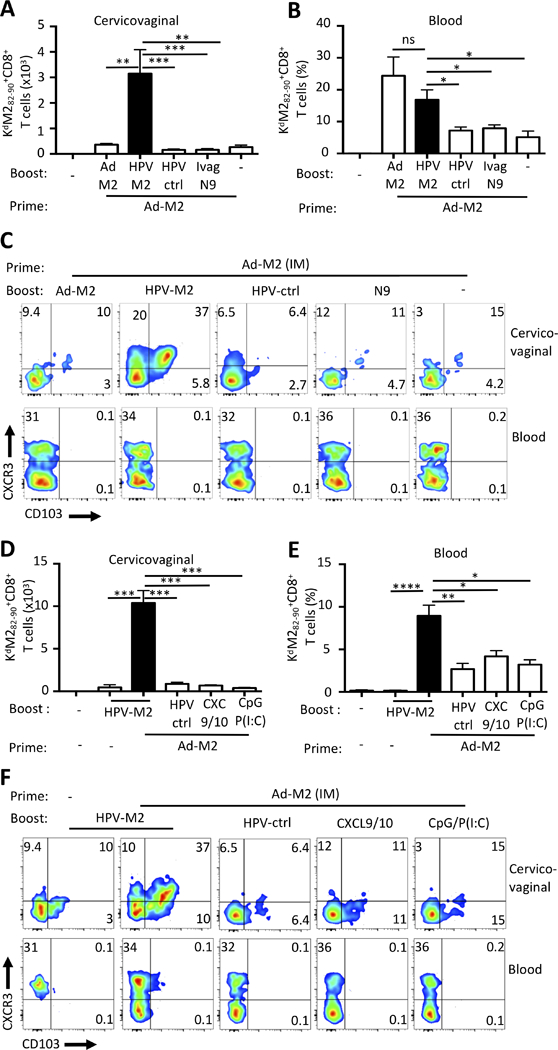
Local antigen expression, but not inflammation, supports the expansion and differentiation of genital CD8+ TRM. BALB/c mice were primed IM with Ad5-MM2 vector and one month later were boosted with Ad5-MM2 IM, HPV16-MM2 Ivag (HPV-M2), HPV16-control (HPV-ctrl), or were treated with N-9, a mixture of CXL9 and CXL10 (CXC9/10), a mixture of CpG and poly(I:C) (CpG P(I:C)) or carboxymethyl cellulose (sham). (A-C) One month or (D-F) one week after the final immunization, cell suspensions were analyzed in by FACS for the presence of H-2Kd/M282–90 tetramer+CD8+ T lymphocytes in cervicovaginal (A, D) and blood (B, E) tissues and (C, F) expression of CD103 and CXCR3 by M2-specific CD8+ T cells was assessed. (A, B, D, E) Histogram bars represent the mean total number or percent of H-2Kd/M282–90 tetramer+CD8+ T cells +/- SEM. (C, F) Representative FACS plot of CD103 and CXCR3 expression by Kd/M282–90 tetramer+CD8+ T cells. Data are representative of two independent experiments (n=4–5 mice per group). P values (*P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001) were determined by one-way ANOVA with post hoc Tukey analysis.
Topical instillation of immunologically active molecules has been used to increase trafficking of circulating CD8+ T cells to peripheral tissues, a strategy referred to as the “prime-and-pull” approach(16, 18). However, the potential of this approach to the induction of bona fide intraepithelial CD103+CD8+ T cells remains to be demonstrated. In Ad5-MM2 IM primed mice, we compared HPV16-MM2 Ivag booster immunization to topical instillation of either chemokine receptor ligands (CXCL9 and CXCL10) or a mixture of TLR3 and 9 agonists (Poly(I:C) and CpG) for their ability to induce cervicovaginal CD8+ TRM responses. The number of cervicovaginal M2-specific CD8+ T cells was 10-fold higher after HPV16-MM2 Ivag booster immunization compared to the instillation of chemokines or TLR agonists (Fig. 2D). The number of blood circulating M2-specific CD8+ T cells was increased by 3-fold after Ivag booster compared to local inflammation (Fig. 2E). Notably, topical chemokine or TLR agonist treatment did not increase the percentage of tetramer-specific CXCR3+CD103+ cells in the cervicovaginal mucosa compared to HPV16-ctrl (Fig. 2F). Therefore, in these experimental settings the recruitment of circulating memory CD8+ T cells triggered by local inflammation does not increase significantly the number of antigen-specific intraepithelial CD8+ TRM.
Induction of cervicovaginal resident memory CD8+ T cells does not require CD4+ T cell help during Ivag booster immunization
The role of CD4+ T cell help in the induction of memory CD8+ T cell responses upon infection or vaccination is well established. However, whether helper CD4+ T cell participate in the induction of secondary CD8+ T cells, particularly secondary CD8 TRM, remains to be established. We assessed the CD4+ T cell help in the induction of secondary CD8+ TRM after Ivag booster immunization. Mice were primed IM with Ad5-MM2 and later treated with either the GK1.5 antibody to deplete CD4+ cells or an isotype control antibody at the time of the HPV16-MM2 Ivag booster immunization (Fig. 3A). CD4-depletion resulted in 95% reduction in CD4+ T cells which was sufficient to abrogate the induction of neutralizing antibodies against the capsid of the HPV16 vectors (Supplemental Fig. 2A, 2B). Ivag booster immunization in CD4-depleted mice or isotype control treated mice induced similar cervicovaginal M2-specific CD8+ T cell responses whereas spleen M2-specific CD8+ T cell responses were reduced in CD4-depleted mice (Fig. 3B-3D). IM/Ivag induced 3-fold higher M2-specific CD8+ T cell number compared to single Ivag immunization (Fig. 3C). Interestingly, recall of circulating M2-specific CD8+ T cells was abrogated in the blood of CD4-depleted mice (Fig. 3E). Cervicovaginal M2-specific CD8+ T cells displayed predominantly a tissue resident phenotype characterized by expression of CD103 and CD69 (Fig. 3F and Supplemental Fig. 2C, 2D) and were induced to a similar degree with or without CD4+ T cell help (Fig. 3G). Interestingly, CD4-depletion did not prevent CD127 expression, nor did it alter the expression the CD69 and CXCR3 by cervicovaginal M2-specific CD8+ T cells (Fig. 3F, and Supplemental Fig. 2C, 2D). Notably, CD127 expression was reduced after a single Ivag immunization compared to IM/Ivag in GK1.5 or isotype control-treated animals (Fig. 3F and Supplemental Fig. 2C, 2D).
FIGURE 3.
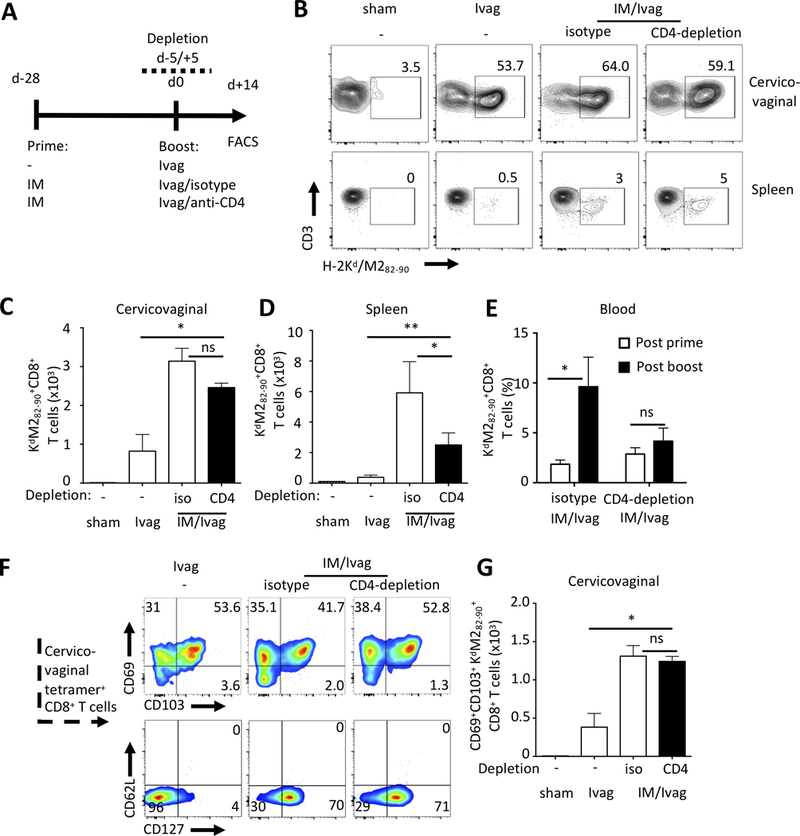
Induction of cervicovaginal resident memory CD8+ T cells does not require CD4+ T cell help. (A) BALB/c mice were primed IM with Ad5-MM2 vector and one month later received a booster Ivag with HP16-MM2 in CD4+ T cells depleted (GK1.5-treated) and isotype control-treated mice. (B-D) One month after booster immunization, cervicovaginal and spleen cell suspensions were analyzed by FACS for the presence of H-2Kd/M282–90 tetramer+CD8+ T lymphocytes. (B) Representative FACS plot of Kd/M282–90+ staining. (E) M2-specific CD8+ T cell responses were analyzed in blood samples after prime and after booster immunization. (F) Representative FACS plots of Kd/M282–90+ staining and CD103, CXCR3, CD127 and CD62L expression by Kd/M282–90+CD8+ T cells and (G) absolute number of M2-specific CD69+CD103+CD8+ T cells. Histogram bars represent the mean total number or percent of H-2Kd/M282–90 tetramer+CD8+ T cells +/- SEM. Data are representative of two independent experiments (n=4–5 mice per group). P values (*P≤0.05, **P≤0.01) were determined by (B, D, G) one-way ANOVA with post-hoc Tukey analysis and (E) two-way ANOVA with post-hoc Sidak analysis.
Similar ranked abundance of TCR-β productive rearrangement between genital and systemic M2-specific CD8+ T cells
Next, we performed a T cell receptor β-chain analysis on cDNA from sorted spleen and cervicovaginal Kd/M82-90 tetramer-positive CD8+ T cells two weeks after IM/Ivag. The TCR-Vβ domain usage of spleen and cervicovaginal M2-specific CD8+ T cells was enriched for Vβ13.2 compared to the repertoire of naïve BALB/c CD8+ T cells, consistent with the TCR-Vβ domain usage by M2-specific CD8+ T cells after RSV pulmonary infection (Fig. 4A and Supplemental Fig. 3A)(35). Overall, the ranked abundance of the most represented TCR-β chain productive rearrangements at the nucleotide and amino acid levels was conserved between spleen and cervicovaginal compartments within the same mouse (Fig. 4B, 4C, Supplemental Fig. 3B, 3C). These results suggest that CD8+ TRM cells induced by the Ivag booster immunization share essentially the same TCR repertoire with their systemic CD8+ T cell memory counterpart. Interestingly, only two productive rearrangements were found in more than one mouse (Supplemental Fig. 3D).
FIGURE 4.
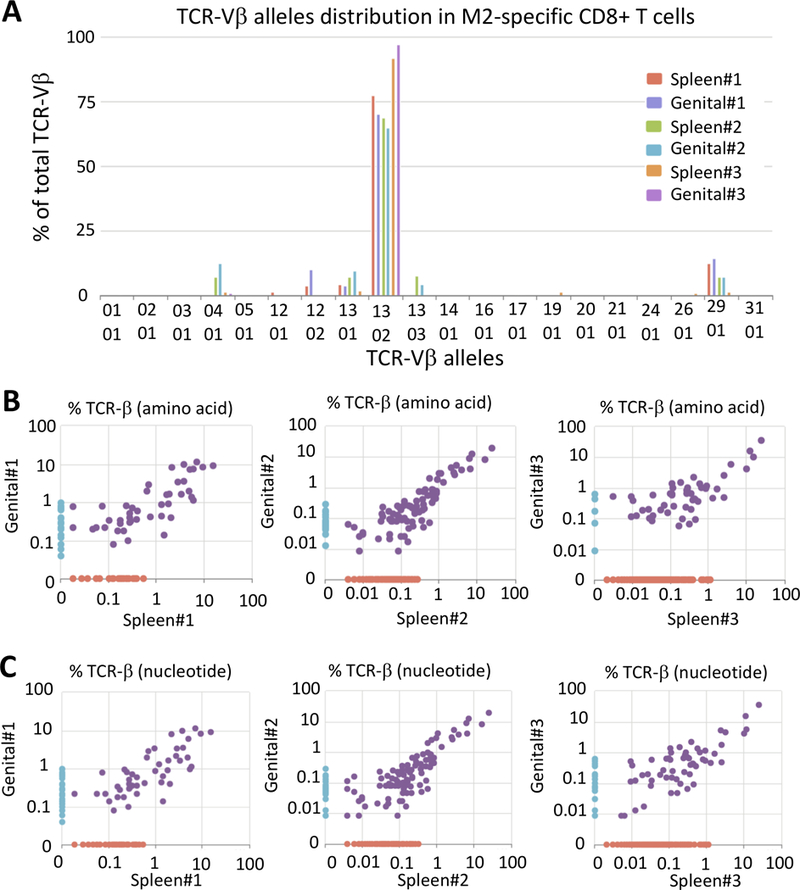
Similar ranked abundance of TCR-β productive rearrangement between genital and systemic M2-specific CD8+ T cells. BALB/c mice were primed IM with Ad5-MM2 vector and one month later received a booster Ivag with HP16-MM2. One month after the final immunization spleen, cervicovaginal and dLN Kd/M282–90 tetramer+CD8+ T cells were sorted, and cDNA were generated for TCR- β repertoire analysis using the ImmunoSEQ technology platform. (A) TCR-Vβ usage by spleen and cervicovaginal Kd/M282–90 tetramer+CD8+ T cells. Scatter plot of TCR-β rearrangement usage at (B) the amino acid and (C) the nucleotide levels between genital and systemic Kd/M282–90 tetramer+CD8+ T cells.
IM prime followed by Ivag booster immunization confers CD8-mediated protection against vaccinia virus and long-term cervicovaginal memory CD8+ T cell responses
We next compared IM/Ivag and IM/IM prime-boost regimen in a vaginal challenge with vaccinia virus expressing the M282-90 epitope (VV-M2). Two months following the boost, mice were challenged Ivag with 107 PFU of VV-M2 and viral titers were measured in cervicovaginal tissue homogenates by plaque assays. In mice immunized IM/Ivag, the median cervicovaginal mucosa viral titer was reduced 1000-fold and 100-fold compared to sham-treated and IM/IM immunized mice, respectively (Fig. 5A). To assess the role of CD8+ T cells in VV-M2 control, mice were treated with a CD8-depleting, which resulted in 99% depletion of systemic and cervicovaginal CD8+ T cells, or with control isotype antibodies (Data not shown). Cervicovaginal VV-M2 titer was reduced by 1000-fold in mice immunized IM/Ivag treated with the control isotype antibody compared to mice treated with CD8-depleting antibody or sham-immunized (Fig. 5B).
FIGURE 5.
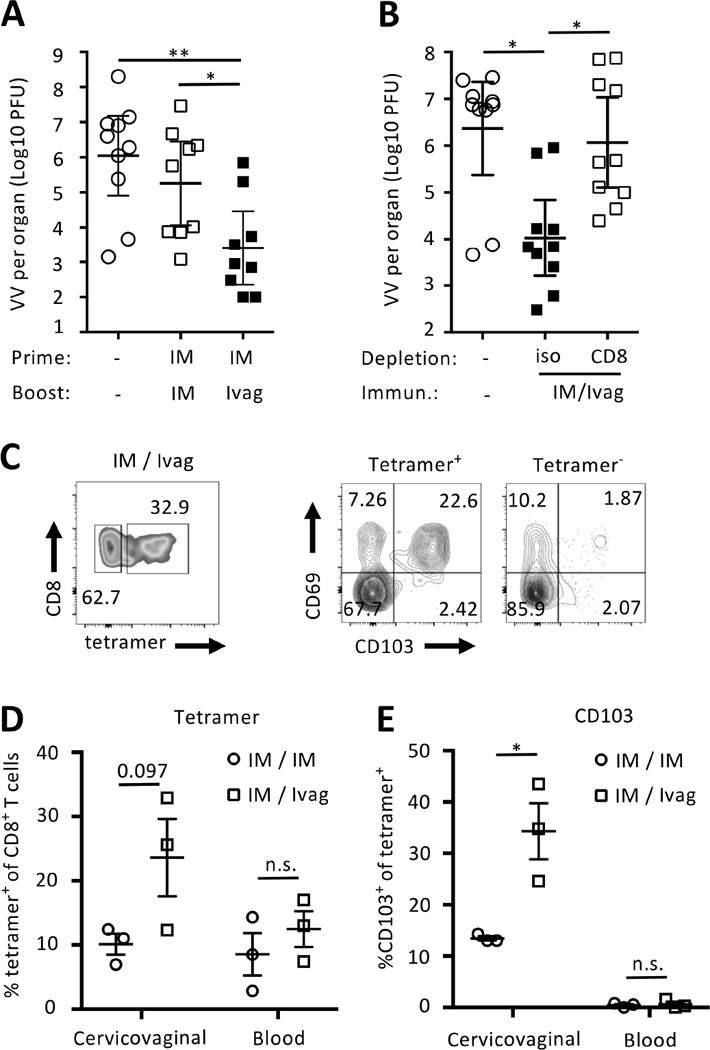
IM prime followed by Ivag booster immunization confers CD8-mediated protection against genital vaccinia virus infection and long-term cervicovaginal memory CD8+ T cell responses. Naïve BALB/c mice were prime-boost immunized IM/IM or IM/Ivag with HPV16-MM2 Psv (Ivag, 5×107 IU) and Ad5-MM2 (IM, 5×106 PFU). (A and B) Two months after the booster immunization, mice were challenged with 107 PFU of recombinant vaccinia virus expressing the minimal peptide epitope M282–90 derived from the RSV M2 protein. Vaccinia virus titers in cervicovaginal tissues was measured by plaque assay. (B) Immune mice were depleted with anti CD8 antibody or isotype control antibodies (100μg) on day -3 and day -1 and day +1 respective to vaccinia Ivag challenge. One year after the booster immunization, cell suspensions from cervicovaginal tissues and blood were analyzed by FACS. Representative FACS plot (C) of tetramer staining and expression of CD103 and CD69 by M2-specific CD8+ T cells in the indicated tissues. (D) Percent of H2-Kd/M282–90 tetramer+ CD8+ T cells and (E) percent of CD103 expression by H2-Kd/M282–90 tetramer+ CD8+ T cells. Data are shown as individual mice and mean percentage +/- SEM of M2-specific CD8+ T cells and % of CD103 expression by M2-specific CD8+ T cells. P values (*P≤0.05) were determined by one-way ANOVA with post-hoc Tukey analysis. Vaccinia challenge experiments are representative of two independent experiments (n=9–10 mice per group). P values were determined (*P≤0.05, **P≤0.01) by one-way ANOVA with post-hoc Tukey analysis.
We assessed the persistence of memory CD8+ T cells in blood and in the cervicovaginal mucosa (Fig. 5C-E) one year after booster immunization. The frequency of tetramer+CD8+ T cells (Fig 5D), and also the frequency of teramer+CD8+ T cells expressing CD103 in the cervicovaginal mucosa (Fig 5E), was increased after IM/Ivag compared to IM/IM immunization. Together, these data indicate that Ivag booster immunization elicits preferentially long-term persistence of cervicovaginal CD8+ TRM and confers CD8-mediated protection against a genital vaccinia infection.
Immunogenicity of HPV PsV expressing a detoxified fusion protein of HPV16 oncoproteins E6 and E7
Current human papillomavirus (HPV) vaccines protect against incident infection but do not act therapeutically(36). Persistent expression of HPV oncoproteins E6 and E7, which are absent from the prophylactic HPV vaccines, is the primary factor in the development of cervical and vaginal intraepithelial neoplasia(37). Therefore, increasing trafficking of CD8+ T cells to the cervicovaginal mucosa after vaccination might be key for effective therapeutic vaccination against HPV-associated neoplasia(38, 39). We generated HPV PsV that expressed a detoxified fusion protein of HPV16 E6 and E7 (E6E7) with or without a signal peptide from HSV-2 gD in order to target secretory pathways (spE6E7) (Fig. 6A). E6E7 fusion protein was detected by Western blot in lysates and supernatants from cells transfected with both constructs but the spE6E7 product displayed a higher apparent molecular weight E6E7 product, suggesting additional posttranslational modifications (Fig. 6B).
FIGURE 6.
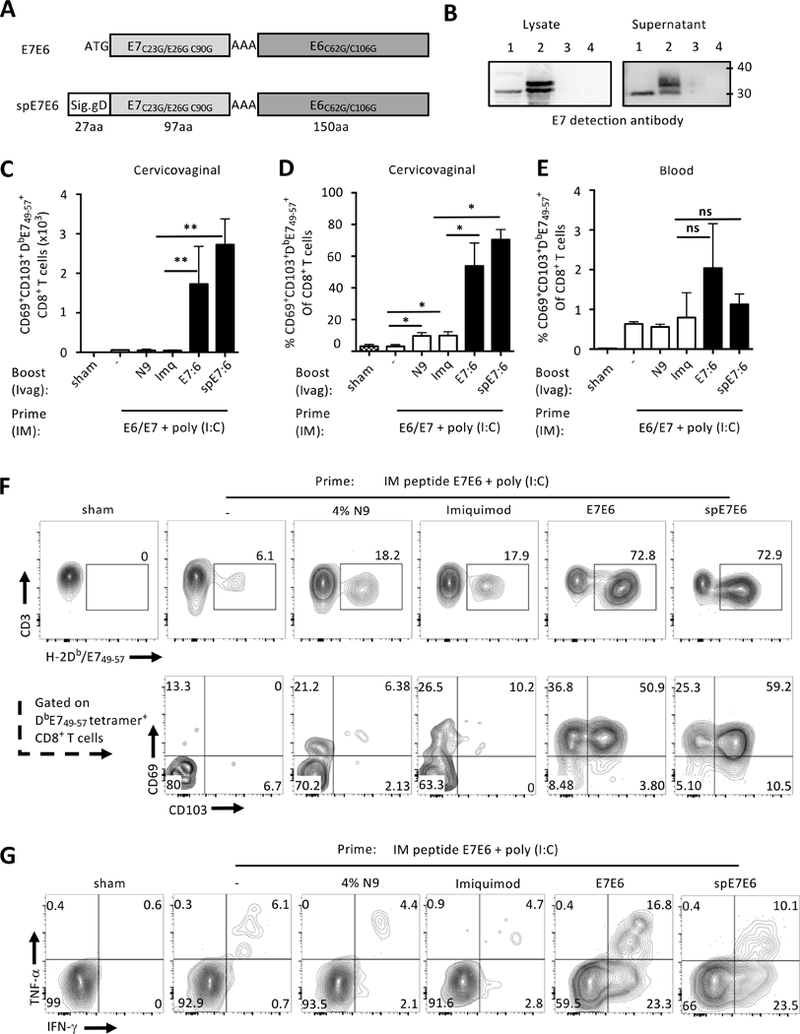
Immunogenicity of HPV PsV expressing a detoxified fusion protein of HPV16 oncoproteins E6 and E7. (A) Two plasmids expressing inactivated E7 and E6 fusion were generated with the signal peptide from HSV2 glycoprotein D (spE6E7) or without (E6E7). (B) Western-blot analysis of E7E6 expression in transfected cell lysate or supernatant with anti E7 antibody. Lane numbers correspond to E6E7 (1), spE6E7 (2), and luciferase (3) plasmids transfection, or untransfected (4). C57BL/6 mice were immunized IM twice 5 days apart with E744–62, E643–60 long peptides and poly(I:C) in saline. Two weeks later, mice were immunized Ivag with 108 IU of HPV PsV expressing the E7E6, 5% Imiquimod, 4% N-9 or remained untreated. (C-F) Two weeks after the booster immunization, genital and blood E7-specific CD8+ T cell responses were assessed by H-2Db/E749–67 tetramer staining. Histogram bars +/− SD represent (C, D) absolute numbers of E7-specific CD8+ T cells in the cervicovaginal mucosa and (E) percent in blood. (F) Representative FACS plots of E7-tetramer staining and expression of CD69 and CD103. (G) Production of IFN-γ and TNF- α by cervicovaginal CD8+ T cells was analyzed by FACS after in vitro stimulation with E7 peptide. Data are representative of three independent experiments (n=5 mice per group). P values (*P≤0.05, **P≤0.01) were determined by one-way ANOVA with post-hoc Tukey analysis
HPV PsV expressing either of the E6E7 constructs induced modest CD8+ T cell responses after Ivag priming (data not shown) and so were used in the IM/Ivag prime-boost regimen. For the IM prime, we used a cluster immunization protocol consisting of short interval IM immunization with long peptides of E6 and E7 admixed with the TLR3 agonist poly(I:C)(40). IM prime followed by HPV-E6E7 or HPV-spE6E7 PsV Ivag boost induced a large increase in the number of cervicovaginal H-2Db/E749-67+ CD8+ T cells compared to untreated mice or those treated with N-9 or the TLR7/8 agonist Imiquimod (Fig. 6C, 6D). An Ivag booster immunization with either of the E6E7 constructs induced a modest increase in blood E7-specific CD8+ T cells compared to sham-, N-9-, and Imiquimod-treated (Fig. 6E). Most cervicovaginal E7-specific CD8+ T cells expressed CD103 and CD69 after Ivag booster with either HPV PsV E6E7 constructs. This suggests that the acquisition of a TRM phenotype was not specific to the model antigens or the systemic priming strategy but rather is a general feature of HPV PsV Ivag booster immunization (Fig. 6F). Interestingly, topical Ivag instillation of Imiquimod or N-9 induced a modest but significant increase in the percentage of cervicovaginal CD8+ T cells and was associated with increased expression of CD69 (Fig. 6D, 6F). Consistent with the tetramer staining data, only Ivag immunization with HPV16 PsV expressing either E6E7 or spE6E7 constructs induced high frequency of IFN-γ and TNF-α producing cervicovaginal CD8+ T cell in response to in vitro stimulation with E749-67 peptide (Fig. 6G).
Antigen presentation by cervicovaginal cells promotes in situ proliferation and upregulation of CD103
We investigated the mechanisms of specific amplification of cervicovaginal CD8 TRM after Ad5-MM2 IM prime/HPV16-MM2 Ivag booster immunization. First, we analyzed the expression of the proliferation marker Ki-67 by CD8+ T cells in cervicovaginal tissue sections from mice immunized IM/Ivag by immunofluorescence. On day 4 post Ivag booster immunization, we observed clusters of CD8+ T cells positively stained for Ki-67 (Fig. 7A). Next, we performed an ex vivo presentation assay with cervicovaginal cells isolated from mice immunized Ivag with HPV16 PsV expressing chicken egg ovalbumin as a model antigen (HPV-OVA) or with control HPV16 PsV (HPV-ctrl). As a readout, we measured proliferation of purified naïve OVA257-264-specific CD8+ T cells (OT-I) labelled with CellTrace Violet proliferation dye (CTV). C57BL/6 mice were Ivag immunized with 4×108 IU HPV-OVA, two days later, cervicovaginal cells were extracted and cultured for 3 days with naïve CTV-labelled OT-1 cells. OT-I cells cultured with cervicovaginal cells from HPV-OVA, but not HPV-ctrl, immunized mice underwent extensive proliferation (Fig. 7B, 7C) and upregulated CD103. This upregulation was apparent as soon as after one cell division (Fig. 7B, 7D). In contrast, OT-I cells stimulated with spleen cells pulsed with the minimal peptide OVA257-264 underwent extensive proliferation but did not upregulate CD103 (Fig. 7B-7D).
FIGURE 7.
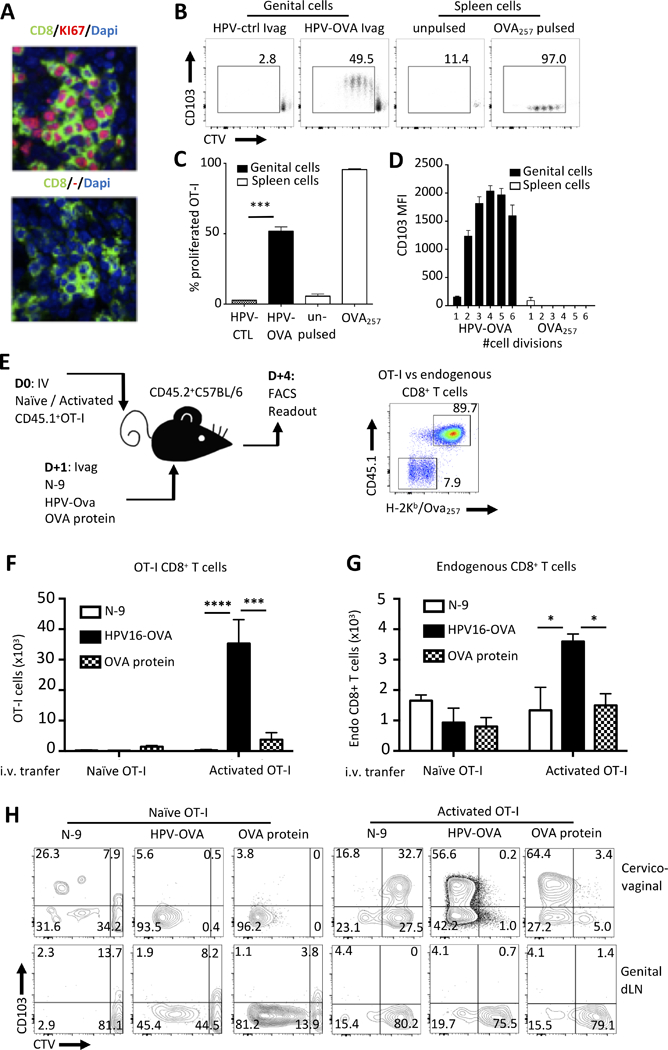
Local antigen presentation promotes in situ proliferation and upregulation of CD103. (A) Naïve BALB/c mice were prime IM with Ad5-MM2 and received boosted Ivag with HPV-MM2. On day 4 after booster, the expression of CD8 (Green) and Ki-67 (red) was assessed by confocal microscopy on frozen cervicovaginal tissue sections were analyzed. (B-D) Naïve C57BL/6 mice were immunized Ivag with HPV PsV expressing OVA or a control plasmid. On day 2 after immunization, genital cell suspensions from immunized mice or spleen antigen-presenting cells pulsed with OVA257–264 peptide or unpulsed were incubated with naïve OT-I CD8+ T cells labeled with CTV. (B) On day 3 after immunization, OT-I CD8+ T cell proliferation and expression of CD103 was analyzed by FACS. (C) The mean percent of proliferated shown as histogram bar ± SD. (D) The mean fluorescence intensity of CD103 per cell division is shown as histogram bars ± SD. (E) naïve or in vitro activated CD45.1+OT-I CD8+ T cells were labeled with CTV and transferred into naïve C57BL/6 mice (5×106/mouse) one day prior to ivag immunization with HPV PsV expressing OVA or a control plasmid. (F, G) On day 4 after immunization, absolute numbers of (F) OT-I and (G) endogenous CD8+ T cells were assessed in cervicovaginal mucosa and dLN. (H) representative FACS plot of CD103 expression and CTV labeling by cervicovaginal and dLN OT-I CD8+ T cells. The mean total number of OT-I or endogenous CD8+ T cells per organs is shown as horizontal bar ± SD. Data are representative of three independent experiments (n=4–5 mice per group). P values (*P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001) were determined by one-way ANOVA with post-hoc Tukey analysis.
Next, we assessed HPV PsV Ivag immunization in C57BL/6 mice that had been adoptively transferred with naïve or activated CTV-labelled OVA257-264-specific OT-I cells expressing the congenic marker CD45.1 (Fig. 7E). Activated OT-I cells were generated by in vitro expansion of spleen OT-I as described in the Methods section. One day after adoptive transfer of naïve or activated OT-I cells, mice were immunized Ivag with 1×108 IU HPV-OVA, 40μg ovalbumin protein or were treated with N-9 only. Four days following this treatment, we assessed the in vivo proliferation of transferred OT-I cells. A single Ivag immunization with HPV-OVA induced a 30-fold and 10-fold increase in cervicovaginal activated OT-I cells number compared to mice that were treated Ivag with N-9 or OVA protein, respectively (Fig. 7F).
Importantly, in mice transferred with activated OT-I CD8+ T cells only, Ivag immunization with HPV-OVA increased the number of cervicovaginal endogenous CD8+ T cells compared to other groups (Fig. 7G). Activated OT-I cells upregulated CD103 and underwent extensive proliferation after Ivag immunization with HPV-OVA or OVA protein (Fig. 7H). Surprisingly, most naive OT-I cells recovered from the cervicovaginal mucosa of mice immunized Ivag with HPV-OVA had proliferated but did not express CD103 (Fig. 7H). Interestingly, in the absence of cognate antigen, CD103 was expressed by cervicovaginal activated OT-I cells, but not naïve, after Ivag treatment with N-9 although at lower level than following HPV-OVA Ivag (Fig. 7H). In the genital draining lymph nodes, naive OT-I underwent extensive proliferation after Ivag immunization with HPV-OVA or OVA protein but not after N-9 treatment alone. In contrast, activated OT-I from the genital draining lymph nodes did not proliferate or upregulate CD103 (Fig. 7H). Together these data highlight striking differences in the response of naïve and activated CD8+ T cells to Ivag immunization. Such differences might result from differential migratory properties of naïve and activated CD8+ T cells.
We next assessed the localization of CD8+ T cells within the cervicovaginal mucosa and the expression of CD103 using the adoptive transfer protocol of in vitro activated OT-I CD8+ T cells (Fig S4). Mice immunized Ivag with HPV-OVA displayed a massive CD8+ T cell infiltration of the cervicovaginal epithelium compared to unimmunized or mock-immunized mice. Sparse CD8+ T cell infiltration of the underlying submucosa was also observed in mice immunized Ivag with HPV-OVA (Fig S4A). In HPV-OVA Ivag immunized mice, CD103 expression was observed in 50% of intraepithelial CD8+ T cells and in 7% of submucosal CD8+ T cells (Fig S4B and C). These data indicate that Ivag immunization with HPV PsV induces preferentially intraepithelial localization of CD8+ T cells and that CD103 is predominantly expressed by vaccine-induced intraepithelial CD8+ T cells.
In situ TCR engagement promotes both recruitment of cognate and non-cognate activated CD8+ T cells
We determined to what extent recruitment of activated CD8+ T cells accounts for the expansion of cognate and non-cognate cervicovaginal CD8+ TRM. To approach this, we transferred either alone or together, in vitro activated OT-I and Pmel-1 TCR transgenic CD45.1+CD8+ T cells specific for two unrelated epitopes OVA and melanoma-associated glycoprotein 100 (gp10025-33), respectively (Fig. 8A, 8B).
FIGURE 8.
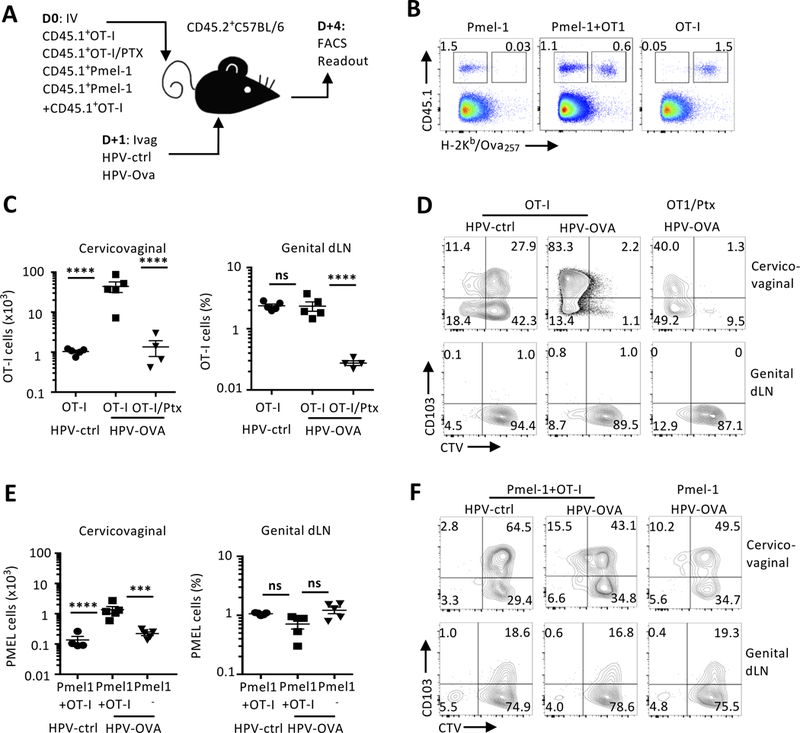
In situ TCR engagement promotes both recruitment of cognate and non-cognate activated CD8+ T cells. (A-D) CTV-labelled activated CD45.1+OT-I were pretreated or not with pertussis toxin and transferred to naïve C57BL/6 mice one day prior to ivag immunization with HPV PsV expressing OVA or a luciferase as a control. (A-B, E-F) CTV-labelled activated CD45.1+OT-I and Pmel-1 CD8+ T cells were co transferred to naïve C57BL/6 mice one day prior to ivag immunization with HPV PsV expressing OVA or a luciferase as a control. (B) In co-transfer condition, OT-I and Pmel-1 CD8+ T cells were distinguished by CD45.1 and H-2Kb/OVA257–264 tetramer staining. (C, E) On day 4 after immunization, absolute numbers and percent of OT-I and Pmel-1 CD8+ T cells were assessed in the cervicovaginal and dLN tissues, respectively. The mean absolute number and percentage of transferred OT-I CD8+ T cells per organs is shown as horizontal bar ± SD, each symbol represents an individual mouse. (D, F) Representative FACS plot of CD103 expression and CTV labeling on transferred OT-I CD8+ T from cervicovaginal and dLN tissues. Data are representative of two independent experiments (n=4–5 mice per group). P values (***P≤0.001, ****P≤0.0001) were determined by one-way ANOVA with post-hoc Tukey analysis.
In vitro activated OT-I cells were treated with pertussis toxin (PTX), to block G-protein signaling and chemotaxis(41). As previously reported by Cyster et al., PTX treatment did not affect the numbers of activated OT-I cells in blood and spleen compared to untreated OT-I cells (data not shown). The number of PTX-treated OT-I cells found in the cervicovaginal mucosa and dLN were greatly reduced compared to untreated OT-I CD8+ T cells after HPV-OVA Ivag (Fig. 8C). The PTX-treated OT-I cells that remained within the cervicovaginal mucosa showed extensive proliferation similar to untreated OT-I cells after HPV-OVA Ivag (Fig. 8D). These results suggest that antigen expression after Ivag immunization triggers the recruitment of the blood precursors of CD8+ TRM.
Next, we assessed the consequence of Ivag immunization on the recruitment, proliferation, and differentiation of non-cognate CD8+ T cells. We co-transferred in vitro activated OT-I and Pmel-1 CD8+ T cells that were identified as CD45.1+H-2Kb/OVA257-264-tetramer+ and CD45.1+H-2Kb/OVA257-264-tetramer-, respectively (Fig. 8B). After Ivag HPV-OVA immunization, the number of cervicovaginal Pmel-1 cells was increased by 10-fold but they did not extensively proliferate or upregulate of CD103 (Fig. 8E, 8F). In contrast, the number of OT-I cells was increased by 100-fold and these cells proliferated extensively and upregulated CD103 after HPV-OVA Ivag as compared to HPV-ctrl Ivag (Fig. 8C, 8D). Genital dLN Pmel-1 or OT-I cells numbers were unchanged between immunization groups (Fig. 8C, 8E) and did not proliferate or express CD103 (Fig. 8D, 8F).
Discussion
This study provides a rationale for heterologous prime-boost immunization using two unrelated non-replicating viral vectors to maximize systemic and genital-resident memory T cell responses. It shows that booster immunization is critical to shape the tissue distribution of memory CD8+ T cell responses whereas the main function of priming is to maximize circulating CD8+ T cell responses. In this context, local antigen expression by basal keratinocytes was a key factor in the recruitment of cognate and bystander circulating CD8+ T cells and the elicitation of in situ proliferation and differentiation of cognate intraepithelial CD8+ TRM.
A major hurdle for mucosal immunization remains a limited efficacy for priming, particularly in tissues devoid of organized secondary lymphoid structures. To be immunogenic, vaccine antigens deposited in non-lymphoid tissues, such as the buccal mucosa, the female reproductive tract or the skin, must be carried to the draining lymph nodes and presented to naïve CD8+ T cells(42). A systemic prime- local boost approach could bypass this hurdle by targeting secondary lymphoid organ with Ad vectors given systemically. Interestingly, preclinical studies indicate that i.m. vaccination with adenovirus-based HIV vaccine promote trafficking of CD8+ T cells to the intestinal and genital mucosa(43). However, numerous vaccine clinical trials against sexually transmitted infections given systemically have shown, at most, partial efficacy in spite of inducing circulating CD8+ T cell responses(44). In this context, HPV vectors or other viral vectors able to transduce the genital mucosa might efficiently attract circulating CD8+ T cells or amplify local CD8+ T cells induced after systemic immunization with existing clinical grade vaccines.
The introduction of strong helper epitopes is critical in the design of vaccines to generate B cell or cytotoxic responses. CD4+ T cells are critical in the induction of primary circulating and resident memory CD8+ T cell response(45). However, CD4 T cell help requirement remained to be evaluated for induction secondary CD8+ TRM. The sequential nature of prime-boost immunization permitted us to specifically assess the role of CD4+ T cell help during recall of secondary CD8+ T cell responses. Our results show that fully differentiated memory CD8+ T cells can be recalled and differentiate into CD8+ TRM in absence of CD4+ T cells, reminiscent of the work of Fraser et al. showing that CD4+ T cells are dispensable for recall of systemic memory CD8+ T cells(46). In addition, Ivag booster immunization was equally efficient for the E6E7 fusion expressed with or without a signal peptide, suggesting that targeting the secretion pathway is not critical at this point, in contrast to its apparent importance in Ivag priming(47). We hypothesize that antigen secretion by cervicovaginal keratinocytes functions primarily to facilitate antigen presentation to CD4+ T cells, which is critical for priming but not for local boosting(45, 48).
TCR-β repertoire analyses of M2-specific CD8+ T cells indicate that after Ivag boosting, systemic and genital-resident CD8+ T cells share their most represented TCR-β rearrangements. This indicates that the ranked abundance of TCR-β rearrangements is established during priming and is perpetuated upon recall. Other groups have shown that CD8+ TRM from different tissues share the same repertoire and arise from common naïve precursors(49). A limitation to our repertoire analysis is that it did not account for putative competition between CD8+ T cells against different antigens as all the analyzed cells were specific to the M282-90 immunodominant epitope(23). Also, it remains to be confirmed whether these results will apply to subdominant epitopes.
HPV PsV are uniquely versatile and amenable to vaccine development. First, they can package bacterial plasmids that lack viral sequence. Second, a recently developed cell-free method of production of HPV PsV is compatible to GMP production(50). Third, a formulation of HPV PsV with N-9 seems practical as it is a commercially available spermicide and induces transient disruption of the cervicovaginal epithelium in women(51). Finally, the thickness of the squamous and simple columnar epithelia of the cervicovaginal mucosa do not vary throughout the cycle in contrast to mice thus making hormonal treatment superfluous(52). Using non-replicating vectors for Ivag booster immunization presents an obvious safety advantage over replication-competent vectors, but the transient nature of antigen expression might require multiple booster immunization to achieve sufficient amplification of CD8+ TRM responses. Our approach based on two unrelated viral vectors might best focus the CD8+ T cell responses toward the antigen of interest. Alternatively to HPV vectors, we have recently shown that Ad type 26 and 35 can also transduce efficiently the intact genital epithelia after Ivag immunization(26). Furthermore, the combination of two unrelated viral vectors such as HPV and adenovirus might advantageously prevent the unnecessary amplification of cross-reactive anti-vector T cells(26).
Adoptive transfer experiments of TCR transgenic CD8+ T cells indicate striking differences between naïve and in vitro activated CD8+ T cells in their respective ability to proliferate in response to Ivag immunization. Such differences might result from intrinsic migratory properties that are distinct between naïve T cells and activated TCR transgenic CD8+ T cells. Therefore, it could explain the propensities of naïve CD8+ T cells to proliferate in the draining lymph nodes and activated CD8+ T cells to migrate and proliferate in the cervicovaginal mucosa(53, 54). Importantly, local TCR engagement triggers the recruitment of cognate and bystander circulating CD8+ T cells, but only cognate cells extensively proliferate. Recruitment of bystander CD8+ T cells has been proposed as an essential mechanism of protection in peripheral tissues(55). Since our study was performed in naïve specific pathogen free animals, it remains to be established in a non-specific pathogen free situation whether bystander reactivation of the anti-microbial resident memory populations might alter the distribution of vaccine induced CD8+ T cells(56).
Of note, adoptively transferred activated TCR transgenic CD8+ T cells upregulate CD103 in the cervicovaginal mucosa but not in the genital dLN, even in absence of cognate antigen expression. This is consistent with the notion that microenvironmental factors suffice to dictate the differentiation of CD8+ TRM in the absence of local antigen expression(8, 21, 57). However, expression of CD103 by polyclonal CD8+ T cells induced by systemic vaccination was modest comparatively to CD103 expression by in vitro activated TCR transgenic CD8+ T cells. This apparent discrepancy could be due to intrinsic differences such as TCR expression and signaling. Also, polyclonal memory CD8+ T cells induced by IM vaccination with adenoviral vectors may be less prone to infiltrate inflamed peripheral tissues than early effectors or in vitro activated CD8+ T cells(17, 58).
The maintenance of CD8+ TRM in the brain, skin and in the upper female reproductive tract has been recently shown to depend on local proliferation(59-61). Our ex vivo antigen presentation experiments extend these findings and indicate that antigen-driven proliferation of CD8+ T cells in the cervicovaginal epithelium is a mechanism of tissue-specific enrichment of cognate CD8+ TRM cells after Ivag booster vaccination. Because HPV PsVs exclusively infect basal keratinocytes of the cervicovaginal mucosa(34), it remains to be established whether direct presentation by basal keratinocytes or cross-presentation by professional antigen-presenting cells control local activation and infiltration of CD8+ T cells. The ability of HPV PsV to recall secondary CD8+ T cells in the cervicovaginal mucosal might result from the direct killing of transduced keratinocytes and the release of additional inflammatory signals to recruit effector cells and antigen presenting cells(62). Finally, the observation that soluble protein also induced CD8+ TRM, although to a lower extent than HPV PsV, suggests that antigen cross-presentation by dendritic cells might be key to the induction of cervicovaginal CD8+ TRM(63).
Our “prime-pull-amplify” strategy would be particularly suited for prophylactic vaccination against HSV-2 where the increased number of CD8+ TRM at the site of viral entry might confer sterilizing immunity(64). In addition, Ivag booster immunization could prove useful therapeutically to reduce HSV-2 recurrence by maintaining or increasing the pool of CD8+ TRM between herpetic episodes. A “prime-pull-amplify” approach might also induce regression of HPV-induced neoplasia(39) in a situation where the antigen is already expressed in the immunosuppressive microenvironment typically induced by HPV-infection(65). Since local engagement of CD8+ TRM in mucosal and cutaneous tissues can act as a tissue immune modifier(10, 11), high level of antigen delivered within or surrounding the lesion could increase effector-to-target ratio and therefore cytotoxicity, thereby inducing a broad inflammatory response.
To conclude, this prime-pull-amplify approach underscores the importance of the delivery route of non-replicating viral vectors in prime-boost immunization regimen. It shows that targeting antigen expression to the portal of pathogen entry can guide preexisting CD8+ T cell responses into the sites where they are most needed. Such an approach could be implemented to improve cervicovaginal CD8+ T cells distribution induced by systemically delivered vaccines and could impact the design of prime-boost vaccines against sexually-transmitted infections and the treatment of HPV-neoplasia.
Supplementary Material
Acknowledgements
We thank the NIH tetramer facility for providing critical reagents and the NCI Flow Cytometry Core for assistance with cell sorting. We thank Dr Rebecca Cerio for helpful comments and critical reading of the manuscript.
This work was supported by the NIH intramural program.
Abbreviations used in this article:
- Ad
adenovirus
- CMC
carboxymethyl cellulose
- CTV
Cell Trace Violet
- dLN
draining lymph node
- HPV
human papillomavirus
- IU
infectious units
- IM
intramuscular
- Ivag
intravaginal
- MM2
M and M2-1 fusion
- N-9
nonoxynol-9
- OVA
ovalbumin
- PFU
plaque-forming unit
- PsV
pseudovirus
- RSV
respiratory syncytial virus
- TRM
tissue resident memory
- VV-M2
vaccinia virus expressing M282-90 epitope
Footnotes
Disclosure
The authors declare that they have no competing financial interests.
References
- 1.Starnbach MN, and Roan NR. 2008. Conquering sexually transmitted diseases. Nat Rev Immunol 8: 313–317. [DOI] [PubMed] [Google Scholar]
- 2.Plotkin SA 2010. Correlates of protection induced by vaccination. Clin Vaccine Immunol 17: 1055–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schiller JT, and Lowy DR. 2015. Raising expectations for subunit vaccine. J Infect Dis 211: 1373–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Picker LJ, Hansen SG, and Lifson JD. 2012. New paradigms for HIV/AIDS vaccine development. Annu Rev Med 63: 95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnston C, Koelle DM, and Wald A. 2014. Current status and prospects for development of an HSV vaccine. Vaccine 32: 1553–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holmgren J, and Czerkinsky C. 2005. Mucosal immunity and vaccines. Nat Med 11: S45–53. [DOI] [PubMed] [Google Scholar]
- 7.Sallusto F, Lenig D, Forster R, Lipp M, and Lanzavecchia A. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401: 708–712. [DOI] [PubMed] [Google Scholar]
- 8.Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, and Kupper TS. 2012. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature 483: 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, and Carbone FR. 2009. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol 10: 524–530. [DOI] [PubMed] [Google Scholar]
- 10.Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, and Masopust D. 2014. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346: 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, Jacobs H, Haanen JB, and Schumacher TN. 2014. T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science 346: 101–105. [DOI] [PubMed] [Google Scholar]
- 12.Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, Southern PJ, and Masopust D. 2015. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 161: 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Freestone D, Braun A, Wynne-Jones E, Behr FM, Stark R, Pellicci DG, Godfrey DI, Belz GT, Pellegrini M, Gebhardt T, Busslinger M, Shi W, Carbone FR, van Lier RA, Kallies A, and van Gisbergen KP. 2016. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352: 459–463. [DOI] [PubMed] [Google Scholar]
- 14.Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, and Jameson SC. 2013. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol 14: 1285–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, and Gebhardt T. 2013. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol 14: 1294–1301. [DOI] [PubMed] [Google Scholar]
- 16.Shin H, and Iwasaki A. 2012. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 491: 463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nolz JC, and Harty JT. 2014. IL-15 regulates memory CD8+ T cell O-glycan synthesis and affects trafficking. J Clin Invest 124: 1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Domingos-Pereira S, Decrausaz L, Derre L, Bobst M, Romero P, Schiller JT, Jichlinski P, and Nardelli-Haefliger D. 2013. Intravaginal TLR agonists increase local vaccine-specific CD8 T cells and human papillomavirus-associated genital-tumor regression in mice. Mucosal Immunol 6: 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chandy AG, Nurkkala M, Josefsson A, and Eriksson K. 2007. Therapeutic dendritic cell vaccination with Ag coupled to cholera toxin in combination with intratumoural CpG injection leads to complete tumour eradication in mice bearing HPV 16 expressing tumours. Vaccine 25: 6037–6046. [DOI] [PubMed] [Google Scholar]
- 20.Soong RS, Song L, Trieu J, Knoff J, He L, Tsai YC, Huh W, Chang YN, Cheng WF, Roden RB, Wu TC, Trimble CL, and Hung CF. 2014. Toll-like receptor agonist imiquimod facilitates antigen-specific CD8+ T-cell accumulation in the genital tract leading to tumor control through IFNgamma. Clin Cancer Res 20: 5456–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, Heath WR, Carbone FR, and Gebhardt T. 2012. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A 109: 7037–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khan TN, Mooster JL, Kilgore AM, Osborn JF, and Nolz JC. 2016. Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med 213: 951–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muschaweckh A, Buchholz VR, Fellenzer A, Hessel C, Konig PA, Tao S, Tao R, Heikenwalder M, Busch DH, Korn T, Kastenmuller W, Drexler I, and Gasteiger G. 2016. Antigen-dependent competition shapes the local repertoire of tissue-resident memory CD8+ T cells. J Exp Med 213: 3075–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu S 2009. Heterologous prime-boost vaccination. Curr Opin Immunol 21: 346–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cuburu N, Graham BS, Buck CB, Kines RC, Pang YY, Day PM, Lowy DR, and Schiller JT. 2012. Intravaginal immunization with HPV vectors induces tissue-resident CD8+ T cell responses. J Clin Invest 122: 4606–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cuburu N, Khan S, Thompson CD, Kim R, Vellinga J, Zahn R, Lowy DR, Scheper G, and Schiller JT. 2017. Adenovirus vector-based prime-boost vaccination via heterologous routes induces cervicovaginal CD8(+) T cell responses against HPV16 oncoproteins. Int J Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cassetti MC, McElhiney SP, Shahabi V, Pullen JK, Le Poole IC, Eiben GL, Smith LR, and Kast WM. 2004. Antitumor efficacy of Venezuelan equine encephalitis virus replicon particles encoding mutated HPV16 E6 and E7 genes. Vaccine 22: 520–527. [DOI] [PubMed] [Google Scholar]
- 28.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, and Restifo NP. 2003. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med 198: 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, and Carbone FR. 1994. T cell receptor antagonist peptides induce positive selection. Cell 76: 17–27. [DOI] [PubMed] [Google Scholar]
- 30.Buck CB, Pastrana DV, Lowy DR, and Schiller JT. 2005. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol Med 119: 445–462. [DOI] [PubMed] [Google Scholar]
- 31.Rodrigo Mora J, and Von Andrian UH. 2006. Specificity and plasticity of memory lymphocyte migration. Current topics in microbiology and immunology 308: 83–116. [PubMed] [Google Scholar]
- 32.Mueller SN, Gebhardt T, Carbone FR, and Heath WR. 2013. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol 31: 137–161. [DOI] [PubMed] [Google Scholar]
- 33.Hickman HD, Reynoso GV, Ngudiankama BF, Cush SS, Gibbs J, Bennink JR, and Yewdell JW. 2015. CXCR3 chemokine receptor enables local CD8(+) T cell migration for the destruction of virus-infected cells. Immunity 42: 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts JN, Buck CB, Thompson CD, Kines R, Bernardo M, Choyke PL, Lowy DR, and Schiller JT. 2007. Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat Med 13: 857–861. [DOI] [PubMed] [Google Scholar]
- 35.Billam P, Bonaparte KL, Liu J, Ruckwardt TJ, Chen M, Ryder AB, Wang R, Dash P, Thomas PG, and Graham BS. 2011. T Cell receptor clonotype influences epitope hierarchy in the CD8+ T cell response to respiratory syncytial virus infection. The Journal of biological chemistry 286: 4829–4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schiller JT, and Lowy DR. 2012. Understanding and learning from the success of prophylactic human papillomavirus vaccines. Nat Rev Microbiol 10: 681–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, and Stanley MA. 2012. The biology and life-cycle of human papillomaviruses. Vaccine 30 Suppl 5: F55–70. [DOI] [PubMed] [Google Scholar]
- 38.Cuburu N, and Schiller JT. 2016. Moving forward with human papillomavirus immunotherapies. Hum Vaccin Immunother: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maldonado L, Teague JE, Morrow MP, Jotova I, Wu TC, Wang C, Desmarais C, Boyer JD, Tycko B, Robins HS, Clark RA, and Trimble CL. 2014. Intramuscular therapeutic vaccination targeting HPV16 induces T cell responses that localize in mucosal lesions. Sci Transl Med 6: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wick DA, Martin SD, Nelson BH, and Webb JR. 2011. Profound CD8+ T cell immunity elicited by sequential daily immunization with exogenous antigen plus the TLR3 agonist poly(I:C). Vaccine 29: 984–993. [DOI] [PubMed] [Google Scholar]
- 41.Cyster JG, and Goodnow CC. 1995. Pertussis toxin inhibits migration of B and T lymphocytes into splenic white pulp cords. J Exp Med 182: 581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, and Carbone FR. 2006. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 25: 153–162. [DOI] [PubMed] [Google Scholar]
- 43.Kaufman DR, Liu J, Carville A, Mansfield KG, Havenga MJ, Goudsmit J, and Barouch DH. 2008. Trafficking of antigen-specific CD8+ T lymphocytes to mucosal surfaces following intramuscular vaccination. J Immunol 181: 4188–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McMichael AJ 2018. Is a Human CD8 T-Cell Vaccine Possible, and if So, What Would It Take? Could a CD8(+) T-Cell Vaccine Prevent Persistent HIV Infection? Cold Spring Harb Perspect Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laidlaw BJ, Zhang N, Marshall HD, Staron MM, Guan T, Hu Y, Cauley LS, Craft J, and Kaech SM. 2014. CD4+ T cell help guides formation of CD103+ lung-resident memory CD8+ T cells during influenza viral infection. Immunity 41: 633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fraser KA, Schenkel JM, Jameson SC, Vezys V, and Masopust D. 2013. Preexisting high frequencies of memory CD8+ T cells favor rapid memory differentiation and preservation of proliferative potential upon boosting. Immunity 39: 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cuburu N, Wang K, Goodman KN, Pang YY, Thompson CD, Lowy DR, Cohen JI, and Schiller JT. 2015. Topical herpes simplex virus 2 (HSV-2) vaccination with human papillomavirus vectors expressing gB/gD ectodomains induces genital-tissue-resident memory CD8+ T cells and reduces genital disease and viral shedding after HSV-2 challenge. J Virol 89: 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun JC, and Bevan MJ. 2003. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 300: 339–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaide O, Emerson RO, Jiang X, Gulati N, Nizza S, Desmarais C, Robins H, Krueger JG, Clark RA, and Kupper TS. 2015. Common clonal origin of central and resident memory T cells following skin immunization. Nat Med 21: 647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cerqueira C, Thompson CD, Day PM, Pang YS, Lowy DR, and Schiller JT. 2017. Efficient Production of Papillomavirus Gene Delivery Vectors in Defined In Vitro Reactions. Mol Ther Methods Clin Dev 5: 165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vincent KL, Stanberry LR, Moench TR, Breitkopf CR, Loza ML, Wei J, Grady J, Paull J, Motamedi M, and Rosenthal SL. 2011. Optical coherence tomography compared with colposcopy for assessment of vaginal epithelial damage: a randomized controlled trial. Obstet Gynecol 118: 1354–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patton DL, Thwin SS, Meier A, Hooton TM, Stapleton AE, and Eschenbach DA. 2000. Epithelial cell layer thickness and immune cell populations in the normal human vagina at different stages of the menstrual cycle. Am J Obstet Gynecol 183: 967–973. [DOI] [PubMed] [Google Scholar]
- 53.Sinclair LV, Finlay D, Feijoo C, Cornish GH, Gray A, Ager A, Okkenhaug K, Hagenbeek TJ, Spits H, and Cantrell DA. 2008. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat Immunol 9: 513–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Masopust D, Vezys V, Marzo AL, and Lefrancois L. 2001. Preferential localization of effector memory cells in nonlymphoid tissue. Science 291: 2413–2417. [DOI] [PubMed] [Google Scholar]
- 55.Schenkel JM, Fraser KA, Vezys V, and Masopust D. 2013. Sensing and alarm function of resident memory CD8(+) T cells. Nat Immunol 14: 509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hand TW, Dos Santos LM, Bouladoux N, Molloy MJ, Pagan AJ, Pepper M, Maynard CL, Elson CO 3rd, and Belkaid Y. 2012. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science 337: 1553–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Masopust D, Vezys V, Wherry EJ, Barber DL, and Ahmed R. 2006. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol 176: 2079–2083. [DOI] [PubMed] [Google Scholar]
- 58.Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, Fraser KA, Webby RJ, Brinkmann V, Butcher EC, Newell KA, and Ahmed R. 2010. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med 207: 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wakim LM, Gebhardt T, Heath WR, and Carbone FR. 2008. Cutting edge: local recall responses by memory T cells newly recruited to peripheral nonlymphoid tissues. J Immunol 181: 5837–5841. [DOI] [PubMed] [Google Scholar]
- 60.Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, Fonseca R, Burbach BJ, Hickman HD, Vezys V, Fife BT, and Masopust D. 2018. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat Immunol 19: 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, Alexandre YO, Gregory JL, Russell TA, Gebhardt T, Carbone FR, Tscharke DC, Heath WR, Mueller SN, and Mackay LK. 2018. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol 19: 183–191. [DOI] [PubMed] [Google Scholar]
- 62.Brewitz A, Eickhoff S, Dahling S, Quast T, Bedoui S, Kroczek RA, Kurts C, Garbi N, Barchet W, Iannacone M, Klauschen F, Kolanus W, Kaisho T, Colonna M, Germain RN, and Kastenmuller W. 2017. CD8(+) T Cells Orchestrate pDC-XCR1(+) Dendritic Cell Spatial and Functional Cooperativity to Optimize Priming. Immunity 46: 205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu CI, Becker C, Wang Y, Marches F, Helft J, Leboeuf M, Anguiano E, Pourpe S, Goller K, Pascual V, Banchereau J, Merad M, and Palucka K. 2013. Human CD1c+ dendritic cells drive the differentiation of CD103+ CD8+ mucosal effector T cells via the cytokine TGF-beta. Immunity 38: 818–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu J, Koelle DM, Cao J, Vazquez J, Huang ML, Hladik F, Wald A, and Corey L. 2007. Virus-specific CD8+ T cells accumulate near sensory nerve endings in genital skin during subclinical HSV-2 reactivation. J Exp Med 204: 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stanley MA 2012. Epithelial cell responses to infection with human papillomavirus. Clin Microbiol Rev 25: 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.