

Abstract
HIV-1 envelope glycoprotein [Env; trimeric (gp160)3 cleaved to (gp120/gp41)3] interacts with primary receptor CD4 and coreceptor (e.g. chemokine receptor CCR5 or CXCR4) to promote viral entry by fusing viral and target cell membranes. Encounter of gp120 with the coreceptor was thought to be the most crucial trigger for unleashing the fusogenic potential of gp41. Here we report a cryo-EM structure, at 3.9Å resolution, of a full-length gp120 complexed with a soluble CD4 and an unmodified human CCR5. The V3 loop of gp120 inserts into the chemokine binding pocket formed by seven transmembrane helices of CCR5, while the N-terminus of CCR5 contacts the CD4-induced bridging sheet of gp120. CCR5 induces no obvious allosteric changes in gp120 that can propagate to gp41, but it brings the Env trimer close to the target membrane. The N-terminus of gp120, gripped by gp41 in the prefusion or CD4-bound Env, flips back in the CCR5-bound conformation and may irreversibly destabilize gp41 to initiate fusion. The coreceptor probably functions by stabilizing and anchoring the CD4-induced conformation of Env near the cell membrane. These results advance our understanding of HIV-1 entry and may guide development of vaccines and therapeutics.
Introduction
HIV-1 envelope glycoprotein (Env) fuses viral and cell membranes, allowing entry of the virus into host cells. Its precursor, gp160, trimerizes to (gp160)3, which is cleaved into two noncovalently associated fragments: gp120 (receptor-binding) and gp41 (fusion)1. Three copies of each fragment constitute the mature viral spike (gp120/gp41)3. Sequential binding of gp120 to primary receptor CD4 and a coreceptor (e.g. CCR5 or CXCR4) are believed to induce conformational changes that trigger likely dissociation of gp120 and refolding of gp411. Structural rearrangements in gp41 bring the two membranes together, promoting membrane fusion. We have detailed structural information for the interaction of HIV-1 Env with CD42-6, but no molecular picture for its interaction with a coreceptor, which continues to be the subject of speculative molecular modeling7-10
The chemokine receptors, CCR5 and CXCR4, were identified as the HIV-1 coreceptors in 199611. They are G protein-coupled receptors (GPCRs) with seven transmembrane-spanning segments (7TMs). Choice of coreceptor is the major determinant for viral tropism11. Viruses using CCR5 (R5 viruses) are generally responsible for viral transmission; those using CXCR4 (X4 viruses) or both (dual-tropic; R5X4 viruses) emerge later during disease progression12,13. Both CCR5 and CXCR4 have an extracellular N-terminal segment, three extracellular loops (ECL), three intracellular loops (ICL) and a cytoplasmic C-terminal tail. Crystal structures have been reported for a C-terminally truncated CXCR4 containing stabilizing mutations and a T4 lysozyme fusion, in complex with different ligands, and for a similarly modified CCR5 containing a rubredoxin fusion in complex with either an anti-HIV drug, maraviroc or a modified chemokine, [5P7]CCL57,10,14,15. These structures show a typical 7TM helical bundle topology seen for other GPCRs (Extended Data Fig. 1). Consistent with the so-called two-site model16, the N-terminal segment of CXCR4 or CCR5 forms “chemokine recognition site 1” (CRS1) to interact with the globular core domain of chemokine, while their 7TM bundle forms a binding pocket (“chemokine recognition site 2”; CRS2) that accommodates the N-terminus of the chemokine. While they elucidate chemokine receptor function, these structures do not explain how CCR5 and CXCR4 function as HIV-1 coreceptors.
Mutagenesis studies have mapped the gp120 binding site to the N-terminal segment and ECL2 for CCR5; and to the N-terminal segment, ECL2, and ECL3 for CXCR417,18. The footprint of the coreceptor on gp120 probably includes the V3 loop and the bridging sheet, a structure accessible only after CD4 binding19,20. The N-terminus of the coreceptor may contact the gp120 bridging sheet, while the tip of the V3 loop may insert into the coreceptor CRS220. Interactions of V3 with CCR5 or CXCR4 have also been modeled by molecular dynamics simulations and free energy calculations7-10. Tyrosine sulfation near the N-terminus only enhances HIV-1 entry for CCR5, but not for CXCR421,22. The C-terminal tail, containing palmitoylation and phosphorylation sites, is required for efficient cell signaling, but not for HIV-1 coreceptor function23. Chemokines such as, MIP-1α, MIP-1β, CCL5/RANTES and CXCL12/SDF-1 block gp120 binding and prevent viral infection11.
Results
To understand how a coreceptor functions, we have determined the structure of an unmodified CCR5 in complex with a full-length HIV-1 gp120 and a 4-domain soluble CD4 by single-particle cryo-electron microscopy (cryo-EM).
Purification of the CCR5 complex and structure determination
To produce functional CCR5, we generated HEK293T or Expi293F cell lines stably expressing wildtype human CCR5 by our published protocol24. CCR5 on these cells was fully active as a chemokine receptor (Extended Data Fig. 2a), and it also formed a tight complex with HIV-1 gp120 in the presence of soluble CD4 (Extended Data Fig. 2b). Moreover, these cells fused efficiently with HIV-1 Env-expressing cells only when activated by soluble CD4 (Extended Data Fig. 2c), suggesting that the expressed CCR5 is a fully functional HIV-1 coreceptor. Finally, neither gp120 alone nor the gp120-CD4 complex could activate the G-protein mediated signaling pathways in these cells (Extended Data Fig. 2d and 2e).
To purify the wildtype CCR5 in its Env-bound conformation, we isolated the CD4-gp120-CCR5 complex from the CCR5-expressing cells (Extended Data Fig. 3a and 3b). The purified CD4-gp120-CCR5 complex eluted from a size-exclusion column as a single sharp peak at the expected volume, confirming its stability and conformational homogeneity (Extended Data Fig. 3c). Coomassie-stained SDS-PAGE and negative stain EM of the peak fractions showed all three components in a stoichiometry of 1:1:1 (Extended Data Figs. 3c and 4a-c). The absence of any G proteins in the purified complex is consistent with the notion that gp120, unlike chemokines, does not require G protein coupling for high affinity binding to CCR525.
We acquired cryo-EM images on a Titan Krios electron microscope with a K2 Summit direct detector (Extended Data Fig. 4d) and used RELION26 for image processing. Two-dimensional (2D) class averages of the particle images showed secondary structural features for both gp120 and CCR5 (Extended Data Fig. 4e). Three-dimensional (3D) classification of the particles was performed (Extended Data Fig. 5a), using the low-pass filtered 3D reconstruction of the complex in negative stain as an initial model (Extended Data Fig. 4c). The particles from the classes with defined structural features were combined and refined to generate a map at 4.6Å resolution, showing all three components (Extended Data Fig. 5a). The structure was determined by rounds of 3D classification, 3D classification with signal subtraction and masked refinement, as described in Methods and Extended Data Fig. 5a-d. The final resolution was 3.9Å, when the last two domains of CD4 were excluded
Overall structure of the CD4-gp120-CCR5 complex
The atomic structures of 4D CD4, the gp120 core complexed with CD4 and the modified CCR5, none of which was used in image processing, all fit as rigid bodies remarkably well to the EM density of the CD4-gp120-CCR5 at 4.5Å resolution (ref10,27,28; Fig. 1a and 1b and Extended Data Fig. 5b). The gp120 core, containing inner and outer domains and the bridging sheet2, occupied excellent density, indicating that it has the same rigid structure in this ternary complex as it does in the CD4-induced conformation2,28. Several N-linked glycans were marked by protruding densities from the protein surfaces, as expected for a fully-glycosylated gp120. All four domains of soluble CD4 were visible, with density for first two (D1-D2) slightly stronger than for last two (D3-D4) domains. There was also density for a N-linked glycan at N271 in D3 (Fig. 1b), suggesting that the hinge region between D2 and D3 may not be very flexible27. All 7TM helices of CCR5 were well resolved in detergent micelle. Its palmitoylated cytoplasmic tail, which presumably interacts with membrane, was disordered in this detergent-solubilized complex. Extra densities between CCR5 and gp120 that cannot be explained by the existing structures define the details of CCR5-gp120 interaction.
Figure 1. Cryo-EM structure of the CD4-gp120-CCR5 complex.
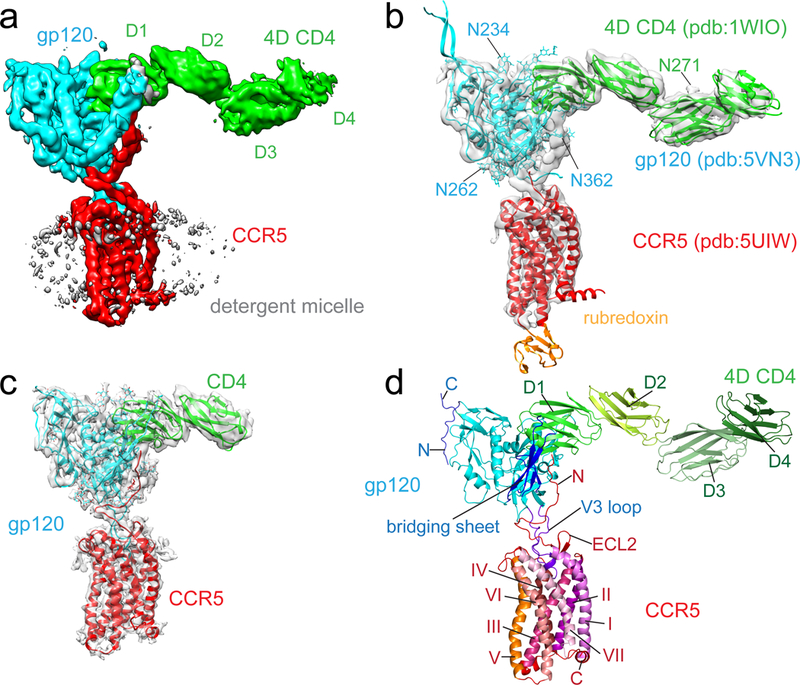
(a) Cryo-EM map of the complex containing HIV-1 gp120 (cyan), CCR5 (red), 4D CD4 (green; D1-4, domain 1-4), and detergent micelle (gray). (b) Fit of structures of gp120 (pdb ID: 5VN328), CCR5 (pdb ID: 5UIW10) and 4D CD4 (pdb ID: 1WIO27) into the EM map in (a). N271 of CD4 (green), N234, N262 and N362 of gp120 in cyan are N-linked glycosylation sites. (c) The structure of the CD4-gp120-CCR5 complex was modeled based on a 3.9Å density map. (d) Overall structure of the 4D CD4-gp120-CCR5 complex shown in ribbon diagram. N, N-terminus; C, C-terminus; ECL2, extracellular loop 2; I, II, III, IV, V, VI, VII, transmembrane helices (TM) 1-7.
Interfaces between gp120 and CCR5
The map from the masked refinement showed good density for the gp120 and CCR5 interfaces (Fig. 1c and Extended Data Fig. 6). As postulated previously7,14, there are indeed two major contacting interfaces between gp120 and CCR5 (Fig. 1d). The V3 loop of gp120 inserts into the CRS2 of CCR5 and contacts with all the 7TM helices. The CCR5 N-terminal segment adopts an extended conformation with several sharp turns and contacts the surface of the bridging sheet of gp120, which forms only after CD4 binding2,28.
gp120 V3 loop and CCR5 CRS2.
The conserved 310GPGR(Q)313 motif at the tip of the V3 loop penetrates by approximately 1/3 of the thickness of lipid bilayer into the CRS2 pocket with the residue P311 reaching most deeply (Fig. 2a). Residues 309-316 of the V3 loop adopt a structure, including a one-turn helix, similar to that of the N-terminus (residues 1-8) of [5P7]CCL5 (ref10; Extended Data Fig. 7a). Both structures have a proline residue (P311 of gp120 and P3 of [5P7]CCL5) that reaches the bottom of the CRS2, packing against the side chains of W86 and Y108 in CCR5 (Fig. 2a and Extended Data Fig. 7a). R313 in V3 appears to be sandwiched between Y251 and E283 of CCR5. The CRS2 of chemokine receptors has minor and major subpockets, formed by TM helices I-III, VII; and III-VII, respectively10. The V3 loop of this HIV-1 strain occupies mainly the minor subpocket and just part of the major subpocket (Fig. 3a), leaving room in the CRS2 to accommodate different V3 sequences from other viral isolates. Like [5P7]CCL5, different V3 loops may also have water-mediated interactions with CCR510. The ECL2 of CCR5 forms a nearly semi-circular grip as it wraps around V3, making contacts with residues in both the V3 stem and crown29. Particularly, E172 in ECL2 and R304 in V3 probably form a salt bridge (Fig. 2a). Overall, the gp120 V3 makes extensive contacts with the CRS2 of CCR5, contributing to the high affinity of HIV-1 Env for its coreceptor.
Figure 2. Interfaces between gp120 and CCR5.
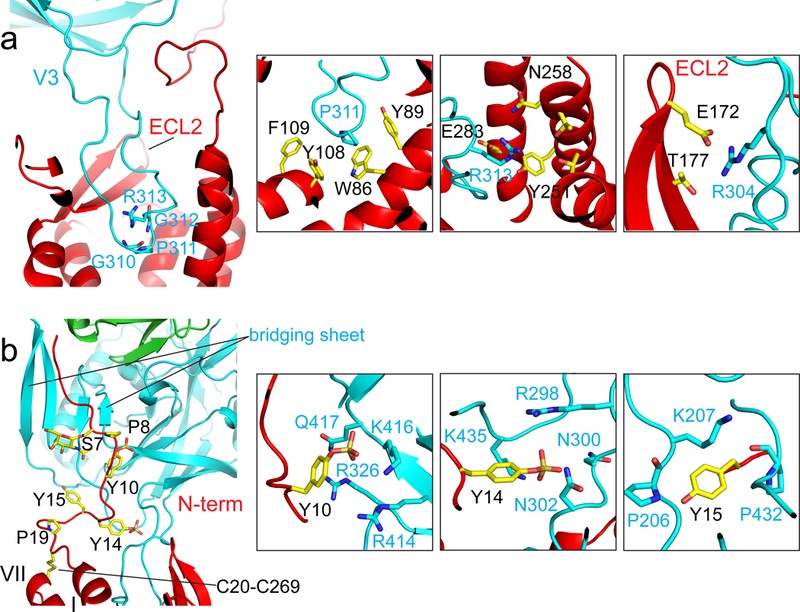
(a) Interactions between the V3 loop of gp120 (cyan) and the CRS2 of CCR5 (red). Left, ribbon diagram of V3 inserting into the CRS2. The GPGR motif of V3 is in stick model. Right, major contacts between residues Pro311, Arg313 and Arg304 of gp120 in cyan and those from CCR5. (b) Interactions between the CCR5 N-terminus (red) and the bridging sheet of gp120 (cyan). Left, overall view of the CCR5 N-terminus attaching to the four-stranded bridging sheet formed by the V1V2 stem and β21-β22 of gp120. Residues Ser7, Pro8, sulfated Tyr10 and Tyr14, Tyr15, Pro19, the O-linked glycan at Ser7, the disulfide between Cys20 and Cys269 of CCR5 are in stick model. Right, major contacts between sulfated Tyr10 and Tyr14, as well as Tyr15 of CCR5 and residues from gp120.
Figure 3. Conformational differences between gp120-bound and other liganded CCR5s.
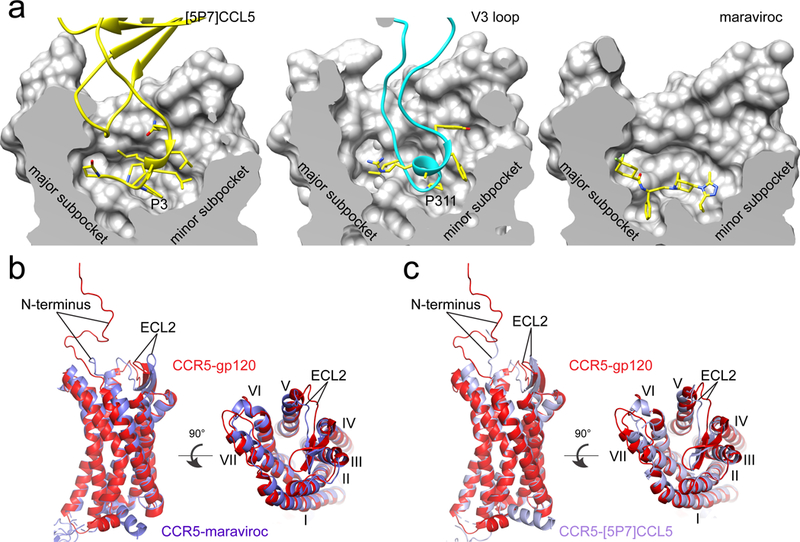
(a) The section of the CCR5 CRS2 (divided into a major and a minor subpocket) is shown in surface representation for the [5P7]CCL5, gp120 and maraviroc complexes, respectively. Interacting residues, including Pro3 from [5P7]CCL5, Pro311 from the V3 loop, and compound maraviroc, are in stick model. (b) Superposition of the structures of the gp120-CCR5 complex (red) and the maraviroc-CCR5 complex (blue). N-terminus, ELC2 and seven TM helices (I, II, III, IV, V, VI, VII) are indicated. (c) Superposition of the structures of the gp120-CCR5 complex (red) and the [5P7]CCL5-CCR5 complex (blue).
gp120 bridging sheet and CCR5 N-terminus.
The second major interface between Env and its coreceptor is formed by the N-terminus of CCR5 and the bridging sheet of gp120. Helices I and VII of CCR5 are locked by a disulfide (Cys20-Cys269), and the N-terminal segment (residues 1-19), connected to the helix I, is largely disordered in the crystal structures of CCR5 complexed with other ligands7,10. In the CD4-gp120-CCR5 complex, the N-terminal segment adopts an extended conformation with several sharp turns, attaching to the surface of the bridging sheet, formed by the V1V2 stem and the β20-β21 hairpin upon CD4 binding (Fig. 2b). Three tyrosine residues, Tyr10, Tyr14, and Tyr15, of CCR5, all of which can be sulfated21, make the most intimate contacts with gp120. There appears to be density and room for sulfate groups on Tyr10 and Tyr14, but not on Tyr15 (Extended Data Fig. 6). The aromatic ring of sulfated Tyr10 packs against the side chain of Arg326 in gp120 probably by a cation-π interaction30, placing the putative sulfate group near Lys416 and Arg414 of gp120. Sulfated Tyr14 wedges between the bases of both the V3 stem and bridging sheet, positioning its sulfate group near the side chains of Arg 298 and Lys 435 of gp120. The side chain of Tyr15 is close to Lys207 at the base of the V1V2 stem, potentially forming another cation-π interaction. Sulfated Tyr10 and Tyr14 mimic the interactions with gp120 of two sulfated tyrosines Tyr100 and Tyr100c in antibody 412d (ref20; Extended Data Fig. 7b). There also appears to be an O-linked glycan at Ser7, a glycosylation site identified previously31, and the carbohydrate may help maintain the configuration of the N-terminal region of CCR5.
One unexpected feature of the complex structure is the orientation of CD4 relative to the 7TM helices of CCR5. The long axis of CD4 lies almost perpendicular to the axis of the CCR5 TM helices and roughly parallel to the plane of lipid bilayer, raising the possibility that formation of the ternary complex could induce a local bend in the membrane. Although binding of an Env trimer to three copies each of CD4 and CCR5 simultaneously is stereochemically possible (Extended Data Fig. 7c), it may be an inefficient process if such a stoichiometry is needed to activate gp41.
Conformational differences between gp120-bound and other liganded CCR5s
There is no published structure for unliganded CCR5, which may be conformationally flexible, sampling a range of conformations even in its native membrane environment32. In a complex with CCR5 stabilized by specific mutations, maraviroc binds in a hydrophobic pocket near the bottom of CRS2; it has been thought to inhibit HIV-1 infection by stabilizing a conformation that Env cannot recognize33. We find, however, that the overall dimension and shape of the gp120-bound CRS2 pocket from the wildtype CCR5 are not very different from those of other liganded CCR5s with stabilizing modifications (Fig. 3a). Comparison of the structures of the CCR5-maraviroc and CCR5-gp120 complexes indicates that the parts of all TM helices near the intracellular side, including TM6, which is critical for GPCR activation34, show few differences, indicating that the gp120-bound CCR5 also adopts an inactive conformation (Extended Data Fig. 7d). Indeed, binding of the gp120-CD4 complex did not activate the G protein-signaling pathways (Extended Data Fig. 2d and 2e). The parts near the extracellular surface move outwards by 1-3Å to accommodate the inserted V3 loop (Fig. 3b). The largest changes are in the long β-hairpin of ECL2, where part of its body moves outwards by 2-3Å while its tip moves inwards by >5Å tightening the grip on the V3 loop. These conformational differences are unlikely to block V3 access to the CRS2 pocket, however. In addition, the binding site of maraviroc partially overlaps with that of the V3 tip, suggesting that the drug blocks gp120 binding by direct competition, not by restricting conformational availability. Moreover, the antagonist ([5P7]CCL5) bound CCR5 with stabilizing mutations also adopts a conformation very similar to the gp120-bound wildtype CCR5, suggesting that the conformational freedom of CCR5 TM helices is limited, at least in the liganded, inactive forms (Fig. 3c).
Conformational differences between CD4- and CCR5-bound gp120s
The most unexpected aspect of the CD4-gp120-CCR5 structure is lack of obvious allosteric changes that can propagate from the CCR5 binding site to gp41, as previously hypothesized. While CD4 and/or CD4i antibody-induced conformational changes in the SOSIP-based Env trimer have been described in great detail recently28, comparison of the CD4- and CCR5-bound gp120 with the CD4-bound gp120 shows no major differences in the gp120 core region (Fig. 4a and Extended Data Fig. 8a). In particular, a ~50Å zone between the CCR5 binding site and the gp120-gp41 interface, including inner and outer domains and bridging sheet, remains almost invariant, suggesting that CCR5 does not induce any major structural changes affecting gp41. There are, however, some differences between the two structures. First, when CCR5 binds, the V3 loop reconfigures in order to fit into the CRS2 pocket (Fig. 3a). This conformation, partially mimicking that of the N-terminus of [5P7]CCL5, has not been seen for either unliganded or antibody-bound V3 loops (ref20,35-38; Extended Data Fig. 8b). Second, a more significant difference is in the region including the N- and C-termini of gp120 near its interface with gp41. In the prefusion SOSIP trimer structure, the gp120 termini are surrounded by a so-called “4-helix collar” of gp41, which closes up by insertion of the side chain of Met530 on one of the helices (α6) into a hydrophobic clasp formed by three tryptophan residues on two other helices (α8 and α9)5. CD4 binding induces a shift of α6, allowing the fusion peptide to pack directly against the gp120 termini (ref28; Fig. 4b). In the CD4-gp120-CCR5 complex, the N- and C-termini bend back at pivot regions containing the highly conserved 40GVP42 and 489PLG491 sequences, respectively. In particular, the N-terminus rotates almost by ~180° to pack against the surface of gp120 and occupies the space of the fusion peptide in the Env trimer (Fig. 4b). There is no obvious density for the rest of the C-terminus, which is probably disordered, although the histidine tag could have influenced its conformation.
Figure 4. Conformational differences between CD4-bound and CCR5-bound gp120s.
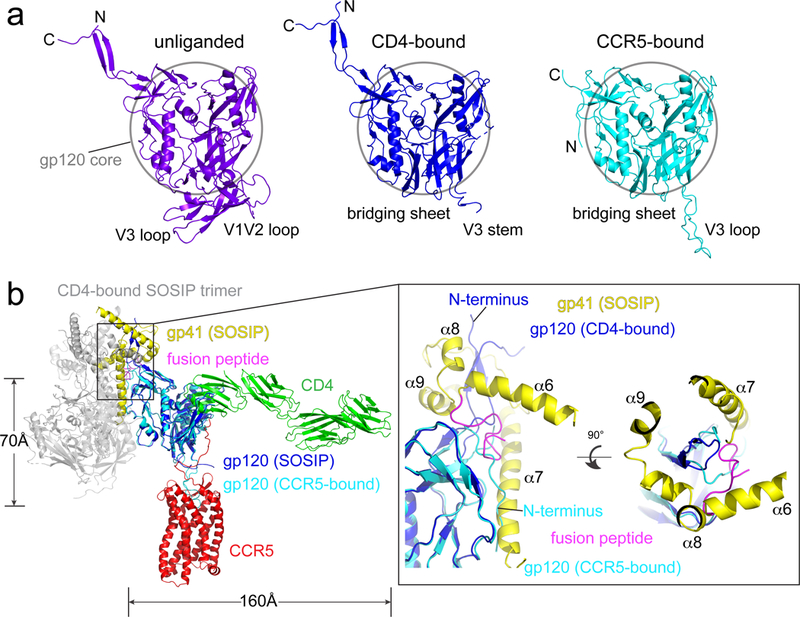
(a) Comparison of structures of gp120 in the unliganded SOSIP Env trimer (pdb ID: 4ZMJ35; purple), in the CD4-bound SOSIP trimer (pdb ID:5VN328; blue) and in complex with CD4 and CCR5 (cyan). A 50Å-circle marks the gp120 core region. (b) Superposition of structures of the CD4-gp120-CCR5 complex and the CD4-bound SOSIP trimer. Left, the two structures are superposed by the gp120 core region and the first two domains of CD4. CCR5-bound gp120 is in cyan, CCR5 in red, CD4 in green; one of the gp120s from the trimer in blue, the corresponding gp41 in yellow, except for its fusion peptide in magenta, the rest of the SOSIP trimer in gray. The distances between the fusion peptide and the TM domains of CD4 and CCR5 are 160Å and 70Å, respectively. Right, close-up views of the gp120 N- and C-terminal region. Four helices (α6, α7, α8 and α9) of gp41 forming the 4-helix collar are indicated. The N-terminus of the CCR5-bound gp120 overlaps with the fusion peptide in the CD4-bound trimer.
Discussion
A model for how CCR5 functions as an HIV-1 coreceptor
Since CCR5 binding does not appear to induce any allosteric changes that can unleash gp41 to fuse membranes, how does it function as an essential coreceptor? Based on the known structures of the HIV-1 Env trimer3-5,28,39, the prefusion gp41, which wraps around the N- and C-termini of gp120, is no longer stable and likely to enter an irreversible refolding process once gp120 dissociates. Thus, gp120 dissociation may be the crucial “trigger” that initiates gp41 refolding events, including insertion of its fusion peptide into the target membrane and formation of the six-helix bundle postfusion conformation. In the prefusion conformation, the gp120 N- and C-termini are gripped by the 4-helix collar of gp415. CD4 binding leads to a large shift of the C-terminus of helix α6 away from the gp120 termini, creating a pocket, filled by the fusion peptide28, which packs against the pivot region (40GVP42 ) of the gp120 N-terminus (Fig. 4b). When intrinsic conformational dynamics cause the fusion peptide to dissociate from the pocket, it will open up one side of the gp41 grip, and the gp120 N-terminus can then bend back to adopt the conformation observed in the CCR5-bound structure (Extended Data Fig. 9). The rearrangements of the gp120 termini, probably independent of CCR5 binding, can prevent the fusion peptide from reoccupying the pocket and effectively weaken gp120-gp41 interactions, possibly leading to complete dissociation. Indeed, spontaneous or CD4-induced gp120 shedding from Env trimers are well documented for many HIV-1 isolates40,41, indicating that gp120 is prone to dissociation from gp41 even in the absence of a coreceptor. We note that the impact of the membranes and Env trimer organization remains unknown and will require further investigation.
If the rearrangement of gp120 termini to activate gp41 does not depend on CCR5 binding, then why would a coreceptor still be needed? First, premature gp120 dissociation, in absence of a coreceptor, would be non-productive; for a virion attached to the target cell surface only through an Env trimer-CD4 contact, the distance between the fusion peptide and membrane surface can be ~160Å (Fig. 4b). If gp120 dissociates, the fusion peptide would be too far from reaching the target membrane. Binding of gp120 to CCR5 can bring the fusion peptide of gp41 within 70Å (Fig. 4b), consistent with the distance needed for the fusion peptide to translocate and reach the target membrane5. Second, gp120-CD4 association, measured by single-molecule force spectroscopy with infectious virions and live host cells, is unstable and rapidly reversible unless CCR5 binding immediately follows42,43. CCR5 is therefore needed to stabilize the CD4-induced conformational changes, which are already competent for promoting fusion. In particular, tucking away V3 by CCR5 will prevent the Env trimer from moving back to the prefusion conformation and help shift the equilibrium towards the irreversible step - gp41 refolding. Third, membrane fusion may require more than one Env trimer to induce fusion pore formation44, as shown for other viral fusion proteins45. Since the number of Env trimers on the virion is low (~14/virion; ref46), a long lifetime of Env-receptor complex would be important for recruiting additional, CD4- and coreceptor-primed trimers. Thus our structure shows how coreceptor can be essential for membrane fusion, even though it does not actively induce gp41 refolding.
Coreceptor switch
The switch from CCR5 to CXCR4 is often associated with an accelerated increase in viral load and decrease in CD4+ T cells, as well as with faster disease progression12,13. Our structure supports a model for how simple mutations in Env can achieve this seemingly complicated transition. First, the coreceptor is required only to stabilize the CD4-induced conformation, not to trigger additional allosteric changes in Env through specific interactions. The switch can thus be accomplished if the V3 loop gains sufficient affinity for CXCR4 and does not release the coreceptor, as no specific mutations in Env are needed to make it “triggerable” by CXCR4. Second, the overall dimensions of the CRS2 pocket in the gp120-bound CCR5 and the liganded CXCR4 are very similar (ref14,15; Extended Data Fig. 1), suggesting that changes of the surface-exposed residues in V3 to make it compatible with CXCR4 binding would be sufficient. Use of both coreceptors by R5X4 isolates further underscores the similarities between the two11. Third, the main contacts between gp120 and the CCR5 N-terminus are electrostatic. CXCR4 has seven acidic residues in its N-terminus (before the first disulfide-forming Cys residue) while CCR5 has four, in addition to two or three sulfotyrosines. No additional mutations in the bridging sheet region would be needed if the extra acidic residues in CXCR4 can replace the two critical sulfated tyrosines in CCR5. Finally, X4 V3 loops generally have more positive charges than those of R5 viruses, consistent with a more negatively charged CRS2 in CXCR4 than CCR5. Evolution from CCR5 to CXCR4 usage indeed can be achieved by multiple mutational pathways, but often with gaining net positive charges in the V3 region47-49.
CCR5 antagonist-based therapeutics
The V3 binding site only partially overlaps with the minor subpocket of the maraviroc binding site (Fig. 3a). Maraviroc-resistant viruses can emerge either in infected individuals under the treatment or by in vitro selection. Major changes in the escaped viruses map to the V3 region, some with a three-residue deletion, but they show no consistent patterns50-52. Some resistant viruses can infect by recognizing the drug-bound CCR550,53. In addition, replication-competent HIV-2 viruses have been selected that lack the entire V3 loop54. They can use both coreceptors for entry by gaining additional positively charged residues near the bridging sheet to enhance interactions with the coreceptor N-terminus. These data indicate that no specific structural determinants in V3 are required for the coreceptor to function, fully consistent with our conclusion that CCR5 does not actively trigger gp41 through specific interactions with gp120
Our structure also suggests a general strategy for how to improve maraviroc-like therapeutics. Since the V3 loop mainly overlaps with maraviroc in the minor subpocket, which is primarily occupied by the triazole group of the compound, additional groups may be added to the triazole ring to enhance its competing power with the V3 loop and increase the barrier to drug resistance.
Methods
Constructs and stable cell lines
The gene of the intact human C-C chemokine receptor type 5 (CCR5; NCBI Reference Sequence: NP_000570.1) was cloned into pCMV-IRES-puro vector (Codex BioSolutions, Inc, Gaithersburg, MD). Genes of HIV-1 gp120 (residues 1-507) from the isolate 92BR020 with a C-terminal 6xhistidine tag, and of 4 domain CD4 (residues 1-388) with a C-terminal twin strep tag [(GGGGS)2WSHPQFEK(GGGGS)2WSHPQFEK)] were synthesized by GenScript (Piscataway, NJ) and cloned into pCMV-IRES-puro vector. 293T cell lines (Thermo Fisher Scientific, Waltham, MA) stably transfected with these constructs were generated either in-house or at Codex Biosolutions. Briefly, 8×105 HEK-293T cells in 2 ml of DMEM containing 10% FBS and no antibiotics were seeded on a 6 well-plate and incubated for overnight. The cells were then transfected with the expression constructs using DNA-In® 293 Transfection Reagent (MTI-GlobalStem, Gaithersburg, MD), following a protocol recommended by manufacturer. 24 hours post-transfection, the cells were transferred to a medium containing DMEM, 10% FBS and 1 μg/ml puromycin for selection. Single colonies were picked in 2-3 weeks, and transferred to 24-well plates in the same selective medium. Protein expression was confirmed by both western blot and a fluorescence-activated cell sorting (FACS) assay (see below). Positive clones were expanded, frozen and stored in liquid nitrogen. To grow cells in large-scale in suspension, we also generated stable cell lines expressing CCR5 with Expi293F cells (Thermo Fisher Scientific). Hybridoma cells for production of an anti-V3 antibody 447-52D55 was kindly provided by Dr. Susan Zolla-Pazner, New York University.
Purification of recombinant proteins
HIV-1 gp120 of the isolate 92BR020.
Cells expressing C-terminal his-tagged 92BR020 gp120 were grown in 250 ml roller bottles with DMEM containing 10% FBS and 1 μg/ml puromycin. The protein was purified by affinity chromatography using Ni-NTA agarose (Qiagen, Hilden, Germany), followed by gel filtration chromatography, as described previously56,57. The peak fractions were pooled and concentrated to 10 mg/ml using a 10 kDa MWCO Millipore filter (MilliporeSigma, Burlington, MA).
Soluble CD4.
Cells expressing strep-tagged CD4 were grown in 250 ml roller bottles with DMEM containing 10% FBS and 1 μg/ml puromycin. Once the cells reached ~70% confluence, the medium was replaced with 293T serum free expression medium. After 5 days, cell supernatants were harvested and loaded onto a Strep-Tactin Sepharose (IBA Lifesciences, Goettingen, Germany) column. The column was then washed with 100 mM Tris-HCl, pH 8.0 and 150 mM NaCl. The protein was eluted with 100 mM Tris-HCl, pH 8.0, 150 mM NaCl and 2.5 mM desthiobiotin (IBA). Eluted fractions were analyzed by SDS-PAGE, and peak fractions containing CD4 were pooled and dialyzed against 100 mM Tris-HCl, pH 8.0 and 150 mM NaCl using a dialysis tubing with 6-8 kDa MWCO (Spectrum Laboratories, Inc., Rancho Dominguez, CA). The protein was further purified by gel filtration chromatography using a Superdex 200 column (GE healthcare, Chicago, IL) in a buffer containing 30 mM Tris-HCl, pH 8.0, and 150 mM NaCl. The peak fractions were pooled and concentrated to 10 mg/ml using 10 kDa MWCO Millipore filters.
Anti-V3 antibody 447-52D.
The hybridoma cells expressing anti-V3 antibody 447-52D were gown in roller bottles with RPMI medium supplemented with ultra low IgG FBS (Thermo Fisher Scientific). The antibody was purified by affinity chromatography using GammaBind Plus Sepharose (GE Healthcare) as described previously57,58. The eluted fractions were analyzed by SDS-PAGE and those containing the antibody were pooled, concentrated, frozen in liquid nitrogen and stored at -80 °C.
Western blot and flow cytometry
Western blot was performed using an anti-CCR5 antibody following a protocol described previously24. To test CCR5 cell-surface expression and its binding to gp120, flow cytometry was carried out as described previously24,59. Briefly, CCR5 was detected by PE Mouse antihuman CD195 (BD Biosciences, San Jose, CA) and bound-gp120 was stained with Anti-His tagged PE conjugated Mouse IgG (R&D Systems, Minneapolis, MN). Extensive washing of the gp120-bound cells with PBS or leaving them in PBS up to 4 hours did not change the fluorescence signals, suggesting that gp120 dissociates very slowly from CCR5 on the cell surfaces. Control experiments were carried out to ensure the binding specificity and all experiments were repeated at least three times with almost identical results.
CCR5 functional assays
When CCR5 is activated by its chemokine ligands, its intracellular regions can interact mainly with the inhibitory heterotrimeric G protein Gαi and coupling of CCR5 to Gαi reduces the intracellular concentration of the secondary messenger cyclic AMP (cAMP)60. We measured changes in the intracellular cAMP concentration in these cells when CCR5 was activated by chemokine RANTES using ACTOne technology (ref61; Codex Biosolutions). Briefly, 293 cells or CCR5 expressing cells were transfected with pcDNA3.2-cyclic nucleotide-gated (CNG) channel gene using Lipofectamine 3000 reagent (Thermo Fisher Scientifics). ~24 hours posttransfection, the cells were transferred to a 384-well black clear plate at the density of 1.2×104 cells/well in 20 μl culture medium. On day 3, the cells were treated with different concentrations of CCL5/RANTES, CD4, gp120 or the gp120-CD4 complex in the presence of 25 μM Ro20-1724 (4-(3-Butoxy-4-methoxybenzyl)imidazolidin-2-one; a phosphodiesterase 4 inhibitor) and 500 nM 5’-N-Ethylcarboxamidoadenosine (NECA; an endogenous adenosine A2b receptor agonist that increases cellular cAMP levels). Changes in the cAMP concentration in live cells directly correlate with the ion flux through CNG channel, which was measured by a fluorescent plate reader Hamamatsu FDSS 7000 after staining with the ACTOne membrane potential dye (Codex Biosolutions). Similar results were obtained for both the 293T-CCR5 and Expi293F-CCR5 stable cell lines.
It has been reported that HIV-1 gp120 or viral particles can induce CCR5 or CXCR4 cell signaling measured as calcium flux in primary unstimulated CD4+ T cells62. To test calcium flux in our 293T-CCR5 stable cells in response to gp120, we seeded the cells in a 384-well black clear plate at the density of 1.5×104 cells/well in 20 μl culture medium. On the second day, 20 μl of 1x Non-Wash Calcium Dye solution (Codex BioSolutions) was added to each well. The plate was then incubated in a CO2 incubator at 37°C for one hour. The different concentrations of various ligands were prepared in 1x Hank’s balanced salt solution (HBSS) with 20 mM HEPES (pH 7.4) (at the 5x of the final concentration). The fluorescent intensity of each well was recorded on the FDSS 7000 reader at a rate of 1 image/sec with an excitation wavelength of 480 nM and an emission wavelength of 540 nM. The base line of each well was recorded for 10 seconds before addition of 10 μl of a prepared ligand at 5x of the final concentration. Fluorescent intensity was recorded for additional 390 seconds.
Purification of the CD4-gp120-CCR5 complex
We first screened many different HIV-1 gp120s for binding to the cell-surface-expressed CCR5 in the presence of soluble CD4 by flow cytometry and small-scale purification in detergent. The 92BR020 gp120 was chosen based on not only the stability of its CCR5 complex on the cell surfaces, but also the stability of the purified complex in detergent. For large-scale protein purification, we grew Expi293F stable cells stably transfected with the CCR5 expression construct in suspension. The cells were harvested at the density of ~5-7×106/ml by centrifugation (4,000 rpm at 4°C for 30 min). The cell pellets were washed with PBS, re-suspended in PBS supplemented with 1% BSA, and incubated with the preformed CD4-gp120 complex for one and half hour at 4°C. The cells were then washed three times with PBS, resuspended in a lysis buffer containing100 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% lauryl maltose neopentyl glycol (w/v) (LMNG, Anatrace, Maumee, OH), 0.2 % cholesteryl hemisuccinate (w/v) (CHS, Anatrace), EDTA-free complete protease inhibitor cocktail (Roche, Basel, Switzerland), and incubated at 4°C for one hour. The supernatants were collected after centrifugation (18,000 rpm at 4°C for 60 min), and then loaded on a strep-tactin column equilibrated with the lysis buffer. The column was washed with 5 column volumes of a washing buffer (100 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.5% LMNG (w/v), 0.04 % CHS (w/v)), followed by 50 column volumes of another washing buffer (100 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.001% LMNG (w/v), 0.1% n-Dodecyl-β-D-Maltopyranoside (DDM, Anatrace) (w/v), 0.04 % CHS (w/v)). The CCR5 complex was eluted by an elution buffer containing 100 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.001% LMNG (w/v), 0.1% DDM (w/v), 0.04 % CHS (w/v), and 5 mM desthiobiotin. Elution fractions were analyzed by SDS-PAGE and those containing the CCR5 complex were pooled.
The anti-V3 antibody 447-52D only binds the CD4-gp120 complex with the V3 loop exposed, but not the CD4-gp120-CCR5 complex with the V3 protected by possible interactions with the coreceptor. To remove extra CD4-gp120 complex, 447-52D was loaded onto the GammaBind Plus column, which was subsequently washed with 50 column volumes of PBS to remove unbound antibody. The column was then equilibrated with the elution buffer for the strep-tactin column. The pooled fractions containing the CCR5 complex from the strep-tag purification were loaded onto the 447-52D column. The flowthrough was collected and then concentrated using an Amicon Ultra Centrifugal Filter (MWCO 100 kDa). The complex was further purified by size-exclusion chromatography on a Sepharose 6 10/300 column (GE Healthcare) in a buffer containing 100 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.001% LMNG (w/v), 0.025% DDM (w/v), and 0.04 % CHS (w/v). Eluted fractions containing the CCR5 complex were pooled and concentrated. The complex could be concentrated at least 3 mg/ml without any aggregation on a size-exclusion column.
Negative stain EM
To prepare grids, 2.5 μl of the freshly purified CCR5 complex was adsorbed to a glow-discharged carbon-coated copper grid, washed with deionized water, and stained with freshly prepared 0.75% uranyl formate. Images were recorded at room temperature with a magnification of 52,000x and a defocus value of 1.5 μm following low-dose procedures, using a Philips Tecnai F20 electron microscope (Thermo Fisher Scientific) equipped with a Gatan US4000 CCD camera and operated at voltage of 200 kV. Particles were picked manually and 2D class averages were generated by using EMAN2 software package63. These 2D averages were used to generate a 3D initial model starting from a density of isotropic Gaussian distribution64.
Cryo-EM sample preparation and data collection
To prepare cryo grids, 3 μl of the freshly purified CD4-gp120-CCR5 complex at 1.0 mg/ml was applied to a 1.2/1.3 Quantifoil grid (Quantifoil Micro Tools GmbH, Großlöbichau, Germany), which had been glow-discharged for 90 sec at 20 mA. Grids were immediately plunge-frozen in liquid ethane using a Vitrobot (Thermo Fisher Scientific) with a blotting time of 4 sec. The grids were first screened for ice thickness and particle distribution using a Talos Arctica transmission electron microscope (Thermo Fisher Scientific) operated at 200 kV and equipped with a K2 Summit direct detector (Gatan). For data collection, images were acquired with selected grids using a Titan Krios transmission electron microscope (Thermo Fisher Scientific) operated at 300 kV with a K2 detector. Automated data collection was carried out using SerialEM65 at a magnification of 130,000× and the K2 detector in super-resolution counting mode (pixel size: 0.529Å) at a dose rate of ~6 electrons/physical pixels/s. Each movie had a total accumulated exposure of ~46 e/Å2 fractionated in 35 frames of 200 ms. Total of four datasets were acquired in different sessions using a defocus range of 1.0-2.8 μm.
Image processing and 3D reconstructions
For cryo-EM data, drift correction was performed using MotionCor266 and images were binned 2×2 by Fourier cropping to a pixel size of 1.059Å. The contrast transfer function (CTF) was estimated by CTFFIND467 using motion-corrected sums without dose-weighting. Motion-corrected sums with dose-weighting were used for all other image processing. RELION 2.126 was used for particle picking, 2D classification, 3D classification and refinement procedures. Around 2,000 particles were manually picked and classified by 2D classification to generate the templates for automatic particle picking. After the manual inspection of auto-picked particles, a total of 1,707,675 particles were extracted from 9,776 selected images (out of 10,530 movie stacks in total collected in four sessions). These particles were subjected to 2D classification in three groups, giving a total of 1,546,032 particles. The low-resolution negative stain reconstruction of the complex was low-pass filtered to 60Å resolution, and used as the initial model for 3D classification. A total of 691,508 particles from 3D classes that show clear structural features were combined and subjected to 3D refinement which led to a reconstruction at 4.6Å resolution. Local refinement with a mask to exclude the last two domains (D3-D4) of CD4 improved the overall resolution to 4.0Å. Cryo-EM particles were then subjected to 3D classification with signal subtraction, focusing on gp120, the first domain of CD4 and half of the CCR5. The best class with the highest resolution contained 307,346 particles and 3D refinement produced a map at 4.5Å resolution. Further local refinement using a mask to exclude the last two domains of CD4 led to a final map at 3.9Å resolution.
Reported resolutions are based on the gold-standard Fourier shell correlation (FSC) using the 0.143 criterion, calculated with “SAM (Simplified Application Managing Utilities for EM Labs) scripts of the Liao lab (https://liao.hms.harvard.edu/samuel). All density maps were corrected for the modulation transfer function (MTF) of the K2 summit direct detector and then sharpened by applying temperature factor that was estimated using post-processing in RELION. Local resolution was determined using LocalRes in RELION with half-reconstructions as input maps.
Model building
The initial model of gp120 was a homology model calculated by I-TASSER68, using the cryo-EM structure of gp120 from the CD4-bound SOSIP trimer (pdb ID: 5VN3) as a template. The crystal structures of CD4 (pdb ID: 1WIO) and CCR5 (pdb ID: 5WIU) were also used as initial templates for model building. Several rounds of manual building were performed in coot69. The model was finalized by refinement in Phenix70 against the 3.9Å cryoEM map. The refinement statistics are summarized in Extended Data Table 1.
Extended Data
Extended Data Figure 1. Known structures of CCR5 and CXCR4.
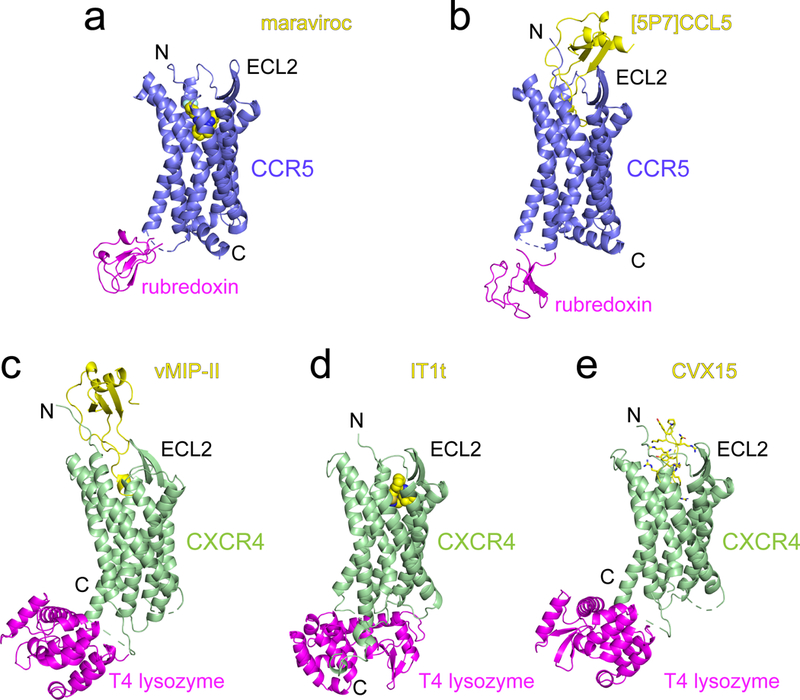
CCR5 and CXCR4 were identified as the coreceptors for HIV-1 entry in 199671-77. (a)-(b) Crystal structures of a modified CCR5 (ΔC224-N226→rubredoxin; ΔF320-L352; and point mutations C58Y, G163N, A233D, K303E) in complex with HIV entry inhibitor maraviroc (pdb ID: 4MBS; ref7) and a modified chemokine [5P7]CCL5 (an antagonist; pdb ID: 5UIW; ref10). CCR5 is shown in ribbon diagram in blue, the internally fused rubredoxin in magenta and the ligands in yellow. N-terminus (N), C-terminus (C) and the second extracellular loop (ECL2) are indicated. (c)-(e) Crystal structures of an engineered CXCR4 in complex with a viral chemokine antagonist vMIP-II (pdb ID: 4RWS; ref15), a small molecule antagonist IT1t (pdb ID: 3ODU; ref14) and a cyclic peptide antagonist CVX15 (pdb ID: 3OE0; ref14). CXCR4 is shown in green, the fused T4 lysozyme in magenta and the ligands in yellow. N, N-terminus; C, C-terminus; and ECL2, the second extracellular loop.
Extended Data Figure 2. Characterization of stable 293 cell lines expressing wildtype human CCR5.
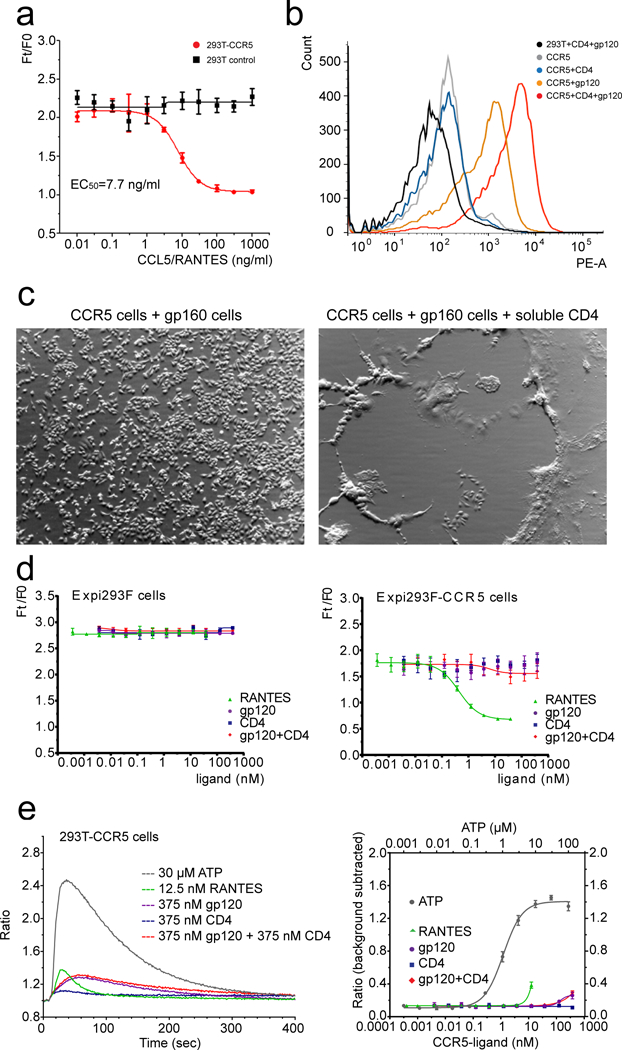
(a) Chemokine receptor assay. 293T and 293T-CCR5 (stable) cells were treated with different concentrations of CCL5/RANTES. Ft/F0 is a fluorescence-signal ratio proportional to that of intracellular cAMP concentration at 40 min post CCL5/RANTES-activation and time 0. The dose response curves were plotted for both 293T (black) and 293T-CCR5 (red) cells. The experiment was carried out in quadruplicate and repeated at least three times with similar results. Error bars indicate the standard deviation calculated by the Excel STDEV function. (b) Flow cytometry histograms of HIV-1 gp120 binding to CCR5 expressed on the cell surfaces in the absence (orange) or presence (red) of soluble CD4. 293T cells (black), CCR5-expressing cells only (gray) and CCR5-expressing cells with soluble CD4 only (blue) were negative controls. The experiment was repeated independently at least twice with similar results. (c) HIV-1 Env mediated cell-cell fusion. 293T cells stably transfected with CCR5 were mixed with HIV-1 Env (gp160) expressing cells in the absence or presence of soluble CD4. The CCR5 cells fuse with CD4-triggered Env cells very efficiently and form large syncytia that almost cover the entire well. The experiment was repeated independently twice with similar results. (d) Chemokine receptor assay by various ligands. As in (a), Expi293F and Expi293F-CCR5 (stable) cells were treated with CCL5/RANTES, gp120, CD4 or the complex of gp120 and CD4. The dose response curves were plotted for both Expi293F (control, left) and Expi293F-CCR5 (right) cells with different ligands as indicated. The experiment was carried out in quadruplicate and repeated at least three times with similar results. Error bars indicate the standard deviation calculated by the Excel STDEV function. (e) Left, kinetic curves of 5 representative wells of 293T-CCR5 cells treated with 5 different ligands as indicated. ATP activates the endogenous Gq coupled GPCR - P2Y receptor, as a positive control. Ratio, fluorescence intensity/baseline intensity. Right, dose response curve of each ligand. The y-axis is a background-subtracted ratio (peak fluorescent intensity ratio - 1). We conclude that our gp120 and gp120-CD4 do not activate G-protein mediated calcium flux at the concentrations tested here. The experiment was carried out in quadruplicate and repeated twice with similar results. Error bars indicate the standard deviation calculated by the Excel STDEV function.
Extended Data Figure 3. Purification of the CD4-gp120-CCR5 complex.
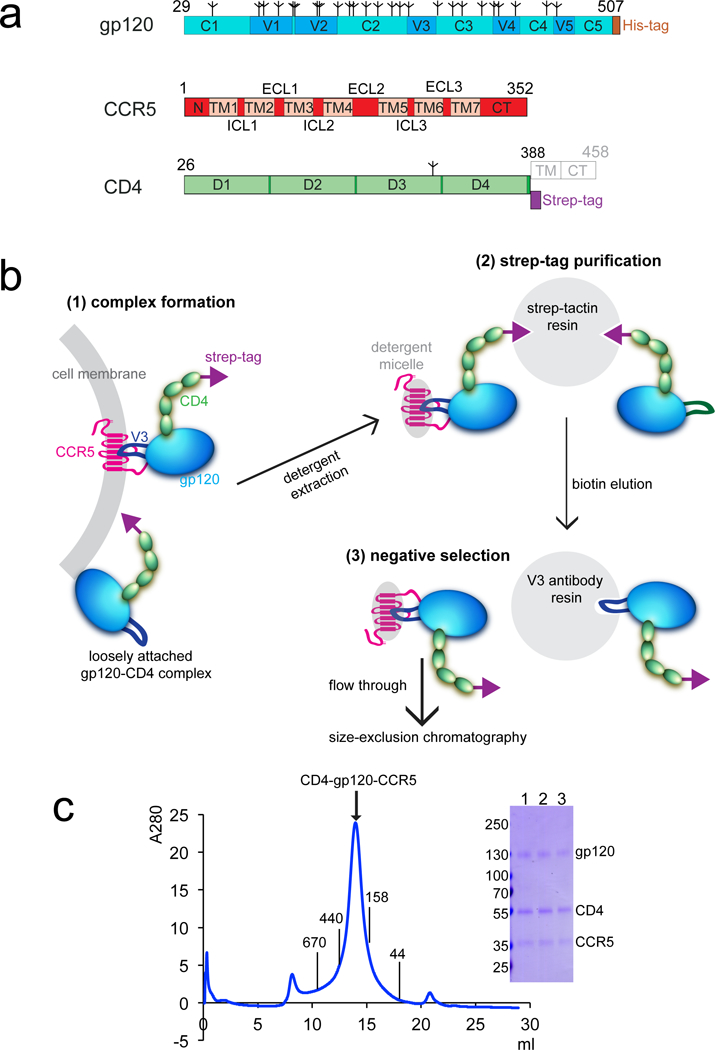
(a) Schematic representation of expression constructs for HIV-1 gp120, human CCR5 and CD4. Segments of gp120 are designated as follows: C1-C5, conserved regions 1-5; V1-V5, variable regions 1-5; and His-tag, a six-histidine tag. Tree-like symbols represent glycans. Those for CCR5 include: N, N-terminus; TM1-7, transmembrane helices 1-7; ECL1-3, extracellular loop 1-3; ICL3, intracellular loop 1-3; and CT, cytoplasmic tail. For CD4, they are: D1-4, immunoglobulin (Ig) domain 1-4; and strep tag, a purification tag. TM (transmembrane segment) and CT (cytoplasmic tail) in gray are truncated in the expression construct. (b) Unmodified human CCR5 in complex with HIV-1 gp120 and 4D CD4 was purified by the following steps: 1) complex formation, HIV-1 gp120 (green) and strep-tagged, 4 domain CD4 (light blue) were incubated with CCR5 (magenta) expressed cells to allow formation of the CD4-gp120-CCR5 complex on cell surfaces; 2) strep-tag purification, the CCR5 complex, as well as some CD4-gp120 complex, were captured to strep-tactin resin via the strep-tagged CD4 (strep tag in yellow). They were eluted by D-desthiobiotin under mild conditions; 3) negative selection by an anti-V3 antibody to remove the CD4-gp120 complex. The CCR5 complex was further purified by size-exclusion chromatography. (c) The purified CD4-gp120-CCR5 complex was resolved by gel-filtration chromatography on a Superose 6 column in the presence of detergent LMNG. The molecular weight standards include thyoglobulin (670 kDa), ferritin (440 kDa), γ-globulin (158 kDa) and ovalbumin (44 kDa). The expected size of the CCR5 complex is ~310 kDa (120 kDa for gp120, 50 kDa for 4D CD4, 40 kDa for CCR5 and ~100 kDa for LMNG micelle). Peak fractions were analyzed by Coomassie stained SDS-PAGE (lanes 1-3). Labeled bands were confirmed by western blot and protein sequencing. The experiment was repeated independently at least 15 times with similar results.
Extended Data Figure 4. Characterization of the CD4-gp120-CCR5 complex by EM.
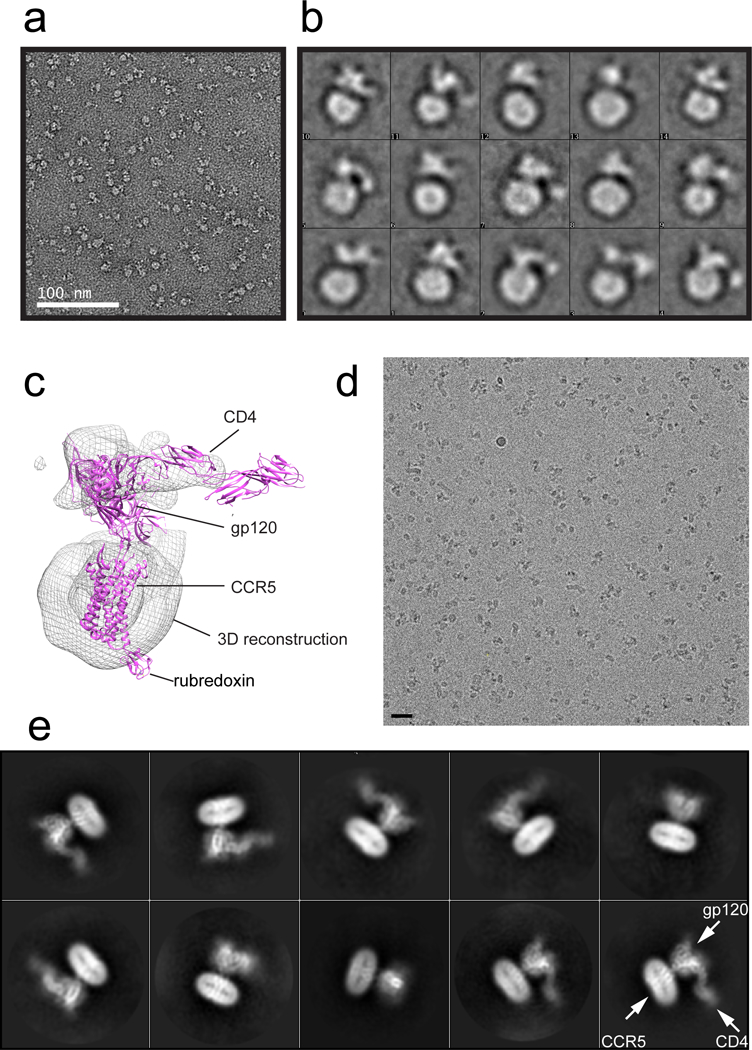
(a) Representative image of the CD4-gp120-CCR5 complex in negative stain. The experiment was repeated independently at least 4 times with similar results. (b) 2D averages of the negatively stained CD4-gp120-CCR5 complex. The box size of 2D averages is ~330Å. (c) 3D reconstruction of the negatively stained CD4-gp120-CCR5 complex, fitted with a gp120 structure containing an extended V3 loop (pdb ID: 2QAD; ref20), 4D CD4 (pdb ID: 1WIO) and CCR5 (pdb ID: 4MBS). (d) A representative cryo-EM image of the 4D CD4-gp120-CCR5 complex. The scale bar represents 25 nm. Five independent large data sets were collected with similar results. (e) 2D averages of the cryo-EM particle images show secondary structural features for both gp120 and CCR5.
Extended Data Figure 5. Single-particle cryo-EM analysis of the CD4-gp120-CCR5 complex.
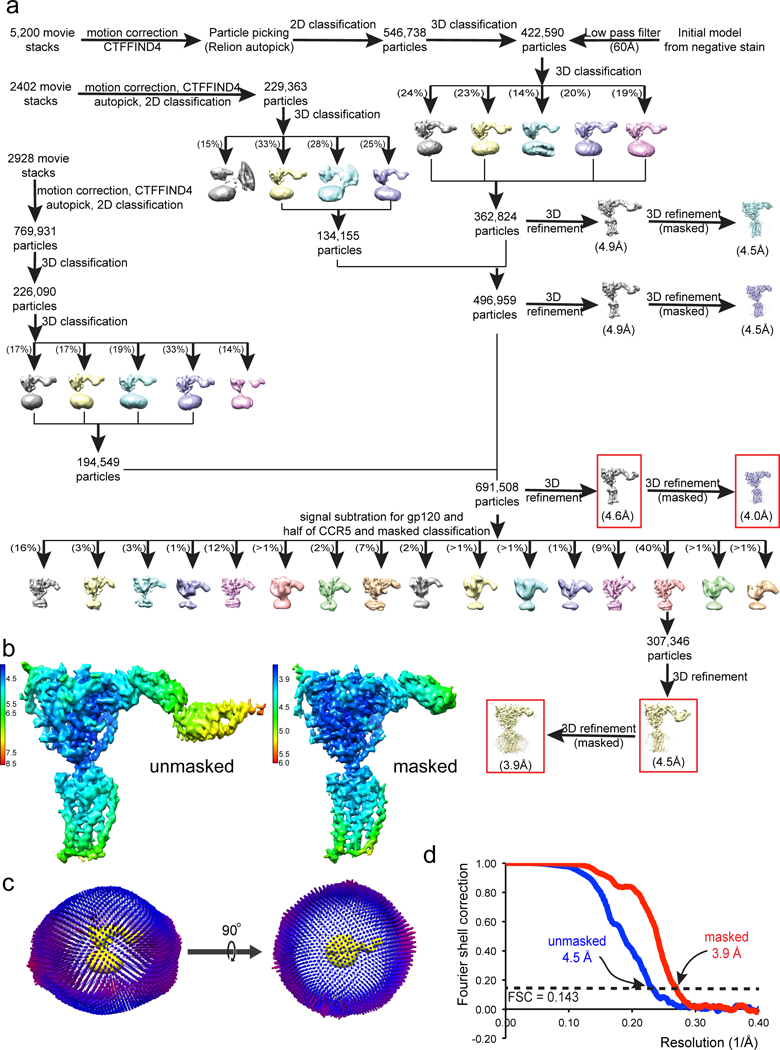
(a) Data processing workflow for the CD4-gp120-CCR5 complex. (b) 3D reconstructions of the CD4-gp120-CCR5 complex refined with no mask at an overall resolution of 4.5Å (left) and with a mask to exclude the last two domains of CD4 at a resolution of 3.9Å (right) are colored according to local resolution estimated by RELION. (c) Angular distribution of the cryo-EM particles used in the reconstruction was also shown in respect to both the side and top views of the EM map. (d) Gold standard FSC curves of the unmasked and masked EM reconstructions shown in (b).
Extended Data Figure 6. Gallery of representative density for the CD4-gp120-CCR5 complex.
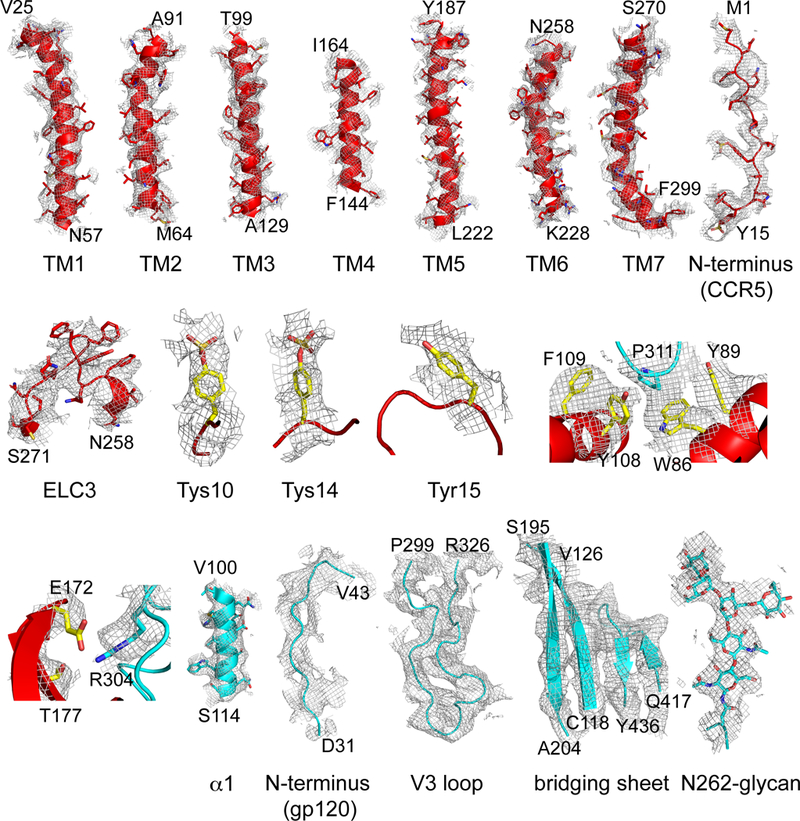
Representative density in gray mesh from the 3.9Å resolution EM map is shown for TM1-7, the N-terminus of CCR5, ELC3 near TM6, Tys10, Tys14 and Tyr15 (red model); two V3 regions; and for helix α1, N-terminus, V3 loop, the bridging sheet and N-linked glycan at Asn262 of gp120 (cyan model).
Extended Data Figure 7. Comparison of conformations of V3 loop and [5P7]CCL5 in complex with CCR5, as well as of gp120-bound CCR5 and G protein-bound β2 adrenergic receptor (β2AR).
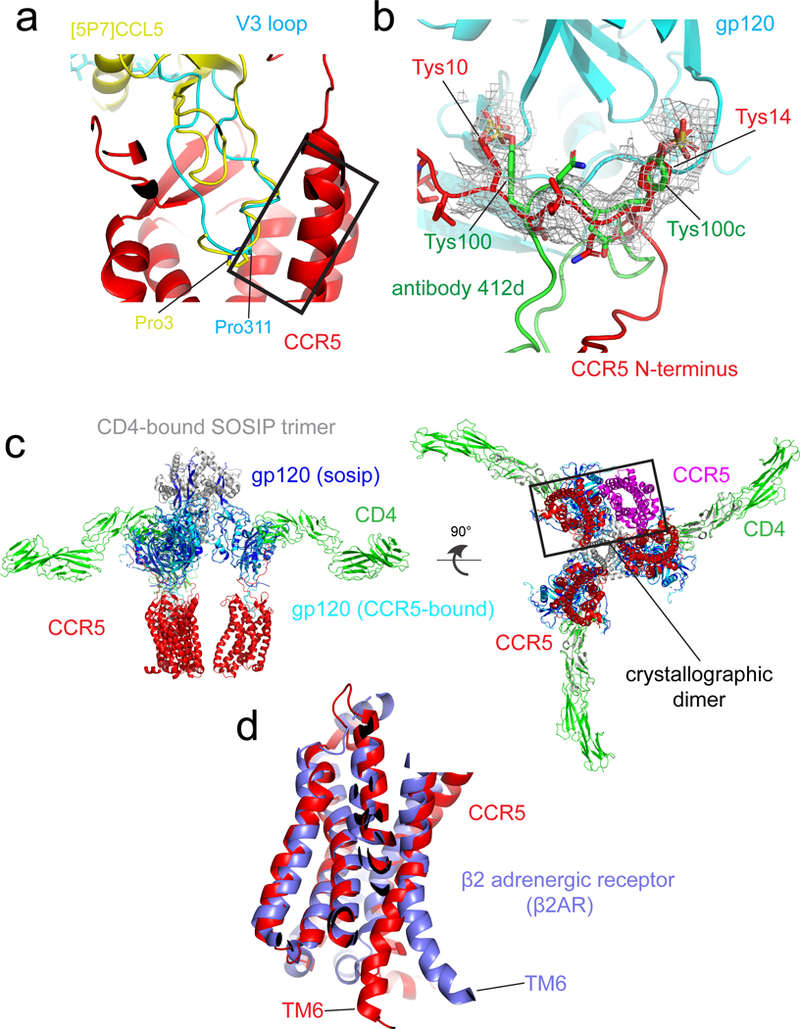
(a) The structures of the CD4-gp120-CCR5 and [5P7]CCL5-CCR5 complexes are superposed on CCR5 (red). Gp120 V3 loop with its Pro311 in stick model is in cyan and [5P7]CCL5 with its Pro3 in stick model in yellow. Residues 309-316 of the V3 loop and residues 1-8 of [5P7]CCL5 adopt a very similar structure are highlighted in a rectangular box. (b) Superposition of the structures of the N-terminus of the gp120-bound CCR5 (red) and the CDR H3 loop of antibody 412d in complex with gp120 core (green). The EM density of the CD4-gp120-CCR5 is shown in gray. The positions of the sulfated tyrosine (Tys) residues, including Tys10 and Tys14 from CCR5; Tys100 and Tys100c from 412d, are indicated. (c) A model for interactions of three CD4 and three CCR5 with the SOSIP Env trimer. The side and bottom views of a composite structure of the CD4-CCR5-SOSIP Env trimer complex are shown. The model was generated using the CD4-bound SOSIP trimer (pdb ID:5VN3) and the structure of the CD4-gp120-CCR5 complex from this study. All the structures were aligned based on the gp120 core region. CCR5 is shown in red, CD4 in green, gp120 in blue and gp120 of SOSIP dark blue; gp41 of SOSIP gray. The crystallographic dimer of CCR5 was also shown on left only in a rectangular box using pdb ID: 4MBS. The observed crystallographic dimer of CCR5 or the TM5-mediated dimer by modeling does not seem to be relevant to binding to either monomeric or trimeric gp1207,78. (d) Superposition of the structures of the gp120-bound CCR5 (red) and the Gs protein-bound β2 adrenergic receptor (blue). The position of TM6, critical for GPCR activation, is indicated.
Extended Data Figure 8. Comparison of conformations of different monomeric gp120s and various V3 loops.

(a) Comparison of structures of an unliganded gp120 core (pdb ID:4OLV; purple), a CD4-bound monomeric gp120 core with the V3 loop (pdb ID:2QAD; blue) and gp120 in complex with CD4 and CCR5 from this study (cyan). The gp120 core region is marked by a circle with a diameter of 50Å. N- and C-termini, V1V2 stem, V3 stem or loop and bridging sheet are indicated. (b) Representative conformations that an HIV-1 V3 loop can adopt. From left to right: V3 loop in the unliganded SOSIP BG505 Env trimer (pdb ID: 4ZMJ); the first V3-containing gp120 core in complex with CD4 and antibody X5 (pdb ID: 2B4C; ref29); CD4- and 412d-bound monomeric gp120 core with V3 (pdb ID: 2QAD); CCR5-bound intact gp120 (this study); and V3 peptide in complex with antibody 447-52D (pdb ID: 3GHB; ref36); antibody 268-D (pdb ID: 3GO1; ref37); antibody 2557 (pdb ID: 3MLV; ref37); antibody 10A37 (pdb ID: 5V6L; ref38). The root-mean-square deviation (RMSD) of each structure, except for 5V6L, relative to the CCR5-bound gp120 monomer is shown at the bottom in parenthesis.
Extended Data Figure 9. Model for HIV-1 Env activation to induce membrane fusion.
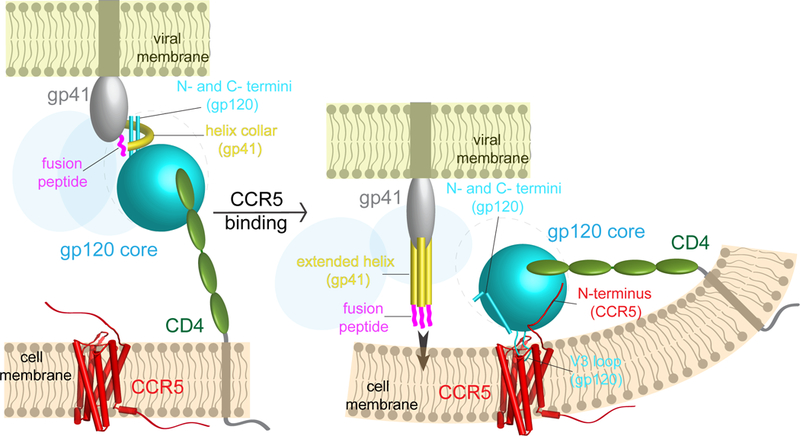
A hypothesis of how cellular receptors CD4 and CCR5 trigger HIV-1 Env trimer to induce membrane fusion and viral entry. Left, virus attaches to the target cell by gp120 (cyan) binding to CD4 (green). Helix collar of gp41, the 4-helix collar gripping the N- and C-termini of gp120. Right, immediate binding by CCR5 (red) prevents rapid dissociation between gp120 and CD4, stabilizes the CD4-induced conformational changes within the Env trimer and also brings the trimer close to the cell membrane. Simultaneous binding of gp120 to both CD4 and CCR5 may require bending in the cell membrane. The fusion peptide (magenta) of gp41 (gray) flips out due to intrinsic conformational dynamics, allowing bending back of the N- and C-termini of gp120, which blocks the fusion peptide from resuming its original position in the trimer. The movements of the fusion peptide and gp120 termini effectively weakens the non-covalent association between the two subunits and may lead to partial or complete dissociation of gp120 and a series of refolding events in gp41 to adopt the prehairpin intermediate conformation with the fusion peptides inserting into the target cell membrane. Extended helix in gp41, three helices in the fusion intermediate conformation of gp41.
Extended Data Table 1.
Cryo-EM data collection, refinement and validation statistics.
| CD4-gp120-CCR5 complex masked (EMDB -9108) (PDB 6MEO) |
CD4-gp120-CCR5 complex overall (EMDB -9109) (PDB 6MET) |
|
|---|---|---|
| Data collection and processing | ||
| Magnification | 130000 | 130000 |
| Voltage (kV) | 300 | 300 |
| Electron exposure (e—/A2) | —46 | —46 |
| Defocus range (pa) | 1-2.8 | 1-2.8 |
| Pixel size (A) | 0.529 | 0.529 |
| Symmetry imposed | Cl | Cl |
| Initial particle images (no.) | 1,707,575 | 1,707,575 |
| Final particle images (no.) | 307346 | 307346 |
| Map resolution (A) | 3.9 | 4.5 |
| FSC threshold | 0.143 | 0.143 |
| Refinement | ||
| Initial model used (PDB code) | 5UIW, 1WIO, 2QAD and 5VN3 |
5UIW, 1WIO, 2QAD and 5VN3 |
| Map sharpening B factor (A2) | −190 | −190 |
| Model composition | ||
| Non-hydrogen atoms | 7462 | 8911 |
| Protein residues | 887 | 1074 |
| Ligands | 32 | 32 |
| R.m.s. deviations | ||
| Bond lengths (A) | 0.004 | 0.009 |
| Bond angles (°) | 0.954 | 1.386 |
| Validation | ||
| MolProbity score | 1.60 | 1.97 |
| Clashscore | 3.51 | 5.84 |
| Poor rotamers (%) | 0.25 | 1.48 |
| Ramachandran plot | ||
| Favored (%) | 92.77 | 90.83 |
| Allowed (%) | 7.23 | 8.51 |
| Disallowed (%) | 0 | 0.66 |
Supplementary Material
Acknowledgments:
We thank S. Harrison and A. Kruse for generous advice, K. Song, J. Chen, R. Martin and W. Chang for technical assistance, N. Grigorieff and T. Grant for discussion at the early stage of the project, and S. Harrison, A. Kruse for critical reading of the manuscript. This work was supported by NIH grants AI141002 (to B.C.), AI106488 (to B.C.), AI129721 (to B.C.), AI127193 (to B.C. and J.J.C.), the Center for HIV/AIDS Vaccine Immunology - Immunogen Design AI-100645 (to Barton F. Haynes), and Collaboration for AIDS Vaccine Discovery (CAVD) grant OPP1169339 (to Dan H. Barouch from the Bill and Melinda Gates Foundation).
Footnotes
Data Availability:
The atomic structure coordinates are deposited in the ProteinDataBank under the accession number 6MEO and 6MET; and the EM maps in the EMDataBank under the accession number EMD-9108 and EMD-9109. All other related data generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References:
- 1.Harrison SC Viral membrane fusion. Nature structural & molecular biology 15, 690–698, doi: 10.1038/nsmb.1456 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwong PD et al. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393, 648–659 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Julien JP et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 342, 1477–1483, doi: 10.1126/science.1245625 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JH, Ozorowski G & Ward AB Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 351, 1043–1048, doi: 10.1126/science.aad2450 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pancera M et al. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature 514, 455–461, doi: 10.1038/nature13808 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H et al. Cryo-EM structure of a CD4-bound open HIV-1 envelope trimer reveals structural rearrangements of the gp120 V1V2 loop. Proc Natl Acad Sci U S A 113, E7151–E7158, doi: 10.1073/pnas.1615939113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan Q et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 341, 1387–1390, doi: 10.1126/science.1241475 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamamis P & Floudas CA Molecular recognition of CXCR4 by a dual tropic HIV-1 gp120 V3 loop. Biophys J 105, 1502–1514, doi: 10.1016/j.bpj.2013.07.049 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamamis P & Floudas CA Molecular recognition of CCR5 by an HIV-1 gp120 V3 loop. PLoS One 9, e95767, doi: 10.1371/journal.pone.0095767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng Y et al. Structure of CC Chemokine Receptor 5 with a Potent Chemokine Antagonist Reveals Mechanisms of Chemokine Recognition and Molecular Mimicry by HIV. Immunity 46, 1005–1017 e1005, doi: 10.1016/j.immuni.2017.05.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berger EA, Murphy PM & Farber JM Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol 17, 657–700 (1999). [DOI] [PubMed] [Google Scholar]
- 12.Connor RI, Sheridan KE, Ceradini D, Choe S & Landau NR Change in coreceptor use coreceptor use correlates with disease progression in HIV-1--infected individuals. J Exp Med 185, 621–628 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verhofstede C, Nijhuis M & Vandekerckhove L Correlation of coreceptor usage and disease progression. Curr Opin HIV AIDS 7, 432–439, doi: 10.1097/COH.0b013e328356f6f2 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Wu B et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071, doi: 10.1126/science.1194396 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin L et al. Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347, 1117–1122, doi: 10.1126/science.1261064 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scholten DJ et al. Pharmacological modulation of chemokine receptor function. Br J Pharmacol 165, 1617–1643, doi: 10.1111/j.1476-5381.2011.01551.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin G, Baribaud F, Romano J, Doms RW & Hoxie JA Identification of gp120 binding sites on CXCR4 by using CD4-independent human immunodeficiency virus type 2 Env proteins. J Virol 77, 931–942 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doranz BJ et al. Two distinct CCR5 domains can mediate coreceptor usage by human immunodeficiency virus type 1. J Virol 71, 6305–6314 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rizzuto CD et al. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 280, 1949–1953 (1998). [DOI] [PubMed] [Google Scholar]
- 20.Huang CC et al. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 317, 1930–1934 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farzan M et al. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 96, 667–676 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Farzan M et al. The role of post-translational modifications of the CXCR4 amino terminus in stromal-derived factor 1 alpha association and HIV-1 entry. J Biol Chem 277, 29484–29489, doi: 10.1074/jbc.M203361200 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Oppermann M Chemokine receptor CCR5: insights into structure, function, and regulation. Cell Signal 16, 1201–1210 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Chen J et al. HIV-1 ENVELOPE. Effect of the cytoplasmic domain on antigenic characteristics of HIV-1 envelope glycoprotein. Science 349, 191–195, doi: 10.1126/science.aaa9804 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colin P et al. HIV-1 exploits CCR5 conformational heterogeneity to escape inhibition by chemokines. Proc Natl Acad Sci U S A 110, 9475–9480, doi: 10.1073/pnas.1222205110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scheres SH RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530, doi: 10.1016/j.jsb.2012.09.006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu H, Kwong PD & Hendrickson WA Dimeric association and segmental variability in the structure of human CD4. Nature 387, 527–530 (1997). [DOI] [PubMed] [Google Scholar]
- 28.Ozorowski G et al. Open and closed structures reveal allostery and pliability in the HIV-1 envelope spike. Nature 547, 360–363, doi: 10.1038/nature23010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang CC et al. Structure of a V3-containing HIV-1 gp120 core. Science 310, 1025–1028 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gallivan JP & Dougherty DA Cation-pi interactions in structural biology. Proc Natl Acad Sci U S A 96, 9459–9464 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bannert N et al. Sialylated O-glycans and sulfated tyrosines in the NH2-terminal domain of CC chemokine receptor 5 contribute to high affinity binding of chemokines. J Exp Med 194, 1661–1673 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berro R et al. Multiple CCR5 conformations on the cell surface are used differentially by human immunodeficiency viruses resistant or sensitive to CCR5 inhibitors. J Virol 85, 8227–8240, doi: 10.1128/JVI.00767-11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moore JP & Kuritzkes DR A piece de resistance: how HIV-1 escapes small molecule CCR5 inhibitors. Curr Opin HIV AIDS 4, 118–124, doi: 10.1097/COH.0b013e3283223d46 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manglik A & Kruse AC Structural Basis for G Protein-Coupled Receptor Activation. Biochemistry 56, 5628–5634, doi: 10.1021/acs.biochem.7b00747 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwon YD et al. Crystal structure, conformational fixation and entry-related interactions of mature ligand-free HIV-1 Env. Nat Struct Mol Biol 22, 522–531, doi: 10.1038/nsmb.3051 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burke V et al. Structural basis of the cross-reactivity of genetically related human anti-HIV-1 mAbs: implications for design of V3-based immunogens. Structure 17, 1538–1546, doi: 10.1016/j.str.2009.09.012 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang X et al. Conserved structural elements in the V3 crown of HIV-1 gp120. Nat Struct Mol Biol 17, 955–961, doi: 10.1038/nsmb.1861 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Pan R et al. Increased epitope complexity correlated with antibody affinity maturation and a novel binding mode revealed by structures of rabbit antibodies against the third variable loop (V3) of HIV-1 gp120. J Virol, doi: 10.1128/JVI.01894-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lyumkis D et al. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science 342, 1484–1490, doi: 10.1126/science.1245627 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore JP, McKeating JA, Weiss RA & Sattentau QJ Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science 250, 1139–1142 (1990). [DOI] [PubMed] [Google Scholar]
- 41.Thali M, Furman C, Helseth E, Repke H & Sodroski J Lack of correlation between soluble CD4-induced shedding of the human immunodeficiency virus type 1 exterior envelope glycoprotein and subsequent membrane fusion events. J Virol 66, 5516–5524 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang MI, Panorchan P, Dobrowsky TM, Tseng Y & Wirtz D Single-molecule analysis of human immunodeficiency virus type 1 gp120-receptor interactions in living cells. J Virol 79, 14748–14755, doi: 10.1128/JVI.79.23.14748-14755.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dobrowsky TM, Zhou Y, Sun SX, Siliciano RF & Wirtz D Monitoring early fusion dynamics of human immunodeficiency virus type 1 at single-molecule resolution. J Virol 82, 7022–7033 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brandenberg OF, Magnus C, Regoes RR & Trkola A The HIV-1 Entry Process: A Stoichiometric View. Trends Microbiol 23, 763–774, doi: 10.1016/j.tim.2015.09.003 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Floyd DL, Ragains JR, Skehel JJ, Harrison SC & van Oijen AM Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci U S A 105, 15382–15387, doi:0807771105 [pii] 10.1073/pnas.0807771105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu P et al. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 441, 847–852, doi:nature04817 [pii] 10.1038/nature04817 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Rosen O, Sharon M, Quadt-Akabayov SR & Anglister J Molecular switch for alternative conformations of the HIV-1 V3 region: implications for phenotype conversion. Proc Natl Acad Sci U S A 103, 13950–13955 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ho SH, Trunova N, Gettie A, Blanchard J & Cheng-Mayer C Different mutational pathways to CXCR4 coreceptor switch of CCR5-using simian-human immunodeficiency virus. J Virol 82, 5653–5656, doi: 10.1128/JVI.00145-08 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edo-Matas D, van Dort KA, Setiawan LC, Schuitemaker H & Kootstra NA Comparison of in vivo and in vitro evolution of CCR5 to CXCR4 coreceptor use of primary human immunodeficiency virus type 1 variants. Virology 412, 269–277, doi: 10.1016/j.virol.2011.01.010 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Westby M et al. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J Virol 81, 2359–2371 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seclen E et al. Primary resistance to maraviroc in a large set of R5-V3 viral sequences from HIV-1-infected patients. J Antimicrob Chemother 65, 2502–2504, doi: 10.1093/jac/dkq381 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Jiang X et al. Characterizing the Diverse Mutational Pathways Associated with R5-Tropic Maraviroc Resistance: HIV-1 That Uses the Drug-Bound CCR5 Coreceptor. J Virol 89, 11457–11472, doi: 10.1128/JVI.01384-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pugach P et al. HIV-1 clones resistant to a small molecule CCR5 inhibitor use the inhibitor-bound form of CCR5 for entry. Virology 361, 212–228, doi: 10.1016/j.virol.2006.11.004 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin G et al. Replication-competent variants of human immunodeficiency virus type 2 lacking the V3 loop exhibit resistance to chemokine receptor antagonists. J Virol 81, 9956–9966 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zolla-Pazner S et al. The cross-clade neutralizing activity of a human monoclonal antibody is determined by the GPGR V3 motif of HIV type 1. AIDS Res Hum Retroviruses 20, 1254–1258 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Frey G et al. A fusion-intermediate state of HIV-1 gp41 targeted by broadly neutralizing antibodies. Proc Natl Acad Sci U S A 105, 3739–3744 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kovacs JM et al. HIV-1 envelope trimer elicits more potent neutralizing antibody responses than monomeric gp120. Proc Natl Acad Sci U S A 109, 12111–12116, doi: 10.1073/pnas.1204533109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Freeman MM et al. Crystal structure of HIV-1 primary receptor CD4 in complex with a potent antiviral antibody. Structure 18, 1632–1641, doi: 10.1016/j.str.2010.09.017 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cai Y et al. Antigenicity-defined conformations of an extremely neutralization-resistant HIV-1 envelope spike. Proc Natl Acad Sci U S A 114, 4477–4482, doi: 10.1073/pnas.1700634114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brady AE & Limbird LE G protein-coupled receptor interacting proteins: emerging roles in localization and signal transduction. Cell Signal 14, 297–309 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Visegrady A, Boros A, Nemethy Z, Kiss B & Keseru GM Application of the BD ACTOne technology for the high-throughput screening of Gs-coupled receptor antagonists. J Biomol Screen 12, 1068–1073, doi: 10.1177/1087057107309035 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Melar M, Ott DE & Hope TJ Physiological levels of virion-associated human immunodeficiency virus type 1 envelope induce coreceptor-dependent calcium flux. J Virol 81, 1773–1785 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tang G et al. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157, 38–46, doi: 10.1016/j.jsb.2006.05.009 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Ru H et al. Molecular Mechanism of V(D)J Recombination from Synaptic RAG1-RAG2 Complex Structures. Cell 163, 1138–1152, doi: 10.1016/j.cell.2015.10.055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. Journal of structural biology 152, 36–51, doi: 10.1016/j.jsb.2005.07.007 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332, doi: 10.1038/nmeth.4193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rohou A & Grigorieff N CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221, doi: 10.1016/j.jsb.2015.08.008 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roy A, Kucukural A & Zhang Y I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols 5, 725–738, doi: 10.1038/nprot.2010.5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta crystallographica. Section D, Biological crystallography 66, 486–501, doi: 10.1107/S0907444910007493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica. Section D, Biological crystallography 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feng Y, Broder CC, Kennedy PE & Berger EA HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272, 872–877 (1996). [DOI] [PubMed] [Google Scholar]
- 72.Wu L et al. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 384, 179–183 (1996). [DOI] [PubMed] [Google Scholar]
- 73.Alkhatib G et al. CC CKR5: A RANTES, MIP-1a, MIP-1b receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272, 1955–1962 (1996). [DOI] [PubMed] [Google Scholar]
- 74.Choe H et al. The b-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85, 1135–1148 (1996). [DOI] [PubMed] [Google Scholar]
- 75.Deng H et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381, 661–666 (1996). [DOI] [PubMed] [Google Scholar]
- 76.Doranz BJ et al. A dual-tropic primary HIV-1 isolate that uses fusin and the b-chemokine receptors CKR-5, CKR-3 and CKR-2b as fusion cofactors. Cell 85, 1149–1158 (1996). [DOI] [PubMed] [Google Scholar]
- 77.Dragic T et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR5. Nature 381, 667–673 (1996). [DOI] [PubMed] [Google Scholar]
- 78.Jin J et al. CCR5 adopts three homodimeric conformations that control cell surface delivery. Sci Signal 11, doi: 10.1126/scisignal.aal2869 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.