

Abstract
Inflammation affects tumor immune surveillance and resistance to therapy. Here, we show that production of IL1 β in primary breast cancer tumors is linked with advanced disease and originates from tumor-infiltrating CD11c+ myeloid cells. IL1 β production is triggered by cancer cell membrane-derived TGFβ. Neutralizing TGFβ or IL1 receptor prevents breast cancer progression in humanized mouse model. Patients with metastatic HER2− breast cancer display a transcriptional signature of inflammation in the blood leukocytes, which is attenuated after IL1 blockade. When present in primary breast cancer tumors, this signature discriminates patients with poor clinical outcomes in two independent public datasets (TCGA and METABRIC).
Significance:
pIL1 β orchestrates tumor-promoting inflammation in breast cancer and can be targeted in patients using an IL1 receptor antagonist.
Introduction
The clinical success of checkpoint inhibitors and adoptively transferred, genetically engineered T cells (1, 2) brought cancer immunotherapy into mainstream oncology. Improved survival has been documented for patients with metastatic melanoma treated with a blocking antibody targeting the CTL-associated protein (CTLA)-4 (1). Objective clinical responses have also been observed in other tumor types with therapies targeting another T-cell checkpoint, programmed death (PD)-1, or programmed death ligand (PDL)-1 (1). However, only a minority of patients responds to current immunotherapies, as is the case for treatmentresistant breast cancer where less than 20% of patients experience durable responses to checkpoint inhibitors (3).
Chronic inflammation, a hallmark of many cancers, is maintained by interplay of intrinsic (oncogenes and tumor suppressor genes) and extrinsic (immune and stromal components) factors (4, 5). Different types of inflammation might have distinct effects on cancer and ultimately treatment outcomes (4). Clinical studies indicate that chronic inflammation increases the risk of both treatment resistance and breast cancer metastasis (6). Yet, there are no clinically proven approaches to decrease cancer-promoting inflammation in breast cancer largely due to an incomplete understanding of associated pathways. Tumor-infiltrating CD4+ T cells producing type 2 cytokines, such as IL4, IL5, IL9, and IL13 (7), have a substantial impact on breast cancer progression in experimental models of metastatic breast cancer (8, 9). Recent study showed a correlation between high levels of IL5 in breast cancer tumor interstitial fluid and shortened patient overall survival (10). In genetically engineered mouse models of metastatic mammary carcinoma, CD4+ T cells accelerated the development of pulmonary metastasis via IL4-dependent mechanisms (8), and blocked the function of anticancer CD8+ T cells (11). Yet, in some scenarios, type 2 cytokines appear to protect against mammary carcinoma development in MMTV-polyoma middle T (PyMttg) mice (12). Furthermore, in patients with breast cancer, CD4+ T cells with follicular helper phenotype and function predict survival (13); possibly by signifying the presence of organized tertiary lymphoid structures supporting T-cell activation rather than disorganized lymphocyte infiltrates (14). Thus, the local context and tissue microenvironment likely dictate the final impact of T-cell immunity measured by tumor progression.
The mechanisms that promote induction and maintenance of the type 2 cytokine microenvironment upon injury to the epithelial barrier include IL25 (15), thymic stromal lymphopoietin (TSLP; ref. 16), as well as members of the IL1 family, including IL1 β and IL33 (17). Here we report that IL1β orchestrates tumor-promoting inflammation in breast cancer, and that it can be targeted in patients with metastatic breast cancer with a soluble IL1 receptor antagonist. Our experimental and clinical findings provide a rationale for investigating IL1β as an immunomodulatory target in treatment-resistant breast cancer.
Materials and Methods
Cell lines and reagents
Breast cancer cell lines, Hs587T and Hs578Bst, were purchased from ATCC; MDA-MB-231 purchased from Xenogen was cultured in nonselecting media. All lines are banked as low-passage stock from which working banks are periodically renewed. All lines were verified by gene microarrays and verified by short tandem repeat analysis from ATCC. The cell lines were tested Mycoplasma free for each experiment.
Cell lines were cultured in complete RPMI [plus glutamine, 2 mmol/L; penicillin, 50 U/mL; streptomycin, 50 mg/mL; minimum essential medium (MEM) nonessential amino acids, 0.1 mmol/L; HEPES buffer, 10 mmol/L; and sodium pyruvate, 0.1 mmol/L] and 10% FCS inT150 flasks at a seed density of 2 × 106 cells/25 mL. At 90% confluence, fresh medium was added, and cells were cultured for an additional 48 hours. Supernatant was centrifuged and stored at −80° C.
IL1β, IL1α, IL18, IL6, andTNFα cytokines were purchased from R&D Systems. Anti-human CD14 antibody (RM052) was obtained from Immunex. Anakinra (Kineret, Amgen Inc.) was purchased through Baylor University Medical Center pharmacy. Phorbol 12-myristate 13-acetate (PMA), ionomycin, and TAK1 inhibitor (5z-7-oxozeaenol) were obtained from Sigma-Aldrich. TGFβR kinase inhibitor was obtained from EMD Millipore. Caspase-1 activity detection kit was obtained from OncoImmunin Inc. Secondary antibody were purchased from Invitrogen (Alexa Fluor 568 anti-rabbit IgG, Alexa Fluor 647 anti-mouse IgG2b, Alexa Fluor 568 goat anti-mouse IgG2a). Anti-human IL1 β (Ab9722) and cytokeratin-19 (A53-B/A2) antibodies were obtained from Abcam. Caspase-1 inhibitor (Z-WEHD-FMK), anti-TGFβ1 antibody (chicken IgY) and anti-TGFβ neutralizing antibody (1D11) were purchased from R&D Systems. For details, see Supplementary Table S4.
Cytokine production and analysis of tumor samples from patients
Exempt primary tissues from patients were obtained from the Baylor University Medical Center (BUMC) Tissue Bank (Institutional Review Board no. 005–145; otherwise discarded tissues). Consecutive postsurgical tumor samples (from patients with in situ, invasive ductal, lobular, and/or mucinous carcinoma of the breast) were collected between years 2006 and 2013. The combined histologic grading data, including nuclear grade, tubule formation and mitotic rate, and staging system, were provided as per pathologists’ report postsurgery. Fresh whole-tissue fragments (4 × 4 × 4 mm, 0.02 g, approximately) were placed in culture medium with 50 ng/mL PMA and 1 μg/mL ionomycin for 16 hours. IL1β, IL1α, IL33, GM-CSF, TSLP, IL4, IL5, and IL13 levels were analyzed in the culture supernatant by Luminex (EMD Millipore). Concentrations of IL18 and IL1RA from tissue culture supernatants were determined by ELISAs (R&D Systems).
Tumor-bearing mice and in vivo experiments
Mouse.
Four-week-old NOD.Cg-Prkdc(scid)b2m(tm1Unc)/J (abbreviated NOD/scid/b2 null) female mice (Jackson Laboratories) were used. Mouse work and protocols were performed accordingly to relevant institutional and national guidelines and regulations. Animal experiments were carried out with permission from the Institutional Animal Care and Use Committee.
Cell line.
Breast cancer cell line, Hs578T, was used. For details, see cell lines and reagents section.
Primary cell culture
Peripheral blood mononuclear cells (PBMC) were obtained by leukapheresis from healthy donors (Institutional Review Board approved). Monocyte-derived DCs (MDDC) were generated from the adherent fraction of PBMCs by culturing with 100 ng/mL GM-CSF and 10 ng/mL IL4 (R&D Systems).
Procedure
Mice were sublethally irradiated (12 cGy/g body weight of 137Cs γ irradiation) the day before tumor implantation. A total of 1 × 107 Hs-578t cells were injected subcutaneously into the flanks. Mice were then reconstituted with 1 × 106 MDDCs and autologous T cells. CD4+ and CD8+ T cells were positively selected from thawed PBMCs using magnetic selection following manufacturer’s instructions (Miltenyi Biotec). The purity was routinely >90%. A total of 1 × 107 CD4+ T cells and 1 × 107 CD8+ T cells were transferred at days 3, 6, and 9 after tumor implantation. Anakinra (2 mg/kg body weight) or PBS were injected daily in peritumor area since day 3 after tumor engraftment. TGFβ blocking antibody was given on days 3, 6, and 9. Tumor size was monitored every 2–3 days. Tumor volume (ellipsoid) was calculated as follows: [(short diameter)2 × long diameter]/2. On day 16, the tumors were harvested. Tissue fragments were cultured in medium with 50 ng/mL of PMA and 1 μg/mL of Ionomycin for 16 hours. IL13, IL4, TNFα, IFNγ, IL17, and IL1 β levels were analyzed in the culture supernatant by Luminex (EMD Millipore).
Pilot clinical trial
Eleven female patients (Supplementary Table S5) with HER2− metastatic breast cancer received nab-paclitaxel (n = 3), eribulin (n = 5), or capecitabine (n = 2) along with anakinra 100 mg/day (FDA-approved dose for adults with rheumatoid arthritis; Kineret, Amgen Inc.) following 2-week treatment with anakinra only. The study was approved by Institutional Review Board (IRB 012–099), conducted in accordance with ethical guidelines (Declaration of Helsinki), and written informed consent was obtained from the patients. Blood was collected prior to anakinra treatment, then at 2 weeks, and then monthly for 6 months. Eleven healthy controls were included (IRB 012–200). Whole blood mRNA was measured using the NanoString nCounter Human Immunology V2 panel.
Isolation of monocytes and culture of MDDCs and macrophages
CD14+ cells were positively selected from PBMCs of healthy donors using magnetic selection following the manufacturer’s instructions (Miltenyi Biotec). The purity was routinely >95%. Macrophages were generated from CD14+ monocytes by culturing with 100 ng/mL M-CSF. MDDCs were generated from the adherent fraction of PBMCs by culturing with 100 ng/mL GM-CSF and 10 ng/mL IL4 (R&D Systems).
Isolation and culture of myeloid dendritic cells
DCs were enriched from PBMCs obtained after Ficoll-Paque Plus density gradient centrifugation by negative selection using human pan-DC pre-enrichment kit (EasySep). Cells from the negative fraction were immunolabeled with anti-human FITC- labeled lineage cocktail (CD3, CD14, CD16, CD19, CD20, and CD56; BD Biosciences); PE-labeled CD123 (mIgG1, clone 9F5; BD Biosciences), APC-efluor780-labeled HLA-DR (mIgG2b, clone LN3, Sigma-Aldrich) and APC-labeled CD11c (mIgG2b, clone S-HCL-3, BD Biosciences). DCs (lin−, CD123−, HLA-DR+, CD11c+) were sorted in a FACS Aria cytometer (BD Biosciences). DCs were seeded at 1 × 105 cells/well in 200 μL of complete RPMI medium supplemented with 10% human AB serum. DCs were cultured with medium alone or in the presence of 40% of cancer-conditioned supernatant or different reagents.
Coculture and transwell experiment
For tumor cell and blood DC coculture experiments, 1 × 104 tumor cells were seeded in a 24-well plate to grow overnight, then media were refreshed and 1 × 104 DCs were added for another 48 hours. For the transwell experiments, 24-well plates with inserts were used (Corning). A total of 2 × 104 tumor cells were seeded in the plate to grow at least overnight, then 2 × 104 DCs were added into inserts. After 48 hours of coculture, supernatants were harvested to determine IL1 β level by ELISA (Duoset, R&D Systems).
Tissue immunofluorescence staining
Cryosections (6 mm) were consecutively treated with Hyaluronidase 0.03% for 15 minutes, Fc Receptor Block (Innovex Bioscience) for 40 minutes + Background Buster (Innovex Bioscience) for an additional 30 minutes. The sections were then stained with primary antibodies, diluted in PBS + 5% BSA 0.1% Saponin for 1 hour at room temperature, washed, and stained with the secondary antibodies at room temperature for 30 minutes. Nuclei were stained with 4’,6-diamidino-2-phenylindole (1 μg/mL) for 2 minutes.
Primary anti-human antibodies.
CD1a (HI149, BioLegend); CD11c (S-HCL-3 APC-conjugated, BD Biosciences or 3.9, eBioscience); CD14 (UCHM1, AbD Serotec); CD15 (HI98, Bio Legend); panCK (CK3–6H5 FITC conjugated, Miltenyi Biotec); Cytokeratin 19 (A53-B/A2, Abcam); HLA-DR (LN3, BioLegend); IL1a (A15032A, BioLegend); IL1b (rabbit polyclonal, Abcam); pro-IL1b (615417, R&D Systems).
Secondary antibodies.
Depending on the primary antibody combinations, species-and isotype-specific secondary antibody conjugated with either Alexa Fluor 488, Alexa Fluor 568, and Alexa Fluor 647 (Molecular Probes) were used in appropriated mixtures. Tissue scans were acquired on a Leica SP8 confocal microscope using a 20×/0.75 NAdry HC PL APO objective lens with a magnification factor of 1.5. Signal was acquired using HyD detectors at room temperature. Stained tissues were mounted in Fluoromount G. Using Leica acquisition software, LAS × individual tiles were stitched and a maximum intensity projected image was then exported as lif. files and processed using Imaris software.
Histo-cytometry
In situ quantitative analysis of breast cancer tissue was based on a published methodology. Briefly, a cryosection was stained by immunofluorescence for each breast cancer tissue to label nuclei, cytokeratin, CD11c, and IL1 β. Whole tissue scans were acquired using a Leica SP8 confocal microscope (Leica Microsystems). Each scan was then analyzed using image analysis software Imaris (Bitplane). Using the “spot” function in Imaris, the images were subdivided into individual cells, defined as having a nucleus diameter equal or larger than 6 mm. The accuracy of the segmentation was manually verified and adjusted if needed for each sample. Finally, for each generated spot, x;y coordinates and the sum intensity values for all channels were exported into an fcs file to be visualized and quantified using FlowJo software (version 10, Flowjo LLC). Quantification was conducted using FlowJo software to gate CD11c+, cytokeratin 19+, and double-positive cells and to quantify the IL1 β expression within each of the gated populations. FlowJo plots illustrate the gating and quantification process in representative breast cancer tumor sample. Far right panel summarizes the percentages of IL1 β colocalization with cytokeratin 19 and/or with CD11c from three different breast cancer tumors.
Real-time PCR
Samples were treated and lysed with RLT buffer and stored at −80°C until RNA extraction. Total RNA was isolated and purified from each sample by using RNeasy Kit and RNase-free DNase (Qiagen) following the manufacturer’s instructions. cDNA was generated from total RNA with iScript cDNA Synthesis Kit. The resulting cDNA was then used for quantitative gene expression analysis on a Sequence Detection System 7,500 (Applied Biosystems). The primers used were as follows: human (h)TSLP, 5’-TAGCAATCGGCCACATTGCC-3’ and 5’-CTGAGTTTCCGAATA-GCCTG-3; (h)IL1p, 5’-TACCTGTCCTGCGTGTTGAA-3’ and 5’-TCTTTGGGTAATTTTGGGATCr-3’; human (h) GAPDH, 5’-AGC-CACATCGCTCAGACAC-3’ and 5’-GCCCAATACGACCAAATCC-3’; human (h) ABL1, 5’-TGACAGGGGACACCTACACA-3’ and 5’ATACTCCAAATGCCCAGACG-3’; human (h) PGK1, 5’-CTTCCT-CCTTAAAACTCCTCTCC-3’ and 5’-CTAAGGTCTCCAACGC-TCTTCT-3’; human (h) PES1, 5’-CATCACCCATCAGATTGTCG-3’ and 5’-AGCTGCACCCCAGAGAAGTA-3’.
Equal amounts of cDNA were used with the iTaq SYBR Green Supermix with ROX (Bio-Rad) and primer mix according to the real-time PCR manufacturer’s protocols Amplification efficiencies were validated against the housekeeping gene, GAPDH, PES1, and ABL1. Data were normalized to GAPDH mRNA level. The relative quantification of target gene expression was done by the comparative cycle threshold (Ct) method. The formula 2−ΔΔCt was used for each run according to the manufacturer’s instructions and published methods for this system.
Chromatin immunoprecipitation
Chromatin for chromatin immunoprecipitation (ChIP) was prepared from MDA-MB-231 cell lines, treated or not with 10 ng/mL IL1 β for 1 hour, by fixing the cells in 1% formaldehyde for 10 minutes, followed by quenching with glycine for 2 minutes on ice. ChIP was performed using anti-RNA polymerase II (8W16; BioLegend) antibody. Reactions were performed using 100 μL of Protein G Dynabeads (Invitrogen) and 10 μg of antibody in each ChIP reaction. The resulting DNA was purified using phenol/ chloroform/isoamyl alcohol. ChIP qPCR was performed with TSLP (forward primer: AGTCAGCGGTGAATCAGAGG; reverse primer: GGAATGTCGAGGGATTCGGG) and cytokeratin19 (forward primer: GACACTCACTTGGTTCGGAAG; reverse primer: TGTGTTGAACTGGTACCAACCT) promoter region.
Flow cytometry analysis
The anti-human TGFβ1 (chicken IgY; R&D Systems) was used. For surface staining, cells were incubated with the antibodies for 30 minutes at 4° C in the dark, then washed three times and fixed with 1% paraformaldehyde to be acquired in a FACSCanto (Becton Dickinson) and analyzed with FlowJo.
RNA in situ hybridization
RNA transcripts were visualized in OCT-embedded breast tumor sections using the QuantiGene ViewRNA ISH tissue assay kit (Affymetrix). Human PTPRC ViewRNA type 6 probe and human IL1B ViewRNA type 1 probe were obtained from Affymetrix. The assay was performed according to the tissue-based ViewRNA assay protocol with a 15-minute formaldehyde fixation and a 20-minute protease treatment (dilution factor 1:100). Probes were detected at 550 nm and 650 nm with Leica SP8 confocal microscope using a 40×/1.30 NA Oil HC PL APO objective lens. Signal was acquired using HyD detectors at room temperature. Stained tissues were mounted in Fluoromount G. Acquisition software LAS × from which lif. image files were exported and processed using Imaris software.
Statistical and bioinformatics analysis
All statistics and graphs were either done with Prism software (GraphPad) or with custom R-scripts. Differences in variables between two groups of samples obtained from the same patient were analyzed using paired Wilcoxon test. Differences between two experimental conditions were analyzed using unpaired t test. Differences between any 3 or more groups were analyzed by ANOVA or with Kruskal-Wallis test.
Normalized gene expression data from the TCGA project was downloaded from the TCGA data portal. The selected genes identified from clinical trial were used to hierarchically cluster patients to test the association between expression profile of the selected genes and samples. Euclidean distance was used as the distance metric and Ward.D2 agglomeration method was used to cluster the samples.
Results
IL1β secretion in primary breast cancers correlates with disease stage
To better understand the cytokine environment in breast cancer, we measured the secretion of several IL1 family cytokines upon short-term ex vivo activation of primary breast cancer tissue samples and surrounding tissue, that appeared macroscopically uninvolved during surgery (nonmalignant tissue), with PMA and ionomycin (Supplementary Table S1). IL1α, IL1β, IL1Ra, IL18, and IL33 were detected in supernatants of breast cancer samples (Supplementary Table S1; Supplementary Fig. S1A). The secretion of IL1 β, IL1Ra, IL18, and IL1α was significantly higher in breast cancer tissues than in nonmalignant tissue (Fig. 1A; Supplementary Fig. S1B and S1C). Furthermore, in 149 analyzed primary breast cancer tissues, larger stage II or stage III-IV tumors produced significantly higher levels of IL1 β than did stage 0-I breast cancers (Supplementary Table S1; Fig. 1B). We found no link between disease stage and differential production of IL1α (Supplementary Fig. S1D).
Figure 1.
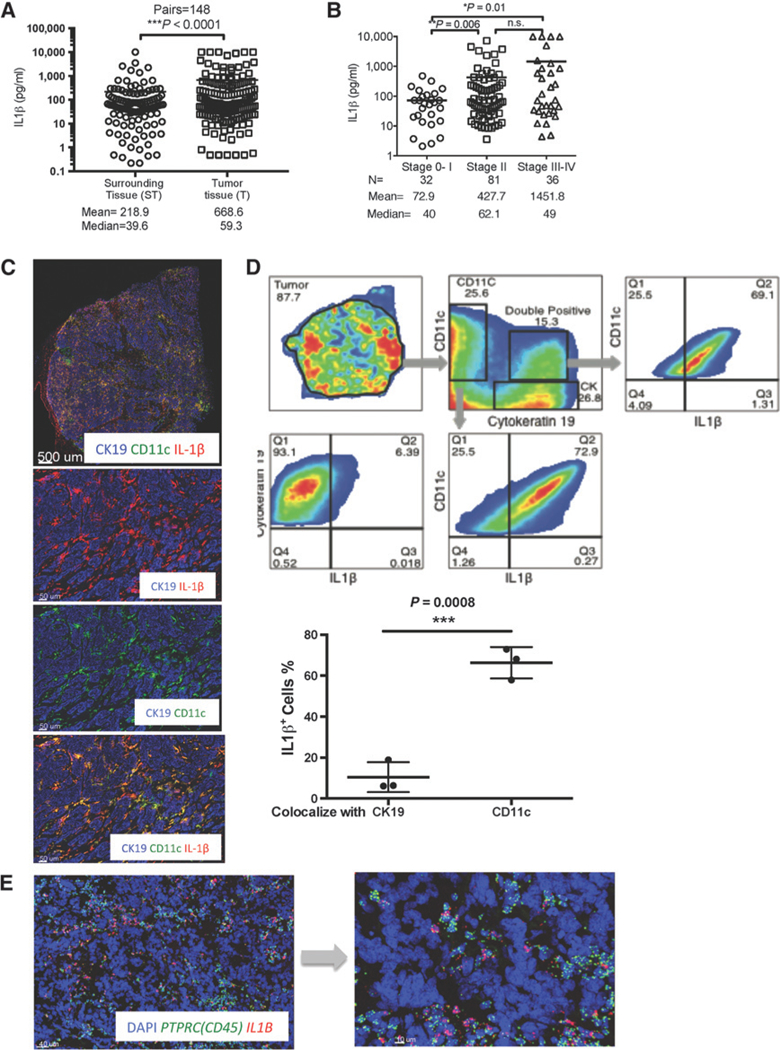
In situ IL1β expression in breast cancer tumors. A, IL1β in supernatants of breast cancer tumor fragments (T) and in macroscopically noninvolved surrounding tissue (ST); mean and median secretion of indicated cytokine in pg/mL. Wilcoxon sign-rank test. B, ILiβ concentrations plotted in relation to histopathologic tumor stage. N, number of tissue samples from different patients with indicated disease stage. Welch t test. C, Top, whole-section scan of representative breast cancer tumor (ER+PR+HER2−). CD11c (green), IL1β (red), cytokeratin (blue) expression. Bottom, zoomed area illustrating coexpression of individual markers as indicated. D, Quantitative histocytometry see (Materials and Methods) analysis of immunofluorescence staining on breast cancer tissue. The nonparametric Wilcoxontest. E, RNAtranscripts visualized in breastcancertumorsectionswith QuantiGeneViewRNA ISH tissueassaykit. Human PTPRC(CD45) and human IL1B are green and red, respectively.
To identify the cellular source of IL1 β in breast cancer tumors, we next analyzed protein and transcript expression in intact breast cancer tissues. An antibody recognizing cleaved/mature IL1β in frozen tissue sections revealed IL1 β reactivity predominantly in tumor-infiltrating CD11c+ cells, as compared with cytokeratin-19 (CK19)-expressing breast cancer cells (Fig. 1C). Quantitative histocytometry revealed that the IL1 β-expressing cells were also CD11c+ (Fig. 1D), and approximately 60% of CD11c+ cells infiltrating breast cancer tissue expressed IL1 β (n = 3, Fig. 1D). Approximately 3%−16% of analyzed cells were double positive CD11c+/CK19+ and included clusters of myeloid and breast cancer cells as well as CD11c+ cells that exhibited cytoplasmic inclusion of CK19. The CD11c+/CK19+ clusters displayed high IL1p expression in CD11c+ myeloid cells, indicating ongoing realtime interactions between cancer and immune cells (Fig. 1D). In situ hybridization (ISH) using ViewRNA methodology confirmed the presence of IL1B transcripts predominantly in CD45+ leukocyte infiltrates, and rarely in CD45 − stromal and breast cancer cells (Fig. 1E).
We next analyzed a set of 870 primary breast cancer samples that had associated clinical annotations from the Cancer Genome Atlas (TCGA) database (Supplementary Table S2; ref. 18). Among genes that significantly correlated with IL1B transcripts (P < 0.0001) (Supplementary Table S2), we found transcripts of myeloid cells ITGAX (CD11c, P < 0.001; Spearman r = 0.49) and CD14 (P < 0.001; Spearman r = 0.52), and the dendritic cell (DC)-expressed CD1c (P < 0.001; Spearman r = 0.29), and CD1a (P < 0.001; Spearman r = 0.38) genes. Thus, expression patterns of IL1B transcripts were similar to those observed at the protein level in primary breast cancer samples, leading us to conclude that CD11c+ myeloid cells, including monocytes and DCs, were the main sources of IL1 β in human primary breast cancer.
IL1β secretion in primary breast cancers correlates with that of T-cell cytokines and TSLP
IL1β positively correlated with T-cell cytokines IL13 and IFNγ (Fig. 2A), but not with IL4 and only marginally with IL5 (Supplementary Table S1). Neither IL18 nor IL33 secretion was linked to production of any type 2 cytokines (Supplementary Table S1). We reported earlier that TSLP might contribute to the inflammatory Th2 microenvironment conducive to breast tumor development (9). Herein, we confirmed in a larger patient cohort that a fraction of primary breast cancers retains the capacity to secrete TSLP on activation, albeit at lower levels than surrounding tissue (similarly to GM-CSF; Supplementary Fig. S1B). We also confirmed in 145 patient samples a positive correlation between the secretion of TSLP and T-cell cytokines IL13 and IFNγ in primary breast cancers (Fig. 2A). The capacity to secrete TSLP on activation significantly correlated to that of IL1 β in a fraction of primary breast cancers (Fig. 2A). To assess the correlation between the four cytokines secretion, we applied cluster analysis (Fig. 2B). The algorithm clustered the 145 patient samples in four groups(Fig. 2B): group 1: secretion of IL1 β, IFNγ, and IL13, but no TSLP (red, n = 47); group 2: low or absent secretion of all cytokines (blue; n = 17); group 3: secretion of IL1β, IFNγ, IL13, and TSLP (green, n = 43); and group 4: secretion of IL1 p, and IFNγ, but no IL13 or TSLP (violet, n = 38; Supplementary Table S1). This clustering was unrelated to disease stage, grade, or breast cancer subset defined by hormone and growth factor receptor expression. Thus, elevated IL1β secretion in breast cancer could explain IL13 production and Th2 signature in breast cancers. To explore potential links between IL1β and TSLP, we first confirmed the expression of TSLP in the intact primary breast cancers by ISH using ViewRNA methodology in conjunction with KRT8 to identify cancer cells (Fig. 2C; Supplementary Fig. S2A). Next, we determined whether IL1β could trigger TSLP production from breast cancer cells. Short-term exposure of MDA-MB-231 breast cancer cells to IL1β resulted in dose- dependent secretion of TSLP at levels similar to that induced by PMA/ionomycin (Supplementary Fig. S1B). Exposure to other inflammatory cytokines had only minor impact (Fig. 2D; Supplementary Fig. S2B). Secreted TSLP was biologically active as determined using reporter cell line (Supplementary Fig. S2C). Induction of protein production was linked with increased TSLP transcription in two different breast cancer cell lines analyzed (Fig. 2D; Supplementary Fig. S2B and S2D). Finally, the initiation of active TSLP transcription in breast cancer cells mediated by IL1β was confirmed with ChIP-qPCR where both the long and short isoforms of TSLP were transcribed upon exposure to IL1β and their transcription was blocked in the presence of IL1β neutralizing antibody or IL1RA (Fig. 2E). These results suggest that breast cancer cells retain their capacity to produce TSLP in approximately 30% of patients and that one of the pathways to Th2 inflammation in breast cancer is dependent on IL1β via TSLP induction.
Figure 2.
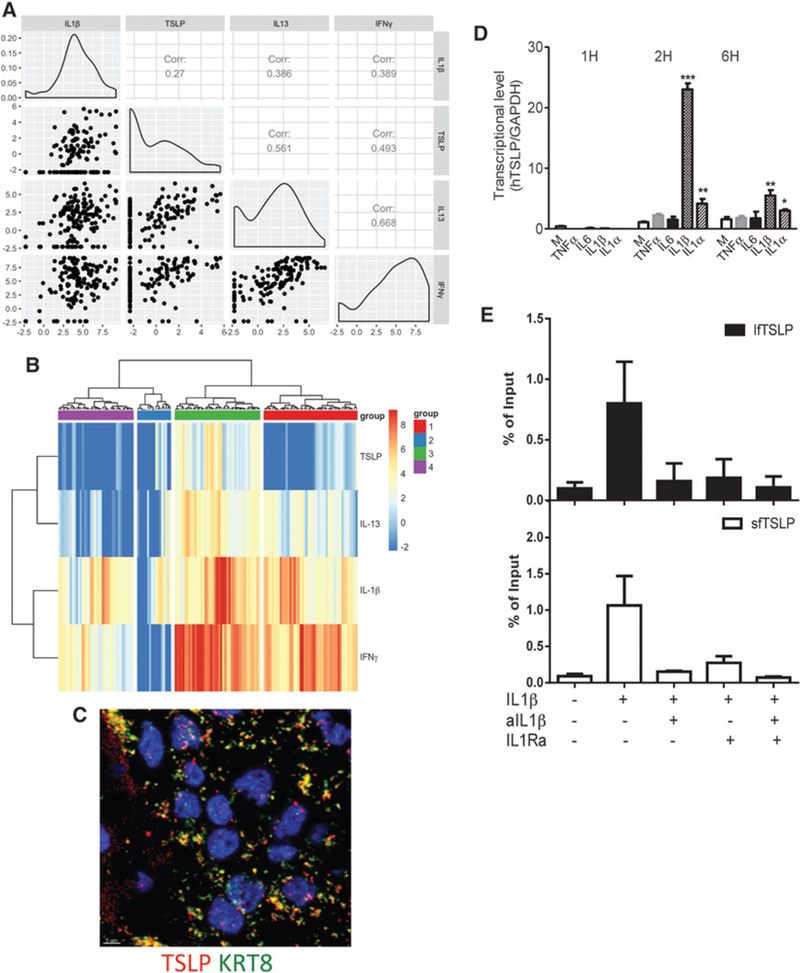
IL1β induces TSLP production from breast cancer cells. A, IL1β, TSLP, IFNγ, and IL13 in supernatants of breast cancer samples. The logarithm of four cytokine measurements (IL1β, TSLP, IL13, IFNγ: left to right at x-axis and top to bottom at y-axis) was taken for total 145 patients and the dependency between any pair of cytokine measurements was tested under the null hypothesis of independence among the four measurements. Log-likelihood and its Bartlett correction were performed and P values for both methods were significantly low (P < 0.001). Histogram plots indicated distribution of the cytokines expression among all patients. Pairwise correlations were tested among 6 pairs of cytokine measurements. Scatter plots indicated the correlation pairwise. Using Pearson and Spearman correlation, all P values were found significant (P < 0.025). B, Heatmap generated on the basis of the log-transformed four cytokine measurements using R package “pheatmap” with “ward.D2” method. Patients were split into four groups based on the “ward.D2” clustering method. The levels of cytokines were color coded as indicated. C, RNA transcripts visualized in breast cancer tumor sections with QuantiGene ViewRNA ISH. Human KRT8 and human TSLP are green and red, respectively. D, MDA-MB-231 cells were treated with medium alone, 10 ng/mL of IL1β, IL1α, TNFα, or IL6 for the indicated time course. TSLP mRNA level by quantitative real-time PCR normalized to internal control GAPDH. Bars show the mean ± SEM for triplicate wells from a representative experiment. *, P < 0.05; **, P < 0.0i; ***, P < 0.000i. E, ChIP by using anti-RNA polymerase II was performed. ChIP-qPCR analysis on lfTSLP (black) and sfTSLP (white) genes from MDA-MB-231 cells with IL1β or IL1β blocking for 1 hour. Percentage of input summarized from three experiments.`
IL1β production by myeloid cells involves inflammasome activation by breast cancer cell-derived factors
IL1p is produced as a precursor protein that requires proteolytic cleavage to liberate its active form (19). This can occur through either canonical caspase-1 (CASP-1)-dependent (20) or noncanonical CASP-8- (21) and CASP-11-dependent (22) pathways. Gene-level analysis from the TCGA dataset revealed a high correlation between CASP1 and IL1B transcripts (Spearman r = 0.50; Fig. 3A), whereas caspase-8 (CASP8) and caspase-11 (SFRS2IP) transcripts were poorly correlated (Spearman r = 0.16 and 0.11, respectively; Fig. 3A). Furthermore, IL1B transcripts correlated with those of the inflammasome complex NLRP3 (Spearman r = 0.56, Fig. 3A) and NLRC4 (Spearman r = 0.51, Fig. 3A).
Figure 3.
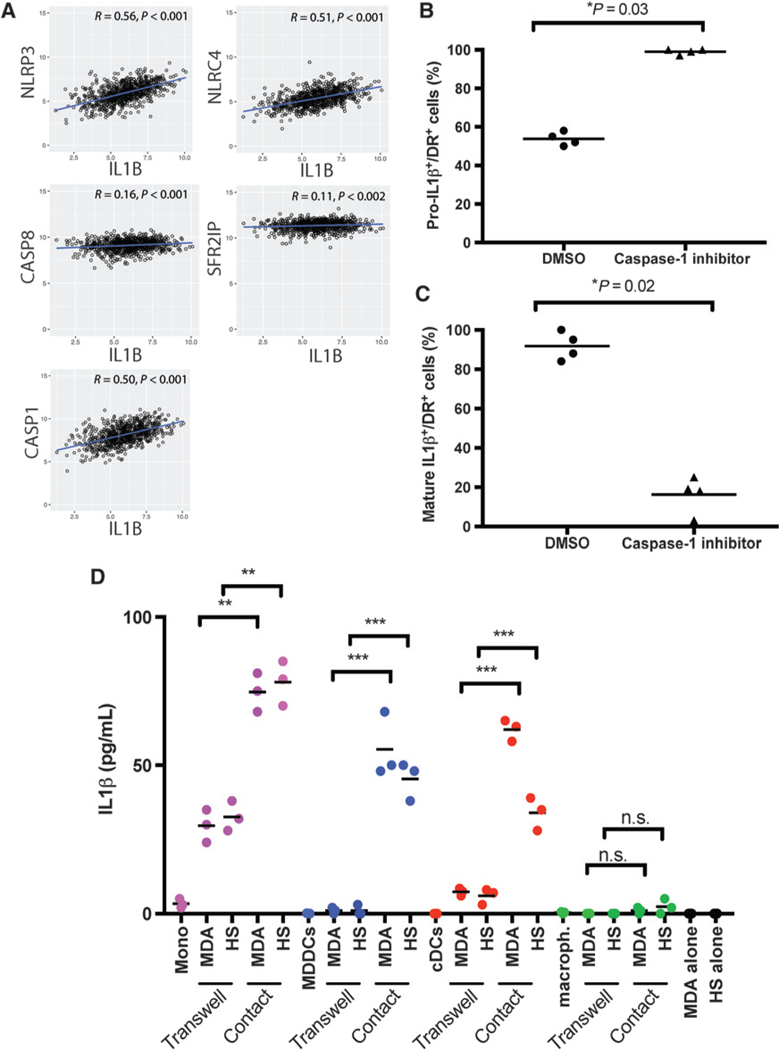
IL1β production in DCs is caspase-1 and contact dependent. A, Scatter plots with line of best fit and Spearman correlation (r) of IL1B with NLRP3, NLRC4, caspase-1 (CASP1), caspase-8 (CASP8), and caspase-11 (SFRS2IP) expression in TCGA dataset (870 patients). B and C, MDA-MB-231 breast cancer cells and cDCs were cocultured in chamber wells for 18 hours, in the presence of caspase-1 inhibitor or DMSO. The percentage of HLA-DR+ cells showing expression of pro-IL1β (B) or that of mature IL1β (C). D, MDA-MB-231 (MDA) or Hs578T (HS) breast cancer cells cocultured with blood monocytes (Mono), MDDCs, cDCs, or monocyte-derived macrophages (macroph.) in regular tissue culture wells (Contact) or Transwell to separate two types of cells in culture for 48 hours. IL1β levels in supernatants by Luminex. Values are plotted as mean ± SEM from triplicate experiments. Welch t test was used. n.s., not significant.
On the basis of this, we examined whether IL1 β production by myeloid cells in vitro required the activation of CASP-1, and how breast cancer cell-derived factors influenced this type of activation. To this end, MDA-MB-231 breast cancer cells and CD11c+ blood circulating DCs (cDC) were cocultured in chamber wells in the presence of 1.0 μmol/L of the CASP-1 inhibitor (Z-WEHD-FMK) (23) or DMSO as a vehicle control. Eighteen hours later, cells were fixed and stained with antibodies specific to either the IL1β propeptide or cleaved/mature IL1β and HLA-DR (Supplementary Fig. S3A and S3B). Treatment with the CASP-1 inhibitor led to the accumulation of IL1β propeptide within HLA-DR+ DCs, while DMSO-treated cells had significantly less propeptide staining (Supplementary Fig. S3A). Microscopy examination revealed that approximately 95% of CASP-1 inhibitor-treated HLA-DR+ DCs displayed intracellular accumulation of IL1β propeptide (n = 4 representative fields of view for each condition, P = 0.03; Fig. 3B). Conversely, the fraction of cells expressing cleaved/mature IL1β dropped significantly (P = 0.02, Fig. 3C). These results indicate canonical CASP-1-mediated cleavage of mature IL1β production in myeloid cells exposed to breast cancer cells.
To determine whether membrane and/or soluble factors triggered IL1 β production, cDCs, monocytes, MDDCs (generated with GM-CSF and IL4), and monocyte-derived macrophages (generated with M-CSF) were cocultured with MDA-MB231 or Hs578T breast cancer cells, separated or not by a transwell membrane with a 0.3-μm pore diameter (Fig. 3D). Breast cancer or myeloid cells cultured alone for 48 hours did not produce detectable IL1 β (Fig. 3D). Coculture of myeloid and breast cancer cells led to significant increases in IL1 β production in cocultures with monocytes, MDDCs, and cDCs, but not in monocyte-derived macrophages (Fig. 3D). Separation of myeloid and breast cancer cell populations by the transwell barrier significantly reduced IL1β production in cocultures with monocytes, MDDCs, and cDCs relative to conditions allowing physical contact between cell populations (Fig. 3D). Thus, production of IL1β is highest when direct physical contact between myeloid cells and breast cancer cells takes place.
Breast cancer cell membrane-associated TGFβ is required for IL1β production by DCs
TGFβ1 can induce IL1B mRNA expression, and in some instances protein production, in human monocytes (24). PLAUR encodes the plasminogen activator, a multidomain glycoprotein tethered to the cell membrane that is involved in proteolytic degradation of the extracellular matrix possibly resulting in activation of latent TGFβ (25). TGFB1 and PLAUR transcripts correlated with IL1B in the TCGA dataset (r = 0.26, P < 0.001 and r = 0.48, P < 0.001, respectively; Supplementary Table S2). To investigate whether breast cancer cell-derived TGFβ1 contributed to IL1β production by myeloid cells, cDCs were cultured with MDA- MB231 and/or Hs578T breast cancer cells, versus Hs578Bst control nonmalignant cells (derived from macroscopically uninvolved breast tissue from the same patient as Hs578T cells). cDCs exposed to breast cancer cells produced IL1 β as determined by flow cytometry (Fig. 4A and B); however, no IL1 β was detected when cDCs were exposed to control nonmalignant Hs578Bst cells (Fig. 4B). We next examined the cancer cell surface for expression of TGFβ1 (26). Both MDA-MB-231 cells and Hs578T cells expressed cell surface TGFβ1, whereas Hs578Bst cells did not (Fig. 4C).
Figure 4.
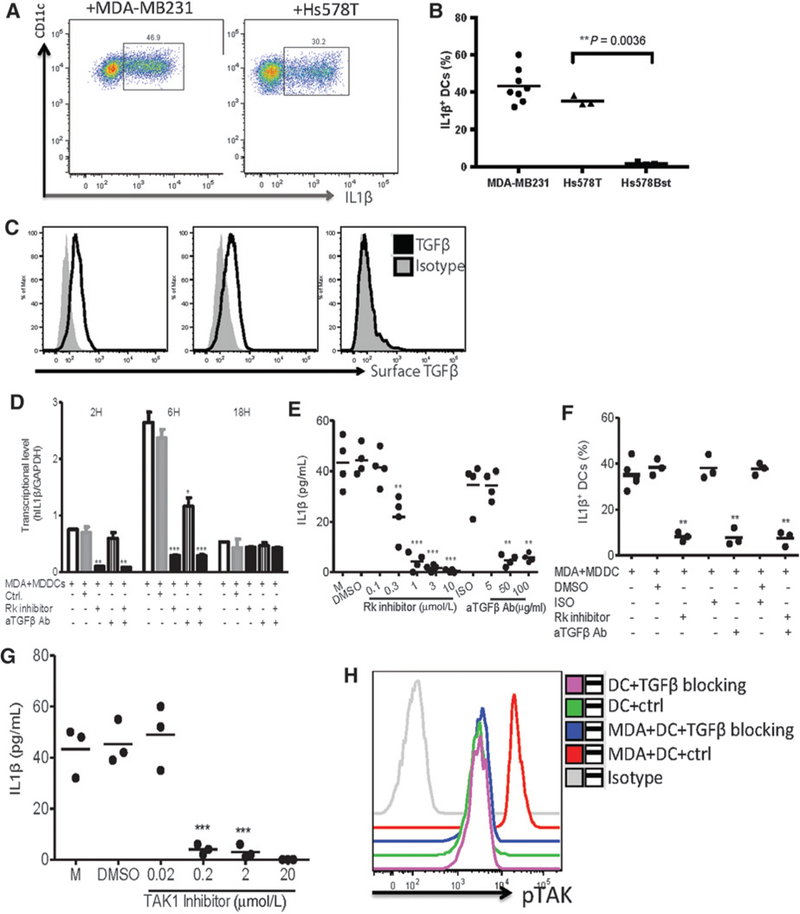
IL1β production in cDCs and monocytes is triggered by TGFβ. A, cDCs were cocultured with MDA-MB-231 breastcancercellsorHs578T breast cancer cells for 16 hours. Intracellular IL1β expression in gated viable cells by FACS. B, Summary of the percentage of IL1β+ cDCs. Each dot represents one experiment. C, Surface expression of activated TGFβ1 by flow cytometry in breast cancer cell lines and nonmalignant cells. D, MDA-MB-231 cells were cocultured with DCs for 48 hours, in presence of different doses of TGFβR kinase inhibitor or anti-TGFβ neutralizing antibody, DMSO, or isotype control, respectively. Histograms of IL1B transcription levels analyzed by quantitative RT-PCR, normalized to GAPDH. Bars show the mean ± SEM for triplicate wells from a representative experiment. Kruskall- Wallis test was used. *, P < 0.05; **, P < 0.01; ***, P < 0.0001. n.s., not significant. E, IL1β level in supernatants by Luminex. Values are plotted as mean ± SEM (independent t test was used). F, Same conditions as in E, but over a 16-hour culture period and with the readout being fraction of total DCs being IL1β+. Intracellular staining of culture with anti-IL1β antibody by flow cytometry. Each dot represents one experiment. G, MDA-MB-231 cells were cocultured with cDCs for 48 hours in presence of different doses of TAK1 inhibitor or DMSO. IL1β levels in the supernatants after 48 hours of coculture by Luminex. Each dot represents one experiment. H, cDCs were cocultured with MDA-MB-231 cells in presence or absence of anti-TGFβ neutralizing antibody plus TGFβR kinase inhibitor (TGFβ blocking) for 60 minutes; pTAK1 was detected by specific staining and analyzed on FACS.
Accordingly, blocking TGFβ1 by adding either TGFβ1 neutralizing antibody or receptor I kinase inhibitor (which inhibits signaling downstream ofTGFβ1) in cocultures with breast cancer cells nearly abolished IL1β secretion by MDDCs, and significantly reduced IL1β production by monocytes (Fig. 4D; Supplementary Fig. S4A-S4C). This was further confirmed by a decrease in frequency of IL1β-expressing MDDCs and monocytes by flow cytometry analysis (Fig. 4E; Supplementary Fig. S4D). Finally, inhibition of TGFβ1 or its downstream signaling decreased IL1B transcript expression, albeit with slightly different kinetics, in MDDCs and monocytes (Fig. 4F; Supplementary Fig. S4C). To determine whether TGFβ 1-mediated induction of IL1β transcription and protein secretion involves inflammasome activation, we used TAK-1 inhibitor (27). Whether used in cultures of DCs or monocytes with breast cancer supernatants, the presence of TAK-1 inhibitor (5Z-7-Oxozeaenol) resulted in the inhibition of caspase-1 activation and IL1β production by DCs as well as by monocytes (Fig. 4G; Supplementary Fig. S4E). Accordingly, the addition of TGFβ 1 neutralizing antibody to cultures of cDCs with breast cancer supernatants prevented the phosphorylation of TAK-1 (Fig. 4H).These findings confirm aroleforTGFβ1/TGFβ1R signaling in priming IL1β production in breast cancer-associated myeloid cells.
Neutralizing TGFβ and IL1 β prevents breast cancer progression and IL13 production in humanized mouse model
To determine the in vivo contribution of TGFβ and IL1 β to breast cancer progression, we utilized a model of IL13-driven breast cancer progression in humanized mouse (9). There, Hs578T breast cancer cells were injected subcutaneously into the flank of irradiated NOD/SCID/β2m–/– mice; MDDCs plus autologous total T cells were injected intratumorally on day 3, 6, and 9 after breast cancer implantation. We analyzed whether local TGFβ or IL1 β blockade could impact breast cancer progression. To this end, we utilized an anti-TGFβ neutralizing antibody, isotype antibody as control, and the IL1R antagonist anakinra, a recombinant soluble nonglycosylated homolog of the human IL1Ra protein that competitively inhibits binding of IL1α and IL1β to IL1R (28).
Anakinra, TGFβ neutralizing antibody and/or controls were each injected intratumorally together with MDDC and T cells (Fig. 5A). Humanized mice that received MDDC+ T cells and isotype control/saline injection exhibited accelerated progression of Hs578T breast cancer tumors, as compared with nonhumanized mice implanted only with Hs578T breast cancer cells and injected with PBS (Fig. 5B). However, treatment with IL1 and/or TGFβ inhibitors resulted in inhibition of breast cancer growth. Thus, at the end of experiment on day 16, the mean tumor volume was 128 mm3 in the PBS group, as compared with 13 mm3 in the anakinra group, and 42.7 mm3 in the TGFβ neutralizing antibody group (P < 0.0001, Fig. 5B).
Figure 5.
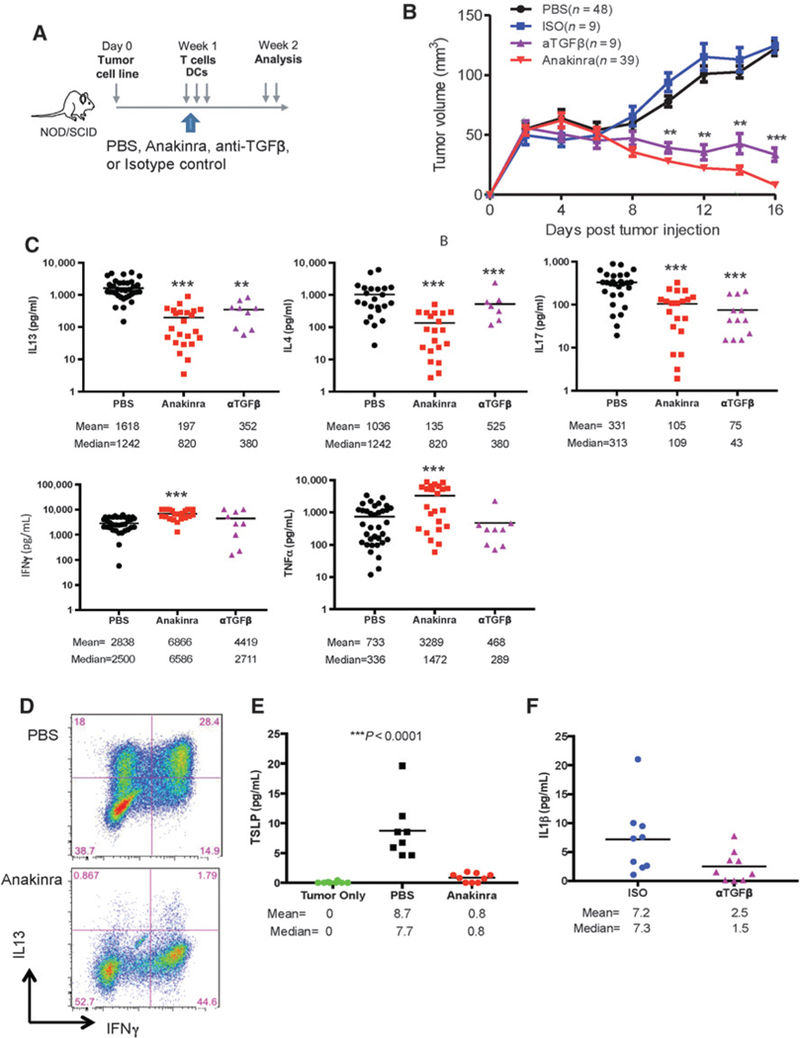
IL1 and TGFβ mediate tumor-promoting type 2 cytokines in humanized mouse model. Hs578T breast tumor-bearing NOD/SCIDβ2–/– mice were reconstituted with MDDCs and autologous T cells. Mice were treated with either: (i) anti-TGFβ neutralizing antibody on days 3, 6, and 9; (ii) anakinra daily starting on day 3; or (iii) controls such as isotype and PBS. A, Scheme of experimental mouse model. B, Kinetics of tumor growth from multiple experiments. Number of mice in each group is indicated. C, Breast tumor fragments were harvested at day 16 after tumor implantation and stimulated for 16 hours with PMA and ionomycin. Cytokines were measured by Luminex. D, Cell suspensions stained for IL13 and IFNγ expression by FACS. Representative plots from three different mice. E, TSLP concentration by Luminex in tumor only versus PBS control versus anakinra group. F, IL1β concentration by Luminex in isotype control versus anti-TGFb neutralizing antibody group.
Small pieces of tumor tissue from mice were subsequently cultured for 16 hours in the presence of PMA and ionomycin, as we did for patient samples, and cytokines measured in culture supernatants. Treatment with anakinra or TGFβ neutralizing antibody resulted in significant decreases in IL13 (P < 0.0001, P < 0.005 respectively), IL4 (P < 0.0001, P < 0.005 respectively), and IL17 production (P < 0.0001, P < 0.0001, respectively) in breast cancer tumors (Fig. 5C). Anakinra, but not TGFβ neutralizing antibody, resulted in significant increase of TNF or IFNγ production (Fig. 5C). Cytokine secretion patterns were confirmed by flow cytometry analysis of tumor-infiltrating lymphocytes that revealed a substantial decrease in the percentage of tumor-infiltrating CD4+ T cells producing IL13 and increase of IFNγ-producing CD4+ T cells after anakinra treatment (Fig. 5D; Supplementary Fig. S5). Tumors from humanized mice produced TSLP, which was blocked by treatment with anakinra (P < 0.0001, Fig. 5E). Furthermore, TGFβ neutralizing antibody treatment decreased IL1p production by breast cancer tumors established in humanized mice (P = 0.07, n = 9, Fig. 5F). Thus, IL1 p controls IL13 production and CD4+ T-cell differentiation in vivo in breast cancer bearing humanized mice. Furthermore, TGFβ acts upstream of IL1β, further supporting the conclusions from our in vitro studies.
Patients with metastatic breast cancer display IL1-dependent transcriptional signature in the blood
Anakinra is highly effective and FDA-approved for the treatment of children with systemic onset juvenile arthritis (29) and other diseases driven by autoinflammation (30).
We conducted a pilot clinical trial (NCT01802970) in which 11 patients with HER2− metastatic breast cancer received treatment with daily subcutaneous anakinra (100 mg/daily, the FDA- approved dose for adults with rheumatoid arthritis) (Supplementary Tables S3 and S5; ref. 48) alone for a two-week run-in treatment period, followed by continuous daily anakinra along with physician’s choice of chemotherapy: weekly nab-paclitaxel (n = 3), eribulin (n = 5), or capecitabine (n = 2) until the development of treatment-limiting toxicity or progressive disease. The median age of the patients was 44 years. The median number of prior cytotoxic regimens for metastatic breast cancer the patients had received was 1, and the organ sites of metastases were: bone 91%; liver 27%; and lung 46%. Patients received anakinra therapy for a median of 4 months (range: 11–179 days). The endpoints were safety of the combination and blood transcriptional signatures. One patient had disease progression prior to beginning chemotherapy and received anakinra for only 14 days. Eight of 11 patients developed grade 1–2 anakinra-related injection site reactions; no other anakinra-related toxicities were observed. Two patients had considerable reduction in tumor volume with chemo/anakinra, 4 had stable disease, 2 stopped anakinra for injection site reactions, and the other 3 had progressive disease. Of the 5 patients who had cancer-associated pain at study entry, three reported improvement in their pain and overall well-being with anakinra plus chemotherapy. All patients were accrued between February 2013 and December 2014, and as of December2017, 3 ofthe 11 patients are alive and continuing antibreast cancer therapy (Supplementary Table S5).
To examine the impact of anakinra on markers of inflammation, the blood leukocytes transcriptome of patients was profiled longitudinally prior to anakinra treatment, after treatment with anakinra alone at 2 weeks (run-in), and then longitudinally (monthly for 6 months) after combined treatment with anakinra and chemotherapy. We also profiled the blood leukocytes transcriptome of 10 healthy age-and gender-matched volunteers. Blood leukocyte mRNA transcripts were hybridized to 579 immune-related probes and 15 housekeeping genes using the NanoString nCounter Human Immunology V2 panel. To identify differentially expressed genes (DEG), we used a linear mixed model (31), which accounted for repeated measures over time and missing observations. First, DEGs between the healthy controls and the patients at any time point in the course of the study were defined. A total of 288 DEGs were detected and their hierarchical clustering enabled identification of transcripts that did or did not change according to anakinra treatment overtime (Fig. 6A and B). Several transcripts were abundant at baseline and sustained after treatment with anakinra combined or not with chemotherapy, including cytokines (IL6, IFNA, IL10), defensins (DEFB1 and DEFB103B), NOS2 and CXCL2 [Gene set (GS)1; Fig. 6A]. Transcripts for IL13 and IL17A were only transiently decreased after anakinra treatment alone and were abundant again after addition of chemotherapy, while several transcripts related to antigen presentation and functions of DCs were not altered (IFNA, XCR1,XCL1, and CD70; GS 1; Fig. 6A). Conversely, several genes that were abundant at baseline were decreased following the two weeks of anakinra alone, including the IL1-related genes IL1B, IL1R1, IL1R2, and IL1RAP (GS3); the myeloid cell-related genes ICAM1, ICAM3, ITGAX, FCGR2A, IL6R, CSF2RB (common subunit of GM-CSF, IL3, and IL5 receptor) and PLAUR; and transcripts encoding innate sensing and downstream signaling molecules such as TLR1, TLR8, CLEC4A, CLEC7A, SYK, and MYD88 (GS 2–4; Fig. 6A and B). Decrease in these genes was sustained over time in the course of treatment with anakinra and chemotherapy (Fig. 6A). The observed changes were not related to fluctuations in blood cell composition, as revealed by longitudinal blood flow cytometry (Supplementary Fig. S6A and S6B).
Figure 6.
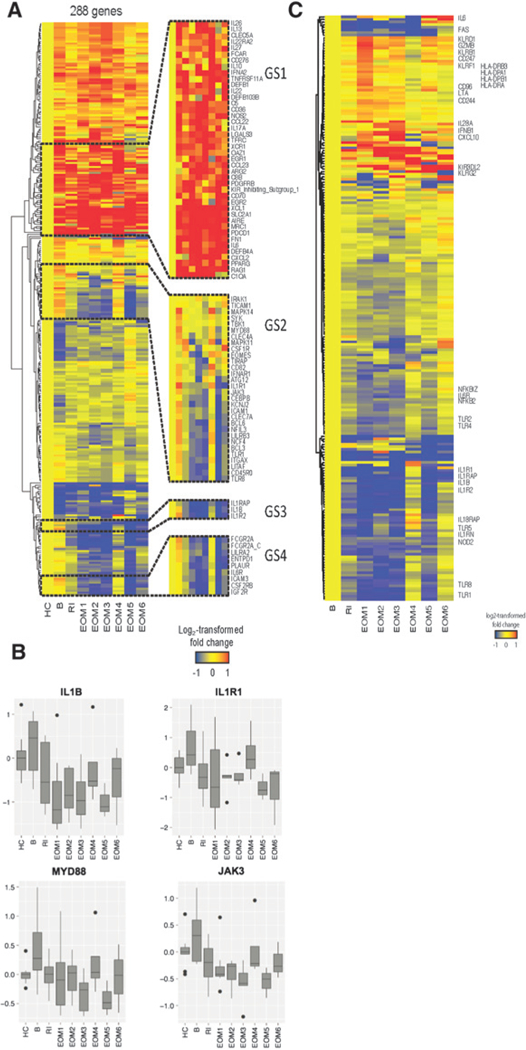
Anakinra modulates transcriptional signature in the blood. A, Hierarchical clustering of the 288 differentially expressed genes identified using mixed model analysis, between healthy controls (HC) and patients with breast cancer at any time point during the study. The expression of genes was averaged from all patients in each time point. B, patient baseline; RI, patient run-in (two weeks after anakinra only); EOM, end of month after anakinra plus chemotherapy. B, Box plots representing the log2-transformed expression of a subset of genes. Horizontal lines, median. Boxes represent the first and third quartiles (25th and 75th percentile). Whiskers extend to the highest or lowest values within 1.5-fold of the interquartile range. C, Genes differentially expressed between any time point posttreatment and patient baseline (B; P <> 0.05).
Next, to further evaluate the effects of anakinra on the blood leukocytes transcriptome of patients over time, we selected the genes differentially expressed between any time point posttreatment and the patient baseline (without the inclusion of HC in the analysis; P < 0.05; Fig. 6C; Supplementary Table S3). Of the 223 DEGs identified, genes related to IL1 signaling (IL1B, IL1R1, IL1R2, IL1RAP, IL1RN, IL6, IL6R), NFkB signaling (NFKB2, NFKBIZ), and innate immunity (TLR1, TLR2, TLR4, TLR5, TLR8, NOD2) were rapidly downregulated following anakinra treatment. Conversely, genes related to NK- and T-cell-mediated cytotoxicity (KLRs, KIRs, GZMB, LTA, CD96, CD244, CD247) were upregulated, especially after 1-month treatment. These results indicate that anakinra effectively downregulated specific components of the systemic inflammatory signature observed in patients with metastatic breast cancer and rescued cytotoxic programs thought to be critical for antitumor activity.
We also compared the blood inflammation-related transcriptome of patients with metastatic breast cancer with those of patients with IL1-driven systemic juvenile idiopathic arthritis (sJIA; ref. 29). From the 288 DEGs identified in the patients with breast cancer compared with healthy controls, we selected the 178 genes that were overexpressed at baseline (prior to anakinra treatment), mapped them to 117 Illumina V2 probes, and analyzed their expression in blood leukocytes from acute, untreated patients with sJIA. Hierarchical clustering revealed overexpression of most of the breast cancer DEGs in patients with sJIA (Supplementary Fig. S6C). Thus, patients with metastatic breast cancer display a blood transcriptional signature that overlaps in part with that of sJIA, a disease whose pathogenesis involves systemic IL1- driven inflammation.
IL1-dependent signature in breast cancer tumors discriminates patients with a subtype of poor prognosis
We then leveraged the TCGA (RNAseq data Supplementary Table S2; ref. 18) and METABRIC datasets (DNA microarrays data (Supplementary Table S2; ref. 32) to examine IL1B expression in breast cancer subsets and to determine whether the blood transcripts modulated by anakinra over time (in comparison with healthy controls) and expressed in primary breast cancers could discriminate patients with poor prognosis. First, in both datasets, IL1B transcripts were significantly higher in samples defined as basal breast cancer, a disease with relatively poor prognosis comparing with other breast cancer subtypes (METABRIC n = 1986 Fig. 7A and TCGA n = 788 Supplementary Fig. S7A). We then defined samples with extreme outcomes: among 870 patients with invasive breast cancer in the TCGA dataset, 30 patients survived more than 10 years after primary surgery (good prognosis), and 70 patients died with breast cancer within 5 years of primary treatment (poor prognosis). Among 1,992 patients with breast cancer in METABRIC dataset, 710 patients had good and 156 patients had poor survival outcomes, respectively. In both cohorts, the samples were hierarchically clustered using the IL1B signature genes using Euclidean distance and ward.D2 agglomeration method. Samples were stratified into two groups based only on the basis of anakinra-modulated signature to test their prognostic significance.
Figure 7.
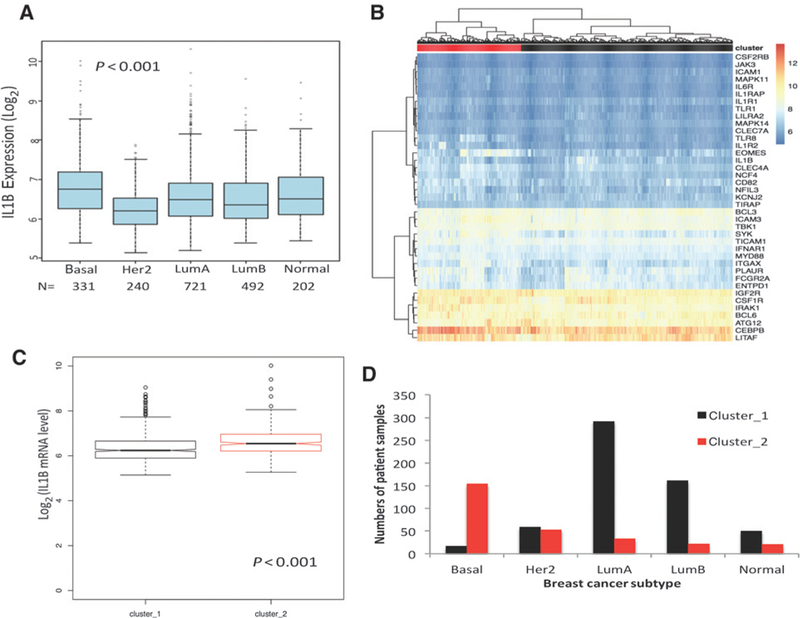
Basal subtype of breast cancer is linked with high IL1β and anakinra-dependent signature. A, IL1B transcript expression in different breast cancer subtypes annotated in the METABRIC database. N, number of samples per subtype. Kruskal–Wallis test was P < 0.001. B, The gene sets controlled by anakinra treatment in the blood were used in unsupervised analyses of transcriptional datasets from invasive BrCa tumors in the METABRIC datasets. Two clusters were formed on the basis of gene expression and assigned as red and black clusters. C, IL1B transcription in two clusters in both datasets. D, Number of patients in each cluster among different subtypes of breast tumors.
In both datasets, the algorithm clustered tumor samples based on the expression level of transcripts that were decreased in the blood following anakinra treatment into those that did not display anakinra-dependent transcriptional signature (cluster 1, black), versus those that did display this signature (cluster 2, red; Fig. 7B; Supplementary Fig. S7B). Accordingly, these clusters showed differential IL1B transcription (P < 0.001 and P < 0.001 in METABRIC and TCGA cohorts, respectively; Fig. 7C; Supplementary Fig. S7C). Further analysis of METABRIC cohort, where the number of patients was high enough to analyze the distribution of anakinra-modulated signature in breast cancer subtypes, revealed that patients whose primary breast cancers had clustered on the basis of higher expression of anakinra-modulated transcripts had predominantly basal breast cancer (Fig. 7D).
Thus, our results point toward an inflammatory signature in primary breast cancers that is associated with a subtype of poor prognosis. Moreover, they identify a subset of patients that could potentially benefit from IL1β-targeted therapies.
Discussion
On the basis of in vitro and in vivo studies, our current results highlight the role of IL1 β in tumor-associated inflammation in human primary and metastatic breast cancer.
First, we demonstrate that primary breast cancer tumors contain high levels of IL1β and that IL1B transcription is elevated in the biologically aggressive basal subtype. Other studies have indicated that IL1 β might play a deleterious role in modulating tumor-associated immunity (33, 35, 35). For example, in syngeneic mouse models, IL1 β can counter antitumor effects, triggered by doxorubicin treatment, by recruiting myeloid-regulatory cells that promote immunosuppression as well as promote cancer invasiveness and metastasis (36); and by conferring a proliferative advantage to the cancer cells (reviewed in ref. 37). Indeed, preclinical studies revealed that blockade of IL1 with anakinra enhanced the antitumor efficacy of 5-FU, leading to tumor regression in about 50% of treated animals (38). In our current study, IL1β production was linked with more advanced disease as defined by pathologic disease stage reflecting tumor size, metastases to regional lymph nodes, and extension to chest wall and/or the skin, and was a measure of tumor invasiveness and virulence, thereby corroborating previous observations (39). It is possible that levels of IL1β reflect enhanced myeloid cell infiltration, which itself is a consequence of larger tumors. Interestingly, levels of secreted IL1α were only marginally affected upon ex vivo breast cancer activation, and did not correlate with disease stage. This was expected because we analyzed supernatants and IL1α is not secreted. Thus, antibodies to IL1α that are used in clinical trials are likely to neutralize intracellular IL1α that is released in hypoxic tumor microenvironment or IL1α expressed as an integral membrane protein (19, 20, 28, 35).
IL1β production by myeloid cells triggered in vitro by breast cancer cell-derived factors is dependent on CASP-1 activation, and IL1B transcription correlates with that of NLRP3. Thus, in addition to the recently described role of NLRC4 in triggering IL1Btranscription and protein production in mammary carcinoma mouse models (40), other inflammasome components, such as NLRP3, may also be involved in triggering IL1B transcription and protein production in humans. Several preclinical models revealed that inflammasome activation resulting in IL1 release promotes different cancer types, while inflammasome-mediated IL18 release was reported to be protective in colitis-associated colorectal cancer (35). Thus, molecular mechanisms and outcomes of inflammasome activation specific to each cancer type must be understood before developing personalized regimens modulating inflammation.
Second, treatment of patients with metastatic breast cancer with the IL1 receptor antagonist anakinra eliminates a systemic transcriptional signature of IL1-associated inflammation in blood cells. Blood transcriptional profiles indicated that in vivo anakinra treatment effectively rescued immune cells’ cytotoxic programs that could contribute to enhanced antitumor activity. In this context, the observed link between TGFβ and IL1 β is of interest based on earlier studies reporting a suppressive role for TGFβ in immunosurveillance against transplantable as well as spontaneous tumors in mice via NKT-cell-derived IL13 (41). In clinical samples, recent meta-analysis of breast cancer tumor expression profiles further underscores the linkage between activation of the TGFp pathway, and poor overall prognosis in breast cancer (42). Interestingly, the interplay between TGFp and IL1 p might be bidirectional as recent studies suggested expression of IL1 p in breast cancer cell lines that is linked with TGFp activity in myeloid cells (43). It will be important, in future studies, to determine how this cross-talk behaves during chemotherapy and radiotherapy and whether the capacity to trigger either direction is patient or breast cancer subtype specific.
A correlative analysis indicates that IL1β might be involved in amplification of type 2 cytokines in patients with breast cancer, although a causative link cannot yet be formally established. This conclusion is also supported by our results revealing that IL1 β controls IL13 production and CD4+ T-cell differentiation in vivo in breast cancer-bearing humanized mice. Furthermore, we identified a subset of patients with breast cancer in whom IL1 β is associated with IL13 and with TSLP, a Th2-inducing cytokine. Indeed, approximately 30% of patient samples produce TSLP on activation. We also demonstrate the presence of TSLP RNA in cancer cells. Thus, our data offer a more nuanced landscape of TSLP and Th2 cytokines in breast cancer than the recently described study, which failed to identify TSLP transcription in breast cancer (44). Future studies will address the prognostic impact of the four different cytokine environments that we identified herein. It is also possible that the contribution of the overall type 2 cytokine inflammation in breast cancer goes beyond CD4+ T cells. Indeed, group 2 innate lymphoid cells (ILC2 cells) are important for type 2 immune responses and can be activated, among others, by IL1 family cytokines including IL1 β (45). IL1 β- driven activation of blood ILC2 has been linked with an atypical chromatin landscape characterized by simultaneous transcriptional accessibility of the locus encoding IFNγ, and the loci encoding IL5 and IL13 (45). This pattern of activation of both type 1 (IFNg) and type 2 cytokines in the same lymphocyte population closely resembles our earlier observations in breast cancer tumors of simultaneous expression of IFNγ and IL13 by CD4+ T cells that promote breast cancer progression in experimental tumors (9).
Future studies are needed to elucidate the impact of IL1 on lymphocyte phenotype and functions in breast cancer tissues, as well as potential links between activation of IL1 family signaling and sensitivity to check-point inhibitor therapy. Indeed, based on broad protumor effects of IL1 such as VEGF production, expansion of immature myeloid cells in periphery, and production of IL8 leading to neutrophil accumulation in tumor among others (19,20,28, 35), it can be envisioned that anakinra might enhance therapeutic effects of other treatment modalities such as chemotherapy and immunotherapy.
Finally, when applied across primary invasive breast cancer tumors in TCGA and METABRIC databases, the anakinra-controlled blood transcriptional signature discriminated patients with basal breast cancer, a subtype with relatively poor prognosis. This further underscores the links between IL1 signaling pathway and high-risk disease in patients with breast cancer.
Anakinra inhibits binding of both the IL1α and IL1β cytokines to IL1 receptors. To further dissect individual contributions of these two cytokines, future studies will need to test specific antibodies. To this end, IL1 β could be inhibited with canakinumab (approved for the treatment of sJIA) or the fusion protein rilonacept, while IL1α could be inhibited with MABp1, which has been recently found to be active in colorectal cancer (46). In line with this concept, the specificblockade of IL1β with canakinumab was recently shown to be associated with decreased incidence of lung cancer (47).
In summary, this study identifies an IL1-associated inflammatory signature in primary breast cancers that, if validated in follow-up clinical studies, could be used to stratify patients at diagnosis and justify use of ILl-directed therapies. Furthermore, blocking IL1 effectively downregulated specific components of the systemic inflammatory signature in patients with breast cancer, and rescued cytotoxic programs that maybe critical for antitumor activity and thereby for enhancement of clinical efficacy of checkpoint inhibitors.(34)
Supplementary Material
Acknowledgments
We thank patients and healthy donors for participation in our studies; BUMC Tissue Bank, clinical teams at BUMC and CCBD, Genomic Core and Flow Cytometry Core at BIIR. This work was supported by BIIR (to A.K. Palucka), Baylor University Medical Center Foundation (to A.K. Palucka, V. Pascual, J. Banchereau), Baylor Charles A. Sammons Cancer Center (to J. O’Shaughnessy), and by The Jackson Laboratory (to A.K. Palucka). This work was partially supported by the National Cancer Institute of the NIH under award number P30CA034196 (to A.K. Palucka).
Footnotes
Disclosure of Potential Conflicts of Interest
K. Palucka is a consultant/advisory board member for Swedish Orphan Biovitrum AB. No potential conflicts of interest were disclosed by the other authors.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
The costs of publication of this article were defrayed in part bythe payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015;348:56–61. [DOI] [PubMed] [Google Scholar]
- 2.Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer 2016;16:566–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, et al. Pembro-lizumab in patients with advanced triple-negative breast cancer: phase Ib KEYNOTE-012 study. J Clin Oncol 2016;34:2460–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palucka AK, Coussens LM. The basis of oncoimmunology. Cell 2016; 164:1233–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008;454:436–44. [DOI] [PubMed] [Google Scholar]
- 6.Pierce BL, Ballard-Barbash R, Bernstein L, Baumgartner RN, Neuhouser ML, Wener MH, et al. Elevated biomarkers of inflammation are associated with reduced survival among breast cancer patients. J Clin Oncol 2009;27:3437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol 1989;7:145–73. [DOI] [PubMed] [Google Scholar]
- 8.DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009;16:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pedroza-Gonzalez A, Xu K, Wu TC, Aspord C, Tindle S, Marches F, et al. Thymic stromal lymphopoietin fosters human breast tumor growth by promoting type 2 inflammation. J Exp Med 2011;208:479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Espinoza JA, Jabeen S, Batra R, Papaleo E, Haakensen V, Timmermans Wielenga V, et al. Cytokine profiling of tumor interstitial fluid of the breast and its relationship with lymphocyte infiltration and clinicopathological characteristics. Oncoimmunology 2016;5:e1248015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeNardo DG, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev 2010;29:309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demehri S, Cunningham TJ, Manivasagam S, Ngo KH, Moradi Tuchayi S, Reddy R, et al. Thymic stromal lymphopoietin blocks early stages ofbreast carcinogenesis. J Clin Invest 2016;126:1458–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu-Trantien C, Willard-Gallo K. Tumor-infiltrating follicular helper T cells: The new kids on the block. Oncoimmunology 2013;2:e26066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prabhakaran S, Rizk VT, Ma Z, Cheng CH, Berglund AE, Coppola D, et al. Evaluation of invasive breast cancer samples using a 12-chemokine gene expression score: correlation with clinical outcomes. Breast Cancer Res 2017;19:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Angkasekwinai P, Park H, Wang YH, Wang YH, Chang SH, Corry DB, et al. Interleukin 25 promotes the initiation of proallergictype 2 responses. J Exp Med 2007;204:1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu YJ, Soumelis V, Watanabe N, Ito T, Wang YH, Malefyt Rde W, et al. TSLP: An epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu Rev Immunol 2007;25:193–219. [DOI] [PubMed] [Google Scholar]
- 17.Pulendran B, Artis D. New paradigms in type 2 immunity. Science 2012;337:431–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity 2013;39:1003–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity 2015;42:991–1004. [DOI] [PubMed] [Google Scholar]
- 21.Antonopoulos C, Russo HM, El Sanadi C, Martin BN, Li X, Kaiser WJ, et al. Caspase-8 as an effector and regulator of NLRP3 inflammasome signaling. J Biol Chem 2015;290:20167–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 2016;352:1232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito T, Hanabuchi S, Wang YH, Park WR, Arima K, Bover L, et al. Two functional subsets of FOXP3+ regulatory T cells in human thymus and periphery. Immunity 2008;28:870–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bogdan C, Nathan C. Modulation of macrophage function by transforming growth factor beta, interleukin-4, and interleukin-10. Ann N Y Acad Sci 1993;685:713–39. [DOI] [PubMed] [Google Scholar]
- 25.Odekon LE, Blasi F, Rifkin DB. Requirement for receptor-bound urokinase in plasmin-dependent cellular conversion of latent TGF-beta to TGF-beta. J Cell Physiol 1994;158:398–407. [DOI] [PubMed] [Google Scholar]
- 26.Blakytny R, Ludlow A, Martin GE, Ireland G, Lund LR, Ferguson MW, et al. Latent TGF-beta1 activation by platelets. J Cell Physiol 2004;199:67–76. [DOI] [PubMed] [Google Scholar]
- 27.Ouyang C, Nie L, Gu M, Wu A, Han X, Wang X, et al. Transforming growth factor (TGF)-beta-activated kinase 1 (TAK1) activation requires phosphorylation of serine 412 by protein kinase A catalytic subunit alpha (PKACalpha) and X-linked protein kinase (PRKX). J Biol Chem 2014;289:24226–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 2012;11:633–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role ofinterleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med 2005;201:1479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ombrello MJ, Kastner DL. Autoinflammation in 2010: expanding clinical spectrum and broadening therapeutic horizons. Nat Rev Rheumatol 2011;7:82–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng RD. Reproducible research in computational science. Science 2011; 334:1226–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drikvandi R, Verbeke G, Khodadadi A, Partovi Nia V. Testing multiple variance components in linear mixed-effects models. Biostatistics 2013;14:144–59. [DOI] [PubMed] [Google Scholar]
- 33.Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486:346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dinarello CA. An expanding role for interleukin-1 blockade from gout to cancer. Mol Med 2014;20 Suppl 1:S43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salcedo R, Cataisson C, Hasan U, Yuspa SH, Trinchieri G. MyD88 and its divergent toll in carcinogenesis. Trends Immunol 2013; 34:379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in car¬cinogenesis and anticancer immune responses. Nat Immunol 2012; 13:343–51. [DOI] [PubMed] [Google Scholar]
- 37.Elaraj DM, Weinreich DM, Varghese S, Puhlmann M, Hewitt SM, Carroll NM, et al. The role of interleukin 1 in growth and metastasis of human cancer xenografts. Clin Cancer Res 2006;12:1088–96. [DOI] [PubMed] [Google Scholar]
- 38.Shurin MR. Dual role of immunomodulation by anticancer chemotherapy. Nat Med 2013;19:20–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med 2013;19:57–64. [DOI] [PubMed] [Google Scholar]
- 40.Soria G, Ofri-Shahak M, Haas I, Yaal-Hahoshen N, Leider-Trejo L, Leibovich-Rivkin T, et al. Inflammatory mediators in breast cancer: coordinated expression ofTNFalpha & IL-1beta with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer 2011; 11:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kolb R, Phan L, Borcherding N, Liu Y, Yuan F, Janowski AM, et al. Obesity- associated NLRC4 inflammasome activation drives breast cancer progres¬sion. Nat Commun 2016;7:13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berzofsky JA, Terabe M. A novel immunoregulatory axis of NKT cell subsets regulating tumor immunity. Cancer Immunol Immunother 2008;57: 1679–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller LD, Chou JA, Black MA, Print C, Chifman J, Alistar A, et al. Immu¬nogenic subtypes of breast cancer delineated by gene classifiers of immune responsiveness. Cancer Immunol Res 2016;4:600–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen Q, Cohen B, Zheng W, Rahbar R, Martin B, Murakami K, et al. Notch shapes the innate immunophenotype in breast cancer. Cancer Discov 2017;7:1320–35. [DOI] [PubMed] [Google Scholar]
- 45.Ghirelli C, Sadacca B, Reyal F, Zollinger R, Michea P, Sirven P, et al. No evidence for TSLP pathway activity in human breast cancer. Oncoimmunology 2016;5:e1178438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohne Y, Silver JS, Thompson-Snipes L, Collet MA, Blanck JP, Cantarel BL, et al. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol 2016;17:646–55. [DOI] [PubMed] [Google Scholar]
- 47.Hickish T, Andre T, Wyrwicz L, Saunders M, Sarosiek T, Kocsis J, et al. MABp1 as a novel antibody treatment for advanced colorectal cancer: a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 2017;18: 192–201. [DOI] [PubMed] [Google Scholar]
- 48.Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017;390: 1833–1842. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.