

Measurements of mutation rates—i.e., how often proliferating cells acquire mutations in their DNA—are essential for understanding cellular processes that maintain genome stability. Many traditional mutation rate measurement assays are based on detecting mutations that cause resistance to a particular drug. Such assays typically work well for laboratory strains but have significant limitations when comparing clinical or environmental isolates that have various intrinsic levels of drug tolerance, which confounds the interpretation of results. Here we report the development and validation of a novel method of measuring mutation rates, which detects mutations that cause loss of fluorescence rather than acquisition of drug resistance. Using this method, we measured the mutation rates of clinical isolates of fungal pathogen Candida glabrata. This assay can be adapted to other organisms and used to compare mutation rates in contexts where unequal drug sensitivity is anticipated.
KEYWORDS: Candida glabrata, GFP, MSH2, mutation rate
ABSTRACT
All evolutionary processes are underpinned by a cellular capacity to mutate DNA. To identify factors affecting mutagenesis, it is necessary to compare mutation rates between different strains and conditions. Drug resistance-based mutation reporters are used extensively to measure mutation rates, but they are suitable only when the compared strains have identical drug tolerance levels—a condition that is not satisfied under many “real-world” circumstances, e.g., when comparing mutation rates among a series of environmental or clinical isolates. Candida glabrata is a fungal pathogen that shows a high degree of genetic diversity and fast emergence of antifungal drug resistance. To enable meaningful comparisons of mutation rates among C. glabrata clinical isolates, we developed a novel fluorescence-activated cell sorting-based approach to measure the mutation rate of a chromosomally integrated GFP gene. We found that in Saccharomyces cerevisiae this approach recapitulated the reported mutation rate of a wild-type strain and the mutator phenotype of a shu1Δ mutant. In C. glabrata, the GFP reporter captured the mutation rate increases caused either by a genotoxic agent or by deletion of DNA mismatch repair gene MSH2, as well as the specific mutational signature associated with msh2Δ. Finally, the reporter was used to measure the mutation rates of C. glabrata clinical isolates carrying different alleles of MSH2. Together, these results show that fluorescence-based mutation reporters can be used to measure mutation rates in microbes under conditions of unequal drug susceptibility to reveal new insights about drivers of mutagenesis.
INTRODUCTION
Acquisition of mutations underlies evolution in all contexts, and in pathogenic microbes this process can result in the emergence of dangerous drug-resistant strains. An organism’s capacity to acquire mutations is typically measured as the rate (per cell, per generation) at which a specific reporter gene accumulates mutations that result in a detectable phenotypic change. Because most genomes are extremely stable and mutation rates are typically very low, the most convenient and widely used mutation measurement assays are set up as selections, where mutation of a reporter gene confers resistance to a particular drug (1). In such an assay, mutations in the reporter gene arise at some low rate in cells proliferating in culture and are then selected by plating the cultures on drug-containing medium, which kills wild-type cells. The CAN1 mutation assay in Saccharomyces cerevisiae is based on this principle and has been used extensively to gain insights into mechanisms controlling genome stability in yeast (2–5). Although drug resistance-based mutation assays have a number of advantages, most particularly in their relative ease and rapidity, their major limitation is that they allow direct comparisons of mutation rates only between strains that have the same level of drug sensitivity. This condition is largely satisfied when one compares the mutation rates of isogenic strains, e.g., a laboratory strain and a DNA repair mutant derived from it. However, this condition does not hold when one wishes to compare mutation rates among nonisogenic strains, e.g., a panel of clinical or environmental isolates, which may have various levels of drug tolerance. In this case, more tolerant strains are expected to survive for a longer period on selection medium, all the while continuing to produce resistance mutations, leading to an artificially high mutation rate estimate, and vice versa. Similar considerations preclude direct comparisons of mutation rates between different species, e.g., one that is highly drug susceptible versus one that is more drug tolerant, or between different growth conditions (e.g., growth in the presence of a stressor that may affect overall stress/drug tolerance). Thus, in order to rigorously measure and compare mutation rates in a way that is not restricted to a small number of laboratory strains and their derivatives, it is necessary to develop a drug resistance-independent method to measure mutation rates.
Candida glabrata is a yeast that is closely related to S. cerevisiae (6) and is also associated with the human microbiome (7). In contrast to S. cerevisiae, however, C. glabrata is an opportunistic pathogen that can cause life-threatening infections in immunocompromised individuals (8, 9). The prevalence of C. glabrata in infections has been increasing, and it is now the second most prevalent cause of invasive candidiasis in the United States and Europe (10, 11). One reason for this increase is that C. glabrata either is intrinsically resistant or acquires resistance relatively quickly to the limited number of antifungal drugs currently in clinical use (9, 12). In C. glabrata, drug resistance is predominantly caused by point mutations, either in transcription factors regulating drug efflux (13–15) or in genes encoding drug targets (16–18). Several studies have also documented the emergence of multidrug-resistant (MDR) C. glabrata infections, for which there are no treatment options and which are associated with extremely high mortality (19, 20).
Comparisons of DNA sequences (both of specific genes and of whole genomes) from different C. glabrata clinical isolates have revealed an exceedingly high level of genetic diversity, in terms of both single nucleotide polymorphisms (SNPs) and chromosomal arrangements (21–26). A multilocus sequencing typing (MLST) scheme based on SNPs at six different loci has identified over 100 distinct sequence types (STs) of C. glabrata, which cluster into seven clades (22, 25; https://pubmlst.org/cglabrata/). However, even strains within the same clade exhibit high genetic diversity (22), which, together with rapid emergence of mutations that cause drug resistance, has led to the hypothesis that C. glabrata may have a highly plastic, or mutable, genome. However, mutation rates in C. glabrata have not been measured or compared to other organisms.
In a previous study, we began to examine the role of DNA mismatch repair (MMR) in maintaining genome stability and emergence of drug resistance in C. glabrata (24). In particular, we found that different STs of C. glabrata are associated with specific SNPs in MMR gene MSH2, some of which result in amino acid changes and, when introduced into an msh2Δ reference strain on a plasmid, do not fully rescue that strain’s hypermutator phenotype. This result suggested that some C. glabrata isolates, e.g., those carrying certain variants of MSH2, may exhibit higher mutation rates and may therefore acquire drug resistance more rapidly. Indeed, in Cryptococcus, naturally occurring mutations in MSH2 have been shown to contribute to microevolution and population diversity (59, 60). Yet, recent clinical studies have not found an association between specific MSH2 alleles and drug resistance (27–29, 61, 62), raising the question of whether clinical isolates carrying these alleles are true mutators. To answer this question, it is necessary to measure and directly compare mutation rates between clinical isolates of C. glabrata. However, as described above, comparisons of different clinical isolates are complicated by the variation in their drug resistance profiles, some of which is due to varying activity of drug efflux pumps (14, 15, 18), which is likely to render any drug resistance-based mutation assay inapplicable.
In this study, we developed and validated a GFP-based mutation reporter that allowed us to measure mutation rates in a drug resistance-independent way. The reporter was shown to recapitulate the mutation rate and spectrum of a DNA mismatch repair mutant and detect DNA damage-induced mutagenesis in C. glabrata, recapitulate the mutation rates of wild-type and mutator strains of S. cerevisiae, and compare spontaneous mutation rates in C. glabrata and S. cerevisiae. Finally, we used this reporter to measure the mutation rates of a number of clinical isolates of C. glabrata, including those carrying a specific MSH2 variant previously suggested to increase mutagenesis.
RESULTS
Developing the GFP-based mutation rate reporter.
To measure mutation rates in C. glabrata, at first we attempted to use traditional drug resistance-based reporters, such as CAN1, which has been used extensively to measure mutation rates in S. cerevisiae (2–5). In that fungus, CAN1 cells are sensitive to the drug canavanine, whereas mutations in the can1 gene cause canavanine resistance and can be selected on canavanine-containing plates. However, although the C. glabrata genome contains several potential CAN1 orthologs (CAGL0J08162g and CAGL0J08184g), commonly used reference strain ATCC 2001 (also known as CBS138) was completely resistant to canavanine up to concentrations of 1 mg/ml (see Fig. S1A in the supplemental material; also data not shown), whereas the typical selection concentration in S. cerevisiae is 60 µg/ml. We also tried using 5-fluoroanthranilic acid (5-FAA), which selects for mutations in the tryptophan biosynthetic pathway (30). Although ATCC 2001 and many clinical isolates of C. glabrata were sensitive to 5-FAA, we discovered that this sensitivity widely varied among different strains (Fig. S1B). Although this variation was not entirely surprising, as different clinical isolates are well known to show different levels of antifungal drug resistance, which is at least in part due to the activity of drug efflux pumps, it also eliminated the possibility of using 5-FAA—or likely any other drug resistance-based approach—to measuring mutation rates in C. glabrata clinical isolates.
Drug resistance-based mutation reporters are not suitable to measure mutation rates in C. glabrata. (A) C. glabrata is highly resistant to canavanine. (B) Different clinical isolates (numbers 1 through 9) show various levels of sensitivity to 5-FAA, a drug that can be used to select for mutations in the tryptophan biosynthesis pathway. Download FIG S1, PDF file, 0.9 MB (928.2KB, pdf) .
Copyright © 2019 Shor et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To enable measurements of mutation rates in a way that was independent of drug resistance, we chose a fluorescence-based approach. We created a cassette where the gene encoding yeast enhanced green fluorescent protein (yEGFP) was driven by the strong constitutive promoter pTEF1 of S. cerevisiae (31), which was also previously shown to strongly induce gene expression in C. glabrata (32) (Fig. 1A). In order to facilitate the chromosomal insertion and subsequent tracking of this construct, the cassette also contained the gene conferring nourseothricin resistance (NAT) driven by its own promoter (Fig. 1A). This cassette was inserted into the right arm of C. glabrata chromosome K between two uncharacterized ORFs (Fig. 1A) and validated by sequencing. The resulting strain was constitutively and strongly fluorescent (Fig. 1B) and was used to measure mutation rates of yEGFP using fluorescence-based cell sorting (FACS) in fluctuation experiments as described below (Fig. 1C; Fig. S2).
FIG 1.

Description of the fluorescence-based mutation reporter. (A) A cassette containing pTEF1-driven yEGFP linked to the gene encoding nourseothricin resistance (NAT) under its own promoter was integrated into the indicated locus on chromosome K in C. glabrata laboratory strain ATCC 2001. (B) The resulting strain became constitutively fluorescent. (C) Schematic describing the fluctuation assay used to measure the mutation rate of yEGFP.
Data from a representative FACS experiment. Cells were first gated based on SSC-W versus SSC-H to identify singlets, then based on PI staining to identify viable cells, and finally based on GFP fluorescence. Although there is a range of fluorescence in both the nonfluorescent strain (bottom left) and the fluorescent strain containing the reporter (bottom right), the two cell populations are well separated in terms of fluorescence intensity, allowing the sorting of rare nonfluorescent cells (bottom right, negative gate) from the fluorescent population. Download FIG S2, PDF file, 1.2 MB (1.2MB, pdf) .
Copyright © 2019 Shor et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Briefly, in a typical experiment, a starter YPD culture was diluted into multiple (e.g., 8 to 12) parallel YPD cultures in a 96-well plate to a starting density of a few cells per well and incubated at 37°C overnight. The following morning, each culture, in its entirety, was diluted severalfold in YPD to ensure that cells collected for FACS analysis several hours later were in log phase, which was found to be necessary to achieve maximum expression of GFP and the optimal resolution between GFP-positive and GFP-negative populations. The cells were then collected by filtration, resuspended in PBS, and analyzed by FACS. Prior to FACS analysis, propidium iodide (PI) was added to each sample to gate out the inviable, PI-positive cell subset (Fig. S2). Although GFP levels of fluorescent cultures varied slightly between experiments (e.g., between the same strain analyzed on different days and between different strains), the overall fluorescence levels of GFP+ cells were always significantly higher than those of GFP− cells (Fig. S3), allowing for efficient sorting of GFP− cells from GFP+ populations.
GFP-positive populations are always clearly separated in terms of fluorescence levels from GFP-negative populations. Although small degrees of variation in fluorescence were observed between different strains with the integrated GFP construct, as well as between the same strain analyzed on different days, GFP-positive populations always fluoresced much more strongly than GFP-negative populations, allowing for efficient sorting of GFP-negative cells from GFP-positive cultures. Download FIG S3, PDF file, 1.7 MB (1.7MB, pdf) .
Copyright © 2019 Shor et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The number of cells per culture (n) was optimized by varying the final volume of the cultures. For strains with lower mutation rates, such as the reference C. glabrata strain ATCC 2001, the optimal n was found to be ≥3 × 106 cells/well, whereas for cells with elevated mutation rates (such as msh2Δ), 1 × 106 cells/well was sufficient to obtain multiple cultures with mutants. Each culture, in its entirety, was analyzed by FACS, and GFP-negative cells were collected, immediately plated onto YPD plates, and allowed to form colonies. These colonies were then validated for (i) the presence of the NAT cassette (by replica plating onto nourseothricin medium) and (ii) reduction of GFP fluorescence (by flow cytometry). Next, yEGFP was sequenced in NAT+ GFP− colonies to identify the mutations responsible for reduced fluorescence. For every strain, 200 GFP+ cells were also collected by FACS and plated on YPD to calculate plating efficiency. Finally, mutation rates and 95% confidence intervals were calculated using the MSS maximum likelihood method (1, 33) based on n (the number of processed cells × PI-negative fraction × plating efficiency) and the number of NAT+ GFP− mutants in every culture (r). Importantly, we found that every NAT+ GFP− colony in which GFP was sequenced contained a single mutation in the yEGFP ORF or, in a few cases, the pTEF1 promoter (see below), indicating that loss of fluorescence is virtually always caused by mutations in yEGFP and that therefore this mutation assay is highly specific to a single locus.
Mutations in GFP do not affect cellular fitness.
A key condition that has to be satisfied by any mutation rate measurement assay is that mutations in the reporter gene should not affect the fitness of the strain, either positively or negatively, as this would result in overestimating or underestimating the mutation rate, respectively (1). To check that this condition is fulfilled in the GFP-based mutation reporter, we isolated two different loss-of-function mutations in yEGFP using FACS and measured their fitness compared to the parent strain over 24 h of growth in YPD, which is the duration of a typical mutation rate experiment. Each strain was mixed 1:1 with the parent strain, producing a coculture where approximately half the population was fluorescent and half was not (Fig. 2, Time 0). Both cocultures were diluted into multiple wells to several hundred cells per well and grown for 24 h at 37°C, mimicking a typical fluctuation experiment. After 24 h, fluorescence measurements showed that the proportions of fluorescent and nonfluorescent cells in the cultures had not significantly changed (Fig. 2, 24 h), indicating that mutations in yEGFP did not affect fitness relative to the parental GFP-positive strain.
FIG 2.

Mutation of yEGFP does not impact fitness relative to the fluorescent parent strain. Two different yEGFP mutants were each cocultured together with the fluorescent parent strain for 24 h. The relative proportions of the strains carrying wild-type and mutant yEGFP genes, quantified and shown as bar graphs, remained constant over the course of the experiment, indicating no difference in relative fitness.
GFP-based mutation reporter recapitulates mutation rates in Saccharomyces cerevisiae.
In addition to inserting the GFP cassette into the C. glabrata genome, we also inserted it at the CAN1 locus in S. cerevisiae (Fig. 3A) in order to test whether this reporter would recapitulate the previously determined mutation rates of two S. cerevisiae strains: a wild-type strain of the W303 background and an isogenic mutant carrying a deletion of the SHU1 gene. SHU1 functions in promoting error-free DNA repair by homologous recombination, and its loss was shown to increase CAN1 mutation rate by approximately 8.5-fold in one study (34) and by 4-fold in another study (35). The FACS-based assay described above (Fig. 1C) was used to measure the mutation rates of yEGFP in wild-ype and shu1Δ strains. The assay recapitulated the increase in mutation rate in the shu1Δ mutant relative to the wild-type strain (4-fold; Fig. 3B). Furthermore, the absolute mutation rate obtained for the wild-type strain was ∼2.7 per 107 cells per generation, which agrees well with typical mutation rates obtained in S. cerevisiae (2, 5, 34).
FIG 3.
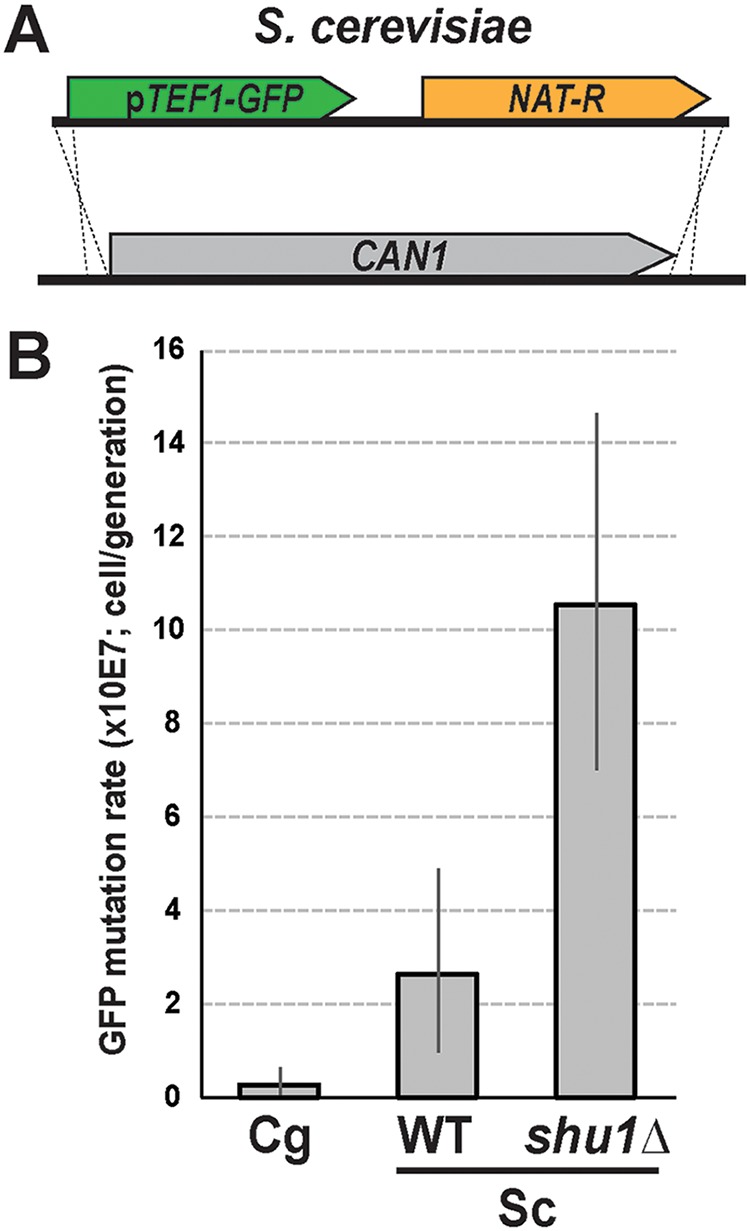
Fluorescence-based mutation reporter recapitulates the mutation rate increase of an S. cerevisiae mutant. (A) The yEGFP-NAT cassette was integrated at the CAN1 locus of S. cerevisiae using homology-based targeting. (B) The assay detected the mutator phenotype of the shu1Δ mutant, previously shown to have an elevated CAN1 mutation rate (34). The assay also showed that spontaneous forward mutation in C. glabrata during standard laboratory growth in YPD is not higher than that in S. cerevisiae. Sc, S. cerevisiae; Cg, C. glabrata. Error bars, 95% confidence intervals.
Finally, using the fluorescence-based mutation reporter, we were able to directly compare spontaneous mutation rates at the C. glabrata locus carrying the cassette (Fig. 1A) and the CAN1 locus of S. cerevisiae. Interestingly, we found that the mutation rate was 9-fold higher at the S. cerevisiae CAN1 locus than at the analyzed locus in C. glabrata (Fig. 3B), suggesting that, at least during unperturbed growth in YPD, the examined strain of C. glabrata (ATCC 2001) does not behave as a spontaneous hypermutator.
GFP-based mutation reporter captures the msh2Δ mutator phenotype and mutation spectrum.
To further validate the GFP-based mutation reporter in C. glabrata, we used CRISPR to insert the cassette into the same chromosomal location (Fig. 1A) in a C. glabrata strain derived from ATCC 2001 but carrying a deletion of DNA mismatch repair (MMR) gene MSH2 (24). MSH2 is the C. glabrata homolog of MutSβ, whose loss has been shown to result in a strong mutator phenotype in all organisms where it has been examined, including C. glabrata (24, 36). Both MSH2 and msh2Δ strains were used in fluctuation experiments to measure their mutation rates as described above (Fig. 1C and Fig. S2). We found that, as expected, msh2Δ resulted in a strong mutator phenotype, increasing the rate of mutation of yEGFP by 40-fold (Fig. 4A), which is very similar to its effect on mutation rates in S. cerevisiae, where the msh2Δ effect on CAN1 mutation rate ranges from a 16- to a 40-fold increase, depending on the study (37–40). Furthermore, sequencing GFP− colonies revealed that the msh2Δ strain produced a very different spectrum of mutations in yEGFP from the MSH2 strain (Fig. 4B and C). Mutations in the wild-type (MSH2) strain were largely comprised of base pair substitutions (bps), whereas the majority of mutations in the msh2Δ mutant were due to single nucleotide deletions or insertions in mononucleotide repeats (e.g., AAAA or TTTT), with the strongest mutation “hot spot” at a run of seven T’s (Fig. 4B). This mutation spectrum recapitulates that of the msh2Δ strain in S. cerevisiae and is thought to be due to DNA polymerase “slippage” errors at mononucleotide repeats, which are normally repaired by MMR (37, 38, 41). Thus, the GFP-based mutation reporter was able to accurately capture both the increase in mutation rate and the change in mutation spectrum of the msh2Δ mutant.
FIG 4.

Fluorescence-based mutation reporter captures the mutator phenotype and mutational signature of the msh2Δ DNA mismatch repair mutant in C. glabrata. (A) Using CRISPR, the yEGFP-NAT cassette was integrated into the same genetic locus in an ATCC 2001-derived strain carrying a deletion of MSH2. The mutation rate of yEGFP was measured and found to be increased 40-fold by msh2Δ relative to the strain carrying wild-type MSH2. Error bars, 95% confidence intervals. (B and C) Sequencing of yEGFP in nonfluorescent mutants isolated by FACS from MSH2 and msh2Δ strains identified a number of mutations throughout the yEGFP ORF. Whereas mutations in the MSH2 strain were mostly base pair substitutions, the majority of mutations in the msh2Δ strain were single nucleotide frameshifts (insertions or deletions) in mononucleotide runs, recapitulating the mutational signature of msh2Δ in S. cerevisiae (37, 38, 41). bps, base pair substitution; ins/del, insertion or deletion.
GFP-based mutation reporter detects DNA-damage induced mutagenesis.
To investigate whether the GFP-based mutation reporter would capture DNA damage-induced mutagenesis, we performed the fluctuation assay on the ATCC 2001 strain grown in the presence of 0.01% methyl methanesulfonate (MMS), an alkylating agent. Indeed, we detected a 48-fold increase in yEGFP mutation rate in cells cultured in the presence of MMS (Fig. 5A). Sequencing mutations in GFP− colonies recovered after growth in MMS showed that the spectrum of mutations did not change significantly from that in cells grown in the absence of MMS (Fig. 5B and C). Thus, MMS caused an overall increase in mutagenesis of yEGFP, as expected, but apparently did not significantly change the cellular pathways by which these mutations were generated. This was consistent with a previous report, where the MMS-induced mutation spectrum in yeast was similar to the spontaneous mutation spectrum (42).
FIG 5.

Fluorescence-based mutation reporter captures the elevated mutation rate in C. glabrata caused by growth in the presence of genotoxic agent MMS. (A) C. glabrata strain ATCC 2001 carrying the fluorescent reporter was cultured in the presence of 0.01% MMS, and its mutation rate was measured as described in the text and in Fig. 1. Error bars, 95% confidence intervals. (B and C) Sequencing of yEGFP in nonfluorescent mutants that formed in the presence of MMS showed a mutation spectrum similar to that produced in the absence of the drug. Several mutations generated during growth in the presence of MMS were in the promoter region (lowercase letters, top row). bps, base pair substitution; ins/del, insertion or deletion.
Mutation rates of C. glabrata clinical isolates.
We used the GFP-based mutation assay to measure mutation rates of six clinical C. glabrata isolates: three that, like ATCC 2001, belonged to sequence type (ST) 15 and carried the corresponding MSH2 sequence and three belonging to ST16 and carrying the variant MSH2E231G/L269F (23, 24). For every clinical isolate, the reporter cassette was integrated into the same chromosomal locus using CRISPR and validated by flow cytometry and DNA sequencing, and the mutation rate was measured using FACS as described above. Interestingly, we found that none of the clinical isolates, including those carrying MSH2E231G/L269F, had elevated mutation rates relative to the reference strain ATCC 2001 (Fig. 6). Thus, even though our previous examination indicated that MSH2E231G/L269F did not fully rescue the mutator phenotype of the msh2Δ mutant in ATCC 2001 (24), a direct, rigorous assessment of the mutation rate of the clinical isolates showed that under standard laboratory conditions this variant, in its native genomic context, is not associated with an elevated spontaneous forward mutation rate.
FIG 6.
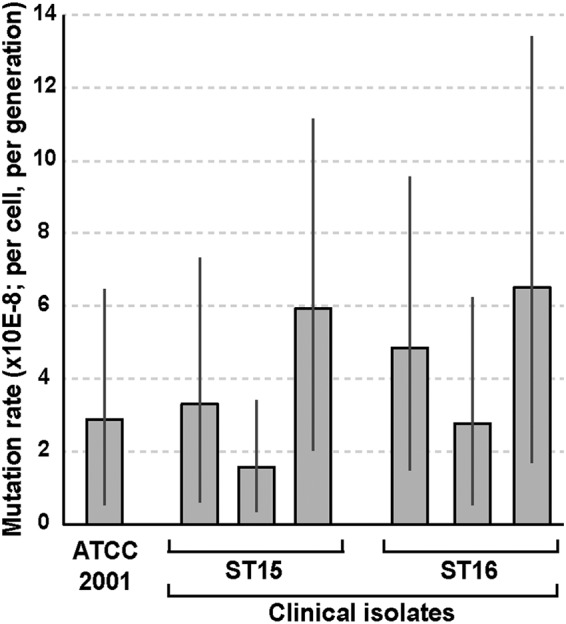
Fluorescence-based mutation reporter reveals similar mutation rates in a panel of clinical C. glabrata isolates. Using CRISPR, the yEGFP-NAT cassette was integrated into the same genomic locus in six clinical isolates of C. glabrata. Three of these isolates, like ATCC 2001, belonged to ST15 and therefore carried the same MSH2 allele, whereas the other three belonged to ST16, which carries the MSH2E231G/L269F variant (23). Mutation rates of yEGFP were measured as described above and found to be similar among all examined isolates irrespective of MSH2 sequence. Error bars, 95% confidence intervals.
DISCUSSION
The goal of this study was to develop, validate, and use a new method for measuring mutation rates in a way that did not rely on drug resistance-based reporters. To this end, we designed a FACS-based scheme to capture and quantify loss-of-function mutations in the gene encoding GFP and used it in fluctuation experiments to measure mutation rates in C. glabrata and S. cerevisiae. We found that this fluorescence-based mutation reporter recapitulated the previously reported mutator phenotype of the S. cerevisiae shu1Δ mutant and captured the expected increase in mutation rates due to loss of MSH2 or treatment with a genotoxic agent in C. glabrata. This reporter also accurately captured the mutational spectrum (a predominance of single nucleotide insertions or deletions in homopolymeric runs) associated with loss of MSH2. Finally, the reporter was used to measure the mutation rates of several clinical C. glabrata isolates, including those carrying the MSH2E231G/L269F variant previously suggested to contribute to increased mutagenesis (24), and showed that all clinical isolates examined had very similar spontaneous mutation rates.
Our mutation assay showed that under standard laboratory growth conditions (YPD, 37°C), C. glabrata clinical isolates carrying the MSH2E231G/L269F variant do not show elevated spontaneous mutation rates relative to ATCC 2001 or to clinical isolates carrying the “wild-type” version of MSH2 (i.e., one identical to that in ATCC 2001). This is consistent with several recent clinical studies that did not see an association between MSH2 genotype and prevalence of drug resistance in C. glabrata (27–29, 61, 62). However, in our previous study, we found that several MSH2 variants, including MSH2E231G/L269F, when introduced on a plasmid into an ATCC 2001-derived msh2Δ mutant, did not fully rescue that strain’s elevated mutation rate (24). There are several non-mutually exclusive possibilities that can reconcile previous data with this present study. First, it is possible that, although the plasmids were maintained by selection, a subpopulation of the culture had lost the plasmid and therefore lacked any copy of MSH2. Second, it is possible that the MSH2 variants carried on a plasmid were present in more than one copy, which, depending on the nature of the mutation, might either help restore function (for a partial loss-of-function mutation) or further exacerbate the associated defects (for a dominant negative mutation). Finally, it is important to consider that in the clinical isolates studied here, the MSH2E231G/L269F variant is present in its normal genomic context, which is that of ST16 (23). ST16 is separated from ATCC 2001 (ST15) by hundreds to thousands of SNPs throughout the genome, including SNPs in genes that encode protein partners of Msh2, such as Msh3 and Msh6 (36). Thus, it is possible that each MSH2 variant has evolved in concert with its partner genes in a way that maintains efficient MMR and low mutation rates. In this scenario, moving a particular variant to a different genomic context would force it to form suboptimal partnerships with noncognate interacting proteins, resulting in less efficient MMR and an increased spontaneous mutation rate. Consistent with this hypothesis, our analyses of MSH3 and MSH6 sequences have revealed that each of these genes has multiple SNPs between ST15 and ST16 strains, including five SNPs in each gene that result in amino acid changes (the ST15 and ST16 whole-genome sequences have been generated by us for a different project [unpublished data]).
The fluorescence-based mutation assay developed in our study has specific advantages and specific limitations that need to be considered when deciding whether to use it or another assay to measure mutation rates in a given experimental system. First, similar to other mutation assays based on a loss of function of a reporter, only mutations that affect GFP function, i.e., its ability to fluoresce in the detectable range, can be identified. Thus, if it is desirable to calculate true mutation rates independently of whether a mutation is expressed, it is more appropriate to use the recently developed whole-genome sequencing (WGS) approaches (43–45). Although this is a very powerful technique, it requires WGS of multiple isolates per strain and is therefore still considerably more expensive and computationally heavy than our method, which requires no computational expertise. One unique hurdle of our assay not shared by other methods is that it requires access to a FACS instrument that can sort millions of cells in minutes; otherwise, the time frame of a single experiment becomes unfeasibly long. Once the sorting is completed, however, the rest of the steps require a standard flow cytometer (or fluorescence microscope) and standard laboratory techniques. As discussed above, this assay is going to be more informative than drug resistance-based assays when the strains under comparison have different drug tolerance profiles, as can be expected for environmental/clinical isolates or different species or when one wishes to measure the mutation rates of strains exposed to different types of environmental stress (e.g., antimicrobial drugs).
We validated our mutation assay by analyzing strains with elevated mutation rates (e.g., shu1Δ in S. cerevisiae and msh2Δ in C. glabrata). It should also be possible to use this assay to identify antimutators—i.e., genes whose loss reduces mutagenesis—which can be extremely informative for identifying cellular pathways that promote mutagenesis and genetic instability (46–48). However, this would require sorting cultures with significantly larger numbers of cells and would therefore take longer, with the sorting time required negatively correlating with the mutation rate of the strain. One potential improvement over the current methodology that would reduce the required time and labor is developing a fluorescence-based assay that uses flow cytometry to count the nonfluorescent cells but skips the sorting and plating steps. In our current setup, this was not possible because the number of GFP-negative cells recorded by the FACS instrument was typically much greater than, and did not correlate with, the number of colonies that grew from the sorted cells. In other words, despite our use of propidium iodide (PI) to gate out membrane-permeable cells, a large subset of PI-negative GFP-negative cells were inviable/nonculturable. Perhaps, with further optimization—e.g., using other fluorescent markers that can be used in conjunction with live/dead dyes—it will be possible to accurately record the number of nonfluorescent live cells using flow cytometry only, without the need for sorting and plating.
We have used the fluorescence-based assay to measure forward mutation rates; however, because the reporter cassette contains two genes (yEGFP and NAT), it can be adapted to measure large deletions by looking for simultaneous loss of both yEGFP and NAT, similar to the CAN1-URA3 loss assay developed in S. cerevisiae (49). Such an assay would be extremely useful in C. glabrata and other fungal pathogens characterized by frequent genomic rearrangements (26, 50, 51). In the current genomic location of the cassette, we did not identify any simultaneous deletions of yEGFP and NAT among the >170 analyzed C. glabrata cultures, including those containing genotoxic agent MMS, indicating that the spontaneous rate of deletions at this locus is extremely low. In future studies, the yEGFP-NAT cassette will be integrated at genomic loci more likely to undergo rearrangements, such as subtelomeric loci containing multiple and variable numbers of genes from the adhesin family (52, 53) and ribosomal DNA (rDNA) (54).
The high degree of genetic diversity in C. glabrata populations and the fast emergence of drug resistance both indicate that at least under some conditions, C. glabrata is able to rapidly mutate and diversify its genome. Our present study indicates that these conditions do not include unperturbed growth in YPD, suggesting that C. glabrata may be subject to stress-induced mutagenesis. Indeed, previously, stress-induced mutator phenotypes have been identified in a majority of natural isolates of Escherichia coli, whereas only 5% were shown to act as constitutive mutators (55). Future studies will examine whether mutagenesis is affected by stress conditions, including those encountered by C. glabrata in the host, such as oxidative stress and exposure to antifungal drugs. The fluorescence-based mutation assay is particularly well suited to address such questions because its outcome is independent of whether the strain’s sensitivity to stress is altered by an exogenous treatment (e.g., by an antifungal drug). This assay can also be adapted and used to address questions regarding mechanisms driving mutagenesis in other clinically relevant microbes, including bacterial pathogens and haploid pathogenic fungi, such as Candida auris, Cryptococcus neoformans, and Candida lusitaniae, where emergence of drug resistance poses a serious public health threat.
MATERIALS AND METHODS
Construction of the GFP reporter cassette and creating fluorescent strains for fluctuation analyses.
The S. cerevisiae TEF1 promoter was amplified using primers CACACCAGAGCTCCAAAATGTTTCTACTCC and CCATTTTGGATCCAAAACTTAGATTAGATTGC and subcloned into the BamHI-SacI sites of pYC54 (56), placing it directly upstream of the YFP ORF. Next, the YFP ORF was replaced by that of yEGFP as follows. yEGFP was amplified from pGRB2.3 (57) using primers ACTAGTGGATCCCCCGGGCTGCAGGAATTCATG and CGAATTGGCTAGCTTTACCTCTATATCGTGTTCG and subcloned into the plasmid using BamHI-NheI sites. The final plasmid contained the pTEF1-yEGFP-NAT cassette (Fig. 1A). This cassette was amplified from the plasmid using primers CCCCTCGAGGACGAAGTTCC and TGTAATACGACTCACTATAGGGCG and transformed into C. glabrata strain ATCC 2001 using nourseothricin resistance as selection.
Because there were no targeting homology sequences on the cassette, it integrated randomly into the C. glabrata genome. Several independent, constitutively fluorescent transformants were chosen and submitted for whole-genome sequencing at the New Jersey Medical School Molecular Resource Facility using the NextSeq (Illumina, San Diego, CA) platform. Libraries were prepared with the Nextera XT kit (Illumina, San Diego, CA) to produce paired-end reads of 150 bp for an approximate minimum coverage of 100×. Data analysis was performed using CLC Genomics Workbench (Qiagen, Hilden, Germany). Each transformant was found to contain a single integration of the cassette. The transformant carrying the cassette integrated between uncharacterized ORFs CAGL0K11132g and CAGL0K11198g on Chr K (strain ESCg36), as shown in Fig. 1A, was chosen for further analysis.
In all other C. glabrata strains reported in this study, the cassette was integrated at the same genomic locus as in ESCg36 using CRISPR-mediated targeted integration. The cassette was amplified from ESCg36 using primers GCAGTCTTTCTTGATCCACATATC and CACAGAATTGGTAGGACGGG, which produced approximately 500-nt 5′ and 3′ homology each to the desired integration locus. CRISPR was performed as in reference 58, except that cells were made competent for electroporation using the Frozen-EZ yeast transformation kit (Zymo Research) according to the manufacturer’s instructions.
To replace the CAN1 ORF of S. cerevisiae with the yEGFP-NAT cassette, the cassette was amplified using primers AAAAGGCATAGCAATGACAAATTCAAAAGAAGACGCCGACATAGAGGACCAGTGAATTGTAATACGACTC and AGGTAATAAAACGTCATATCTATGCTACAACATTCCAAAATTTGTCCTGGTACCGGGCCCCCCCTCGAG, which introduced 48-nt regions of homology directly 5′ upstream and 3′ downstream of the CAN1 ORF. This PCR product was transformed into S. cerevisiae strains W4069-4C (wild-type W303 MATa) and W4220-15A (shu1Δ::HIS3 MATa) using the Frozen-EZ yeast transformation kit (Zymo Research) according to the manufacturer’s instructions. Transformants were selected on nourseothricin plates as described above and validated by sequencing of the CAN1 locus and by flow cytometry to verify the acquisition of green fluorescence.
All primers were ordered from Integrated DNA Technologies, and all Sanger sequencing of the above-described constructs was done by Genewiz.
Measuring yEGFP mutation rates using fluctuation analysis and FACS.
A starter YPD culture of the strain whose mutation rate was being measured was diluted into multiple (e.g., 8 to 12) parallel YPD cultures in a 96-well plate to a starting density of several hundred cells per well and incubated overnight at 37°C (C. glabrata) or 30°C (S. cerevisiae). The following morning, each culture was diluted severalfold in YPD and grown at the same temperature to ensure that when the cells were collected for FACS several hours later, they were in log phase, which was found to be necessary to achieve maximum expression of GFP and the best resolution between GFP-positive and GFP-negative populations. The cells were then collected by filtration using 0.45-µm mixed cellulose ester membrane filters (Millipore), resuspended in PBS, and sorted using the BD FACSAria II (BD Biosciences) at the New Jersey Medical School Flow Cytometry and Immunology Core Laboratory. Ten to 15 min before sorting, each sample was stained with 10 µg/ml propidium iodide (PI; ThermoFisher) to identify and gate out inviable cells. FSC and SSC parameters were set on log scale. Cells were gated for singlets (SSC-W versus SSC-H) followed by live gating on PI negative. Finally, cells were sorted based on GFP expression. GFP-negative cells from each entire culture were collected into microcentrifuge tubes containing 250 µl YPD and then plated immediately onto YPD agar plates. Two hundred GFP-positive cells were also sorted and plated to calculate plating efficiency. The plates were incubated at 37°C (C. glabrata) or 30°C (S. cerevisiae) to allow the sorted cells to form colonies.
The resulting colonies were checked for the presence of the NAT marker by replica plating or patching onto plates containing 100 µg/ml nourseothricin (Jena Bioscience) and then checked for the level of green fluorescence using a BD Accuri C6 flow cytometer (BD Biosciences). Mutation rates and 95% confidence intervals were calculated using the MSS maximum likelihood method (1, 33) based on the number of NAT+ GFP− mutants in every culture (r) and the average number of viable cells per culture (n). To identify the mutations responsible for loss of GFP fluorescence, the yEGFP ORF and promoter were sequenced using primers CTCTTTCGATGACCTCCCATTG and TGTAATACGACTCACTATAGGGCG, respectively.
ACKNOWLEDGMENTS
We thank Puneet Dhawan of the New Jersey Medical School Molecular Resource Facility for assistance with whole-genome sequencing and Sukhwinder Singh and Tammy Mui of the New Jersey Medical School Flow Cytometry and Immunology Core Laboratory for assistance with FACS. We thank the Rothstein laboratory (Columbia University) for the gift of S. cerevisiae strains.
This research was supported by the NIH grant AI109025 to D.S.P.
Footnotes
Citation Shor E, Schuyler J, Perlin DS. 2019. A novel, drug resistance-independent, fluorescence-based approach to measure mutation rates in microbial pathogens. mBio 10:e00120-19. https://doi.org/10.1128/mBio.00120-19.
Contributor Information
Joseph Heitman, Duke University.
Judith Berman, Tel Aviv University.
Susan Rosenberg, Baylor College of Medicine.
REFERENCES
- 1.Rosche WA, Foster PL. 2000. Determining mutation rates in bacterial populations. Methods 20:4–17. doi: 10.1006/meth.1999.0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lang GI, Murray AW. 2008. Estimating the per-base-pair mutation rate in the yeast Saccharomyces cerevisiae. Genetics 178:67–82. doi: 10.1534/genetics.107.071506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lippert MJ, Kim N, Cho JE, Larson RP, Schoenly NE, O’Shea SH, Jinks-Robertson S. 2011. Role for topoisomerase 1 in transcription-associated mutagenesis in yeast. Proc Natl Acad Sci U S A 108:698–703. doi: 10.1073/pnas.1012363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harrington JM, Kolodner RD. 2007. Saccharomyces cerevisiae Msh2-Msh3 acts in repair of base-base mispairs. Mol Cell Biol 27:6546–6554. doi: 10.1128/MCB.00855-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buckland RJ, Watt DL, Chittoor B, Nilsson AK, Kunkel TA, Chabes A. 2014. Increased and imbalanced dNTP pools symmetrically promote both leading and lagging strand replication infidelity. PLoS Genet 10:e1004846. doi: 10.1371/journal.pgen.1004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roetzer A, Gabaldon T, Schuller C. 2011. From Saccharomyces cerevisiae to Candida glabrata in a few easy steps: important adaptations for an opportunistic pathogen. FEMS Microbiol Lett 314:1–9. doi: 10.1111/j.1574-6968.2010.02102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huffnagle GB, Noverr MC. 2013. The emerging world of the fungal microbiome. Trends Microbiol 21:334–341. doi: 10.1016/j.tim.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antinori S, Milazzo L, Sollima S, Galli M, Corbellino M. 2016. Candidemia and invasive candidiasis in adults: a narrative review. Eur J Intern Med 34:21–28. doi: 10.1016/j.ejim.2016.06.029. [DOI] [PubMed] [Google Scholar]
- 9.Glockner A, Cornely OA. 2015. Candida glabrata—unique features and challenges in the clinical management of invasive infections. Mycoses 58:445–450. doi: 10.1111/myc.12348. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen MH, Peacock JE Jr, Morris AJ, Tanner DC, Nguyen ML, Snydman DR, Wagener MM, Rinaldi MG, Yu VL. 1996. The changing face of candidemia: emergence of non-Candida albicans species and antifungal resistance. Am J Med 100:617–623. doi: 10.1016/S0002-9343(95)00010-0. [DOI] [PubMed] [Google Scholar]
- 11.Guinea J. 2014. Global trends in the distribution of Candida species causing candidemia. Clin Microbiol Infect 20(Suppl 6):5–10. doi: 10.1111/1469-0691.12539. [DOI] [PubMed] [Google Scholar]
- 12.Morio F, Jensen RH, Le Pape P, Arendrup MC. 2017. Molecular basis of antifungal drug resistance in yeasts. Int J Antimicrob Agents 50:599–606. doi: 10.1016/j.ijantimicag.2017.05.012. [DOI] [PubMed] [Google Scholar]
- 13.Vale-Silva L, Ischer F, Leibundgut-Landmann S, Sanglard D. 2013. Gain-of-function mutations in PDR1, a regulator of antifungal drug resistance in Candida glabrata, control adherence to host cells. Infect Immun 81:1709–1720. doi: 10.1128/IAI.00074-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, Rohde B, Bauser C, Bader O, Sanglard D. 2009. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog 5:e1000268. doi: 10.1371/journal.ppat.1000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai HF, Krol AA, Sarti KE, Bennett JE. 2006. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob Agents Chemother 50:1384–1392. doi: 10.1128/AAC.50.4.1384-1392.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Effron G, Lee S, Park S, Cleary JD, Perlin DS. 2009. Effect of Candida glabrata FKS1 and FKS2 mutations on echinocandin sensitivity and kinetics of 1,3-beta-d-glucan synthase: implication for the existing susceptibility breakpoint. Antimicrob Agents Chemother 53:3690–3699. doi: 10.1128/AAC.00443-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park S, Kelly R, Kahn JN, Robles J, Hsu MJ, Register E, Li W, Vyas V, Fan H, Abruzzo G, Flattery A, Gill C, Chrebet G, Parent SA, Kurtz M, Teppler H, Douglas CM, Perlin DS. 2005. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob Agents Chemother 49:3264–3273. doi: 10.1128/AAC.49.8.3264-3273.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cowen LE, Sanglard D, Howard SJ, Rogers PD, Perlin DS. 2015. Mechanisms of antifungal drug resistance. Cold Spring Harb Perspect Med 5:a019752. doi: 10.1101/cshperspect.a019752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexander BD, Johnson MD, Pfeiffer CD, Jimenez-Ortigosa C, Catania J, Booker R, Castanheira M, Messer SA, Perlin DS, Pfaller MA. 2013. Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin Infect Dis 56:1724–1732. doi: 10.1093/cid/cit136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arendrup MC, Patterson TF. 2017. Multidrug-resistant Candida: epidemiology, molecular mechanisms, and treatment. J Infect Dis 216:S445–S451. doi: 10.1093/infdis/jix131. [DOI] [PubMed] [Google Scholar]
- 21.Bader O, Schwarz A, Kraneveld EA, Tangwattanachuleeporn M, Schmidt P, Jacobsen MD, Gross U, De Groot PWJ, Weig M. 2012. Gross karyotypic and phenotypic alterations among different progenies of the Candida glabrata CBS138/ATCC2001 reference strain. PLoS One 7:e52218. doi: 10.1371/journal.pone.0052218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrete L, Ksiezopolska E, Pegueroles C, Gomez-Molero E, Saus E, Iraola-Guzman S, Loska D, Bader O, Fairhead C, Gabaldon T. 2018. Patterns of genomic variation in the opportunistic pathogen Candida glabrata suggest the existence of mating and a secondary association with humans. Curr Biol 28:15–27.e7. doi: 10.1016/j.cub.2017.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Healey KR, Jimenez Ortigosa C, Shor E, Perlin DS. 2016. Genetic drivers of multidrug resistance in Candida glabrata. Front Microbiol 7:1995. doi: 10.3389/fmicb.2016.01995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Healey KR, Zhao Y, Perez WB, Lockhart SR, Sobel JD, Farmakiotis D, Kontoyiannis DP, Sanglard D, Taj-Aldeen SJ, Alexander BD, Jimenez-Ortigosa C, Shor E, Perlin DS. 2016. Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance. Nat Commun 7:11128. doi: 10.1038/ncomms11128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lott TJ, Frade JP, Lockhart SR. 2010. Multilocus sequence type analysis reveals both clonality and recombination in populations of Candida glabrata bloodstream isolates from U.S. surveillance studies. Eukaryot Cell 9:619–625. doi: 10.1128/EC.00002-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polakova S, Blume C, Zarate JA, Mentel M, Jorck-Ramberg D, Stenderup J, Piskur J. 2009. Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata. Proc Natl Acad Sci U S A 106:2688–2693. doi: 10.1073/pnas.0809793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh A, Healey KR, Yadav P, Upadhyaya G, Sachdeva N, Sarma S, Kumar A, Tarai B, Perlin DS, Chowdhary A. 2018. Absence of azole or echinocandin resistance in Candida glabrata isolates in India despite background prevalence of strains with defects in the DNA mismatch repair pathway. Antimicrob Agents Chemother 62:e00195-18. doi: 10.1128/AAC.00195-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delliere S, Healey K, Gits-Muselli M, Carrara B, Barbaro A, Guigue N, Lecefel C, Touratier S, Desnos-Ollivier M, Perlin DS, Bretagne S, Alanio A. 2016. Fluconazole and echinocandin resistance of Candida glabrata correlates better with antifungal drug exposure rather than with MSH2 mutator genotype in a French cohort of patients harboring low rates of resistance. Front Microbiol 7:2038. doi: 10.3389/fmicb.2016.02038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou X, Xiao M, Wang H, Yu SY, Zhang G, Zhao Y, Xu YC. 2018. Profiling of PDR1 and MSH2 in Candida glabrata bloodstream isolates from a multicenter study in China. Antimicrob Agents Chemother 62:e00153-18. doi: 10.1128/AAC.00153-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toyn JH, Gunyuzlu PL, White WH, Thompson LA, Hollis GF. 2000. A counterselection for the tryptophan pathway in yeast: 5-fluoroanthranilic acid resistance. Yeast 16:553–560. doi:. [DOI] [PubMed] [Google Scholar]
- 31.Partow S, Siewers V, Bjorn S, Nielsen J, Maury J. 2010. Characterization of different promoters for designing a new expression vector in Saccharomyces cerevisiae. Yeast 27:955–964. doi: 10.1002/yea.1806. [DOI] [PubMed] [Google Scholar]
- 32.Enkler L, Richer D, Marchand AL, Ferrandon D, Jossinet F. 2016. Genome engineering in the yeast pathogen Candida glabrata using the CRISPR-Cas9 system. Sci Rep 6:35766. doi: 10.1038/srep35766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hall BM, Ma CX, Liang P, Singh KK. 2009. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 25:1564–1565. doi: 10.1093/bioinformatics/btp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shor E, Weinstein J, Rothstein R. 2005. A genetic screen for top3 suppressors in Saccharomyces cerevisiae identifies SHU1, SHU2, PSY3 and CSM2: four genes involved in error-free DNA repair. Genetics 169:1275–1289. doi: 10.1534/genetics.104.036764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang ME, Rio AG, Nicolas A, Kolodner RD. 2003. A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc Natl Acad Sci U S A 100:11529–11534. doi: 10.1073/pnas.2035018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schofield MJ, Hsieh P. 2003. DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol 57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- 37.Huang ME, Rio AG, Galibert MD, Galibert F. 2002. Pol32, a subunit of Saccharomyces cerevisiae DNA polymerase delta, suppresses genomic deletions and is involved in the mutagenic bypass pathway. Genetics 160:1409–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ni TT, Marsischky GT, Kolodner RD. 1999. MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-guanine in S. cerevisiae. Mol Cell 4:439–444. doi: 10.1016/S1097-2765(00)80346-9. [DOI] [PubMed] [Google Scholar]
- 39.Gammie AE, Erdeniz N, Beaver J, Devlin B, Nanji A, Rose MD. 2007. Functional characterization of pathogenic human MSH2 missense mutations in Saccharomyces cerevisiae. Genetics 177:707–721. doi: 10.1534/genetics.107.071084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marsischky GT, Filosi N, Kane MF, Kolodner R. 1996. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev 10:407–420. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- 41.Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA. 1997. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol 17:2859–2865. doi: 10.1128/MCB.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee GS, Blonsky KS, Van On DL, Savage EA, Morgan AR, von Borstel RC. 1992. Base alterations in yeast induced by alkylating agents with differing Swain-Scott substrate constants. J Mol Biol 223:617–626. doi: 10.1016/0022-2836(92)90978-S. [DOI] [PubMed] [Google Scholar]
- 43.Lee H, Popodi E, Tang H, Foster PL. 2012. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci U S A 109:E2774–E2783.doi: 10.1073/pnas.1210309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Long H, Behringer MG, Williams E, Te R, Lynch M. 2016. Similar mutation rates but highly diverse mutation spectra in ascomycete and basidiomycete yeasts. Genome Biol Evol 8:3815–3821. doi: 10.1093/gbe/evw286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lynch M, Sung W, Morris K, Coffey N, Landry CR, Dopman EB, Dickinson WJ, Okamoto K, Kulkarni S, Hartl DL, Thomas WK. 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci U S A 105:9272–9277. doi: 10.1073/pnas.0803466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Layton JC, Foster PL. 2003. Error-prone DNA polymerase IV is controlled by the stress-response sigma factor, RpoS, in Escherichia coli. Mol Microbiol 50:549–561. doi: 10.1046/j.1365-2958.2003.03704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liefshitz B, Steinlauf R, Friedl A, Eckardt-Schupp F, Kupiec M. 1998. Genetic interactions between mutants of the ‘error-prone’ repair group of Saccharomyces cerevisiae and their effect on recombination and mutagenesis. Mutat Res 407:135–145. doi: 10.1016/S0921-8777(97)00070-0. [DOI] [PubMed] [Google Scholar]
- 48.Ragheb MN, Thomason MK, Hsu C, Nugent P, Gage J, Samadpour AN, Kariisa A, Merrikh CN, Miller SI, Sherman DR, Merrikh H. 2019. Inhibiting the evolution of antibiotic resistance. Mol Cell 73:157–165.e5. doi: 10.1016/j.molcel.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Srivatsan A, Putnam CD, Kolodner RD. 2018. Analyzing genome rearrangements in Saccharomyces cerevisiae. Methods Mol Biol 1672:43–61. doi: 10.1007/978-1-4939-7306-4_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun S, Xu J. 2009. Chromosomal rearrangements between serotype A and D strains in Cryptococcus neoformans. PLoS One 4:e5524. doi: 10.1371/journal.pone.0005524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Forche A, Magee PT, Selmecki A, Berman J, May G. 2009. Evolution in Candida albicans populations during a single passage through a mouse host. Genetics 182:799–811. doi: 10.1534/genetics.109.103325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gabaldon T, Martin T, Marcet-Houben M, Durrens P, Bolotin-Fukuhara M, Lespinet O, Arnaise S, Boisnard S, Aguileta G, Atanasova R, Bouchier C, Couloux A, Creno S, Almeida Cruz J, Devillers H, Enache-Angoulvant A, Guitard J, Jaouen L, Ma L, Marck C, Neuveglise C, Pelletier E, Pinard A, Poulain J, Recoquillay J, Westhof E, Wincker P, Dujon B, Hennequin C, Fairhead C. 2013. Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genomics 14:623. doi: 10.1186/1471-2164-14-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Timmermans B, De Las Peñas A, Castaño I, Van Dijck P. 2018. Adhesins in Candida glabrata. J Fungi (Basel) 4:60. doi: 10.3390/jof4020060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takeuchi Y, Horiuchi T, Kobayashi T. 2003. Transcription-dependent recombination and the role of fork collision in yeast rDNA. Genes Dev 17:1497–1506. doi: 10.1101/gad.1085403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bjedov I, Tenaillon O, Gerard B, Souza V, Denamur E, Radman M, Taddei F, Matic I. 2003. Stress-induced mutagenesis in bacteria. Science 300:1404–1409. doi: 10.1126/science.1082240. [DOI] [PubMed] [Google Scholar]
- 56.Yáñez-Carrillo P, Orta-Zavalza E, Gutiérrez-Escobedo G, Patrón-Soberano A, De Las Peñas A, Castaño I. 2015. Expression vectors for C-terminal fusions with fluorescent proteins and epitope tags in Candida glabrata. Fungal Genet Biol 80:43–52. doi: 10.1016/j.fgb.2015.04.020. [DOI] [PubMed] [Google Scholar]
- 57.Zordan RE, Ren Y, Pan SJ, Rotondo G, De Las Penas A, Iluore J, Cormack BP. 2013. Expression plasmids for use in Candida glabrata. G3 (Bethesda) 3:1675–1686. doi: 10.1534/g3.113.006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grahl N, Demers EG, Crocker AW, Hogan DA. 2017. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere 2:e00218-17. doi: 10.1128/mSphere.00218-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boyce KJ, Wang Y, Verma S, Shakya VPS, Xue C, Idnurm A. 2017. Mismatch repair of DNA replication errors contributes to microevolution in the pathogenic fungus Cryptococcus neoformans. mBio 8:e00595-17. doi: 10.1128/mBio.00595-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Billmyre RB, Clancey SA, Heitman J. 2017. Natural mismatch repair mutations mediate phenotypic diversity and drug resistance in Cryptococcus deuterogattii. Elife 6:e28802. doi: 10.7554/eLife.28802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bordallo-Cardona MA, Agnelli C, Gomez-Nunez A, Sanchez-Carrillo C, Bouza E, Munoz P, Escribano P, Guinea J. 2019. MSH2 gene point mutations are not antifungal resistance markers in Candida glabrata. Antimicrob Agents Chemother 63:e01876-18. doi: 10.1128/AAC.01876-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Biswas C, Marcelino VR, Van Hal S, Halliday C, Martinez E, Wang Q, Kidd S, Kennedy K, Marriott D, Morrissey CO, Arthur I, Weeks K, Slavin MA, Sorrell TC, Sintchenko V, Meyer W, Chen SC. 2018. Whole genome sequencing of Australian Candida glabrata isolates reveals genetic diversity and novel sequence types. Front Microbiol 9:2946. doi: 10.3389/fmicb.2018.02946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Drug resistance-based mutation reporters are not suitable to measure mutation rates in C. glabrata. (A) C. glabrata is highly resistant to canavanine. (B) Different clinical isolates (numbers 1 through 9) show various levels of sensitivity to 5-FAA, a drug that can be used to select for mutations in the tryptophan biosynthesis pathway. Download FIG S1, PDF file, 0.9 MB (928.2KB, pdf) .
Copyright © 2019 Shor et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data from a representative FACS experiment. Cells were first gated based on SSC-W versus SSC-H to identify singlets, then based on PI staining to identify viable cells, and finally based on GFP fluorescence. Although there is a range of fluorescence in both the nonfluorescent strain (bottom left) and the fluorescent strain containing the reporter (bottom right), the two cell populations are well separated in terms of fluorescence intensity, allowing the sorting of rare nonfluorescent cells (bottom right, negative gate) from the fluorescent population. Download FIG S2, PDF file, 1.2 MB (1.2MB, pdf) .
Copyright © 2019 Shor et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
GFP-positive populations are always clearly separated in terms of fluorescence levels from GFP-negative populations. Although small degrees of variation in fluorescence were observed between different strains with the integrated GFP construct, as well as between the same strain analyzed on different days, GFP-positive populations always fluoresced much more strongly than GFP-negative populations, allowing for efficient sorting of GFP-negative cells from GFP-positive cultures. Download FIG S3, PDF file, 1.7 MB (1.7MB, pdf) .
Copyright © 2019 Shor et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.