

Abstract
Controllable exchange of molecules between the interior and the external environment of vesicles is critical in drug delivery and micro/nano‐reactors. While many approaches exist to trigger release from vesicles, controlled loading remains a challenge. Herein, we show that gigahertz acoustic streaming generated by a nanoelectromechanical resonator can control the loading and release of cargo into and from vesicles. Polymer‐shelled vesicles showed loading and release of molecules both in solution and on a solid substrate. We observed deformation of individual giant unilamellar vesicles and propose that the shear stress generated by gigahertz acoustic streaming induces the formation of transient nanopores, with diameters on the order of 100 nm, in the vesicle membranes. This provides a non‐invasive method to control material exchange across membranes of different types of vesicles, which could allow site‐specific release of therapeutics and controlled loading into cells, as well as tunable microreactors.
Keywords: controlled loading, controlled release, gigahertz acoustic streaming, transient nanopores, vesicles
Current challenges in biomedical therapies lie mostly at its interface with physics, chemistry, and engineering. Control over matter at various length and time scales is targeted, by drawing on concepts such as compartmentalization,1 smart materials,2 and process triggers,3 with the aim to design, for example, artificial cells.4, 5 With biomimetic bilayer structures, artificial vesicles have been applied as a general tool to achieve compartmentalization for various biomedical applications, such as drug carriers and micro/nanoscale reactors.6, 7, 8, 9, 10, 11
One of the key challenges in the development of vesicles as reservoirs for localized storage and nano‐vessels for reactions is the controllable exchange of compounds between the interior and the exterior of the vesicles, that is, the loading or release of cargo into or from the vesicles. Numerous studies have used triggers to quantitatively control the release of molecular cargo from vesicles, which were internal such as pH12, 13, 14 and redox state15, 16, 17 or external such as light,2, 18, 19 temperature,20, 21, 22 ultrasound,23, 24, 25 and magnetic field.26, 27, 28 In these triggered systems, the release rates have important implications, for example, for the therapeutic activities of various types of drug delivery systems.29, 30, 31
In contrast with the advancements in controlled release, it is still highly challenging to achieve controlled loading. In most cases, substances are preloaded into vesicles when they are prepared by extrusion or sonication methods,32, 33 so that the primary loading concentration is predetermined and cannot be modified. However, in applications such as micro/nanoscale reactors or sustained‐release carriers, control over loading—preferentially remotely—is necessary to change the amount or dosage of a reagent at will.34 Examples exist in which pH‐sensitive materials have been included into polymer capsules to control the loading efficiency by tuning proton gradients or temperature.35, 36, 37 However, these methods are either limited by specific types of chemistry or are restricted to cargos with specific properties.38 Thus, controlled loading and unloading of vesicles require general tools that depend on the chemistry of neither the vesicles nor the cargo.
So far, ultrahigh frequency acoustofluidics has rarely been studied owing to the lack of such high‐frequency acoustic devices. In this work, we report a method for controlling both loading and release of materials into and from vesicles without damaging their structures using the gigahertz (GHz) acoustic streaming generated by a thin film‐based nanoelectromechanical (NEMS) resonator. Such resonators have recently been reported by us to generate high‐speed (>m s−1) acoustic streaming with strong forces (>nn), which has been applied to enhance the solution mixing in microfluidic chips39 and to remove nonspecific binding at solid–liquid interfaces.40 Since acoustic streaming can exert mechanical forces on cells that are immobilized at the solid–liquid interface,41 we envisaged that vesicles, which are soft and hollow structures, would also be affected and could experience mechanical deformation under such acoustic stimulation. We hypothesized that owing to the fluidic nature of the lipid membranes, mechanical deformation of the vesicles might induce transient pores in the membrane, which would change the membrane permeability and facilitate materials exchange between the interior and exterior of the vesicles (Scheme 1).
Scheme 1.
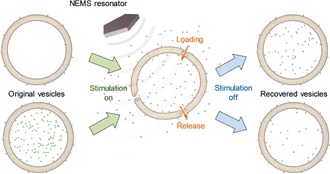
Schematic of the acoustically controlled loading (top) and release (bottom) of empty and pre‐filled vesicles, respectively, through the use of a NEMS‐based acoustic resonator of 2.5 GHz.
Polymer‐shelled vesicles (PSVs)42, 43 were used to demonstrate the controlled loading and release of cargo both in solution and on a solid substrate. The loading and unloading rates were determined by fluorescence measurements. Real‐time deformation of giant unilamellar vesicles (GUVs) was investigated to obtain insights into the mechanism of the acoustic‐controlled materials exchange, which was further evaluated by finite element modeling (FEM) simulations. Polystyrene nanoparticles (PS NPs) of different sizes were loaded into the GUVs to test the uptake limits, thus to estimate the pore size generated by the GHz acoustic streaming. Since our approach is purely physical, using hydrodynamic forces induced by acoustic streaming, it provides a non‐invasive way to control the loading and release of various substances into or from vesicles with different sizes and compositions.
To demonstrate the tunable loading and release of cargo, we employed PSVs as a model system because of their reported use as a highly stable nanocontainer for intracellular delivery.42, 44 As shown in Figure 1 a, PSVs were prepared from cyclodextrin vesicles, onto which adamantyl‐functionalized poly(acrylic acid) (Ad‐PAA) was attached by host–guest recognition, followed by the cross‐linking of the carboxylic acid groups and conjugation with biotin to the PSV surface to allow the specific immobilization of the vesicles on a streptavidin (SAv)‐coated glass substrate through biotin‐SAv recognition. To facilitate the fluorescence characterization of the loading and release, multiple PSVs were patterned on a glass substrate using micromolding in capillaries (MIMIC).45
Figure 1.
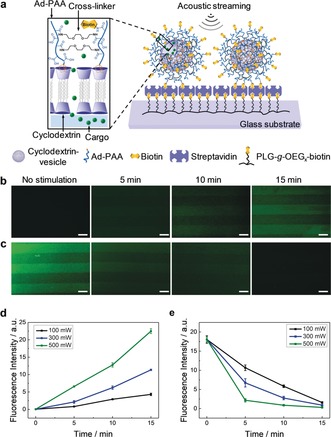
Controlled loading into and release from immobilized PSVs under acoustic streaming. a) Schematic of the biotin–SAv binding motif used for the immobilization of the biotinylated PSVs. The chemical structure of the PSVs is shown in the zoomed‐in cartoon. Fluorescence imaging of b) the loading of empty line‐patterned PSVs in a solution of 5 mm CF and c) release from CF‐encapsulated (0.1 mm) PSVs into water, without or with the acoustic stimulation at 100 mW for different durations. Scale bars=10 μm. Time‐dependent changes of fluorescence intensity with d) the loading (from experiments shown in (b) and Figure S1) and e) the release of the CF dye (from experiments shown in (c) and Figure S2) stimulated at different power levels (100, 300, and 500 mW).
We first tested the controlled loading process. The carboxyfluorescein (CF) dye was used as the cargo to be loaded into PSVs to facilitate fluorescence imaging. Time‐dependent fluorescence images of the PSVs without and with the acoustic stimulation are shown in Figure 1 b. The surface‐patterned and empty PSVs were incubated in a 5 mm CF solution. Without stimulation, the empty PSVs did not show any fluorescence changes even after prolonged incubation (15 min). However, the empty PSVs stimulated by acoustic streaming at 100 mW exhibited an increased green fluorescence. By gradually extending the duration from 5 to 15 min, a higher fluorescence intensity was observed in the patterned areas, which indicates that the CF dye was successfully loaded into the vesicles and the loaded amount is dependent on the stimulation time. The loading kinetics extracted from the fluorescence measurements at different power levels (Figure S1) show that more CF dye is loaded into the vesicles at higher power (Figure 1 d and Figure S1 e) and that the loading rate is approximately linear to the applied power (Figure S1 f). Controls in the absence of hypersound (Figure S1 a–d) indicate no fluorescence intensity increase, supporting the conclusion that the intensities observed in the presence of hypersound are caused by uptake into the vesicles. The data shown in Figure S1 e indicates that at the applied CF concentration and power level, the loading is not saturating within 15 min. The experiments suggest that control over the loaded amount of cargo is feasible by variation of the power and loading time.
To test the controlled release, immobilized PSVs that were pre‐loaded with CF were subjected to acoustic streaming at 100 mW (Figure 1 c). Compared to the sample without stimulation, the acoustic streaming induced an obvious decrease of fluorescence intensity in the patterned areas, indicating a successful release of the CF dye. Similarly, by increasing the power from 100 to 500 mW (Figure S2), the green fluorescence of the patterned PSVs decreased faster (Figure 1 e) whereas the fluorescence intensity of the CF released into the sealed chamber increased accordingly (Figure S3 a–d). The release kinetics derived from the fluorescence changes in the sealed chamber show that the release rate is approximately linearly dependent on the power of the acoustic streaming (Figure S3 e). As expected, the intensity curves shown in Figure 1 e follow an exponential decay trend (as fitted in Figure S2 e), which is also linearly dependent on power (Figure S2 f), and complete release is achieved within 10–30 min.
We note that the relative fluorescence changes occurring within the initial 5 min of the release process (Figure 1 e) are larger than those observed during loading (Figure 1 d), which can be related to the different kinetics induced by the highly different CF concentrations.
To evaluate possible damage to the vesicle structure induced by the acoustic streaming, the cyclodextrin amphiphiles in the PSVs were covalently labeled with rhodamine B. The red fluorescence intensity of rhodamine B‐labeled PSVs did not change after 20 min of acoustic stimulation at 500 mW (Figure S4), confirming that the immobilized PSVs remained intact and did not detach from the surface along the acoustically generated vortex. Instead, the encapsulated dye was released under the acoustic streaming.
These results demonstrate that the acoustic streaming‐triggered exchange of cargo from PSVs is a bi‐directional process and that the (un)loading rates can be controlled by the input power of the acoustic streaming.
In many applications, control over release is required when the vesicles are suspended in a solution. For this purpose, we investigated the dye release from PSVs in solution by using a home‐built dialysis system (Figure 2 a). The resonator was placed at the bottom of the system to generate the acoustic streaming. CF‐encapsulated PSVs were preloaded in the bottom chamber and separated by a dialysis membrane from the top chamber. Pure buffer was placed in the top chamber before any stimulation was applied.
Figure 2.
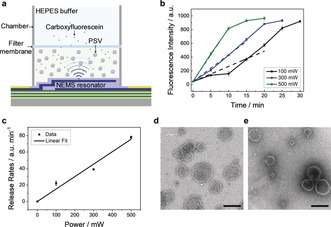
Controlled release from PSVs in solution under acoustic streaming. a) Chamber‐based release system. A filter membrane (molecular weight cut‐off=12–15 kDa) was sandwiched between two PDMS chambers to keep the PSV‐encapsulated CF dye in the lower chamber, while the liberated CF dye can pass through the filter membrane. Thus, the amount of the released dye can be determined by measuring the fluorescence intensity in the top chamber. b) The fluorescence intensities of the released CF dye as a function of time, and linear fits of the initial parts of these curves. The fluorescence intensity at 0 min was obtained by measuring the buffer solution in the upper chamber without stimulation after waiting for 20 min (Figure S6). c) Initial release rates as a function of power. TEM images of the PSVs d) before and e) after the acoustic stimulation (500 mW for 20 min). Scale bars=100 nm.
The fluorescence intensity of the released CF was measured in the top chamber upon a release from the PSVs stimulated at 100, 300, and 500 mW (Figure S5) and plotted as a function of time. The release curves show a continuously increasing trend that gradually levels off to a plateau, the height of which does not depend on the applied power (Figure 2 b). The observed lag time, most apparent at high power, is attributed to the diffusion time needed for the released dye to reach the upper chamber. For assessing the initial rates, this effect is ignored, and the initial sections of the curves were fitted linearly. These initial slopes are regarded as the release rates, which show a linear dependence on power (Figure 2 c). These results suggest that the acoustic streaming induces the release of dye to a level at which the concentration is balanced between the two chambers. The final concentration is not dependent on power; however, a faster release of the dye is achieved at higher power.
The release in solution is more complex than that on a surface. The high‐velocity acoustic steaming accelerates the motion of the suspended PSVs, which may enhance the frequencies of vesicle–vesicle and vesicle–interface collisions. Moreover, the high‐speed mixing owing to the strong vortex may facilitate the diffusion of the dye through the filter membrane. Such combined effects will enhance the translocation of the dye across the dialysis film.
To evaluate whether the acoustic streaming caused any structural changes of the suspended PSVs, transmission electron microscopy (TEM) was performed. Before the acoustic stimulation, PSVs appeared as circular objects as typically observed for vesicles (Figure 2 d), and no apparent difference was observed after the acoustic treatment at 500 mW for 20 min (Figure 2 e). This suggests that the high‐speed collisions and mixing do not damage the vesicle structures and that the release of cargo can be turned on and off, which can be attributed to the formation of transient nanopores in the membrane of the vesicle. The intact shape and minimally affected size of the PSVs, which was also confirmed by dynamic light scattering (DLS) measurements (Figure S7), could indicate a relatively stable re‐loading/release capacity of PSVs under this switchable acoustic trigger, but repeated loading/release was not attempted in this study.
To study mechanical deformations of vesicles under acoustic streaming, GUVs were immobilized on a supported lipid bilayer (SLB) by the specific biotin–SAv interaction pairs (Figures S9). GUVs showed a clear deformation under acoustic streaming but recovered their initial shape instantly once the power was turned off. This dependence on the input power (Figure S10 and Videos S1–S4) and the vertical distance (Figure S11 and Videos S5–S8) were attributed, respectively, to the velocity and the spatial distribution of the acoustic streaming.
To test the hypothesis that the mechanical deformation of vesicles creates pores in the membrane and estimate the size of these pores, we tried to load polystyrene nanoparticles (PS NPs) of various sizes into GUVs under acoustic streaming. Upon application of continuous acoustic streaming at 300 mW from a fixed vertical distance of 100 μm (Figure S8 a) to the GUVs for 10 min, we observed (Figure 3 b) the blue fluorescence from 50 nm PS NPs inside the vesicles. As a control (Figure 3 a), vesicles were incubated with PS NPs for 10 min without stimulation and blue fluorescence was not observed inside the GUVs. This indicates that the acoustic streaming is necessary to allow the transport of 50 nm PS NPs into the vesicles. The power dependence and reproducibility of the loading are shown in Figures S14 and S15, respectively.
Figure 3.
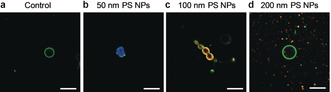
Loading of PS NPs into GUVs under acoustic streaming. CLSM images of TopFluor‐labeled GUVs, immobilized on a SLB, loaded with a,b) PS NPs of 50 (blue), c) 100 (orange) and d) 200 nm (red fluorescence) without (control, (a)) and with (b,c,d) acoustic streaming (300 mW, 10 min) applied from a fixed distance of 100 μm. Before imaging, the samples were rinsed three times to remove the remaining PS NPs from the surrounding solution. Scale bars=10 μm.
To evaluate the size of the pores formed by acoustic streaming, we performed similar experiments with PS NPs of 100 and 200 nm (Figure 3 c,d and Figure S14), which indicated that larger particles were progressively more difficult to incorporate. When the power of the acoustic streaming was increased to 500 mW, both the blue (50 nm PS NPs) and the orange fluorescence (100 nm PS NPs) intensities increased, while the 200 nm PS NPs still stayed outside the vesicle (Figure S14). Therefore, the loading of nanoparticles is power dependent, which we assume is related to the dynamic pore formation process generated during the deformation of vesicles. As a result, the combination of vesicles with the NEMS resonator can be used as a size‐based filter to exchange particles or other materials of specific sizes.
To understand the mechanism of vesicle deformation induced by the acoustic streaming, we used a 3D finite‐element model (FEM) simulation (details are provided in the Supporting Information). The results show that the displacement across the surface of the vesicle is non‐uniformly distributed under the acoustic streaming, which indicates the deformation of the vesicle (Figure 4).
Figure 4.
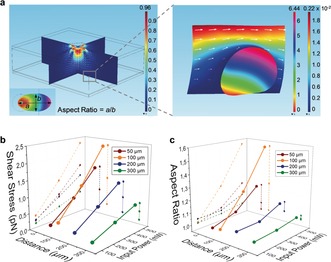
FEM simulations of vesicle deformation under acoustic streaming. a) Simulated patterns of the acoustic streaming distributed around the resonator. The vesicle was represented by an elastic and hollow sphere (inside medium, water) surrounded by water. The model resonator was located above the vesicle. The 10‐μm vesicle was located at coordinates of (45 μm, 0 μm, −100 μm) relative to the resonator center (Figure S16). The frequency of the resonator was 2.5 GHz. Simulated patterns of the displacement of the vesicle are shown in the zoomed‐in image (right). The right color bar indicates the magnitude of the streaming velocity outside the vesicle from min (blue) to max (red), while the left color bar indicates the magnitude of the displacement of the vesicle from min (red) to max (pink). 3D plots of b) the total shear stress in the x direction and c) the mechanical deformation in terms of aspect ratio of the vesicle at different power levels (100, 300, and 500 mW) and different vertical distances (50, 100, 200, and 300 μm). The total shear stress in the x direction was obtained by the integration of the x‐partial shear stresses across the surface of the vesicle.
We have demonstrated that GHz acoustic streaming can be used as a general tool to control the transfer of cargo into and out of vesicles. PSVs, either immobilized on a surface or suspended in solution, were used to study the acoustically triggered loading and release processes. The kinetics of loading and release can be tuned through the applied power and potentially by other parameters such as cargo concentration and vesicle type. An increased frequency of the device can also possibly enhance the (un)loading process, by accelerating the velocity of the acoustic streaming. For quantitative prediction of the obtained cargo concentration after a loading or release experiment, calibration may be needed, as done in Figures 1 and 2. The mechanical deformation of individual GUVs was analyzed by simulation and experiment to understand the materials exchange across the vesicle membrane. We have proposed that transient nanopores are generated in the membrane when vesicles are deformed by the acoustic streaming. The formation of nanopores increases the membrane permeability and allows the transport of materials both into and out of the vesicles. The size of these pores is such that it allows transport of 100 nm, but not of 200 nm, PS NPs into the GUVs. Both GUVs and PSVs stay intact during the acoustic streaming, which confirms the non‐invasive nature of this acoustic approach. We showed that the GHz acoustic poration effect can be applied to vesicles of different sizes and compositions. Thus, it can be used as a versatile tool to control the release and uptake of different materials for different types of vesicles.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Acknowledgements
X.D. acknowledges financial support from the Natural Science Foundation of China (NSFC No. 61674114, 91743110, 21861132001), National Key R&D Program of China (2017YFF0204600), and the 111 Project (B07014). N.J.O. and J.H. acknowledge financial support from the Volkswagen Foundation (FlapChips project, 91‐056). W.C.V. acknowledges a fellowship of the Fonds der Chemischen Industrie. W.C.V. and B.J.R. sincerely thank the Deutsche Forschungsgemeinschaft (DFG SFB858) for funding. Matthias Tesch is acknowledged for providing assistance with the synthesis of Ad‐PAA, and Nadja Möller for assistance with TEM imaging. M.A. Abolghassemi Fakhree (Nanobiophysics group, University of Twente) is thanked for assistance with the GUV synthesis. Pieter H. Hamming is thanked for the MatLab script for image analysis.
Y. Lu, W. C. de Vries, N. J. Overeem, X. Duan, H. Zhang, H. Zhang, W. Pang, B. J. Ravoo, J. Huskens, Angew. Chem. Int. Ed. 2019, 58, 159.
Contributor Information
Prof. Dr. Xuexin Duan, Email: xduan@tju.edu.cn
Prof. Dr. Bart Jan Ravoo, Email: b.j.ravoo@uni-muenster.de.
Prof. Dr. Jurriaan Huskens, Email: j.huskens@utwente.nl.
References
- 1. Peyret A., Ibarboure E., Pippa N., Lecommandoux S., Langmuir 2017, 33, 7079. [DOI] [PubMed] [Google Scholar]
- 2. Yavuz M. S., Cheng Y., Chen J., Cobley C. M., Zhang Q., Rycenga M., Xie J., Kim C., Song K. H., Schwartz A. G., Nat. Mater. 2009, 8, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Esser-Kahn A. P., Odom S. A., Sottos N. R., White S. R., Moore J. S., Macromolecules 2011, 44, 5539. [Google Scholar]
- 4. Buddingh B. C., van Hest J. C., Acc. Chem. Res. 2017, 50, 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Takinoue M., Takeuchi S., Anal. Bioanal. Chem. 2011, 400, 1705. [DOI] [PubMed] [Google Scholar]
- 6. Allen T. M., Cullis P. R., Adv. Drug Delivery Rev. 2013, 65, 36. [DOI] [PubMed] [Google Scholar]
- 7. Tanner P., Baumann P., Enea R., Onaca O., Palivan C., Meier W., Acc. Chem. Res. 2011, 44, 1039. [DOI] [PubMed] [Google Scholar]
- 8. Soussan E., Cassel S., Blanzat M., Rico-Lattes I., Angew. Chem. Int. Ed. 2009, 48, 274; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 280. [Google Scholar]
- 9. Nourian Z., Roelofsen W., Danelon C., Angew. Chem. Int. Ed. 2012, 51, 3114; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3168. [Google Scholar]
- 10. Christensen S. M., Bolinger P.-Y., Hatzakis N. S., Mortensen M. W., Stamou D., Nat. Nanotechnol. 2012, 7, 51. [DOI] [PubMed] [Google Scholar]
- 11. Bolinger P.-Y., Stamou D., Vogel H., J. Am. Chem. Soc. 2004, 126, 8594. [DOI] [PubMed] [Google Scholar]
- 12. Siegel R. A., Falamarzian M., Firestone B. A., Moxley B. C., J. Controlled Release 1988, 8, 179. [Google Scholar]
- 13. Borchert U., Lipprandt U., Bilang M., Kimpfler A., Rank A., Peschka-Süss R., Schubert R., Lindner P., Förster S., Langmuir 2006, 22, 5843. [DOI] [PubMed] [Google Scholar]
- 14. Chen W., Meng F., Cheng R., Zhong Z., J. Controlled Release 2010, 142, 40. [DOI] [PubMed] [Google Scholar]
- 15. Ma N., Li Y., Xu H., Wang Z., Zhang X., J. Am. Chem. Soc. 2010, 132, 442. [DOI] [PubMed] [Google Scholar]
- 16. Chang Y., Yang K., Wei P., Huang S., Pei Y., Zhao W., Pei Z., Angew. Chem. Int. Ed. 2014, 53, 13126; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13342. [Google Scholar]
- 17. Huo M., Yuan J., Tao L., Wei Y., Polym. Chem. 2014, 5, 1519. [Google Scholar]
- 18. Peng K., Tomatsu I., Kros A., Chem. Commun. 2010, 46, 4094. [DOI] [PubMed] [Google Scholar]
- 19. Liu J., Yang G., Zhu W., Dong Z., Yang Y., Chao Y., Liu Z., Biomaterials 2017, 146, 40. [DOI] [PubMed] [Google Scholar]
- 20. Qin S., Geng Y., Discher D. E., Yang S., Adv. Mater. 2006, 18, 2905. [Google Scholar]
- 21. Mura S., Nicolas J., Couvreur P., Adv. Mater. 2013, 12, 991. [DOI] [PubMed] [Google Scholar]
- 22. Qiu F., Mhanna R., Zhang L., Ding Y., Fujita S., Nelson B. J., Sens. Actuators B 2014, 196, 676. [Google Scholar]
- 23. Chen W., Du J., Sci. Rep. 2013, 3, 2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rizzitelli S., Giustetto P., Cutrin J. C., Castelli D. D., Boffa C., Ruzza M., Menchise V., Molinari F., Aime S., Terreno E., J. Controlled Release 2015, 202, 21. [DOI] [PubMed] [Google Scholar]
- 25. Huang L., Yu C., Huang T., Xu S., Bai Y., Zhou Y., Nanoscale 2016, 8, 4922. [DOI] [PubMed] [Google Scholar]
- 26. Ji X., Dong S., Wei P., Xia D., Huang F., Adv. Mater. 2013, 25, 5725. [DOI] [PubMed] [Google Scholar]
- 27. Oliveira H., Pérez-Andrés E., Thevenot J., Sandre O., Berra E., Lecommandoux S., J. Controlled Release 2013, 169, 165. [DOI] [PubMed] [Google Scholar]
- 28. Zhou J., Chen M., Diao G., ACS Appl. Mater. Interfaces 2014, 6, 18538. [DOI] [PubMed] [Google Scholar]
- 29. Allen T. M., Cullis P. R., Science 2004, 303, 1818. [DOI] [PubMed] [Google Scholar]
- 30. Gupta P., Vermani K., Garg S., Drug Discovery Today 2002, 7, 569. [DOI] [PubMed] [Google Scholar]
- 31. Dash S., Murthy P. N., Nath L., Chowdhury P., Acta Pol. Pharm. 2010, 67, 217. [PubMed] [Google Scholar]
- 32. Sou K., Naito Y., Endo T., Takeoka S., Tsuchida E., Biotechnol. Prog. 2003, 19, 1547. [DOI] [PubMed] [Google Scholar]
- 33. Hauschild S., Lipprandt U., Rumplecker A., Borchert U., Rank A., Schubert R., Förster S., Small 2005, 1, 1177. [DOI] [PubMed] [Google Scholar]
- 34. Yoo J.-W., Irvine D. J., Discher D. E., Mitragotri S., Nat. Rev. Drug Discovery 2011, 10, 521. [DOI] [PubMed] [Google Scholar]
- 35. Choucair A., Lim Soo P., Eisenberg A., Langmuir 2005, 21, 9308. [DOI] [PubMed] [Google Scholar]
- 36. Yu B., Wang D. A., Ye Q., Zhou F., Liu W., Chem. Commun. 2009, 6789. [DOI] [PubMed] [Google Scholar]
- 37. Ma Z., Jia X., Hu J., Zhang G., Zhou F., Liu Z., Wang H., Langmuir 2013, 29, 5631. [DOI] [PubMed] [Google Scholar]
- 38. Duncanson W. J., Arriaga L. R., Ung W. L., Kopechek J. A., Porter T. M., Weitz D. A., Langmuir 2014, 30, 13765. [DOI] [PubMed] [Google Scholar]
- 39. Qu H., Yang Y., Chang Y., Tang Z., Pang W., Wang Y., Zhang H., Duan X., Sens. Actuators B 2017, 248, 280. [Google Scholar]
- 40. Pan S., Zhang H., Liu W., Wang Y., Pang W., Duan X., ACS Sens. 2017, 2, 1175. [DOI] [PubMed] [Google Scholar]
- 41. Zhang Z., Wang Y., Zhang H., Tang Z., Liu W., Lu Y., Wang Z., H, Yang , Pang W., Zhang H., Zhang D., Duan X., Small 2017, 13, 1602962. [Google Scholar]
- 42. Samanta A., Tesch M., Keller U., Klingauf J., Studer A., Ravoo B. J., J. Am. Chem. Soc. 2015, 137, 1967. [DOI] [PubMed] [Google Scholar]
- 43. de Vries W. C., Tesch M., Studer A., Ravoo B. J., ACS Appl. Mater. Interfaces 2017, 9, 41760. [DOI] [PubMed] [Google Scholar]
- 44. de Vries W., Grill D., Tesch M., Ricker A., Nüsse H., Klingauf J., Studer A., Gerke V., Ravoo B. J., Angew. Chem. Int. Ed. 2017, 56, 9603; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9732. [Google Scholar]
- 45. Kim E., Xia Y., Whitesides G. M., Nature 1995, 376, 581. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary