

Abstract
Gold catalysis has experienced a tremendous development over the past decades, and is nowadays widely used in organic synthesis to perform chemical transformations of π‐bond‐containing molecules. Catalyst development has been based mostly on ligand development and counter‐ion strategies. More recently, the encapsulation of gold catalysts in (supra)molecular cages was explored as a new way to control selectivity and reactivity of gold catalysts. In this review, we describe the cages that have been employed as hosts for gold complexes, along with their impact on the catalytic performance. Covalent and supramolecular approaches to encapsulate single metal complexes will be described and the impact on the catalytic performance will be discussed. Also, recent strategies to pre‐organize multiple metal centers will be discussed.
Keywords: Gold, Supramolecular Chemistry, Host-guest, Catalysis, Self-assembly
Introduction
For a long time, gold was considered solely as a noble metal that provides metal complexes that are inactive in homogeneous catalysis. However, research reported in the last two decades demonstrates that gold complexes can have remarkable reactivity, partly due to relativistic effects, providing reactivity that is complementary to that displayed by typical palladium and rhodium catalysts.1,2 Gold(I) catalysts exhibit strong π‐acidity, leading to activation of alkynes and allenes and even alkenes, while being rather stable to air and water and other functional groups that may be present in the substrate. Numerous reports on homogeneous gold catalysis have been published showing the versatility of gold catalysis3 and recent application in total synthesis of natural products has been reported.4, 5, 6
Current strategies in catalyst development in this area are dominated by ligand variation and counter‐ion strategies. This has resulted in substantial progress in the field, yet several challenges in the area remain. For example, catalyst loadings are generally high, typically up to 5 mol%, and control of catalyst properties by steric variation of the ligand is limited because of the preferred linear coordination geometry of gold(I). As a consequence it is challenging to develop chiral complexes that provide the products with high enantioselectivity.7
Supramolecular approaches in catalysis went through an important development during the last decades. Inspired by Nature's most efficient catalyst, enzymes, much effort has been devoted to modulating catalyst properties via the second coordination sphere by placing catalysts in molecular cages.8,9 Such enzyme mimics can be obtained by encapsulation of catalysts through covalent binding within cages or by supramolecular binding using complementary interactions with the capsule. In these systems the transition metal complex is (partly) isolated from the bulk phase which may already lead to changed reactivity. In addition, substrate preorganization by bringing it in close proximity to the catalyst, sometimes in a specific conformation induced by the steric constraints of the cage, also may lead to different selectivity and reactivity compared to reactions that take place in bulk solution.10, 11, 12, 13 Furthermore, isolation of the complex from the bulk enhances the stability of the catalyst providing catalyst systems that can reach high turn‐over.
This Minireview describes the different strategies that have been used to encapsulate gold complexes and discusses the impact on the catalytic properties. Several advances in the field including selective transformations, substrate selectivity, switching reactivity and application in tandem reactions will be outlined. This review is divided in three parts, based on the different encapsulation strategies that were used. The first part covers systems in which the gold center is incorporated by covalent strategies, for example, in the wall of a capsule (Figure 1a). The second describes cages that encapsulate gold complexes by means of supramolecular interactions (Figure 1b), whereas the last part focusses on cages that can host multiple gold complexes (Figure 1c). In all systems, the gold complex is at least partly isolated from the bulk solution, giving rise to for example rate enhancement and increased selectivity. Stabilization of gold nanoparticles by supramolecular assemblies as well as gold embedded in solid supports are beyond the scope of this review and will not be discussed.
Figure 1.
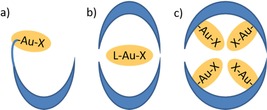
Different strategies to encapsulate a gold complex. a) Metal coordination of the gold complex to the cage. b) The gold complex is encapsulated inside the capsule by means of non‐covalent interactions. c) Multiple gold centers are encapsulated in one cage.
Covalently Functionalized Catalysts
One strategy to create a well‐defined cage around a gold complex is the covalent attachment of a cavitand to the ligand that coordinates to the gold complex. This has been demonstrated for resorcin[4]arene‐based cavitands and cyclodextrins, but also for phosphine ligands that contain very bulky groups remote from the donor atom, coined holey ligands. These will be discussed in this section.
Holey Phosphine Ligands
Sawamura and co‐workers developed a series of ethynyl phosphine ligands with very bulky substituents (Figure 2) which can be considered as ligands in confined space. This is clear upon inspection of the gold complexes as a conical cavity around the gold atom is formed as demonstrated by the space filling model compared to PPh3. These gold complexes were used in several gold catalyzed cyclization reactions.14 Surprisingly, no reaction of the alkyne groups of the ligand was observed, which is explained by the confinement of the gold catalyst which prevents the activation of alkyne groups of other ligands. Importantly, the use of the holey ligands resulted in an important rate enhancement in the 6‐exo‐dig cyclization reaction compared to [PPh3AuNTf2] and [P(OPh)3AuNTf2], the latter complex having comparable electronic properties.15 The influence of the cavity was evident by using similar ethynyl phosphine ligands without the bulky substituents, which all gave lower conversion.15 This demonstrated the importance of the steric effect from the cavity formed by the bulky substituents in the rate enhancement of the reaction. The substrate scope could be extended to a variety of 7‐membered rings16, 17, 18 and to 8‐membered rings for which the carbon‐analogue of the ligand was required (see Figure 2).19,20
Figure 2.
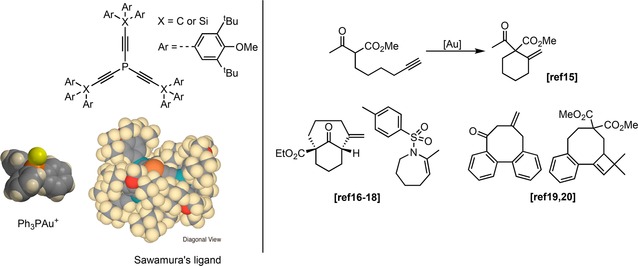
Left: PPh3Au+ and Sawamura's holey ligand (X=Si or C) and the corresponding space‐filling model (beige=hydrogen, black=carbon, red=oxygen, cyan=silicon, orange=phosphorus and yellow=gold) for X=Si (Adapted with permission from Org. Lett. 2010, 12, 4380–4383. Copyright 2010 American Chemical Society). Right: various transformations to form cyclic products for which an increased reaction rate using holey phosphine was observed.
Resorcin[4]arene cavitands
Iwasawa and co‐workers developed a resorcin[4]arene‐based cavitand, containing a phosphorous atom that can function as a ligand that coordinates to a metal center.21 The lone pair of the P atom is oriented toward the cavity space, so a coordinating metal ion is directed into the cavity.22 This strategy was previously described by Rebek and co‐workers using palladium,23 and coordination complexes with rhodium24 and platinum25 have been reported as well. Iwasawa and co‐workers attached a gold center to the resorcin[4]arene cavitand (Figure 3).21 The crystal structure showed that the gold center pointed inward and was surrounded by the walls of the cavity. After dehalogenation by AgOTf, the gold‐cavitand complex was successfully employed in the hydration of terminal alkynes and the Conia‐ene reaction using 5 mol% catalyst loading. This demonstrated the catalytic activity of the complex; however differences between this system and free gold catalysts were not described.
Figure 3.
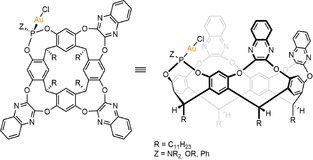
A resorcin[4]arene‐based cavitand that functions as a ligand for the gold atom.
A dinuclear version of the cavitand with two gold atoms opposite to each other was published shortly after (Figure 4).26,27 This dinuclear gold catalyst enabled dimerization of terminal alkynes to form conjugated enynes, whereas the mononuclear gold‐cavitand was unreactive in this reaction. The dinuclear gold‐cavitand complex showed high selectivity for the intermolecular dimerization.
Figure 4.
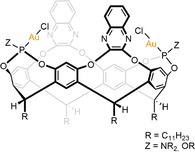
A dinuclear resorcin[4]arene cavitand with two gold complexes.
The exact function of the cavity was addressed in a follow‐up paper, where the catalytic perfomance of the dinuclear gold cavity was compared to cavitands with smaller ‘walls’.28 The quinoxaline walls were found to play an essential role, because they increased the interaction between the two alkyne substrates and created a strong π‐cloud environment that limited reaction pathways and stabilized the desired intermediates.
Cyclodextrins
Cyclodextrins have been extensively studied as enzyme mimics due to their accessibility and low toxicity, and they are well known host molecules that bind hydrophobic guest molecules in water.9 Sollogoub and co‐workers developed a series of cyclodextrins with an N‐heterocylic carbene (NHC) gold complex attached to the small rim pointing inside the cavity (Figure 5).29,30 Consequently, coordination of metals to the NHC leads to systems in which the metal atom is deeply buried inside the cavity, as proven by extensive NMR studies.
Figure 5.
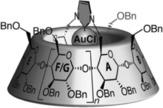
An NHC‐capped cyclodextrin, with a gold‐center deeply buried in the cavity. Adapted with permission from Angew. Chem. Int. Ed. 2013 , 52, 7213–7218. Copyright 2013 John Wiley and Sons.
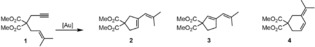
A series of NHC functionalized CD's has been prepared (α‐, β‐ and γ‐cyclodextrins (CD) (n=1, n=2, n=3 respectively in Figure 5), and the effect of the available space on the gold‐catalyzed cycloisomerization of compound 1 was investigated (Table 1). The reaction catalyzed by the normal carbene‐based gold catalyst [Au(IPr)X] (IPr=1,3‐bis(2,6‐diisopropylphenyl)imidazol‐2‐ylidene) gives products 2 and 3 in a 1 : 0.65 ratio. The small α‐CD did not alter the selectivity of the gold catalyst, but the slightly bigger β‐CD changed the product distribution drastically. Product 3 was not formed, and next to some product 2 the 6‐membered ring compound 4 was the major product. The γ‐CD on the other hand, also changed the product distribution but to a lesser extent. These results clearly show that in this gold‐catalyzed transformation the product distribution can be changed by just changing the second coordination sphere.
Table 1.
The influence of cyclodextrins on the selectivity in a gold‐catalyzed cycloisomerization reaction.
| |||
|---|---|---|---|
| [Au] | Conversion [%] | Yield [%] | Selectivity (2 : 3 : 4) |
| IPr−Au−X | 100 | 88 | 1 : 0.65 : 0 |
| α‐CD−Au−X | 100 | 52 | 1 : 0.65 : 0 |
| β‐CD−Au−X | 100 | 63 | 1 : 0 : 3.3 |
| γ‐CD−Au−X | n.r. | 80 | 1 : 0.56 : 0.61 |
The influence of the cyclodextrins was also investigated in an asymmetric cycloisomerization (Scheme 1). Interestingly, the enantiomeric excess of the product could be increased to values varying between 7 and 48 % using α‐ and γ‐CD, and even reached up to 80 % using the β‐CD. It is interesting to note that the CD was the only source of chirality in these complexes.
Scheme 1.
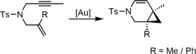
Gold‐catalyzed asymmetric cycloisomerization.
A reasonable explanation for the differences in catalytic outcome between α‐ and β‐CD was presented in a report on cyclodextrins containing a copper center.31 Both cyclodextrins contain a groove wherein the metal center is located, but the bigger β‐CD also accommodates a pit situated next to the metal center (Figure 6). The authors suggested that the β‐CD can pre‐organize a substrate next to the metal center in the cavity, whereas there is no space for substrates in the pocket of the α‐CD. This structural difference has a major impact on the linear to branched (L/B) ratio of the copper‐catalyzed hydroboration of alkynes, and it can be imagined that a similar effect also applies to gold‐catalyzed transformations using these cyclodextrin appended complexes.
Figure 6.
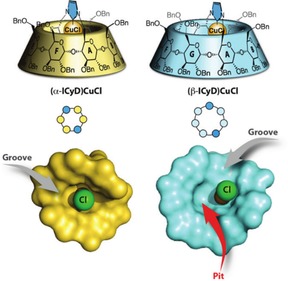
α‐CD and β‐CD contain a groove, the latter also has a pit next to the metal center. Reprinted with permission from Angew. Chem. Int. Ed. 2017, 56, 10821–10825. Copyright 2017 John Wiley and Sons.
Non‐Covalent Host‐Guest Systems
The hexameric resorcin[4]arene and an anionic M4L6 cage have thoroughly investigated during the last decade as two different host systems for gold catalysis. Both capsules stabilize and/or strongly bind cations and thus are suited for the encapsulation of cationic gold complexes which are active in catalysis.
Hexameric Resorcin[4]arene Capsule
A single resorcin[4]arene is a bowl‐shaped molecule and can encapsulate small cationic guests.32,33 In 1997 Atwood discovered that when dissolved in apolar wet solvents, six of these resorcin[4]arene molecules and eight water molecules form a hexameric self‐assembled structure that is held together by hydrogen bonds (Scheme 2).34 Various guest molecules can be bound in these self‐assembled cages.
Scheme 2.
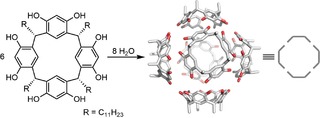
Six resorcin[4]arene molecules form a hydrogen‐bonded hexameric structure in water‐saturated apolar solvents.
Scarso, Strukul and Reek showed that the hexameric structure can encapsulate a cationic gold NHC complex by non‐covalent, attractive weak interactions.35 Encapsulation of the catalyst strongly influenced the outcome of the catalysis (Scheme 3). In the hydration reaction of 4‐phenyl‐1‐butyne, the free catalyst gave the ketone quantitatively as the sole product within 30 minutes reaction time. In contrast, when the catalyst was encapsulated in the hexameric resorcin[4]arene, the reaction was slowed down and only 5 % conversion was obtained after 30 minutes. Interestingly, new products were formed: next to the ketone also a cyclization product and the aldehyde were formed. The decrease in reaction rate can be explained by the barrier caused by the capsule for the substrate to reach the catalyst. The reason for the change in product distribution was not studied in detail, but a likely explanation is that the supramolecular cage supports folding of the substrate‐metal complex to promote the cyclization reaction.
Scheme 3.
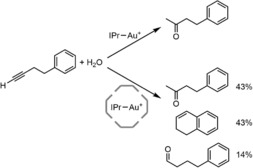
Effect of the hexameric resorcin[4]arene on the gold‐catalyzed hydration of 4‐phenyl‐1‐butyne.
Scheme 4.
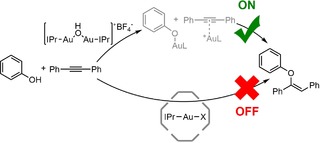
On/off‐switching of the dual gold‐catalyzed hydrophenoxylation reaction using the hexameric resorcin[4]arene cage.
In follow‐up work, it was shown that this hexameric cage can also be used for substrate selective catalysis, in particular for gold‐catalyzed alkyne hydration reactions.36 The free catalyst [Au(IPr)(OTf)] converted the aliphatic cyclic alkyne ethynylcyclohexane 1.5 times faster than linear ones (1‐octyne and 1‐dodecyne); inside the cage, the cyclic substrate reacted even 3 times faster than the linear ones. The explanation is that a cyclic substrate can be easily accommodated in the cage, whereas a linear analogue has to fold itself to be encapsulated which is entropically unfavorable and as such this leads to a lower reactivity.
Substrate selectivity was also achieved based on size. In a competition experiment, the hydration of three rigid aromatic alkynes was evaluated (Figure 7). In absence of cage the more electron‐rich para‐substituted substrates were converted about 1.5 times faster than the non‐substituted substrate. In contrast, when the encapsulated catalyst was applied, the smaller phenyl acetylene was converted 1.6 times faster than the bigger 4‐tert‐butylphenylacetylene. The steric confinement of the cage was responsible for this preference for smaller substrates.
Figure 7.
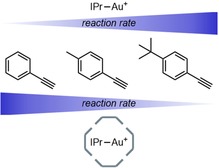
Effect of the hexameric resorcin[4]arene on the relative reaction rate of the hydration of aryl‐alkynes.
Echavarren and Ballester did a thorough investigation on the encapsulation of the catalysts [Au(IPr)(tmbn)]SbF6 (tmbn=2,4,6‐trimethoxybenzonitrile) and [Au((2‐biphenyl)di‐t‐butylphosphine)(NCMe)]SbF6 ([JohnPhos‐Au(MeCN)]SbF6) in the hexameric resorcin[4]arene cage.37 Evaluation of the encapsulation processes by NMR and DOSY NMR reveal that the gold complex encapsulated in the cage is thermodynamically favorable and kinetically accessible. Bulky coordinating counter‐ions on the gold complex have to dissociate before encapsulation, and inside the cage this counter‐ion is replaced by a small solvent molecule or H2O. This process is facilitated by an excess of water, and indeed the use of extra water resulted in rapid encapsulation at room temperature. However, an excess of water also favored the formation of μ‐OH bridged dinuclear complexes. The dinuclear JohnPhos complex could be encapsulated, but the analogous complex based on IPr carbene ligands did not fit in the cavity of the hexameric cage. Instead, the equilibrium between the mononuclear aqua complex and the μ‐OH bridged dinuclear complex is shifted in favor of the mononuclear complex as a result of the encapsulation.
A similar equilibrium was also explored by Reek and co‐workers and they used this as a basis to generate a switchable catalyst system.38 They applied the dual‐gold catalyst [{Au(IPr)}2(μ‐OH)]BF4 and took advantage of the difference in reactivity of the mononuclear and the dinuclear complex: the mononuclear gold catalyst reacts via π‐activation of substrates only, whereas the dinuclear complex operates via σ,π‐activation. Substrates like phenol and terminal alkynes are more nucleophilic when σ‐activated. As a result, the reactivity of the catalyst system can be controlled by encapsulation events using the hexameric resorcin[4]arene cage, as the hexameric resorcin[4]arene cage breaks the dinuclear complex into monomers that are bound in the cage. The dual‐gold catalyst was employed in the dual‐gold‐catalyzed hydrophenoxylation reaction, for which two gold centers are needed (scheme 4). After encapsulation, the monomeric complex is inactive in this reaction,39 and as a consequence, the catalyst activity is switched off. Switching the activity on again can be achieved by the addition of competing guest that releases the gold complex, which when released forms the dinuclear species again.
Cage‐induced switching between a mononuclear and dinuclear reaction pathway was also achieved in the reaction with 4‐phenyl‐1‐butyne (Scheme 5). When free in solution, the dual gold complex activates the terminal alkynes in a dual way, giving dimerization products. In contrast, when the complex is encapsulated as mononuclear species this pathway is not possible leading to the formation of ketone and cyclization product. Full control over the selectivity was not attained as under the conditions applied the ketone product was also formed outside the cage.
Scheme 5.
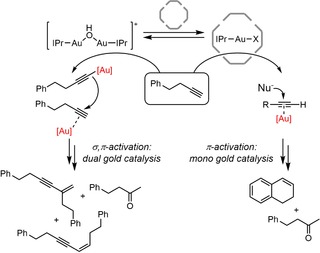
Switching between a dual‐ and mono‐activation pathway using a strategy that involves selective encapsulation of mononuclear gold complexes in the hexameric resorcin[4]arene cage.
M4L6 cages
Raymond and coworkers developed an anionic, water‐soluble, tetrahedral M4L6 cage (Figure 8). The C2‐symmetric ligands each bear a −4 charge and coordinate to four Fe(III) or Ga(III) ions, resulting in an overall charge of −12.40 The cage is water‐soluble with a hydrophobic cavity that allows encapsulation of cationic species and (cationic) transition metal complexes of small size.
Figure 8.
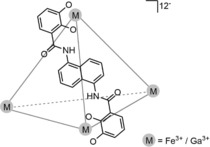
Anionic, water‐soluble M4L6 tetrahedral cage.
In a collaboration, Bergman, Raymond and Toste used the anionic Ga4L6 cage to encapsulate the gold phosphine complex [Au(PMe3)Br].41 As the cage encapsulates cationic species, the bromide ion dissociates upon inclusion, leaving the more active complex [Au(PMe3)]+ inside the cage. This encapsulated species catalyzed the intramolecular hydroalkoxylation of an allenol in 48 % yield in 18 hours, whereas the non‐encapsulated [Au(PMe3)Br] gave only 11 % yield after this time (Scheme 6). Based on the initial rates, encapsulation of the catalyst increases by 8‐fold the reaction rate resulting in a TON of 67. This is a nice example of cage‐induced halide dissociation by catalyst stabilization in a supramolecular cage, thereby activating the gold catalyst. In addition, the reaction can be performed in water.
Scheme 6.
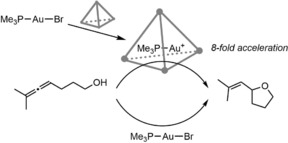
The intramolecular hydroalkoxylation is faster when catalyzed by encapsulated [Au(PMe3)]+ compared to the free [Au(PMe3)Br].
Further investigation of the system showed that the Ga4L6 cage excludes water from the cavity, preventing the reaction of water with reactive intermediates.42 In accordance, the cycloisomerization reaction of substrate 5 yielded product 7 as the main product in the cage, while the free catalyst in water only gave H2O addition product 6 (Scheme 7).
Scheme 7.
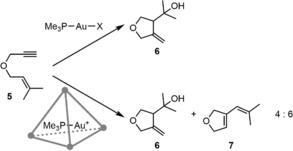
The Ga4L6 cage excludes water from its cavity, leading to a different product distribution in gold catalysis.
A beautiful example of how cages can be used to protect catalysts, which is important in tandem catalysis, appeared in 2013. The team of Bergman, Raymond and Toste used a gold catalyst encapsulated in the Ga4L6 cage to apply this in a cascade reaction in combination with enzymes.43 Usually, enzymes lose their activity in the presence of organometallic complexes as a result of binding of the complex to the enzyme. In this work, this issue was elegantly solved by the site‐isolation of the gold catalyst by means of encapsulation. As a consequence, both the life‐time of the enzymes and of the gold complexes improved, making these catalyst systems compatible for cascade reactions.
The tandem reaction that was studied is shown in Scheme 8. The first step is an enzymatic hydrolysis of an allenic acetate by esterase, followed by an intramolecular hydroalkoxylation reaction catalyzed by the encapsulated gold catalyst. The yield of the final product dropped significantly when the gold catalyst was not encapsulated in the Ga4L6 cavity, underlining the crucial role of the cage to prevent inhibition of the enzyme.
Scheme 8.
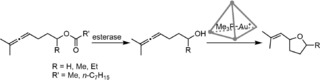
Tandem reaction catalyzed by esterase and subsequently an encapsulated [Au(PMe3)]+.
In the systems discussed so far, the aim was to encapsulate the catalyst throughout the catalytic cycle, but in an interesting paper in 2015 the team of Bergman, Raymond and Toste showed a new strategy in which the complex is only encapsulated during a specific part of the catalytic cycle, thereby acting as a catalyst for a specific step.44 In this particular example, encapsulation of the complex facilitates alkyl‐alkyl reductive elimination from gold(III) complexes. Although relevant for catalytic cross‐coupling reactions, this mechanistic step is usually slow for transition metals, as the low‐coordinate, cationic transition metal complex is not stable. The authors argued that the Ga4L6 cage could stabilize these species by encapsulation in the cavity.
The impact of the cage was studied in the reductive elimination of ethane from complex 8 (R=Me). A catalytic amount of cage accelerated the reaction 4000‐fold, based on the initial reaction rates (Scheme 9). After long reaction times, an unreactive metal complex (9) formed, which was strongly bound by the cage. This inhibits binding of other complexes in the cavity and as such this hampered further reactivity. The use of a slightly bigger phosphine ligand (PEt3) could prevent deactivation, as the resulting bisphosphine complex is too sterically demanding for the interior of the cage. As a consequence, the reaction is even 80,000 times faster compared to the reaction in absence of cage, and a TON of over 300 for the cage was achieved. Analysis of the kinetic profiles demonstrated that the reaction is first‐order in cage and follows Michaelis‐Menten‐type kinetics, which is commonly observed for enzymes (Scheme 10). The mechanism involves a pre‐equilibrium comprising halide dissociation and subsequent reversible encapsulation of the cationic complex. The subsequent rate constant k cat was found to be 3.3×10−2 s−1.
Scheme 9.
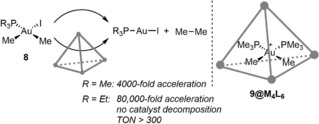
The Ga4L6 cage accelerates the reductive elimination of ethane from a gold(III) complex.
Scheme 10.
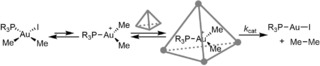
The reductive elimination of ethane from [Au(PR3)(Me)2I] catalyzed by the Ga4L6 cage follows Michaelis‐Menten‐type kinetics.
The reductive elimination reaction inside the Ga4L6 cage was studied in more detail in a follow‐up paper.45 The influence of the spectator ligand PR3 appeared not only to be of influence in the binding process of the decomposition product, but also the transition state of the reductive elimination was stabilized in the cavity to a larger extend when using PEt3 instead of PMe3. Possible explanations include the increased steric compression of the complex, thereby destabilizing the bound ground state, increased hydrophobicity, stabilization of the cationic complex due to more electron donation from the ligand, and better shape complementarity to the cage. The scope of the reductive elimination reaction could be extended to the formation of butane from [Au(PMe3)(Et)2I]; no other attempts to make additional products are described. Most likely, the scope of the reaction is limited by the restricted size of the interior of the cavity and further extension of the scope of this reaction will require larger cages.
For the gold complexes, only the stoichiometric reductive elimination was described, however, for the platinum analogue the cage was used in a catalytic application. A dual catalytic system with a platinum(IV) complex and tetramethyltin, to reoxidize the reduced complex after reductive elimination, was reported. Again, the reductive elimination to form ethane was greatly accelerated by performing this step in the cage. The authors pointed out that this strategy may be extended to other catalyst systems. Importantly, this work shows for the first time that the complex needs to be encapsulated in only one of the steps of the catalytic cycle. In principle, different microenvironments around the active center can be specifically designed for each step of the catalytic cycle with the note that catalyst encapsulation/release are new steps that may become rate limiting.
Multiple Gold Centers in One Cage
In the examples described in the previous sections, the focus was on the encapsulation of single metal complexes in cages. However, the encapsulation of multiple gold centers in one host can also affect the catalytic properties. Indeed, aurophilic interactions have been proven to have beneficial effect on enantioselective catalysis.46 Developing strategies in which multiple gold complexes are pre‐organized in confined space may therefore lead to new interesting reactivity.
An M4L6 Cage with Six Gold Centers
Nitschke presented an M4L6 cage in which the ligands contained an NHC moiety that is coordinated to a gold chloride complex (Figure 9).47 The resulting cage structure contains six gold complexes that each closely resemble the established gold catalyst [Au(IPr)Cl]. However, removal of the six chlorides using silver salts resulted in decomposition of the cage and the formation of gold nanoparticles, making this cage unsuitable for catalysis.
Figure 9.
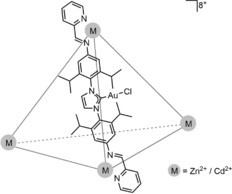
A cationic M4L6 cage containing six gold centers attached to the edges of the tetrahedron.
M12L24 Cages
Reek and co‐workers explored the use of M12L24 cages, based on extensive fundamental work by Fujita and co‐workers (Scheme 11),48 to pre‐organize gold complexes in nano‐spheres through ligand template strategies.49 These type of cuboctahedral cages typically form by self‐assembly of 24 ligands containing two 4‐pyridyl groups that coordinate to palladium or platinum in a variety of polar organic solvents such as DMF, DMSO, and acetonitrile. Functionalization of the ditopic pyridyl ligands can lead to endo‐ or exo‐functionalized nanospheres; the example in the figure shows functionalized ligands that lead to nanospheres with 24 functional groups residing in the cavity.
Scheme 11.
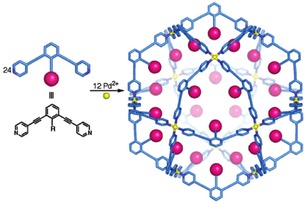
The cuboctahedral M12L24 cage. Reprinted with permission from J. Am. Chem. Soc., 2005, 127, 11950–11951. Copyright 2005 American Chemical Society.
The group of Reek used ditopic ligands with a gold chloride complex attached via a covalent linker (Scheme 12a, L−Au−Cl), leading to a Pd12L24 cage with 24 gold complexes at the inside.50 The local concentration of gold chloride is estimated to be around 1 M. In comparison, the catalyst concentration typically used in catalysis is about 10−6 to 10−3 M, and in this context 1 M is extremely high. Interestingly, M12L24 nanospheres could also be generated using a mixture of building blocks, using gold‐containing ligands combined with unfunctionalized ligands L‐empty (Scheme 12a). By varying the L−Au−Cl : L‐empty ratio between 0 : 24 and 24 : 0, a series of nanospheres with local concentrations of gold chloride ranging from 0.05 to 1.1 M were prepared. With this series the effect of the local concentration of catalyst can be nicely studied.
Scheme 12.
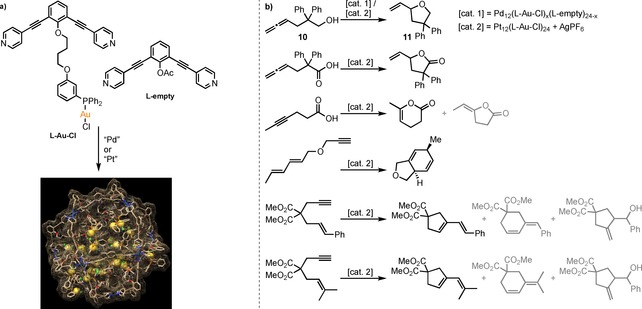
M12L24 cages with multiple gold centers on the inside can be used to do catalytic transformations. a) Gold‐containing and ‘empty’ ligand for the M12L24 spheres. b) Reactions performed inside the gold‐functionalized M12L24 cages, where the gold center(s) catalyze the reaction. Main products are depicted in black; other products that were formed are shown in grey.
The performance of the M12L24 cages was studied in the intramolecular hydroalkoxylation reaction of allenol 10 (Scheme 12b, upper reaction). In the experiments, the overall concentration of gold chloride was kept constant, but the local concentration was gradually increased by changing the L‐Au : L‐empty ratio. Generally, [AuCl] complexes are inactive in catalysis and dehalogenation is required to activate the complex, especially when using phosphines as a ligand.51, 52, 53, 54 Surprisingly, using the M12L24 nanospheres, conversion of the substrate was seen at local concentrations of 0.27 M and higher. Moreover, it was observed that the activity of the catalyst increases with the local gold catalyst concentration. UV/Vis experiments demonstrated the existence of d10‐d10 aurophilic interactions at these high local concentrations. This result implied that at these high local concentrations Au−Au interactions lead to the formation of multinuclear complexes that are active catalysts without the need for activation by dehalogenation. Dehalogenation of the gold complexes with a silver salt led to degradation of the palladium based Pd12L24 nanosphere, a problem that was not encountered for the more stable platinum based Pt12L24 nanospheres.55 The more stable Pt12L24 cages also provided a higher tolerance towards functional groups, so the substrate scope for gold‐catalyzed transformations could be enlarged (Scheme 12b). The catalytic activity increased when higher local gold concentrations were present in the spheres, and higher yields in [4+2] cycloaddition reactions were obtained compared to the free mononuclear gold catalyst [Au(PPh3)X]. Although absolute selectivity towards one product could not be achieved in all cases, the selectivity displayed by the caged catalysts was generally higher than that induced by the monomeric analogue.
To bind catalysts in a non‐covalent manner, M12L24 cages functionalized with guanidinium moieties at the inside were prepared (Scheme 13).56 These binding sites strongly bind sulfonate functionalized guests (Ka>105 M−1), even in polar solvents such as in acetonitrile, because they are hydrogen bonded to two guanidine moieties. Carboxylate groups are also bound to these guanidinium groups but more weakly (∼103 M−1) as they are bound in a monotopic fashion. As a result, 4 gold complexes with sulfonated phosphine ligands can be strongly bound in the M12L24 cage, whereas the remaining guanidinium sites can be used to pre‐organize carboxylate‐containing substrates (Scheme 13). The pre‐organization effect in the resulting nanosphere enhanced the efficiency in the cyclization of 4‐pentynoic acid to the corresponding enol lactone as it led to a 40‐fold increase in activity for the encapsulated gold complex compared to the free analogue. The substrate was deprotonated with a catalytic amount of base leading to binding in the cage. The product of the reaction lacked a carboxylate group, preventing product inhibition. The nanosphere was also used in substrate selective catalysis, converting substrates that have carboxylate functions while leaving the alcohol‐containing substrates untouched, whereas the free gold complex converted both substrates with similar rates.
Scheme 13.
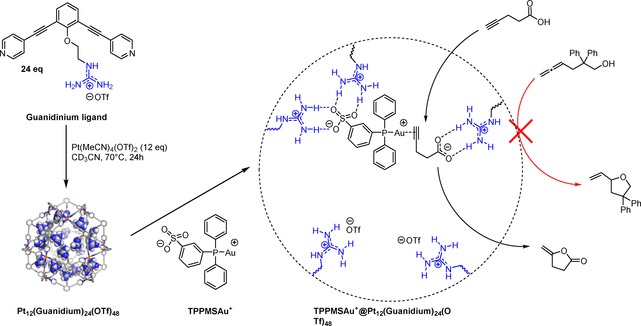
Preparation of the nano‐concentrator based on a guanidinium‐containing building block and a schematic representation of the pre‐organization effect within the nano‐concentrator during the cyclization of 4‐pentynoic acid. Substrate selectivity is based on recognition. Reprinted with permission from Acc. Chem. Res. 2018, 51, 2115–2128. Copyright 2018 American Chemical Society.
Summary and Outlook
Whereas traditional approaches to develop new gold catalysts rely on ligand modification, more recent strategies include the control via the second coordination sphere by catalyst confinement in molecular cages. The encapsulation of gold catalysts inside (supra)molecular capsules has been explored using various concepts. The gold complex can be covalently attached to a cavitand, or can be bound in a supramolecular fashion in self‐assembled cages, such as an M4L6 cage, resorcin[4]arene based cage or M12L24 cage. Mostly cationic complexes are bound in cages that bind cationic guest molecules. In general, the specific steric environment created around the gold complex can lead to a change in product selectivity induced by the gold complex. For systems where the binding relies on supramolecular interactions, reaction enhancements can be observed because of stabilization of the transition state or just by creation of the active species upon catalyst binding. Catalyst encapsulation can also stabilize the gold complex by isolation from the bulk solution, which is a particular advantage when applied in tandem reactions in combination with enzymes. Encapsulation has also been a tool to shift the equilibrium between dinuclear gold complexes and their (encapsulated) monomeric analogues, which is the basis for a switchable catalytic system. Encapsulation of multiple complexes in nanospheres also has been demonstrated to lead to new reactivity, and gold chloride complexes are active at extremely high local concentrations even without the need for activation through dehalogenation. Overall, we can conclude that in general gold catalysis is well compatible with supramolecular chemistry, and catalyst encapsulation offers a variety of new tools to control activity and selectivity in gold‐catalyzed transformations. Although currently these examples are clearly at the proof of principle stage, we foresee that these strategies will be useful in applied catalysis in the nearby future further expanding the potential of gold catalysis. For example, the extension of the use of cages in enantioselective gold catalysis that, as a result of steric confinement, allow to control enantioselectivity issues that cannot be addressed with currently available catalyst. Furthermore, recently catalytic cycles involving Au(I)/Au(III) catalysis have been reported and it is anticipated that these reaction may also benefit from encapsulation in cages, as the stabilization of these complexes will be different. Finally, we see also a lot of potential for other catalytic reactions that are catalyzed by silver or copper analogues, and clearly on a small fraction of the available cage structures has been explored so far.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Anne Jans studied Chemistry at the VU University Amsterdam and received her MSc degree in 2014. Her Master thesis was on supramolecular bidentate ligands for rhodium catalyzed asymmetric hydrogenation reactions under supervision of Joost Reek. In 2014, she joined this group for her PhD research. Her current research interests are supramolecular cages and their effect on gold‐catalyzed reactions.

Biographical Information
Xavier Caumes, born in 1988, received his education in chemistry from university Paris Descartes and his doctoral degree from university Pierre and Marie Curie in 2016 under the supervision of L. Bouteiller and M. Raynal. He joined the group of Joost Reek in 2017 to pursue research on chirality amplification in supramolecular cages and their application in transition metal catalysis.

Biographical Information
Joost Reek obtained his PhD at the University of Nijmegen under supervision of Prof. R.J.M. Nolte in the area of supramolecular chemistry. After a post‐doc with Prof. Crossley in Sydney, he moved to the University of Amsterdam in 1998, where he was promoted to full professor in 2006, and faculty professor in 2017. His research interests include homogeneous catalysis and supramolecular chemistry, and he is exploring new research on the border of these research topics. In addition, he has an interest in develping solar to fuel devices based on molecular components, with a focus on the catalytic processes involved.

Acknowledgements
We thank the European Research Council (ERC‐AG 339786‐NAT_CAT) for financial support.
A. C. H. Jans, X. Caumes, J. N. H. Reek, ChemCatChem 2019, 11, 287.
References
- 1. Gorin D. J., Toste F. D., Nature 2007, 446, 395–403. [DOI] [PubMed] [Google Scholar]
- 2. Pyykkö P., Angew. Chem. Int. Ed. 2004, 43, 4412–4456; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 4512–4557. [Google Scholar]
- 3. Li Z., Brouwer C., He C., Chem. Rev. 2008, 108, 3239–3265. [DOI] [PubMed] [Google Scholar]
- 4. Hashmi A. S. K., Rudolph M., Chem. Soc. Rev. 2008, 37, 1766–1775. [DOI] [PubMed] [Google Scholar]
- 5. Sugimoto K., Matsuya Y., Tetrahedron Lett. 2017, 58, 4420–4426. [Google Scholar]
- 6. Zhang Y., Luo T., Yang Z., Nat. Prod. Rep. 2014, 31, 489–503. [DOI] [PubMed] [Google Scholar]
- 7. Gorin D. J., Sherry B. D., Toste F. D., Chem. Rev. 2008, 108, 3351–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Raynal M., Ballester P., Vidal-Ferran A., Leeuwen P. W. N. M. van, Chem. Soc. Rev. 2014, 43, 1660–1733. [DOI] [PubMed] [Google Scholar]
- 9. Raynal M., Ballester P., Vidal-Ferran A., Leeuwen P. W. N. M. van, Chem. Soc. Rev. 2014, 43, 1734–1787. [DOI] [PubMed] [Google Scholar]
- 10. Zarra S., Wood D. M., Roberts D. A., Nitschke J. R., Chem. Soc. Rev. 2015, 44, 419–432. [DOI] [PubMed] [Google Scholar]
- 11. Leenders S. H. A. M., Gramage-Doria R., Bruin B. de, Reek J. N. H., Chem. Soc. Rev. 2015, 44, 433–448. [DOI] [PubMed] [Google Scholar]
- 12. Otte M., ACS Catal. 2016, 6, 6491–6510. [Google Scholar]
- 13. Catti L., Zhang Q., Tiefenbacher K., Chem. Eur. J. 2016, 22, 9060–9066. [DOI] [PubMed] [Google Scholar]
- 14. Iwai T., Sawamura M., Bull. Chem. Soc. Jpn. 2014, 87, 1147–1160. [Google Scholar]
- 15. Ochida A., Ito H., Sawamura M., J. Am. Chem. Soc. 2006, 128, 16486–16487. [DOI] [PubMed] [Google Scholar]
- 16. Ito H., Ohmiya H., Sawamura M., Org. Lett. 2010, 12, 4380–4383. [DOI] [PubMed] [Google Scholar]
- 17. Ito H., Harada T., Ohmiya H., Sawamura M., Beilstein J. Org. Chem. 2011, 7, 951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ito H., Harada A., Ohmiya H., Sawamura M., Adv. Synth. Catal. 2013, 355, 647–652. [Google Scholar]
- 19. Iwai T., Okochi H., Ito H., Sawamura M., Angew. Chem. Int. Ed. 2013, 52, 4239–4242; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4333–4336. [Google Scholar]
- 20. Iwai T., Ueno M., Okochi H., Sawamura M., Adv. Synth. Catal. 2018, 360, 670–675. [Google Scholar]
- 21. Schramm M. P., Kanaura M., Ito K., Ide M., Iwasawa T., Eur. J. Org. Chem. 2016, 2016, 813–820. [Google Scholar]
- 22. Iwasawa T., Tetrahedron Lett. 2017, 58, 4217–4226. [Google Scholar]
- 23. Gibson C., Rebek J., Org. Lett. 2002, 4, 1887–1890. [DOI] [PubMed] [Google Scholar]
- 24. Chavagnan T., Bauder C., Sémeril D., Matt D., Toupet L., Eur. J. Org. Chem. 2017, 70–76. [Google Scholar]
- 25. Chavagnan T., Sémeril D., Matt D., Toupet L., Eur. J. Org. Chem. 2017, 2017, 313–323. [Google Scholar]
- 26. Endo N., Kanaura M., Schramm M. P., Iwasawa T., Eur. J. Org. Chem. 2016, 2016, 2514–2521. [Google Scholar]
- 27. Endo N., Kanaura M., Schramm M. P., Iwasawa T., Tetrahedron Lett. 2016, 57, 4754–4757. [Google Scholar]
- 28. Kanaura M., Endo N., Schramm M. P., Iwasawa T., Eur. J. Org. Chem. 2016, 2016, 4970–4975. [Google Scholar]
- 29. Guitet M., Zhang P., Marcelo F., Tugny C., Jiménez-Barbero J., Buriez O., Amatore C., Mouriès-Mansuy V., Goddard J.-P., Fensterbank L., Angew. Chem. Int. Ed. 2013, 52, 7213–7218; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7354–7359. [Google Scholar]
- 30. Zhang P., Tugny C., Suárez J. Meijide, Guitet M., Derat E., Vanthuyne N., Zhang Y., Bistri O., Mouriès-Mansuy V., Ménand M., Chem 2017, 3, 174–191. [Google Scholar]
- 31. Zhang P., Suárez J. Meijide, Driant T., Derat E., Zhang Y., Ménand M., Roland S., Sollogoub M., Angew. Chem. Int. Ed. 2017, 56, 10821–10825; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10961–10965. [Google Scholar]
- 32. Schneider H.-J., Güttes D., Schneider U., Angew. Chem. Int. Ed. Engl. 1986, 25, 647–649. [Google Scholar]
- 33. Schneider H.-J., Güttes D., Schneider U., J. Am. Chem. Soc. 1988, 110, 6449–6454. [Google Scholar]
- 34. MacGillivray L. R., Atwood J. L., Nature 1997, 389, 469–472. [Google Scholar]
- 35. Cavarzan A., Scarso A., Sgarbossa P., Strukul G., Reek J. N. H., J. Am. Chem. Soc. 2011, 133, 2848–2851. [DOI] [PubMed] [Google Scholar]
- 36. Cavarzan A., Reek J. N. H., Trentin F., Scarso A., Strukul G., Catal. Sci. Technol. 2013, 3, 2898–2901. [Google Scholar]
- 37. Adriaenssens L., Escribano-Cuesta A., Homs A., Echavarren A. M., Ballester P., Eur. J. Org. Chem. 2013, 2013, 1494–1500. [Google Scholar]
- 38. Jans A. C. H., Gómez-Suárez A., Nolan S. P., Reek J. N. H., Chem. Eur. J. 2016, 22, 14836–14839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oonishi Y., Gómez-Suárez A., Martin A. R., Nolan S. P., Angew. Chem. Int. Ed. 2013, 52, 9767–9771; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9949–9953. [Google Scholar]
- 40. Caulder D. L., Powers R. E., Parac T. N., Raymond K. N., Angew. Chem. Int. Ed. 1998, 37, 1840–1843; [Google Scholar]; Angew. Chem. 1998, 110, 1940–1943. [Google Scholar]
- 41. Wang Z. J., Brown C. J., Bergman R. G., Raymond K. N., Toste F. D., J. Am. Chem. Soc. 2011, 133, 7358–7360. [DOI] [PubMed] [Google Scholar]
- 42. Hart-Cooper W. M., Clary K. N., Toste F. D., Bergman R. G., Raymond K. N., J. Am. Chem. Soc. 2012, 134, 17873–17876. [DOI] [PubMed] [Google Scholar]
- 43. Wang Z. J., Clary K. N., Bergman R. G., Raymond K. N., Toste F. D., Nat. Chem. 2013, 5, 100–103. [DOI] [PubMed] [Google Scholar]
- 44. Kaphan D. M., Levin M. D., Bergman R. G., Raymond K. N., Toste F. D., Science 2015, 350, 1235–1238. [DOI] [PubMed] [Google Scholar]
- 45. Levin M. D., Kaphan D. M., Hong C. M., Bergman R. G., Raymond K. N., Toste F. D., J. Am. Chem. Soc. 2016, 138, 9682–9693. [DOI] [PubMed] [Google Scholar]
- 46. Zi W., Toste F. Dean, Chem. Soc. Rev. 2016, 45, 4567–4589. [DOI] [PubMed] [Google Scholar]
- 47. Ramsay W. J., Foster J. A., Moore K. L., Ronson T. K., Mirgalet R. J., Jefferson D. A., Nitschke J. R., Chem. Sci. 2015, 6, 7326–7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tominaga M., Suzuki K., Murase T., Fujita M., J. Am. Chem. Soc. 2005, 127, 11950–11951. [DOI] [PubMed] [Google Scholar]
- 49. Jongkind L. J., Caumes X., Hartendorp A. P. T., Reek J. N. H., Acc. Chem. Res. 2018, 51, 2115–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gramage-Doria R., Hessels J., Leenders S. H. A. M., Tröppner O., Dürr M., Ivanović-Burmazović I., Reek J. N. H., Angew. Chem. Int. Ed. 2014, 53, 13380–13384; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13598–13602. [Google Scholar]
- 51. Oliver-Meseguer J., Cabrero-Antonino J. R., Dominguez I., Leyva-Perez A., Corma A., Science 2012, 338, 1452–1455. [DOI] [PubMed] [Google Scholar]
- 52. Kronig S., Theuergarten E., Daniliuc C. G., Jones P. G., Tamm M., Angew. Chem. Int. Ed. 2012, 51, 3240–3244; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3294–3298. [Google Scholar]
- 53. Belger K., Krause N., Eur. J. Org. Chem. 2015, 2015, 220–225. [Google Scholar]
- 54. Li F., Wang N., Lu L., Zhu G., J. Org. Chem. 2015, 80, 3538–3546. [DOI] [PubMed] [Google Scholar]
- 55. Leenders S. H. A. M., Dürr M., Ivanović-Burmazović I., Reek J. N. H., Adv. Synth. Catal. 2016, 358, 1509–1518. [Google Scholar]
- 56. Wang Q.-Q., Gonell S., Leenders S. H. A. M., Dürr M., Ivanović-Burmazović I., Reek J. N. H., Nat. Chem. 2016, 8, 225–230. [DOI] [PubMed] [Google Scholar]