

Abstract
There is an urgent need to identify new, non‐traditional antimicrobials. The discovery of new polymeric antimicrobials is limited by current low‐throughput synthetic tools, which means that limited chemical space has been explored. Herein, we employ photochemical “in‐air” reversible addition–fragmentation chain‐transfer (RAFT) polymerization with microwell plates, using liquid‐handling robots to assemble large libraries of cationic polymers, without the need for degassing or purification steps, facilitating transfer to screening. Several lead polymers were identified including a co‐polymer with propylene glycol side chains with significantly enhanced antimicrobial activity and increased therapeutic window. Mechanistic studies showed that this polymer was bacteriostatic, and surprisingly did not lyse the cell membranes, implying an alternative mode of action. This versatile method using simple robotics will help to develop new biomaterials with emergent properties.
Keywords: antimicrobials, bacteria, biomaterials, combinatorial chemistry, polymers
Combinatorial methods are widely employed in small‐molecule chemistry to identify previously unknown leads against well‐characterized targets, and includes concepts, such as fragment‐based design.1, 2 Commercial compound libraries are available with >5000 members, and repurposing of known drugs is underpinned by screening.3 In the discovery of polymer biomaterials, there are the additional variables of monomer, molecular weight, and architecture. This provides vast chemical space to be explored, presenting a challenge and opportunity.4 Polymers for gene delivery have been successfully identified using combinatorial condensation polymerization,5, 6 but there was molecular‐weight heterogeneity. Alexander et al. have developed automated high‐throughput screens for polymer surfaces enabling discovery of polymer surfaces for resisting bacterial attachment7 or the culture of stem cells.8 However, for soluble polymers intended to interface with cells/proteins, well‐defined materials are required with control of MW to enable selection and tuning of the final properties.9, 10 Controlled radical (CRP) or ionic polymerization requires inert atmospheres and sealed vials, and in the case of ionic polymerizations—rigorously anhydrous conditions, adding complexity and time due to processing. Schubert and co‐workers have used automated synthesizers for polymerizations, but such protocols require a precipitation/isolation step limiting the potential of the libraries.4, 11 To truly use combinatorial polymer methods to discover “drug‐like” materials, the synthetic and handling methods should be compatible with the industry standard, 96‐, 384‐, and 1536‐well plates used in biomedical screening with liquid‐handling robotics.12
To address the combinatorial challenge, air‐tolerant CRP methods are emerging. Chapman et al. used glucose oxidase for in situ degassing in 96‐well plate format reversible addition–fragmentation chain‐transfer (RAFT) polymerizations,13 and this approach has also been applied to ATRP formulations.14 Light‐mediated polymerizations15 enable the trapping/removal of oxygen species by using organic16 and inorganic17 photoredox catalysts. Trithiocarbonates can also be used as intrinsic photoredox catalysts in RAFT, without the need for supplemental catalysts which is appealing for biomedical screening.18 Recently, Boyer and co‐workers used photo‐RAFT in 96‐well plates to screen star polymers for binding to a model lectin, facilitating the design of new binders.19 However, there are limited examples of application to urgent biomedical materials screening challenges, such as new antimicrobials to combat resistance.20 Cationic polymers have been employed as antimicrobial agents, inspired by antimicrobial peptides21 with broad spectrum activity and slow emerging resistance.22 The most active antimicrobial polymers are not homopolymers, but require a complex balance of charge and hydrophobicity/‐philicity by incorporation of co‐monomers.23, 24, 25, 26 Their rational design is typically based on targeting membrane lysis, but it is becoming apparent that bacteria aggregation and hence interruption of signaling27, 28 pore‐formation,29 DNA‐binding,30 and interrupting metabolic processes31 are associated with polycations. Structure–function maps to phenotype (bacteria killing), but also to understand mechanism, are needed to generate data sets to enable ab initio materials design.7
Herein, we present combinatorial cationic photopolymer screening for new antimicrobial biomaterials. The intrinsic photo‐RAFT method18 is adapted to enable automation, scalability and ease of use in “open” reaction vessels of a 96 well plate, using liquid handling robots, Figure 1 A. A photo‐RAFT agent 2‐cyano‐2‐propyl dodecylthiocarbonate is used with a tertiary amine (triethanolamine (TEOA)), to degas the solvent (DMSO) enabling polymerization to proceed under a blue LED light. 2‐(Dimethylamino)ethyl methacrylate (DMAEMA) was chosen as the cationic component based on our previous work showing it has potent anti‐mycobacterial activity. Herein, there did not appear to be a molecular‐weight effect of the DPs tested (between 10 and 100) therefore DP 75 was chosen.32, 33 Figure 1 B and Table 1 show results of three parallel DMAEMA polymerizations in 96‐well plates targeting degrees of polymerization of 25, 50, and 100. Each achieved >95 % conversation and comparable molecular‐weight distributions confirming reproducible synthesis in the small reaction volumes (<200 μL). The procedure was validated further by running 60 parallel in‐air polymerizations of DMAEMA within a single plate. Five wells were then chosen by an independent party for SEC analysis, Figure 1 C. Comparable molecular weights and distributions were obtained, confirming control over the reaction and homogeneity across all the mini‐reaction vessels (wells).
Figure 1.
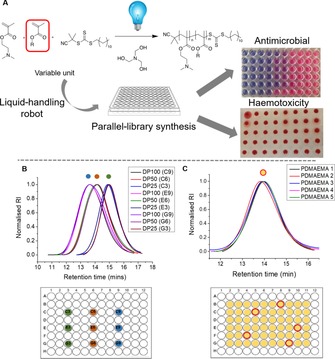
A) Concept of in‐air combinatorial photo‐RAFT discovery. B) SEC of 3×3 DP polymerizations of DMAEMA. C) SEC of five randomly selected (red circles) polymers produced from 60×DMAEMA polymerizations within a single plate.
Table 1.
Characterization of three repeats of three DPs of PDMAEMA.
| Well code |
[M]: [CTA] |
Conv. [%][a] |
M
n(theor)
[g mol−1][b] |
M
n(SEC)
[g mol−1][c] |
M
w/ M n [c] |
|---|---|---|---|---|---|
| C3 | 100 | 95 | 15 300 | 22 900 | 1.66 |
| C6 | 50 | 96 | 7900 | 17 200 | 1.57 |
| C9 | 25 | 98 | 4200 | 9500 | 1.33 |
| E3 | 100 | 96 | 15 400 | 22 100 | 1.63 |
| E6 | 50 | 95 | 7800 | 16 800 | 1.60 |
| E9 | 25 | 95 | 4100 | 9900 | 1.37 |
| G3 | 100 | 96 | 15 400 | 23 200 | 1.61 |
| G6 | 50 | 97 | 8000 | 17 300 | 1.49 |
| G9 | 25 | 98 | 4200 | 9400 | 1.33 |
[a] Determined by 1H NMR analysis against an internal mesitylene standard. [b] Determined by the [M]:[CTA] ratio and conversion, assuming 100 % CTA efficiency. [c] Determined by SEC in DMF; reported values are relative to PMMA standards.
Traditional polymerization methods are limited in their chemical and compositional space meaning the “sweet spots” in co‐polymer libraries can be overlooked. Here, eight co‐monomers were chosen to be co‐polymerized with DMAEMA, including a mixture of hydrophobic and hydrophilic substituents, at four densities (5, 10, 15, 20 mol %) with three repeats, within 96‐well plates to give a combinatorial library of 108 distinct polymers in DMSO, Figure 2 (left column) prepared in a single day. Drug screening was routinely conducted in 1–5 % DMSO to aid solubilization;34 herein, sampling followed by dilution in appropriate buffer/media resulted in [DMSO] <5 wt %, which controls showed did not affect assays.
Figure 2.
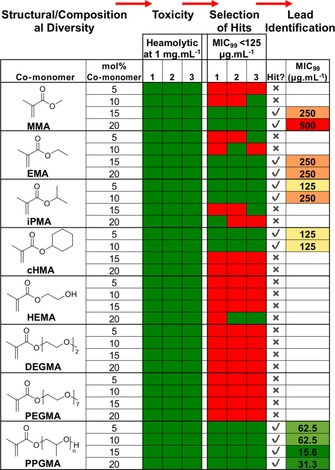
Library structure, haemolysis at 1 mg mL−1 and antimicrobial activity against E. coli at 125 μg mL−1 (0.5 × MIC99 of homopolymer (PDMAEMA)).
A series of functional screens were undertaken and results indicated as a heat map (Figure 2; green indicates desirable outcome, red indicates sample is excluded). To eliminate toxic materials, ovine red blood cell haemolysis was conducted at 1 mg mL−1 (Figure 1 A). All 108 polymers had haemolysis below 2 % and no haemagglutination, hence all passed. To screen for antimicrobial activity, the resazurin reduction assay was used, which gives a colorimetric output (blue to pink, Figure 1 A). Escherichia coli and Mycobacteria smegmatis were used to represent Gram negative and Mycobacteria (which includes M. tuberculosis). The MIC99 (minimum concentration to stop growth of 99 % of organisms) of homo‐PDMAEMA is 250 and 31.3 μg mL−1 against E. coli and M. smegmatis, respectively.32, 33 Co‐polymers were added to the bacteria at 0.5×MIC99 of PDMAEMA to enable selection of co‐polymers that were at least two‐fold more active. Against M. smegmatis, there were few “hits”, potentially due to the complex mycobacterial cell walls, which are rich in mycolic acids and glycans which can “shield” the membrane.32 However, the E. coli screen identified several “hits” with co‐polymers of MMA, iPMA, cHMA, and PPGMA inhibiting E. coli growth at 0.5xMIC99 of the parent homopolymer.
These hits were tested across a wider concentration range to establish their MIC99 (Figure 2, right column). Hydrophobic co‐monomers tended to lower the MIC99. MMA co‐polymers had a sweet spot for activity at 15 wt % with more/less reducing all antimicrobial activity. Similarly, iPMA/cHMA co‐polymers were active at 5 and 10 wt % but not at higher incorporation levels. Several of the hits appeared to not give lower MIC99 values than the homopolymer once tested in full dilution series, justifying the hit‐to‐lead approach. These observations highlight a key benefit of screening to identify non‐linear trends that can be missed in low‐throughput testing. The most active co‐polymer contained 15 wt % poly(propylene glycol)methacrylate (PPGMA) with an MIC99 of 15 μg mL−1, compared to 250 μg mL−1 for homo‐PDMAEMA. Interestingly, this is not the most hydrophobic comonomer (see logP values in the Supporting Information) suggesting that a membrane insertion/disruption mechanism might not be operating. This would not have been predicted based on logP values alone.
To validate these findings, P(DMAEMA(85 %)‐co‐PPGMA(15 %)) hits were resynthesized to various degrees of polymerization (DP30‐240) to give a panel of “pure”, well‐defined polymers (SEC traces, Figure 3 A). Similar MIC99 values were obtained as in the initial screen, but the shortest polymers (DP30) were identified to be least active, Figure 3 B. Membrane‐integrity assays were undertaken to probe the for the greater co‐polymer activity compared to PDMAEMA homopolymer; it is assumed that more hydrophobic units promotes insertion into bacterial cell membranes, leading to lysis and cell death.24 The assay employs a pair of dyes, SYTO 9 (green fluorescence) that enters all cells and is associated with intact bacteria and propidium iodide (red fluorescence) that can only enter membrane‐compromised cells to probe if membrane lysis has occurred. Figure 3 C–H shows confocal microscopy images of E. coli incubated under various conditions. PDMAEMA at 2×MIC99 shows only red bacteria, consistent with the “dead” control (Figure 3 D) and at 0.5×MIC99 a mixture of red/green are seen supportive of PDMAEMA homopolymers killing E. coli by a lytic mechanism. However, P(DMAEMA(85 %)‐co‐PPGMA(15 %)) at a concentration above (2×) MIC99 gave a mixture of red and green bacteria, showing that there is less membrane lysis than the PDMAEMA homopolymers even though these are more active (lower MIC99). This shows that the co‐monomer is not simply increasing activity by more membrane lysis. Confocal microscopy suggested increased bacterial aggregation in response to the co‐polymer, but not the homopolymer. Aggregation is known to modulate bacterial responses in their environment, and the co‐polymers might be influencing their colonizing behaviour to limit growth by a feedback mechanism.27, 35
Figure 3.
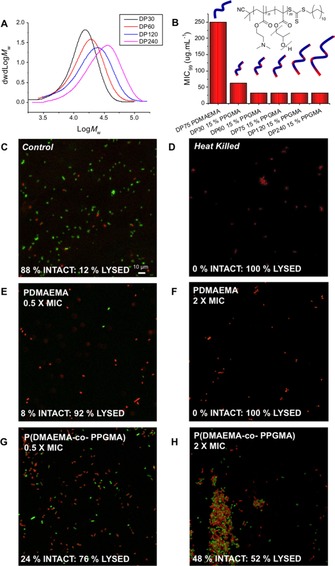
A) SEC of P(DMAEMA(85 %)‐co‐PPGMA(15 %) co‐polymers. B) MIC99 of PDMAEMA compared to P(DMAEMA(85 %)‐co‐PPGMA(15 %) co‐polymers. C–H) Fluorescence microscopy of E. coli upon exposure to varying concentrations of PDMAEMA and P(DMAEMA(85 %)‐co‐PPGMA(15 %)). Green channel shows intact membranes, red is damaged membranes.
To determine if the bacteria were being killed by the co‐polymers, or if their growth was being inhibited, the minimum bactericidal concentration (MBC) was determined. For PDMAEMA homopolymers, the MBC is the same as the MIC99 suggesting membrane lysis is the mode of action as would be expected for traditional cationic polymers. For the co‐polymer, the MBC actually increased to >1000 μg mL−1, showing it was less effective at killing and lysing bacteria membranes than the homopolymer. This suggested that we have identified a mechanism, in which a unique co‐polymer that inhibits E. coli growth potentially due to aggregation was detected, and not physical damage of the cell membrane. Bactericidal and bacteriostatic mechanisms are both valid in terms of antimicrobial therapy with front lines drugs having one or both of these properties.36 The polymers were also evaluated for cytotoxicity against a mammalian cell line (A549; see the Supporting Information). Incorporation of PPGMA co‐monomers slightly decreased cell viability relative to the PDMAEMA after 24 hours. However, due to the increased antimicrobial activity, the PPGMA co‐polymers have a larger window of activity.
In summary, we have developed a rapid, scalable, and simple approach to identify emergent antimicrobial properties of co‐polymer libraries through the use of in‐air polymerization coupled to liquid‐handling robots in 96‐well plates. A screening and selection process enabled identification of hits within a 108‐member co‐polymer library resulting in co‐polymers of oligo(propylene glycol) being identified with 16‐fold increased activity compared to PDMAEMA homopolymers. Crucially, PPGMA was not the most hydrophobic co‐monomer tested, and non‐linear relationships were observed between co‐monomer composition and activity. This material was shown to have a distinct mechanism of action, inhibiting bacterial growth rather than lysing the cell membranes. Such a material would not have been identified by using conventional 1‐vial/1‐polymer methods; furthermore, this process accelerates the discovery of new complex materials with emergent biological interactions.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful for the polymer characterization RTP for size‐exclusion chromatography. M.I.G. holds an ERC Starter Grant (CRYOMAT 638661). EPSRC are thanked for studentship for R.T. (EP/L015307/1) and also INTEGRATE (EP/M027503/1). The microscopy facilities are funded by UoW Advanced BioImaging Research Technology Platform BBSRC ALERT14 award BB/M01228X/1. I. Galpin is acknowledged for assistance with confocal microscopy.
S.-J. Richards, A. Jones, R. M. F. Tomás, M. I. Gibson, Chem. Eur. J. 2018, 24, 13758.
References
- 1. Murray C. W., Rees D. C., Nat. Chem. 2009, 1, 187–192. [DOI] [PubMed] [Google Scholar]
- 2. Kennedy J. P., Williams L., Bridges T. M., Daniels R. N., Weaver D., Lindsley C. W., J. Comb. Chem. 2008, 10, 345–354. [DOI] [PubMed] [Google Scholar]
- 3. Ashburn T. T., Thor K. B., Nat. Rev. Drug Discovery 2004, 3, 673–683. [DOI] [PubMed] [Google Scholar]
- 4. Meier M. A. R., Hoogenboom R., Schubert U. S., Macromol. Rapid Commun. 2004, 25, 21–33. [Google Scholar]
- 5. Brocchini S., James K., Tangpasuthadol V., Kohn J., J. Am. Chem. Soc. 1997, 119, 4553–4554. [Google Scholar]
- 6. Lynn D. M., Anderson D. G., Putnam D., Langer R., J. Am. Chem. Soc. 2001, 123, 8155–8156. [DOI] [PubMed] [Google Scholar]
- 7. Hook A. L., Chang C. Y., Yang J., Luckett J., Cockayne A., Atkinson S., Mei Y., Bayston R., Irvine D. J., Langer R., Anderson D. G., Williams P., Davies M. C., Alexander M. R., Nat. Biotechnol. 2012, 30, 868–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mei Y., Saha K., Bogatyrev S. R., Yang J., Hook A. L., Kalcioglu Z. I., Cho S. W., Mitalipova M., Pyzocha N., Rojas F., Van Vliet K. J., Davies M. C., Alexander M. R., Langer R., Jaenisch R., Anderson D. G., Nat. Mater. 2010, 9, 768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boyer C., Bulmus V., Davis T. P., Ladmiral V., Liu J., Perrier S., Chem. Rev. 2009, 109, 5402–5436. [DOI] [PubMed] [Google Scholar]
- 10. Cobo I., Li M., Sumerlin B. S., Perrier S., Nat. Mater. 2015, 14, 143–159. [DOI] [PubMed] [Google Scholar]
- 11. Becer C. R., Hahn S., Fijten M. W. M., Thijs H. M. L., Hoogenboom R., Schubert U. S., J. Polym. Sci. Part A 2008, 46, 7138–7147. [Google Scholar]
- 12. MacArron R., Banks M. N., Bojanic D., Burns D. J., Cirovic D. A., Garyantes T., Green D. V. S., Hertzberg R. P., Janzen W. P., Paslay J. W., Schopfer U., Sittampalam G. S., Nat. Rev. Drug Discovery 2011, 10, 188–195. [DOI] [PubMed] [Google Scholar]
- 13. Chapman R., Gormley A. J., Stenzel M. H., Stevens M. M., Angew. Chem. Int. Ed. 2016, 55, 4500–4503; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4576–4579. [Google Scholar]
- 14. Enciso A. E., Fu L., Russell A. J., Matyjaszewski K., Angew. Chem. Int. Ed. 2018, 57, 933–936; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 945–948. [Google Scholar]
- 15. Chen M., Zhong M., Johnson J. A., Chem. Rev. 2016, 116, 10167–10211. [DOI] [PubMed] [Google Scholar]
- 16. Xu J., Shanmugam S., Duong H. T., Boyer C., Polym. Chem. 2015, 6, 5615–5624. [Google Scholar]
- 17. Corrigan N., Rosli D., Jones J. W. J., Xu J., Boyer C., Macromolecules 2016, 49, 6779–6789. [Google Scholar]
- 18. Fu Q., Xie K., McKenzie T. G., Qiao G. G., Polym. Chem. 2017, 8, 1519–1526. [Google Scholar]
- 19. Gormley A. J., Yeow J., Ng G., Conway Ó., Boyer C., Chapman R., Angew. Chem. Int. Ed. 2018, 57, 1557–1562; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1573–1578. [Google Scholar]
- 20.J. O'Neill, Review on Antimicrobial Resistance: Tackling drug-resistant infections globally https://amr-review.org; accessed May 17, 2018.
- 21. Seo M.-D., Won H.-S., Kim J.-H., Mishig-Ochir T., Lee B.-J., Molecules 2012, 17, 12276–12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lam S. J., O'Brien-Simpson N. M., Pantarat N., Sulistio A., Wong E. H. H., Chen Y.-Y., Lenzo J. C., Holden J. A., Blencowe A., Reynolds E. C., Qiao G. G., Nat. Microbiol. 2016, 1, 16162. [DOI] [PubMed] [Google Scholar]
- 23. Ilker M. F., Nüsslein K., Tew G. N., Coughlin E. B., J. Am. Chem. Soc. 2004, 126, 15870–15875. [DOI] [PubMed] [Google Scholar]
- 24. Kuroda K., DeGrado W. F., J. Am. Chem. Soc. 2005, 127, 4128–4129. [DOI] [PubMed] [Google Scholar]
- 25. Kuroda K., Caputo G. A., DeGrado W. F., Chem. Eur. J. 2009, 15, 1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tew G. N., Liu D., Chen B., Doerksen R. J., Kaplan J., Carroll P. J., Klein M. L., DeGrado W. F., Proc. Natl. Acad. Sci. USA 2002, 99, 5110–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leire E., Amaral S. P., Louzao I., Winzer K., Alexander C., Fernandez-Megia E., Fernandez-Trillo F., Biomater. Sci. 2016, 4, 998–1006. [DOI] [PubMed] [Google Scholar]
- 28. Lui L. T., Xue X., Sui C., Brown A., Pritchard D. I., Halliday N., Winzer K., Howdle S. M., Fernandez-Trillo F., Krasnogor N., Alexander C., Nat. Chem. 2013, 5, 1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang L., Gordon V. D., Trinkle D. R., Schmidt N. W., Davis M. A., DeVries C., Som A., Cronan J. E., Tew G. N., Wong G. C. L., Proc. Natl. Acad. Sci. USA 2008, 105, 20595–20600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chindera A. K., Mahato M., Sharma A. K., Horsley H., Kloc-muniak K., Kamaruzzaman N. F., Kumar S., Mcfarlane A., Stach J., Bentin T., Good L., Sci. Rep. 2016, 6, 23121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brogden K. A., Nat. Rev. Microbiol. 2005, 3, 238–250. [DOI] [PubMed] [Google Scholar]
- 32. Phillips D. J., Harrison J., Richards S.-J., Mitchell D. E., Tichauer E., Hubbard A. T. M., Guy C., Hands-Portman I., Fullam E., Gibson M. I., Biomacromolecules 2017, 18, 1592–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Richards S.-J., Isufi K., Wilkins L. E., Lipecki J., Fullam E., Gibson M. I., Biomacromolecules 2018, 19, 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maes J., Verlooy L., Buenafe O. E., de Witte P. A. M., Esguerra C. V., Crawford A. D., PLoS One 2012, 7, e43850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Louzao I., Sui C., Winzer K., Fernandez-Trillo F., Alexander C., Eur. J. Pharm. Biopharm. 2015, 95, 47–62, (Pt A). [DOI] [PubMed] [Google Scholar]
- 36. Nemeth J., Oesch G., Kuster S. P., J. Antimicrob. Chemother. 2015, 70, 382–395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary