

Abstract
The core N−H units of planar porphyrins are often inaccessible to forming hydrogen‐bonding complexes with acceptor molecules. This is due to the fact that the amine moieties are “shielded” by the macrocyclic system, impeding the formation of intermolecular H‐bonds. However, methods exist to modulate the tetrapyrrole conformations and to reshape the vector of N−H orientation outwards, thus increasing their availability and reactivity. Strategies include the use of porpho(di)methenes and phlorins (calixphyrins), as well as saddle‐distorted porphyrins. The former form cavities due to interruption of the aromatic system. The latter are highly basic systems and capable of binding anions and neutral molecules via N−H⋅⋅⋅X‐type H‐bonds. This Review discusses the role of porphyrin(oid) ligands in various coordination‐type complexes, means to access the core for hydrogen bonding, the concept of conformational control, and emerging applications, such as organocatalysis and sensors.
Keywords: calixpyrroles, hydrogen bonds, molecular engineering, porphyrinoids, porphyrins
1. Introduction
1.1. Porphyrins
Porphyrins 1 are often discussed as macrocyclic compounds par excellence. They are heteroaromatic systems with a rich metal coordination chemistry (2) and high functional versatility (3, Figure 1, top). As a result of their conformational flexibility, manipulation of the macrocycle conformation allows a fine‐tuning of their physicochemical characteristics, including binding properties and chemical reactivity in general.1 This manifests itself, for example, in tetrapyrrole‐containing proteins, one of the most fundamental classes of enzymes in nature, where a multitude of chemically distinct reactions involves the same porphyrin cofactor, which is largely attributed to protein‐induced macrocyclic distortion.2, 3 Enhancement of the capability of porphyrins to undergo non‐covalent interactions (i.e. hydrogen bonding) in enzymes or artificial systems is to a significant extent an effect of macrocycle nonplanarity. As such, nonplanar free‐base tetrapyrroles can form hydrogen‐bonded complexes N−H⋅⋅⋅X (5) under participation of the pyrrolic N−H groups in the core and suitable substrates X while often, the planar counterparts (4) remain inert (Figure 1, bottom).3
Figure 1.
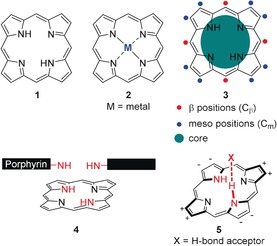
Top: (Metallo)porphyrin macrocycles 1 and 2 and functionalities of a free‐base porphyrin 3. Bottom: N−H‐orientation in a planar porphyrin 4 and the N−H⋅⋅⋅X‐type H‐binding motif in a nonplanar porphyrin 5 (+ and − indicate displacements above and below the 24‐atom least‐squares plane (Δ24), respectively).
Research on (nonplanar) porphyrins has exploded in recent decades and these days, they are used to test and illustrate advances in almost any area of chemistry, such as analytics,4 physical chemistry,5 biomedicine,6 as well as optics and materials science.7 Their organic chemistry has given rise to a bewildering multitude of porphyrinoid macrocycles, which are isomeric, expanded, or contracted in relation to the parent 18 π‐electron system.8 Total synthesis of symmetrical porphyrins has finally made it to a scale suitable for practical applications and unsymmetrical substituted porphyrins are now available en gros in many cases via short syntheses or through functionalization reactions.9 The size of oligoporphyrins has reached the realm of polymer chemistry10 with the field coming to a point where more publications use scanning tunneling microscopy (STM) rather than classical CHN analysis for characterization.11 As a result of the many papers in these areas, this cannot be a classic review; rather we will use illustrative examples to highlight the points we aim to make.
One could ask “what is left to do in porphyrin chemistry?” Well, one area, which has escaped significant attention relates to the coordination chemistry of porphyrins. Interest in porphyrins has been driven to a large extent by their ability to chelate almost any metal in the core. Many of the metal complexes are catalytically active and/or exhibit rich axial coordination chemistry.12, 13 This is most prominently exemplified in nature, recalling, for example, hemes (iron complexes)14 and their role in respiration in the electron transport chain and in a plethora of catalytic reactions or the chlorophylls (magnesium complexes) as the photoactive pigments of photosynthesis.15 Furthermore, the cobalt complex vitamin B12 is essential for the functioning of the brain and nervous system and the formation of red blood cells.16 As a result, there is a rich coordination chemistry involving the central metals and/or peripheral substituents.12, 17
In this context, the pyrrole N−H units in the core of so‐called free‐base porphyrins often appear in discussions solely as the precursor for metal insertion (1→2). The pyrrole N−H moieties are most often thought of as “hidden” in the core and inaccessible for any meaningful use in supramolecular chemistry (e.g., as shown in formula 4, Figure 1). Detailed studies involving these groups have mostly been physical organic chemistry investigations on the N−H tautomerism, that is, studies on the behavior of the N−H units within the porphyrin plane.18
While traditionally in chemistry, a ligand is considered an ion or neutral molecule that binds to metal centers, we will here specifically include and discuss scenarios more akin to the situation and definition of ligands and receptors used in biochemistry.19 Therein, the tetrapyrrole is usually bound to a much larger unit, which qualifies all porphyrins in pigment protein complexes and metalloproteins as ligands. However, we will focus here mainly on an emerging functional porphyrin chemistry where the targeted use of weak interactions, that is, (out‐of‐plane) H‐bonding (of the N4 porphyrin core) is utilized.
1.2. Coordination Types in Porphyrins
1.2.1. Peripheral H‐bonding
Hydrogen bonds are a type of attractive electrostatic interaction (weak interaction) between two polar groups, that is, covalently bound and polarized hydrogen atoms and electronegative atoms or groups.20 They are common and important non‐covalent forces in biological systems, such as proteins, enzymes, and nucleic acids and utilized as structural and functional principles in biomolecules and to control the microenvironment around metal centers in tetrapyrrole‐containing enzymes. Additionally, H‐bonds are partly responsible for the secondary and tertiary structures of proteins and nucleic acids and play an important role in the structure of natural and synthetic polymers. Given the porphyrin motif's pronounced role both in nature and artificial systems, it is unavoidable to highlight the role of hydrogen bonding in porphyrin‐based assemblies.21
In this context, it seems rational to define two major categories of H‐bonds: those involving the tetrapyrrole core and those involving peripheral groups. Both types are vital but found often in substantially different contexts. Peripheral H‐bonding is a major driving force in supramolecular chemistry since the resulting frameworks are usually highly organized and exploit the structural flexibility and diversity that lies in the tunable hydrogen‐bonding strength.22 On the other hand, hydrogen bonds of the tetrapyrrole core are often discussed where porphyrins act as ligands. While peripheral H‐bonding will be introduced briefly at this point, central (N−H⋅⋅⋅X‐type) hydrogen bonding is discussed later as the essential element of this Review. As we will show in this chapter, the former and the latter may also interplay, producing unique porphyrinic architectures.
Hydrogen bonds play an important role in the self‐assembly and stabilization of porphyrin J‐aggregates; for example, where peripheral hydroxyl groups interact with the central nitrogen atoms of an adjacent macrocycle (6, Figure 2).23
Figure 2.
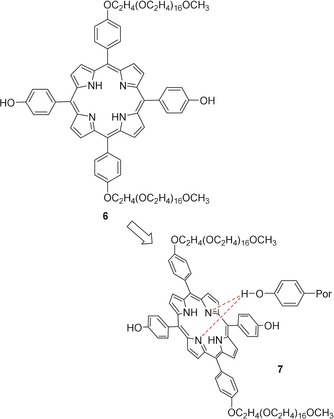
Supramolecular porphyrin complexes formed through peripheral H‐bonding and core interactions. J‐aggregate of 6, 7, where the possibility of hydrogen bonds forming between the porphyrin core and the hydrogen of the O−H group of the adjacent macromolecule has been suggested by molecular mechanics (MM) calculations.23
Further examples of the directing and reinforcing characteristics of hydrogen bonds are found in porphyrinic solids,24 self‐assembled monolayers (SAMs),25 nanofibers and nanorods,26 and nanochannels of 2,3,5,7,8,10,12,13,15,17,18,20‐dodecaphenylporphyrin (H2DPP, 64) derivatives.27 It has also been shown that protoporphyrin IX (PPIX, 71) adsorbed on a Cu surface at low temperature forms adlayers stabilized by tetragonal H‐bonds between the nitrogen atoms of the macrocycle core and peripherally bound carboxyl groups.28
Looking at examples in nature, the malaria pigment hemozoin, a disposal product from the digestion of blood by malaria parasites, is an insoluble, peripherally hydrogen‐bonded dimer of β‐hematin.29 And in chlorosomes, photosynthetic antenna complexes found in some anaerobic bacteria, hydrogen bonding can have a critical influence on exciton dynamics and as such, the light‐harvesting process itself.30 In the future, these findings may motivate new ventures into bionic supramolecular chemistry.
Applications of peripherally hydrogen‐bonded tetrapyrroles are found in material science, molecular electronics, nanotechnology, and solar technology. Representative examples are hydrogen‐bonded organic frameworks (HOFs) based on porphyrins for selective gas separation31 and H‐bonding mediated reversible self‐assembly of porphyrin on a surface for the construction of dye‐sensitized solar cells (DSSCs).32
1.2.2. Peripheral Covalent Bonding/Porphyrin Ligands in Biochemistry
In addition to weaker non‐covalent bonds, nature also utilizes covalent bonding to fix and regulate porphyrin cofactors in defined arrangements. The archetypical case is heme proteins, an indispensable class of porphyrin cofactors involved in a wide range of functions in nature, such as oxygen storage and transport, electron transfer, catalysis, gas sensing, and gene regulation.33 Moreover, they pose an interesting case study to deduce the effects of different coordination types (covalent linkage to proteins: e.g., heme c in cytochrome c 34 vs. axial coordination: e.g., heme a in cytochrome c oxidase, heme b in hemoglobin and myoglobin) in tetrapyrroles.
A comparison of hemes in various binding situations underlines the functional and physicochemical differences originating from the several binding modes, for example, robustness,35 fine‐tuning of reduction potentials over a wide range,36 interaction with proximal amino acids, metal–ligand interactions, metal spin state37 and oxidation state,38 and, potentially, kinetics and thermodynamics of electron transfer reactions itself. Notably, covalent attachment of a protein often results in heme c undergoing conformational changes (i.e. nonplanarity),36b, 39 which effects, for example, the tetrapyrrole's redox properties (see Section 3.3).37, 40 As such, nature highlights the critical role of binding types in porphyrins and provides one of the most interesting case studies on molecular engineering of tetrapyrroles.
Historically, many tetrapyrroles were involved in studies on the biological role of nonplanar porphyrin/porphyrinoid conformations41 and conformational control in enzymes.3a, 42 To date, numerous protein crystal structures were solved in which the tetrapyrroles exhibit distorted macrocycles43 along with considerable movement and flexibility.44 As such, a rich landscape of structural studies, including our own,45 revolves around porphyrin–protein complexes with a focus on weak interactions and tunable properties.
2. Nonaromatic Porphyrinoids
Before focusing on “true” porphyrins, a look must be taken at a number of nonaromatic tetrapyrroles in which the N−H units have been utilized extensively to bind ions and small molecules through hydrogen bonds.46 Macrocyclic conjugation in such systems is interrupted due to sp3‐hybridized meso‐carbon atoms, giving rise to macrocycles with isolated pyrrole units that form what can be considered as cavities. The isolated pyrrole units can easily tilt out of the mean‐plane and are therefore accessible as hydrogen‐bond donors, as opposed to the situation in more rigid and often planar porphyrins. Overall, this is a main prerequisite for the rich coordination chemistry that revolves around these ligands.
2.1. Calix[4]pyrroles
Since their first description by Baeyer (“acetonepyrrole”) in which a tetrapyrrole with four sp3‐hybridized meso‐carbon atoms, octamethylcalix[4]pyrrole (8), was synthesized from acetone and pyrrole (Figure 3),47 calix[4]pyrroles (porphyrinogens) and their analogues, including metal complexes, have been extensively studied and stand now as versatile and often used ligands for the complexation of ions and neutral molecules through H‐bonding with the central pyrrole moieties.48
Figure 3.
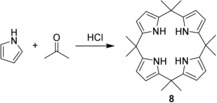
The first synthesis of a calixpyrrole 8 by Baeyer via acid‐catalyzed condensation of acetone and pyrrole.47
However, it was more than a century after the initial discovery that a “gold rush” of using calixpyrroles and related compounds in ligand‐based applications emerged, which yielded hundreds of analogues and a multitude of sensing methods.49 Given the presence of pyrrolic hydrogen‐bond donor groups, they were considered as “old yet new” anion‐binding agents and have been found to frequently complex halides, dihydrogen phosphate, carboxylate, and others, both in solution and in the solid state.50, 51, 52 This is accompanied by a preference towards complexation of small fluoride anions over other guests, which is attributed to the spatial demands of the central cavity (i.e. size exclusion).51, 53 Notably, ligation can occur even in the solid state.54
Unlike most porphyrins where conformational flexibility is limited,55 calix[4]pyrroles modify their shape to accommodate guests. They often adopt a 1,2‐alternate conformation (↑↑↓↓) in the absence of substrates and switch to cone‐like shapes (↑↑↑↑) when bound to anions51a or 1,3‐ (↑↓↑↓) and 1,2‐alternate forms with neutral substrates (Figure 4).56 The result is the formation of aromatic voids defined by the four pyrrole rings within the host molecules and may be considered as a conformational response towards substrates.57
Figure 4.
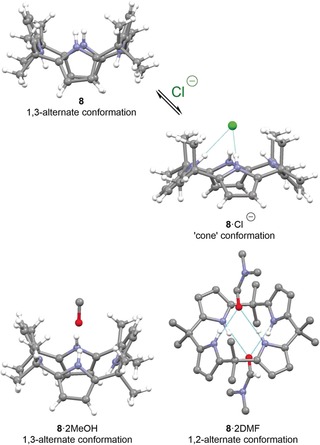
Typical conformations of calix[4]pyrroles and their crystal structures. Top: while the discrete ligand 8 (CCDC: VUSFIY01) is 1,3‐alternate with regard to the pyrrole rings, the tetrapyrrole switches to a “cone” in 8⋅Cl− (CDDC: TEQKIJ) to accommodate anions.51a Bottom: the complexes 8⋅2 MeOH (RECPEU) and 8⋅2 DMF (RECPIY; noncritical hydrogen atoms omitted) with neutral ligands are 1,3‐ and 1,2‐alternate, respectively.56a, 58 DMF=dimethylformamide.
Many qualitative, quantitative, and theoretical studies have been performed on such tetrapyrroles. Examples include compounds 9–15 to compare binding constants and their dependence on electronic properties and the stereochemistry, resulting in a better mechanistic insight into the host–guest chemistry involved (Figure 5).51a, 59
Figure 5.
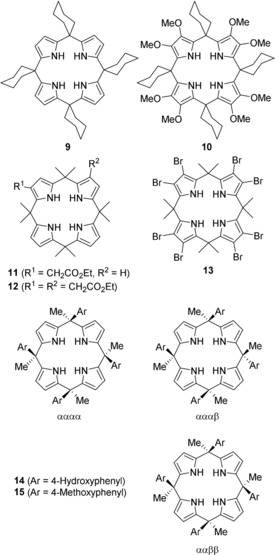
Some functionalized and stereoisomeric calix[4]pyrroles used in binding studies to compare the effects of both Cm‐ and Cβ‐substitution.51a, 59 The tags α and β describe the orientation of the aryl substituents (“up” or “down”).
At the same time, complexation of neutral molecules is usually more challenging due to modest association constants.56a Nevertheless, a large number of neutral receptor–substrate H‐bonded complexes of calix[4]pyrroles were described; for example, with alcohols, amides, a broad range of oxygen‐containing species, and pyridyl‐N‐oxides.56, 60 Based on relatively simple calix[4]pyrroles, numerous more sophisticated receptors have been prepared through an ever‐growing toolbox of functionalization techniques (e.g., 16–19, Figure 6).53b,53c, 56b, 60, 61, 62, 63, 64
Figure 6.
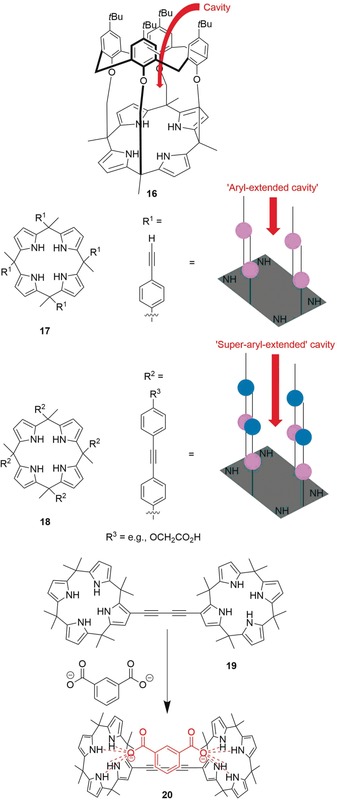
Selected examples of structurally advanced tetrapyrrole receptors: calix[4]arene‐calix[4]pyrrole pseudo dimer 16,[64] “aryl‐extended” and “super‐aryl‐extended” systems 17 and 18,60 and the dimeric anion‐binding complex 19 when binding an isophthalate dianion to form 20.62c
The various calixpyrrole‐dependent recognition techniques were reviewed extensively by Gale and Sessler.65 Optical sensors are often based on covalently linked colorimetric or fluorescent reporter groups where perturbation of the electronic properties upon complexation results in a visual or fluorescence‐based signal (fluorescence quenching/“turn off” or “turn on”).52, 66 A second approach involves a displacement‐assay where an initial host–guest complex dissociates upon addition of a more strongly coordinating analyte, which results in a color change (Figure 7).53a On the other hand, electrochemical sensing utilizes ion‐selective electrodes,67 discrete redox‐active molecular receptors,68 and chemically modified electrodes.69
Figure 7.
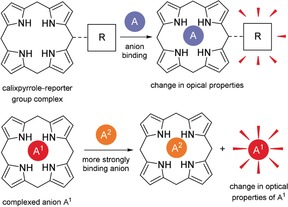
Illustration of common calixpyrrole‐based optical sensors. Top: covalently attached tetrapyrrole‐reporter conjugate; bottom: displacement assay.
H‐bonding calix[4]pyrroles have also been utilized as solid high‐performance liquid chromatography (HPLC) supports in the form of modified silica gels for the separation of anions and neutral substrates such as amino acids and (oligo)nucleotides.70 As potent Lewis acidic multi‐hydrogen bond donors, they possess organocatalytic properties and activate substrates, as illustrated by Diels–Alder reactions and diastereoselective vinylogous additions (Figure 8).71 Furthermore, calix[4]pyrroles can aid regioselective O‐alkylations and ‐acylations.72
Figure 8.
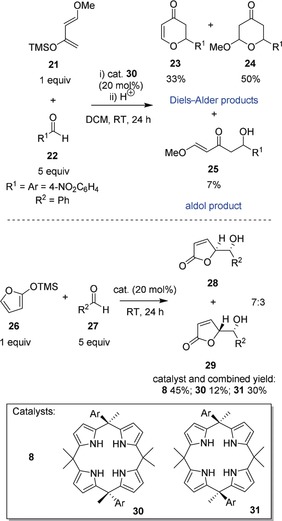
Calix[4]pyrrole organocatalysts. Top: hetero Diels–Alder reaction of Danishefsky's diene 21 and aromatic aldehyde 22 in the presence of 30, yielding a mixture of 23–25.[71a] Bottom: synthesis of γ‐butenolides 28 and 29 via vinylogous addition of 26 and 27 in the presence of calixpyrroles 8, 30, and 31, respectively.[71b] DCM=dichloromethane; TMS=trimethylsilyl.
A recent Review by Kim and Sessler discusses calix[4]pyrroles as molecular containers for ion transport, recognition, and molecular switching functions73 that are often based on weak interactions. Used in this capacity, they stand as selective receptors and extractants for anions and ion pairs across phase boundaries and membranes and as building blocks for stimulus‐responsive materials.74 Due to their remarkable non‐covalent binding properties and their ability to form dimers, trimers,[62b, 75] and aggregates of higher order for selective encapsulation76 and allosteric binding of guests, such as nitroaromatic explosives and fullerenes,77 they are now well‐established in the realms of supramolecular chemistry (Figure 9 A). Furthermore, calix[4]pyrroles were transformed into drug‐delivery vehicles, enzyme mimics, and made an entry as potential anti‐tumor agents when it was observed that such synthetic ion carriers can trigger cell death by facilitating chloride anion transport into cells.78 Even the simple calix[4]pyrrole 8 has been shown to act as an agonist to the G‐protein‐coupled estrogen receptor (GPER‐1).79
Figure 9.
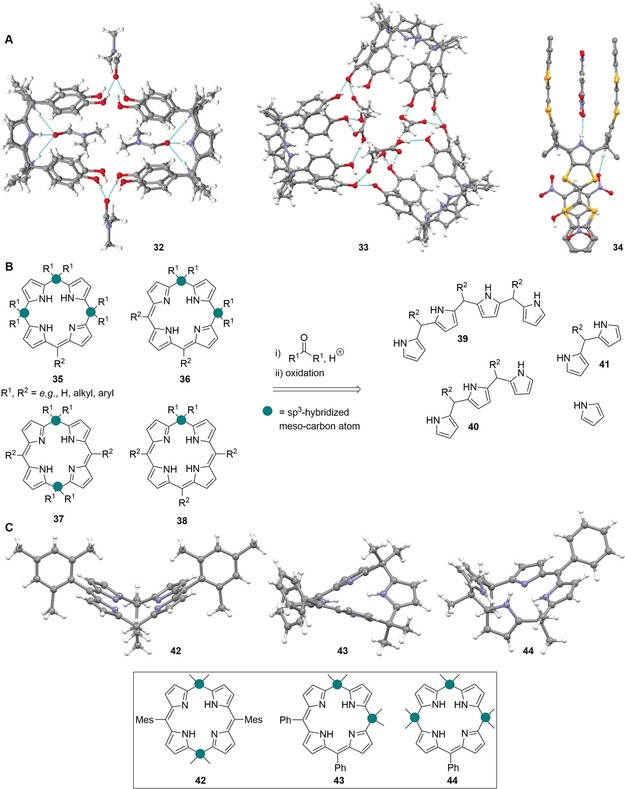
A: Views58 of selected calix[4]pyrrole‐based supramolecules in the crystal. Dimer 32 (CCDC: MAVZIS) linked by hydrogen bonds with DMF. Trimer 33 (CCDC: MAVZOY) linked by hydrogen bonds with AcOH.[62b] Homotropic allosteric calix[4]pyrrole receptor 34 (GUNDUP) with hydrogen‐bonded 2,4,6‐trinitrophenol.[72] B: Acid‐catalyzed condensation of pyrrole and pyrrolic synthons 39–41 with ketones followed by partial oxidation as an efficient sequence to synthesize calixphyrins 35–38. The differentiation of calix[4]phyrins into porphomethenes 35, 5,10‐porphodimethenes 36, 5,15‐porphodimethenes 37, and phlorins 38, is also highlighted. C: Structures[58] of nonplanar calix[4]phyrins in the crystal: 42 (CCDC: LISSIP), 43 (QENDOC), and 44 (QENDIW).81
2.2. Calix[4]phyrins: Phlorins, Porphomethenes, and Porphodimethenes
Formal two‐, four‐, or six‐electron oxidation of these nonaromatic macrocycles leads to calix[4]phyrins, which bear analogy to both porphyrins and calix[4]pyrroles by containing a mixture of sp2‐ and sp3‐hybridized meso‐like positions. The partial interruptions in the conjugation introduce a number of unique structural features since the sp3‐carbon atoms perturb the π‐system and significantly modify the molecular shape and flexibility. Likewise, far‐range electronic induction effects still exist so that functional groups may be used for fine‐tuning of conformational and chemical properties.80
Early examples of rational calixphyrin syntheses include acid‐catalyzed cyclization reactions between ketones and pyrrolic precursors followed by partial oxidation (Figure 9 B)81 and Buchler's reductive methylation to yield metalloporphodimethenes82 or substitution reactions.83 The products are usually distorted and feature structural cavities and accessible inner nitrogen atoms (Figure 9 C), which may aid the binding of substrates.
However, it should be noted that the calixphyrin core, like porphyrins, consists of both amine and imine groups and effectively has a lower number of N−H units to interact with guest molecules as compared to the analogous calixpyrroles. This may account for a less pronounced receptor chemistry revolving around this class of macrocycles, leaving the option of core protonation to introduce more N−H motifs and to increase the anion‐binding capabilities.84 Nevertheless, a number of H‐bonding complexes between (expanded) calixphyrins and various substrates has been observed (Figure 10).85 As such, they provide a potential avenue towards a rich—but as of yet underdeveloped—receptor chemistry up to enantiorecognition.86
Figure 10.
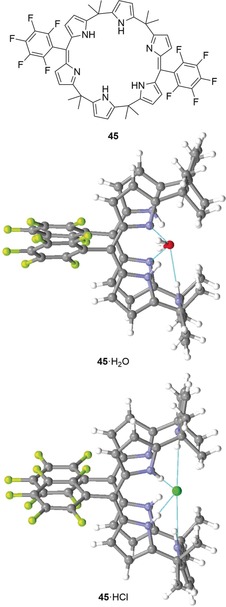
Expanded calixphyrin 45 and the structures of two of its hydrogen‐bonding complexes in the crystal. 45⋅H2O (CCDC: XETYUQ) and 45⋅HCl (XETZAX).58, 85
Much like porphyrins, calixphyrins are basic and bind protons at their imine functions but, intriguingly, the corresponding dications are more conjugated and thus more stable than the neutral species.86b This again highlights the diverse character of calixphyrins as macrocycles with partial traits of both calixphyrins and porphyrins. In practical terms, core‐protonated calix[4]phyrins form in the presence of acids where acid anions are subsequently hydrogen‐bonded to the (protonated) core. In such complexes, for example, 46, the pyrrole cycles are tilted significantly out of the mean‐plane and, depending on the molecular geometry, the meso‐hydrogen atoms can participate in further stabilization of those salts (Figure 11).87
Figure 11.
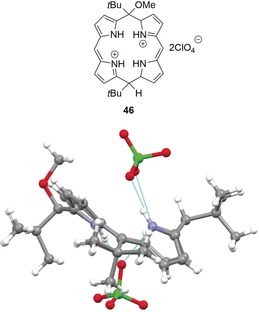
View of the molecular structure of calix[4]phyrin salt 46 in the crystal (CCDC: HETDEP).58, 87
This identifies tetrapyrrole core protonation as an opportunity to enhance the molecules’ receptor potential through out‐of‐plane distortion and exposure of the central nitrogen atoms. Hence, hereafter we will discuss in detail how this promising strategy has been utilized to exercise conformational control on porphyrins.
3. Accessing N−H Units in Porphyrins
A brief look at nonaromatic porphyrin analogues shows that they are to a large extent involved in many applications that are desirable for porphyrins, too; including organocatalysis and a rich receptor chemistry. However, due to the porphyrin motif's planarity, rigidness, and the presence of “only” two N−H donors, as opposed to up to four in tetrapyrrolic porphyrinoids, it is traditionally more challenging to apply them in the same manner and to the same extent. But an increased availability—and therefore reactivity—of the core amine and imine moieties can be achieved through rationally altering their orientation within the mean‐plane via molecular engineering. While various methods to deform the molecular skeleton in such a way exist (Figure 12),1, 88 herein we will focus on a case study of two of the most feasible and widely used strategies, namely core protonation and high peripheral substitution. Additionally, these approaches will be compared to the mode of action of the natural chelatases; enzymes that can induce porphyrin ring distortion.
Figure 12.
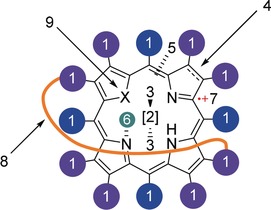
Possibilities to alter the macrocycle's conformation: 1) introduction of sterically demanding substituents; 2) metallation; 3) axial ligands; 4) degree of reduction; 5) interruption of the conjugated system (see Section 2); 6) N‐alkylation, ‐arylation, or protonation; 7) cation radical formation; 8) “strapping” of the macrocycle via covalent linkage of the meso‐ or β‐pyrrole positions; 9) heteroatom substitution. Core and skeleton transformations are possible, too.
Note, that corroles, expanded‐, N‐confused porphyrins, and various other analogues show an ample (chemo)receptor chemistry, too, paralleling that of “true” porphyrins. However, they have recently been reviewed4a and will therefore not be discussed subsequently.
3.1. Porphyrin (Di)cations
Porphyrin cations are readily produced in the form of core diacids through protonation of the internal imine groups (Figure 13 A). This is accompanied by distortion of the macrocycle, which increases the vector of the N−H orientation outwards (Figure 13 B). At the same time, such conformational manipulation dramatically boosts the amine groups’ capacity to contact appropriate small molecules and ions. Accordingly, porphyrin cations can be considered as large anion sensors due to their capability to form hydrogen bonds with counterions (Figure 13 C).3
Figure 13.
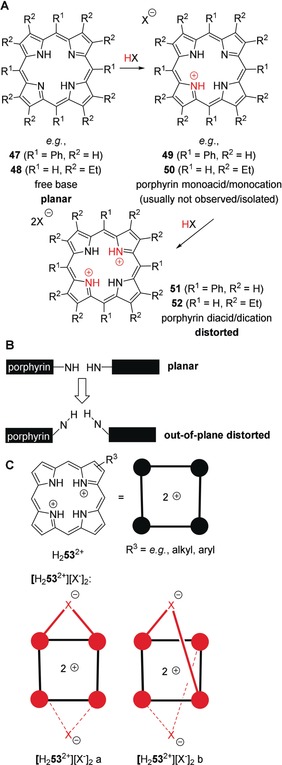
A: Protonation of free base‐porphyrins, for example, 5,10,15,20‐tetraphenylporphyrin (H2TPP, 47) or 2,3,7,8,12,13,17,18‐octaethylporphyrin (H2OEP, 48) by acids HX results in the formation of porphyrin dications and their salts, for example, [H4TPP2+][X−]2 (51) or [H4OEP2+][X−]2 (52). This proceeds via a monocation, for example, 49 or 50, which cannot usually be isolated. X−=acid anion. B: Illustration of pyrrole out‐of‐plane distortion in porphyrins/porphyrin core acids. C: Porphyrin acid salts, for example, [H2 53 2+][X−]2 are H‐bonding complexes where various conformations, such as [H2 53 2+][X−]2 a and b are conceivable.
Indeed, crystal structures of porphyrin diacid salts indicate severely nonplanar geometries of the macrocycles as a result of both steric interactions of the crowded core and electrostatic repulsion of the partially positive pyrrole nitrogen atoms.89 In most cases, protonation results in the formation of saddle‐distorted (sad‐type) porphyrins with 1,3‐alternating pyrrole tilts (“up and down”, ↑↓↑↓, e.g., in 54).90 However, individual cases of 1,2‐alternating forms (↑↑↓↓) have also been observed, for example, in 2,3,7,8,12,13,17,18‐octaalkylporphyrin diacid salts such as 55 (Figure 14).90a, 91
Figure 14.
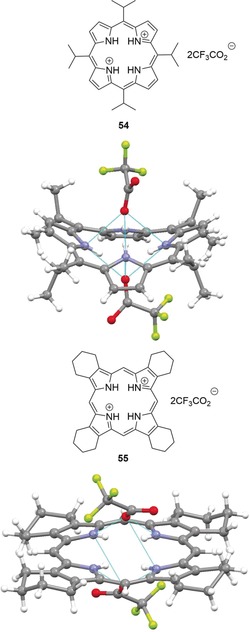
Top: View of the molecular structure of porphyrin salt 54 in the crystal (CCDC: KIBMEN).[58, 90b] Bottom: View of the molecular structure of 55 in the crystal (YEVKAL).58, 91
The question of how much the porphyrin macrocycle can be distorted by this means or whether there is a “breaking point” has been thoroughly discussed and an understanding of this approach is vital for the design of sensors based on weak interactions with the core. Accordingly, it was elaborated that peripheral (see Section 3.3) and core steric strain88 (i.e. protonation and N‐substitution)91, 92 are some of the most important “adjusting screws” to exert conformational control.1 One case study indicated that the ditrifluoroacetate salt of 56, 57, is one of the most nonplanar porphyrins described so far.91 A comparison with the analogous salt of 47, 58 that is less distorted, clearly points at the additional distorting effects of peripheral substituents. These are involved in repulsive peri‐interactions, which cause a highly substituted porphyrin like 56 to be “pre‐distorted” even without protonation. Thus, by comparing dodecasubstituted free‐base porphyrins (e.g., 56) with their dications (e.g., 57), an increase in nonplanarity of only 13–25 %, depending on the individual substituents, was noted. At the same time, this effect is significantly stronger in sterically unhindered systems (e.g., 47) where protonation can result in distortion of up to 300 % (47→58), suggesting that there is a maximum level of nonplanarity for porphyrins (Figure 15).
Figure 15.
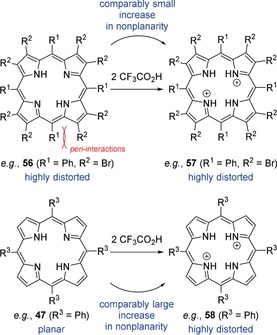
A dodecasubstituted porphyrin like 56 is “pre‐distorted” due to repulsive peri‐interactions of the meso‐ and β‐substituents. As such, core protonation results in a comparably small increase in nonplanarity in 57. At the same time, sterically unhindered 47 undergoes a significantly large increase in distortion upon formation of 58.
Webb and Bampos investigated the dynamics and the complexation behavior of porphyrin diacids in solution and offered insight into the mechanisms of both porphyrin protonation and acid‐accelerated metallation.93 They also showed the effects of varying both the acid and the porphyrin on proton transfer and anion recognition and put an emphasis on the role of macrocyclic conformational control in intramolecular proton transfer. In addition, the effects of saddling, meso‐phenyl twisting, and different hydrogen‐bonded counterions on the optical properties of [H4TPP2+][X−]2 (where X−=F−, Cl−, Br−, I−) were elaborated theoretically via density functional theory (DFT) and time‐dependent density functional theory (TDDFT) by Rosa and co‐workers.94
Next to this fundamental research, a broad landscape of applications emphasizing the H‐bonding and anion‐binding properties of porphyrin dications exists: The diprotonated form of octaalkylporphyrin 59 acted as a bromide‐selective sensor in the system 59‐Et4NBr‐HClO4‐CH3CN (Figure 16).95 Under these conditions, the stable H‐bonding complexes 60 and 61 formed preferably, which was considered as a step towards halogen‐selective molecular anion receptors of good detectability. A later study investigated the binding of various halides and alkali metal cations by diprotonated porphyrin hosts 59 and 62, respectively (Figure 16).96 Upon titration of a 59‐HClO4‐CH3CN system with various halide salts, stable 1:1 and 2:1 hydrogen‐bonding complexes with the halogens formed. While the complexation constants decreased in the order Cl− > Br− > I−, strong fluorescence quenching occurred in the presence of iodide. On the other hand, alkali metal ions could be trapped with monoarylporphyrin dication 62 containing a complexing polyether fragment that was both peripherally attached and hydrogen‐bonded to the core through a pyridyl moiety. Note, that a high binding selectivity of 62 for K+ over Li+ and Na+ was observed.
Figure 16.
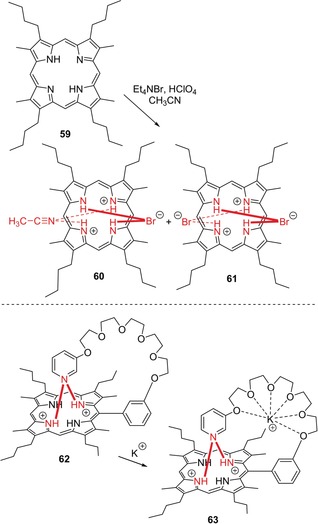
Top: Selective formation of hydrogen‐bonding complexes 60 and 61 in situ from the system 59‐Et4NBr‐HClO4‐CH3CN.95 Bottom: Dicationic hydrogen bonding complex 62 for complexation of potassium, yielding 63.96
Other efforts focused on nonplanar porphyrin diacids in supramolecular assemblies.97 In one report, Honda and co‐workers constructed a series of hydrogen‐bonded supramolecular complexes of saddle‐distorted diprotonated 64 and electron donors with carboxylic acids.98 These were then investigated in terms of photo‐induced electron transfer dynamics. In such studies, the role of the H‐bound counterions is often crucial. For example, 5,10,15,20‐tetrakis(4‐sulfonatothienyl)porphyrin dihydrochloride showed chloride‐specific aggregation in aqueous solution.99 The presence of Cl− induced H‐aggregation followed by conversion into J‐aggregates with increasing chloride concentration. Nakanishi et al. investigated the photoconductivity of nanochannels composed of sad‐type porphyrin dications and electron donors.100 Specifically, the dihydrochloride salt of 64, [H4DPP2+][Cl−]2, gave supramolecular architectures by self‐assembly based on intermolecular π—π interactions that could host tetrathiafulvalene and p‐aminophenol. Additionally, porphyrin diacid–polyelectrolyte supramolecules formed via electrostatic self‐assembly in aqueous solution were used as effective photocatalytic systems for iodide oxidation.101 Herein, the cationic porphyrin architectures showed a significantly higher catalytic activity than aggregates under neutral conditions. Porphyrin dications have also generated some interest, for example, in nonlinear optics (NLO), where laser‐induced protonation of free‐base porphyrin in chloroform gave a positive nonlinear absorption (NLA) due to conformational distortion.102
Often, the more elusive porphyrin monocations are also saddle‐distorted, although tendentially less than the corresponding diacids103 and stand as a “missing species” in porphyrin chemistry since they are generally difficult to produce, characterize, and isolate. Porphyrin monoacids are usually less stable than their diprotonated counterparts; it is likely that a large saddle‐distortion destabilizes the monoacids because their out‐of‐plane distortion significantly reduces the steric crowding of the remaining unprotonated nitrogen atom. This leads to the uptake of a second proton being more energetically favorable than the first.89 As a result, most observations of these species are limited to theoretical methods and spectroscopic sightings, often in equilibria with the respective dication.104 Nevertheless, the crystal structure of the H‐bonded complex [H3TPP+][I3 −] has been solved105 and in another study, Kojima and Fukuzumi obtained the stable monocation complexes 66 and 68 of saddle‐distorted 64 by reaction with anthracene sulfonic acids 65 and 67.106 This was possible presumably due to the only weakly hydrogen‐binding character of the conjugate anthracene bases (Figure 17 A).
Figure 17.
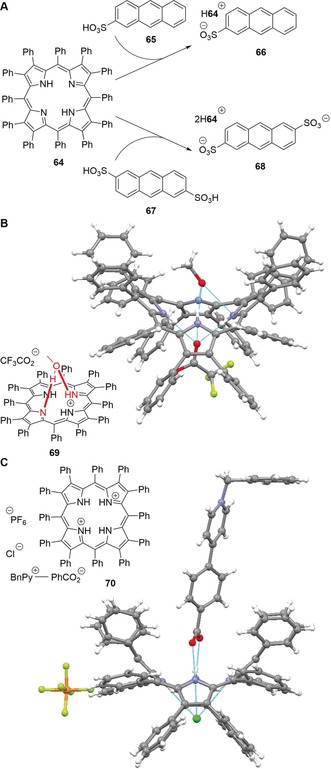
A: Stable H3DPP+ H‐bonding complexes 66 and 68 made from 64 in the presence of anthracene sulfonic acids 65 and 67, respectively.[106] B: Structure of 69 in the crystal (CCDC: RATXOC) where the monoacid H3DPP+ is stabilized by hydrogen bonding with methanol.[58, 107] C: Supramolecular hetero‐triad 70 and its structure in the crystal (RARQEJ).58, 108
H2DPP (64) also formed a stable monoacid complex 69 in the presence of methanol and trifluoroacetic acid in dichloromethane.107 The crucial role of methanol is highlighted as stabilizing the monoacid through hydrogen bonding with an out‐of‐plane N−H group and an imine moiety at the same time, preventing the second acid–base reaction of the macrocycle (Figure 17 B). Furthermore, Almarsson and co‐workers were the first to observe a monoprotonated meso‐tetraphenylporphyrin, which was capped on one face so that the access of a trifluoroacetate anion to the second protonation site was restricted.109 In another notable study, the group of Kojima formed supramolecular hetero‐triads of diprotonated 64, for example, 70.108 In this context, formation of [H3DPP+][X−] species was correlated with destabilization of [H4DPP2+][X−]2 due to decreasing pK a values of the protonating acids used. This occurred as stronger acids provided weaker corresponding bases that were weaker H‐bond acceptors (Figure 17 C).
Still, while anion binding, material science, and other applications of such salts are of contemporary interest, we are ultimately interested in the use of neutral free‐base porphyrins as receptors and organocatalysts. As illustrated in Figure 13 B, this requires nonplanar macrocycle conformations with accessible N−H and imine donors/receptors. As such, methods to achieve significant distortion while retaining the tetrapyrrole's characteristics will be discussed in the following.
3.2. Nature's Way—Chelatases
Here—as so often—nature provides inspiration when taking a closer look at how conformational control in tetrapyrroles is achieved by chelatases. As was surmised for a long time, these natural enzymes incorporate a range of metals (such as iron, magnesium, nickel, and cobalt) into porphyrins by a distortion‐mediated mechanism. This commences by deforming the macrocycle, thus rendering the core more accessible, followed by metal insertion and relaxation of the system.110 The product, a metalloporphyrin, is then released from the protein to fulfill its biological function (Figure 18).
Figure 18.
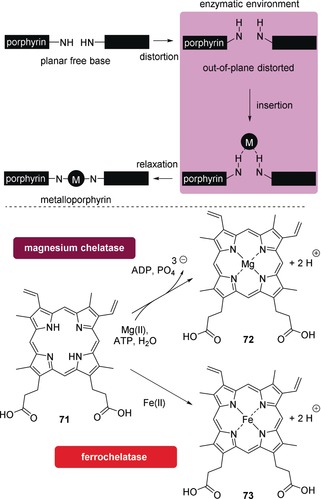
Top: Schematic illustration of metal insertion into porphyrins by chelatases. Bottom: Net reactions of the biocatalyzed MgII and FeII complexation by PPIX (71) to produce (protoporphyrinato IX)magnesium (72) and (protoporphyrinato IX)iron(II) (heme b, 73), respectively. ADP=adenosine diphosphate; ATP=adenosine triphosphate.
This mechanism was initially proposed on the basis of kinetic studies, chemical modifications, and the enzyme's strong inhibition by N‐alkylporphyrins111 and supported by the finding that antibodies elicited to a distorted N‐methylporphyrin (=analogue of ferrochelatase–substrate transition state) could catalyze metal‐ion chelation.112 Further indications were obtained through spectroscopic analyses,103, 113 and crystal structural studies yielded final proof.110
In the case of ferrochelatase, the terminal enzyme in heme biosynthesis that inserts FeII into the macrocycle 71, the energetics of the accompanying ring‐distortion process have been calculated.114 Therein, it was argued that once the metal is inserted, the porphyrin becomes stiffer and flatter, resulting in a lower binding affinity to a site designated to bind its nonplanar form. This would ultimately result in release of the metalloporphyrin 73 from the enzyme. It was also suggested that the protein may increase the basicity of the pyrrole nitrogen atoms by macrocyclic deformation. Furthermore, the structure of the PPIX (71) substrate bound to ferrochelatase was estimated: all pyrrole rings were tilted out of the mean‐plane, most towards the putative binding site of the metal ion.
This brief overview illustrates how nature uses an efficiently “designed” method to achieve what is tedious, maybe even unimitable, in a synthetic setting. Accordingly, researchers have yet to find a synthetic methodology that mimics all the conveniences of these natural enzymes in less time than nature used through evolution.
3.3. Highly Substituted, Nonplanar Porphyrins
Uncharged nonplanar porphyrins (i.e. opposed to core dications in Section 3.1) are essential for biological functions and frequently found in, for example, photobiological systems115, 116 and other proteins.21a However, the first experimental proof of such nonplanar conformations was provided in the early 1960s, namely with the tetragonal forms of H2TPP (47) and CuIITPP.117 The historical development and classic cases of tetrapyrrole nonplanarity have been discussed elsewhere88 and nowadays, the family of dodecasubstituted porphyrins stands as a typical workhorse for studies in this area as they are often accessible by rational syntheses towards conformationally designed targets.55, 118 Therefore, it is important that we give a brief introduction to the basic characteristics (e.g., conformation–properties relationships) of such compounds here before deducing their potential as nonplanar receptors.
The various nonplanar distortion modes for highly substituted porphyrins were defined and categorized by Medforth et al. (Figure 19, top).12, 119 However, in H2OETPP (74) and its derivatives, a well‐understood and frequently used class of saddle‐distorted free‐base porphyrins that are structural hybrids of H2TPP (47) and H2OEP (48), the central N−H donors are severely forced out‐of‐plane. More precisely, a main structural consequence of the repulsive peri‐interactions between neighboring meso‐ and β‐substituents is an alternating “up and down” tilt of the individual pyrrole rings in 74, which are rotated ca. 30° out of the mean‐plane.120 At the same time, meso‐aryl groups in nonplanar porphyrins show increasing in‐plane rotation. Altogether, the sad‐type conformation of OETPPs and other 2,3,7,8,12,13,17,18‐octaalkyl‐5,10,15,20‐tetraarylporphyrins causes the amine functions to be oriented significantly towards the sphere of the tetrapyrrole rather than in‐plane and increases the chances to interact with surrounding H‐bond acceptors. On the other hand, a similar effect may be expected for the imine groups in the form of both increased basicity and H‐bond acceptor potential (Figure 19, bottom).
Figure 19.
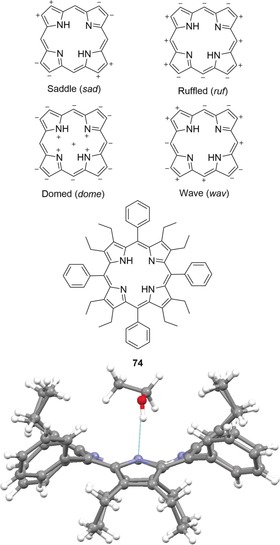
Top: Representation of the four most common distortion modes for porphyrins. Only the most significant displacements are shown. Bottom: H2OETPP (74)⋅EtOH and its structure in the crystal (CCDC: SATQOU).58, 120b, 121
Nonplanarity results in a range of measurable physicochemical effects besides changes in the electronic absorption spectra (i.e. a bathochromic shift).122 Their increased basicity was noted during the initial synthesis of 2,3,7,8,12,13,17,18‐octamethyl‐5,10,15,20‐tetraphenylporphyrin (H2OMTPP, 75) by Dolphin.123 The product was protonated even by water and also showed broad absorption bands, which were attributed to steric interactions between the meso‐aryl and β‐methyl functions. And metallation experiences a significant rate enhancement in nonplanar tetrapyrroles—an effect that is likely to benefit the distortion‐mediated metal insertion by chelatases.124 These and more effects of macrocyclic deformation have been summarized by Shelnutt et al. with an emphasis on new functional properties and their significance in biological systems.21a Another early observation was that nonplanar porphyrins have altered oxidation and reduction potentials compared to flat analogues and that this effect can be as powerful to the extent that electronic substituent effects are surpassed.37, 40, 125
Various other studies have evaluated the nature and effects of nonplanarity in tetrapyrroles, including investigations into (dynamic) photophysical and excited state properties,120b, 126 the oxidation to π‐cation radicals,127 spectroelectrochemistry,128 and conformational flexibility129 of “stereotypical” representatives, such as 2,3,7,8,12,13,17,18‐octaalkyl‐5,10,15,20‐tetraarylporphyrins 76, 2,3,7,8,12,13,17,18‐octahalogeno‐5,10,15,20‐tetraarylporphyrins 77, ruffled 5,10,15,20‐tetraalkylporphyrins 78, 2,3,5,7,8,10,12,13,15,17,18,20‐dodecaarylporphyrins 79, and their metal complexes (Figure 20). Notably, almost all photophysical parameters are directly affected by macrocycle distortion: nonplanar porphyrins have significantly lower fluorescence yields, large Stokes shifts, and shorter lifetimes of the lowest excited state due to faster intersystem crossing and internal conversion.1, 130 This qualifies highly substituted, distorted porphyrins as a good starting point to engineer artificial photosynthetic chromophores.
Figure 20.

Selected porphyrins and their metal complexes discussed in various studies on distorted systems.
An illustrative example of how substituent bulk and their interactions may be utilized for conformational control on the macrocycles is the series of cycloalkenyl‐substituted porphyrins 81–83 “enclosing” the β‐positions that was prepared by Medforth et al.118b These became increasingly nonplanar with larger peripheral cycloalkenyl rings by forcing the methylene groups into closer contact with the meso‐phenyl substituents (Figure 21 A). In line with that are X‐ray studies of conformationally designed nonplanar NiII porphyrins by Barkigia et al.131 and a comparative study on the synthesis and stereochemical properties of 47, 74, and tetraphenylporphyrins with graded degrees of β‐ethyl substitution 84–87 (“H2EtxTPPs”) plus their metal complexes.132
Figure 21.
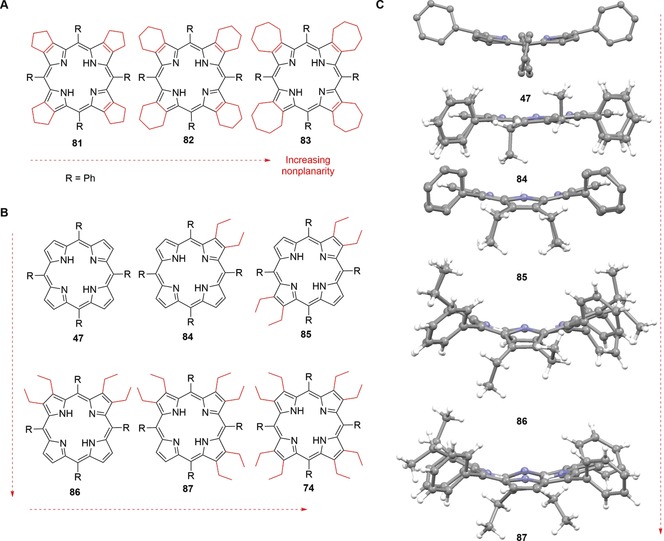
A: Gradually distorted cycloalkenyl‐substituted porphyrins 81–83.[118b] B: Distorted “H2EtxTPPs” 84–87 as well as 47 and 74 with graded degrees of β‐ethyl substitution and incrementally increasing nonplanarity.[132] C: Structures[58] of 47 (CCDC: TPHPOR10)[117a] and 84–87 (TATPOT01, TATPUZ01, TATQAG01, TATQEK01) in the crystal.132 For comparison see Figure 19 for a crystal structure of 74.
Macrocycle distortions lead to the formation of cavities on both sides of the tetrapyrroles where small molecules can bind. As a result, solvents can often be found incorporated within the crystal lattice of nonplanar porphyrins. For example, saddle‐shaped tetrapyrroles often produce tunnel‐like structures in the solid state, which are commonly filled with solvate molecules, as in the case of CuIIOETPP (80)⋅2 DCM (Figure 22; DCM=dichloromethane).133
Figure 22.
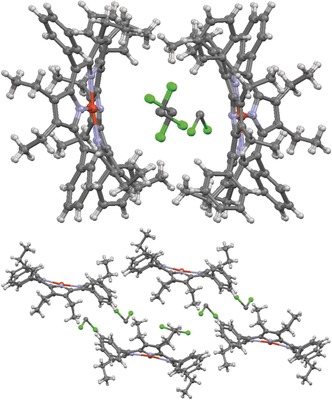
CuIIOETPP (80)⋅2 DCM and excerpts of its crystal structure lattice diagram at different visual angles (CCDC: WADROI).58, 133
Although conformationally close to OETPPs, the structural chemistry of sad‐type DPPs, including weak interactions with guests, appears to be more variable. This is best illustrated by 64 itself, which can undergo macrocycle inversion in solution, as shown by variable temperature (VT) NMR spectroscopy.55 The broad conformational landscape of DPPs is also represented by its various crystal structures: First reported was that of the free‐base 64 in an orthorhombic modification,119 then another orthorhombic form (albeit in a different space group) with a more symmetric saddle‐distortion than previously seen and three water molecules incorporated within the unit cell.134, 135 Similarly to 64, ethanol has also been found within the crystal structure of 74 where it was coordinating to the central nitrogen atoms (Figure 19).121 It should also be noted that multiple conformations in a single dodecaarylporphyrin have been observed, including sad, ruf, wav modes, and mixtures thereof, which is once again ample evidence for their remarkable flexibility, if not adaptability and points at utilization as selective H‐bonding sensors.119
It should be highlighted that due to the wealth of literature on distorted porphyrins, any synoptic view can only be a screenshot of this fast‐developing and diverse landscape, with their applications spanning from medicinal, optical, and technical uses to crystal engineering, methods development,136 catalysis,137 and sensing.138 As we have shown, only the introduction of saddling results in significant N−H exposure and as such, this approach is the basis of a number of new and up‐and‐coming methods for the binding and activation of small molecules.
4. Applications
Surprisingly, distortion as a method to achieve nonplanar tetrapyrrole conformations for use as sensors and organocatalysts has remained mostly unexplored. This leaves the high potential of the inner nitrogen atoms—the primary structural motif of all free‐base porphyrins—to bind and recognize analytes and to activate small molecules unexploited. Therefore, our goal is to explore and discuss the opportunities that conformational design provides as a novel tool to access porphyrin sensors and organocatalysts with an exposed core.
4.1. Porphyrins as Sensors: Anion Binding, Explosive Detection, “Chemical Nose”
When porphyrins applied as sensors, the properties of the porphyrin macrocycle change upon interaction (e.g., metal coordination, H‐bonding, π–π interactions, irreversible chemical reactions) with a substrate, resulting in a detectable response such as a color change, fluorescence quenching/“turn off” or “turn on”, or an electric signal, which mirrors the presence of the analyte (Figure 23).
Figure 23.
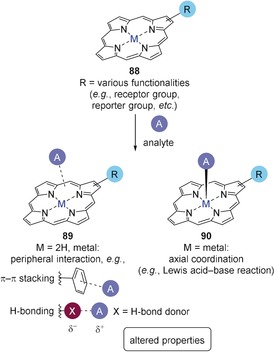
Porphyrins 88 for use as sensors and schematic representation of peripheral (in 89) and axial binding (in 90).
The development of such molecular probes benefits from the distinct relationship between the porphyrins’ physicochemical features and structure, which provides diverse opportunities to design sensors with tailored characteristics. Such methods involve covalent synthetic modification (i.e. the introduction of additional functions, including receptor and reporter groups), metal insertion, or altering the molecular skeleton itself, to name but a few. As outlined above, macrocycle conformation and size can be tuned to a great extent by the number and nature of peripheral substituents,1, 3 thus modulating ion selectivity and sensitivity. What qualifies porphyrin–substrate complexes in particular for straightforward optical and fluorescence‐based detection are their excellent photophysical properties, for instance intense absorption in the Soret and Q bands and red to near‐infrared emission. On a different note, they often mimic biological functions, such as reversible binding of gaseous compounds or catalytic activation as methods of action.139
In practical terms, since there are often scenarios where multiple analytes compete, it is crucial to engineer receptor platforms of high selectivity that specifically emphasize desired interactions while neglecting interferers. This is where the broad and tunable landscape of macrocyclic conformations may take a critical position: as we have elaborated above, porphyrins that have similar substitution patterns can have significantly different conformations. As such, next to say, modulation of the peripherally attached groups and electronic effects, three‐dimensional distortion could benefit the specificity of a recognition process. That is because in a sense, nonplanarity may be seen as an additional “adjusting screw” to produce a situation comparable to the lock‐and‐key model on a molecular level: synthesizing a family of deformed porphyrins based on differing stereochemical parameters (e.g., specific volume and diameter of the binding pocket, chirality) would result in a diverse toolbox of conformationally designed porphyrins to serve as a close fit to specific substrates while disregarding others (Figure 24). This could also be achieved in liaison with surface science, as it has been evidenced that porphyrins undergo structural transformations when anchored to a surface.140 One approach by the group of Aida that relied on such molecular geometry‐based considerations for sensing fabricated a D 2‐symmetric, dodecasubstituted, sad‐type porphyrin where the absolute configuration of chiral acids could be recognized and “memorized” through formation of asymmetric core hydrogen‐bonding complexes and circular dichroism (CD) as an analytical tool.141 The “chirality memory” could also be “released” and “retrieved” in response to stimuli such as heat and light.
Figure 24.
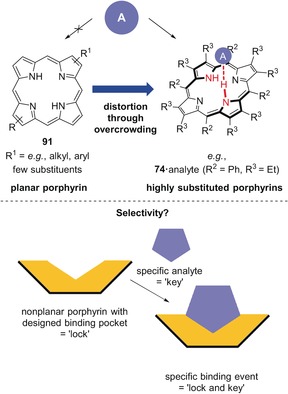
Top: Concept of distortion‐dependent porphyrin sensors. The core of planar porphyrins 91 is unable to complex an analyte, while highly substituted, distorted analogues (e.g., 74) are able to do so. Bottom: One conceptual example of new sensors based on engineered nonplanar porphyrins.
While this approach would mainly depend on N−H⋅⋅⋅X hydrogen bonding as the type of interaction between substrate and sensor and has yet to be translated into a first case study, metalloporphyrins can facilitate multiple interactions and allow for tailoring their physicochemical properties through alteration of the macrocycle, the complexed metal, and the functional groups. Similarly, free‐base porphyrins equipped with particular functional moieties acting as peripheral reporter groups or binding sites are important classes of receptors already at hand. As such, typical (metallo)porphyrin‐ and porphyrinoid‐based chemical sensors can spot gaseous substrates, such as NO2, CO2, and volatile organic compounds (VOCs), as well as analytes in liquid phase; for instance, many common anions, NO in cells, H2O2, dopamine and other neurotransmitters, explosives, pollutants, pharmaceutical analytes, ammonia and amines, metal ions, protons, ascorbic acid, glucose, ion pairs, and reactive oxygen species (ROS).4a,4b, 6d, 139, 142 Moreover, porphyrins are of interest for military and security‐related applications and can counteract explosives (as in the case of 92–95) and hazardous biological, chemical, and radiological/nuclear materials (Figure 25).138, 143, 144, 145, 146 However, currently this rarely involves the core of free‐base porphyrins, leaving plenty of room for new developments and to exploit the high potential of the N−H⋅⋅⋅X binding motif for probing.
Figure 25.
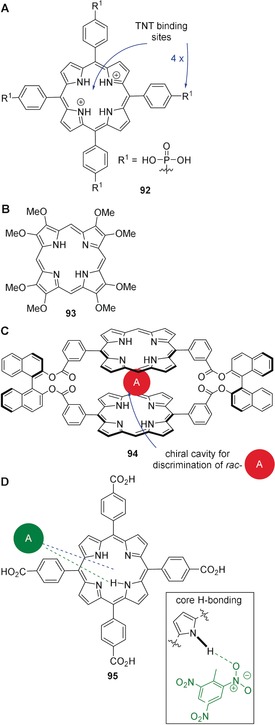
Selected porphyrin‐based sensors for nitroaromatic compounds. A: Porphyrin diacid 92 for efficient recognition of ≥5 ppb 2,4,6‐trinitrotoluene (TNT) through emission “turn off” following intermolecular hydrogen bonding and π—π stacking.143 B: Macrocycle 93 for detection of, for example, TNT, 2,3‐dimethyl‐2,3‐dinitrobutane (DMNB), 1,3,5,7‐tetranitro‐1,3,5,7‐tetrazocane (HMX), and 1,3,5‐trinitro‐1,3,5‐triazinane (RDX).[144] C: Chiral dimer 94 for naked‐eye recognition of 1,3,5‐trinitrobenzene and chiral discrimination of racemic mixtures of nitroaromatics by guest intercalation into a structural cavity via cooperative π—π stacking.145 D: Complex of 95 and TNT (green dashed line=N−H⋅⋅⋅X‐type hydrogen bond, blue‐dashed line=π–π stacking).146
Selective chemical sensors that sense gaseous and liquid analytes based on molecular interactions can be compared to the receptors of the olfactory and gustatory systems. This insight inspired research into artificial sensory organs,147 with the result that today, a range of “chemical noses” and “tongues” are at hand,139, 148 often based on porphyrins. The current trends in such tetrapyrrole‐based applications have been reviewed by Paolesse et al. who engaged in studies on “chemical noses”,139 spanning from basic research149 to applications as far as breath testing and food analysis.150
At the same time, others have merged the same molecular interactions with intelligent, novel analytical methods, resulting, for example, in “optoelectronic noses” based on chemoresponsive dyes or fluorophores. Introduced by Suslick and co‐workers, “optoelectronic noses” can utilize (metallo)porphyrins as platforms for odor visualization151 and since then, their scope has broadened continually142a to recognize explosives,152 pathogens,153 toxic industrial chemicals,154 and even to monitor foods freshness.155 For this purpose, simplest, a colorimetric sensor assay (CSA) is digitally imaged before and during contact with a substrate to produce a difference map via digital subtraction, that is, pixel by pixel of the image of the array before and after contact with regards to red, green, and blue (RGB) values (Figure 26).156 An overview on Suslick's and other groups’ work in this area, including phthalocyanine/porphyrin metal complexes157 and doped materials157a for “smell seeing” applications151b was given in 2013.142a
Figure 26.
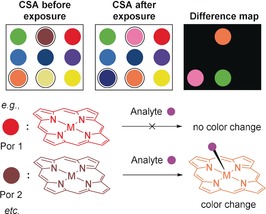
Illustration of a CSA where each spot represents a different chemoresponsive dye (e.g., (metallo)porphyrins) before and after analyte exposure. Following testing, a difference map was generated. Some ligands showed a color change upon contact with the substrate.156
While these reports often depend on metallation of the core, this has the drawbacks of 1) decreased conformational flexibility of the macrocycle (core) and 2) essentially rendering the central nitrogen atoms inert towards the formation of intermolecular hydrogen bonds. However, we propose that the availability of the central amine and imine motifs is critical for novel applications of free‐base nonplanar porphyrins as shape‐selective hydrogen‐bonding sensors. In planar porphyrins, the N−H units are “buried” by the macrocyclic system but distortion, for example, through core protonation or the introduction of a high number of peripheral substituents, is an opportunity to prompt them for such bonds. In line with this is a study on aryl phosphonate‐ and phosphonic acid‐functionalized porphyrins for molecular recognition of ≥5 ppb TNT where macrocyclic distortion upon protonation increased selectivity towards the explosive at nanomolar levels.143 Additionally, quantitative complexation of TNT (0.46 ppm) by a complex of carboxylporphyrin 95 and a metal–organic framework (MOF) occurred in aqueous media and involved hydrogen binding with the porphyrin's central N−H groups.146 Our proposed concept of distortion‐dependent sensing is also supported by early studies on porphyrin core acids used for anion sensing, for example, where protonated 59 was used96, 149e and the binding of neutral substrates, for example, by 80,133 H2OETPP (74),121 and H2DPP (64)88 (see Section 3.3).
4.2. Catalysis
We and others have recently reviewed how (metallo)porphyrins are particularly suited for sensing applications4a,4b, 138, 139, 142 and their catalytic activity has been proven in numerous contributions, including our own.137 However, applying free‐base counterparts in a similar way remains a tedious task, particularly when the bifunctional properties of the core itself (i.e. basic imine functions for deprotonation, which, in turn, once protonated, become H‐bond donors, and acidic pyrroles for hydrogen‐bond donation) are to be exploited for probing and catalysis. As mentioned several times, this is because the inner nitrogen atoms are “hidden” for steric reasons and cannot usually be contacted by suitable reaction components (Figure 27).
Figure 27.
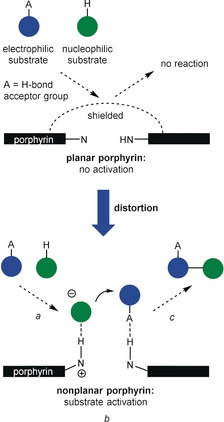
Concept of distortion‐dependent bifunctional porphyrin organocatalysts. Due to the “shielded” core, planar porphyrins cannot activate the electrophilic reaction component through hydrogen bonding or the nucleophilic reagent through deprotonation followed by hydrogen bonding. However, distortion is a method to render the porphyrin catalytically active (a: binding of substrates; b: activation of both substrates and reaction; c: release of the product).
And while free‐base porphyrins, thanks to their various (optical) properties that change upon alteration of the microenvironment of the tetrapyrroles, were indeed used as probes for, for example, sensing of VOCs,158 water and ethanol,159 gases,160 and furthermore, as was recently summarized,142d ions and ion pairs, ROS, chiral discrimination, and as thermosensitive probes, specific tailoring of the free‐base macrocycle skeleton as a form of molecular engineering towards catalysis, improved substrate binding/recognition, and probing appears underrepresented, leaving, in turn, plenty of room for innovation.
Comparable to sensing, during catalysis the substrates bind to the active center and with this being the case, molecular engineering of the porphyrin macrocycle can principally be used to increase selectivity and sensitivity. Having conducted studies on conformational design of distorted porphyrins,1, 3a, 119, 132, 161 and taking into account the accessibility of their nitrogen atoms for hydrogen bonding,18d, 88 we elaborated a model of bifunctional substrate activation as a potential avenue towards porphyrin‐based organocatalysis.3b Therein, sad‐type macrocycles were necessary to deliver catalytically active free‐base porphyrins. Following this proposal, conformational control was exerted to produce tetrapyrroles with enhanced basicity and a core that was available for hydrogen bonding (distortion‐dependent hydrogen bonding).137 Moreover, while previously, all natural and synthetic catalytically active porphyrins were dependent on metallation, this allowed us to apply highly substituted (e.g., 74) and N‐methylated compounds (e.g., 99) as bifunctional organocatalysts. Altogether, this opened a new functional role and points at future perspectives for sensor design based on conformational control.
Specifically, H2OETPP (74) gave the best results and catalyzed a sulfa‐Michael addition of tert‐butyl benzylmercaptan (96) to phenyl vinyl sulfone (97) to afford adduct 98 quantitatively; a reaction susceptible to bifunctional catalysis (Figure 28, top). This was also compared to the performance of various common bases, including 4‐dimethylaminopyridine (DMAP), triethylamine (TEA), and 1,8‐diazabicyclo[5.4.0]undec‐7‐ene (DBU). While the use of weak amine bases failed to promote the reaction, the conversion using 74 was comparable to that of TEA. However, we are confident that in the future, conformationally designed porphyrins will surpass the performance of standard bases due to their greater tunability and superior potential for functionalization.
Figure 28.
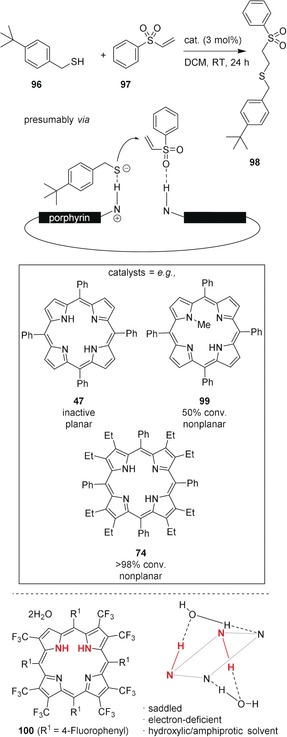
Top: Porphyrin‐catalyzed sulfa‐Michael reaction of 96 and 97.137 Bottom: Structure of cis‐NH‐porphyrin 100⋅2 H2O.162
To illustrate this prospect, one may also take a look at the recently isolated cis‐NH‐porphyrins:18d, 162 Ghosh and co‐workers laid the foundation for new applications of nonplanar porphyrins when they achieved stabilization of cis‐NH‐tautomers (Figure 28, bottom). Depending on solvent, substituents, and conformational effects, the core itself was altered substantially without losing its bifunctionality and the stable cis‐tautomers could show unique binding, sensing, and catalytic properties.
5. Summary and Outlook
This Review shows that porphyrins and their nonaromatic analogues offer rich opportunities for the development of ligands with multiple functions. Due to the diverse properties that are tunable over a wide range of parameters, porphyrinoids are legitimately one of the most important classes of macrocyclic ligands altogether. Thanks to their ability to engage in various covalent and non‐covalent binding modes, including metal and axial coordination, as well as peripheral covalent and hydrogen bonds, very similar tetrapyrrolic systems may fulfill versatile natural and artificial functions. They have a pronounced biological role and omnipresence in nature and many examples show that porphyrin ligands were successfully utilized for the development of, for example, new (electroactive) materials, supramolecular systems, biomimics, and beyond, ranging from surface technology to DSSCs to medicinal research, to name but a few examples.
While planar (metallo)porphyrins are indeed used as sensors, catalysts, etc.,—applications that are highly sought after by scientists—only few cases exist where specific modulation of the macrocyclic skeleton itself was used to tune the binding properties of the ligands.
Prior to discussing “true” porphyrins, we reached out to nonaromatic porphyrinoids—calixpyrroles and calixphyrins—and highlighted their conformational flexibility and unique stereochemical features, which granted them a pole position as N−H⋅⋅⋅X‐type H‐bonding‐based ligands, sensors, assays, and catalysts. Next, approaching “true” porphyrins, we presented two methods to access their otherwise “hidden” bifunctional core and to utilize them in N−H⋅⋅⋅X bonds, specifically protonation and peripheral substitution with sterically demanding substituents to create (core) acids and highly substituted macrocycles, respectively. These alterations can lead to saddle‐type out‐of‐plane distortion and outwards orientation of the central amine and imine groups, which allows them to be contacted by surrounding small molecules. As such, diacids have been widely used in supramolecular chemistry and in some cases as anion sensors. On the other hand, highly substituted porphyrins are neutral free bases that take deformed shapes as a result of repulsive peri‐interactions. This results in the formation of binding pockets that allow them to incorporate and bind guest molecules.
Ultimately, a novel approach yielded the first example of bifunctional porphyrin organocatalysts of tunable basicity,137 which, in turn, is but a screenshot of the many opportunities that conformational design and molecular engineering of free‐base tetrapyrroles towards, for example, distortion‐dependent sensors and catalysts offer. In this context, we introduced a concept of tailoring free‐base tetrapyrroles of specific three‐dimensional conformations as a promising avenue towards shape‐selective ligands with increased conformational flexibility of the macrocycle (core) and central nitrogen atoms. Therein, free bases readily engage in the formation of intermolecular hydrogen bonds, a feature that would otherwise be near to inaccessible. Based on this principle, we propose that the N−H⋅⋅⋅X binding motif in tetrapyrrole ligands can be exploited to a previously unknown extent by means of conformational control to produce improved receptors, sensors, catalysts, or even drugs, advanced (supramolecular) materials, etc. Hopefully, this will open new functional roles and lead to a rethinking, if not renaissance, of the use of natural and synthetic porphyrins, particularly their metal‐free derivatives, with a plenitude of new and up‐and‐coming applications on offer.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Marc Kielmann, M.Sc., studied chemistry (B.Sc.) and medicinal and natural product chemistry (M.Sc.) at the Leibniz University of Hannover. During his master's degree, he did research on ferrocene/ferrocenium chemistry under the supervision of Prof. Dr. Holger Butenschön. He is currently a final year Ph.D. student in the group of Prof. Senge at Trinity College Dublin, working on the synthesis of nonplanar porphyrins for use as organocatalysts and sensors.

Biographical Information
Mathias O. Senge studied chemistry and biochemistry in Freiburg, Amherst, Marburg, and Lincoln. After a Ph.D. from the Universität Marburg (1989) for work with Prof. Horst Senger and postdoctoral studies with Prof. K. M. Smith at UC Davis he received his habilitation in 1996 from the Freie Universität Berlin. Following a Heisenberg fellowship he was appointed Professor of Organic Chemistry at the Universität Potsdam and since 2005 holds the Chair of Organic Chemistry at Trinity College Dublin. His interests are synthetic organic chemistry, the (bio)chemistry of tetrapyrroles, photobiology and photomedicine, photoactive materials, structural chemistry, and history of science.

Acknowledgements
This work was supported by a grant from Science Foundation Ireland (SFI IvP 13/IA/1894) and previous funding over many years by Science Foundation Ireland and the Deutsche Forschungsgemeinschaft is gratefully acknowledged.
M. Kielmann, M. O. Senge, Angew. Chem. Int. Ed. 2019, 58, 418.
Dedicated to Professor Everly B. Fleischer
References
- 1. Senge M. O., Chem. Commun. 2006, 243–256. [DOI] [PubMed] [Google Scholar]
- 2. Huber R., Eur. J. Biochem. 1990, 187, 283–305. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Senge M. O., MacGowan S. A., O'Brien J. M., Chem. Commun. 2015, 51, 17031–17063; [DOI] [PubMed] [Google Scholar]
- 3b. Senge M. O., ECS Trans. 2015, 66, 1–10. [Google Scholar]
- 4.
- 4a. Ding Y., Zhu W.-H., Xie Y., Chem. Rev. 2017, 117, 2203–2256; [DOI] [PubMed] [Google Scholar]
- 4b. Biesaga M., Pyrzynska K., Trojanowicz M., Talanta 2000, 51, 209–224; [DOI] [PubMed] [Google Scholar]
- 4c. Zhang Z., Kim D. S., Lin C.-Y., Zhang H., Lammer A. D., Lynch V. M., Popov I., Miljanić O. Š., Anslyn E. V., Sessler J. L., J. Am. Chem. Soc. 2015, 137, 7769–7774; [DOI] [PubMed] [Google Scholar]
- 4d. Minegishi S., Yumura A., Miyoshi H., Negi S., Taketani S., Motterlini R., Foresti R., Kano K., Kitagishi H., J. Am. Chem. Soc. 2017, 139, 5984–5991. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Hayashi T., Ogoshi H., Chem. Soc. Rev. 1997, 26, 355–364; [Google Scholar]
- 5b. Zawadzka M., Wang J., Blau W. J., Senge M. O., J. Phys. Chem. A 2013, 117, 15–26; [DOI] [PubMed] [Google Scholar]
- 5c. Harsha Vardhan Reddy M., Al-Shammari R. M., Al-Attar N., Kennedy E., Rogers L., Lopez S., Senge M. O., Keyes T. E., Rice J. H., Phys. Chem. Chem. Phys. 2014, 16, 4386–4393; [DOI] [PubMed] [Google Scholar]
- 5d. Liu J., Zhou W., Liu J., Howard I., Kilibarda G., Schlabach S., Coupry D., Addicoat M., Yoneda S., Tsutsui Y., Sakurai T., Seki S., Wang Z., Lindemann P., Redel E., Heine T., Wöll C., Angew. Chem. Int. Ed. 2015, 54, 7441–7445; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7549–7553; [Google Scholar]
- 5e. Haupt S., Lazar I., Weitman H., Senge M. O., Ehrenberg B., Phys. Chem. Chem. Phys. 2015, 17, 11412–11422; [DOI] [PubMed] [Google Scholar]
- 5f. Richert S., Bullard G., Rawson J., Angiolillo P. J., Therien M. J., Timmel C. R., J. Am. Chem. Soc. 2017, 139, 5301–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Ethirajan M., Chen Y., Joshi P., Pandey R. K., Chem. Soc. Rev. 2011, 40, 340–362; [DOI] [PubMed] [Google Scholar]
- 6b. Moylan C., Scanlan E. M., Senge M. O., Curr. Med. Chem. 2015, 22, 2238–2348; [DOI] [PubMed] [Google Scholar]
- 6c. Zou Q., Abbas M., Zhao L., Li S., Shen G., Yan X., J. Am. Chem. Soc. 2017, 139, 1921–1927; [DOI] [PubMed] [Google Scholar]
- 6d. Callaghan S., Senge M. O., Photochem. Photobiol. Sci. 2018, 10.1039/C8PP00008E. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Senge M. O., Fazekas M., Notaras E. G. A., Blau W. J., Zawadzka M., Locos O. B., Ni Mhuircheartaigh E. M., Adv. Mater. 2007, 19, 2737–2774; [Google Scholar]
- 7b. Rawson J., Stuart A. C., You W., Therien M. J., J. Am. Chem. Soc. 2014, 136, 17561–17569; [DOI] [PubMed] [Google Scholar]
- 7c. Zawadzka M., Wang J., Blau W. J., Senge M. O., Photochem. Photobiol. Sci. 2013, 12, 996–1007. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Roznyatovskiy V. V., Lee C.-H., Sessler J. L., Chem. Soc. Rev. 2013, 42, 1921–1933; [DOI] [PubMed] [Google Scholar]
- 8b. Shimizu S., Chem. Rev. 2017, 117, 2730–2784; [DOI] [PubMed] [Google Scholar]
- 8c. Sarma T., Panda P. K., Chem. Rev. 2017, 117, 2785–2838; [DOI] [PubMed] [Google Scholar]
- 8d. Szyszko B., Białek M. J., Pacholska-Dudziak E., Latos-Grażyński L., Chem. Rev. 2017, 117, 2839–2909; [DOI] [PubMed] [Google Scholar]
- 8e. Mack J., Chem. Rev. 2017, 117, 3444–3478. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Senge M. O., Chem. Commun. 2011, 47, 1943–1960; [DOI] [PubMed] [Google Scholar]
- 9b. Lindsey J. S., Acc. Chem. Res. 2010, 43, 300–311; [DOI] [PubMed] [Google Scholar]
- 9c. Hiroto S., Miyake Y., Shinokubo H., Chem. Rev. 2017, 117, 2910–3043. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Aratani N., Osuka A., Kim Y. H., Jeong D. H., Kim D., Angew. Chem. Int. Ed. 2000, 39, 1458–1462; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 1517–1521; [Google Scholar]
- 10b. Cho H. S., Jeong D. H., Cho S., Kim D., Matsuzaki Y., Tanaka K., Tsuda A., Osuka A., J. Am. Chem. Soc. 2002, 124, 14642–14654; [DOI] [PubMed] [Google Scholar]
- 10c. Shinokubo H., Osuka A., Chem. Commun. 2009, 1011–1021; [DOI] [PubMed] [Google Scholar]
- 10d. Jiang H.-W., Tanaka T., Mori H., Park K. H., Kim D., Osuka A., J. Am. Chem. Soc. 2015, 137, 2219–2222; [DOI] [PubMed] [Google Scholar]
- 10e. O'Sullivan M. C., Sprafke J. K., Kondratuk D. V., Rinfray C., Claridge T. D. W., Saywell A., Blunt M. O., O'Shea J. N., Beton P. H., Malfois M., Anderson H. L., Nature 2011, 469, 72–75; [DOI] [PubMed] [Google Scholar]
- 10f. Liu P., Hisamune Y., Peeks M. D., Odell B., Gong J. Q., Herz L. M., Anderson H. L., Angew. Chem. Int. Ed. 2016, 55, 8358–8362; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8498–8502. [Google Scholar]
- 11.
- 11a. Auwärter W., Écija D., Klappenberger F., Barth J. V., Nat. Chem. 2015, 7, 105–120; [DOI] [PubMed] [Google Scholar]
- 11b. Gottfried J. M., Surf. Sci. Rep. 2015, 70, 259–379. [Google Scholar]
- 12. Scheidt W. R., Lee Y. J., Struct. Bonding 1987, 64, 1–70. [Google Scholar]
- 13.
- 13a. Song W. J., Seo M. S., George S. D., Ohta T., Song R., Kang M.-J., Tosha T., Kitagawa T., Solomon E. I., Nam W., J. Am. Chem. Soc. 2007, 129, 1268–1277; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Khade R. L., Zhang Y., J. Am. Chem. Soc. 2015, 137, 7560–7563; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Kuijpers P. F., Tiekink M. J., Breukelaar W. B., Broere D. L. J., van Leest N. P., van der Vlugt J. I., Reek J. N. H., de Bruin B., Chem. Eur. J. 2017, 23, 7945–7952; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13d. Liu Y., Xu W., Zhang J., Fuller W., Schulz C. E., Li J., J. Am. Chem. Soc. 2017, 139, 5023–5026. [DOI] [PubMed] [Google Scholar]
- 14. Tetrapyrroles: Birth, Life and Death (Eds.: M. J. Warren, A. G. Smith), Landes Bioscience, Austin, Texas, 2009. [Google Scholar]
- 15. Senge M. O., Ryan A. A., Letchford K. A., MacGowan S. A., Mielke T., Symmetry 2014, 6, 781–843. [Google Scholar]
- 16. Kräutler B., Biochem. Soc. Trans. 2005, 33, 806–810. [DOI] [PubMed] [Google Scholar]
- 17. Beletskaya I., Tyurin V. S., Tsivadze A. Y., Guilard R., Stern C., Chem. Rev. 2009, 109, 1659–1713. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Wehrle B., Limbach H.-H., Köcher M., Ermer O., Vogel E., Angew. Chem. Int. Ed. Engl. 1987, 26, 934–936; [Google Scholar]; Angew. Chem. 1987, 99, 914–917; [Google Scholar]
- 18b. Wacker P., Dahms K., Senge M. O., Kleinpeter E., J. Org. Chem. 2008, 73, 2182–2190; [DOI] [PubMed] [Google Scholar]
- 18c. Gawinkowski S., Orzanowska G., Izdebska K., Senge M. O., Waluk J., Chem. Eur. J. 2011, 17, 10039–10049; [DOI] [PubMed] [Google Scholar]
- 18d. Thomas K. E., McCormick L. J., Vazquez-Lima H., Ghosh A., Angew. Chem. Int. Ed. 2017, 56, 10088–10092; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10222–10226. [Google Scholar]
- 19.International Union of Pure and Applied Chemistry (IUPAC), 2014, Compendium of Chemical Terminology, Gold Book, p. 835, retrieved from http://goldbook.iupac.org/pdf/goldbook.pdf, accessed March 2018.
- 20. Steiner T., Angew. Chem. Int. Ed. 2002, 41, 48–76; [Google Scholar]; Angew. Chem. 2002, 114, 50–80. [Google Scholar]
- 21.
- 21a. Shelnutt J. A., Song X.-Z., Ma J.-G., Jia S.-L., Jentzen W., Medforth C. J., Chem. Soc. Rev. 1998, 27, 31–42; [Google Scholar]
- 21b. Barkigia K. M., Renner M. W., Senge M. O., Fajer J., J. Phys. Chem. B 2004, 108, 2173–2180. [Google Scholar]
- 22. Desiraju G. R., Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327; [Google Scholar]; Angew. Chem. 1995, 107, 2541–2558. [Google Scholar]
- 23. Villari V., Mineo P., Scamporrino E., Micali N., RSC Adv. 2012, 2, 12989–12998. [Google Scholar]
- 24. Bhyrappa P., Wilson S. R., Suslick K. S., J. Am. Chem. Soc. 1997, 119, 8492–8502. [Google Scholar]
- 25.
- 25a. Lei S. B., Wang C., Yin S. X., Wang H. N., Xi F., Liu H. W., Xu B., Wan L. J., Bai C. L., J. Phys. Chem. B 2001, 105, 10838–10841; [Google Scholar]
- 25b. Zhang Z., Imae T., Nano Lett. 2001, 1, 241–243. [Google Scholar]
- 26. Radivojevic I., Likhtina I., Shi X., Singh S., Drain C. M., Chem. Commun. 2010, 46, 1643–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ishizuka T., Sankar M., Yamada Y., Fukuzumi S., Kojima T., Chem. Commun. 2012, 48, 6481–6483. [DOI] [PubMed] [Google Scholar]
- 28. Garcia-Lekue A., González-Moreno R., Garcia-Gil S., Pickup D. F., Floreano L., Verdini A., Cossaro A., Martín-Gago J. A., Arnau A., Rogero C., J. Phys. Chem. C 2012, 116, 15378–15384. [Google Scholar]
- 29.
- 29a. Pagola S., Stephens P. W., Bohle D. S., Kosar A. D., Madsen S. K., Nature 2000, 404, 307–310; [DOI] [PubMed] [Google Scholar]
- 29b. Senge M. O., Hatscher S., ChemBioChem 2000, 1, 247–249. [DOI] [PubMed] [Google Scholar]
- 30. Oostergetel G. T., van Amerongen H., Boekema E. J., Photosynth. Res. 2010, 104, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang W., Li B., Wang H., Alduhaish O., Alfooty K., Zayed M. A., Li P., Arman H. D., Chen B., Cryst. Growth Des. 2015, 15, 2000–2004. [Google Scholar]
- 32. Zeininger L., Lodermeyer F., Costa R. D., Guldi D. M., Hirsch A., Chem. Commun. 2016, 52, 8842–8845. [DOI] [PubMed] [Google Scholar]
- 33. Paoli M., Marles-Wright J., Smith A., DNA Cell Biol. 2002, 21, 271–280. [DOI] [PubMed] [Google Scholar]
- 34. Yeh S.-R., Han S., Rousseau D. L., Acc. Chem. Res. 1998, 31, 727–736. [Google Scholar]
- 35. Bowman S. E. J., Bren K. L., Nat. Prod. Rep. 2008, 25, 1118–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.
- 36a. Berghuis A. M., Brayer G. D., J. Mol. Biol. 1992, 223, 959–976; [DOI] [PubMed] [Google Scholar]
- 36b. Jentzen W., Song X.-Z., Shelnutt J. A., J. Phys. Chem. B 1997, 101, 1684–1699. [Google Scholar]
- 37. Hodge J. A., Hill M. G., Gray H. B., Inorg. Chem. 1995, 34, 809–812. [Google Scholar]
- 38.
- 38a. Arnold P. A., Benson D. R., Brink D. J., Hendrich M. P., Jas G. S., Kennedy M. L., Petasis D. T., Wang M., Inorg. Chem. 1997, 36, 5306–5315; [Google Scholar]
- 38b. Cowley A. B., Kennedy M. L., Silchenko S., Lukat-Rodgers G. S., Rodgers K. R., Benson D. R., Inorg. Chem. 2006, 45, 9985–10001. [DOI] [PubMed] [Google Scholar]
- 39.
- 39a. Ma J.-G., Laberge M., Song X.-Z., Jentzen W., Jia S.-L., Zhang J., Vanderkooi J. M., Shelnutt J. A., Biochemistry 1998, 37, 5118–5128; [DOI] [PubMed] [Google Scholar]
- 39b. Ma J.-G., Vanderkooi J. M., Zhang J., Jia S.-L., Shelnutt J. A., Biochemistry 1999, 38, 2787–2795. [DOI] [PubMed] [Google Scholar]
- 40. Senge M. O., Renner M. W., Kalisch W. W., Fajer J., J. Chem. Soc. Dalton Trans. 2000, 381–385. [Google Scholar]
- 41.
- 41a. Perutz M. F., Nature 1970, 228, 726–734; [DOI] [PubMed] [Google Scholar]
- 41b. van Grondelle R., Dekker J. P., Gillbro T., Sundstrom V., Biochim. Biophys. Acta Bioenerg. 1994, 1187, 1–65. [Google Scholar]
- 42. Freer A., Prince S., Sauer K., Papiz M., Hawthornthwaite-Lawless A., McDermott G., Cogdell R., Isaacs N. W., Structure 1996, 4, 449–462. [DOI] [PubMed] [Google Scholar]
- 43.
- 43a. Kendrew J. C., Dickerson R. E., Strandberg B. E., Hart R. G., Davies D. R., Phillips D. C., Shore V. C., Nature 1960, 185, 422–427; [DOI] [PubMed] [Google Scholar]
- 43b. Fenna R. E., Matthews B. W., Nature 1975, 258, 573–577; [Google Scholar]
- 43c. Matthews B. W., Fenna R. E., Acc. Chem. Res. 1980, 13, 309–317; [Google Scholar]
- 43d. Tronrud D. E., Schmid M. F., Matthews B. W., J. Mol. Biol. 1986, 188, 443–454. [DOI] [PubMed] [Google Scholar]
- 44.
- 44a. Ladner R. C., Heidner E. J., Perutz M. F., J. Mol. Biol. 1977, 114, 385–413; [DOI] [PubMed] [Google Scholar]
- 44b. Deatherage J. F., Loe R. S., Moffat K., J. Mol. Biol. 1976, 104, 723–728. [DOI] [PubMed] [Google Scholar]
- 45.
- 45a. Senge M. O., Smith K. M., Photochem. Photobiol. 1991, 54, 841–846; [Google Scholar]
- 45b. Senge M. O., Smith N. W., Smith K. M., Inorg. Chem. 1993, 32, 1259–1265; [Google Scholar]
- 45c. MacGowan S. A., Senge M. O., Inorg. Chem. 2013, 52, 1228–1237; [DOI] [PubMed] [Google Scholar]
- 45d. MacGowan S. A., Senge M. O., Chem. Commun. 2011, 47, 11621–11623; [DOI] [PubMed] [Google Scholar]
- 45e. MacGowan S. A., Senge M. O., Biochim. Biophys. Acta Bioenerg. 2016, 1857, 427–442. [DOI] [PubMed] [Google Scholar]
- 46.
- 46a. Beer P. D., Gale P. A., Angew. Chem. Int. Ed. 2001, 40, 486–516; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 502–532; [Google Scholar]
- 46b. Kim S. K., Sessler J. L., Chem. Soc. Rev. 2010, 39, 3784–3809; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46c. Busschaert N., Caltagirone C., Rossom W. V., Gale P. A., Chem. Rev. 2015, 115, 8038–8155; [DOI] [PubMed] [Google Scholar]
- 46d. Vargas-Zúñiga G. I., Sessler J. L., Coord. Chem. Rev. 2017, 345, 281–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baeyer A., Ber. Dtsch. Chem. Ges. 1886, 19, 2184–2185. [Google Scholar]
- 48.
- 48a. Jacoby D., Floriani C., Chiesi-Villa A., Rizzoli C., J. Chem. Soc. Chem. Commun. 1991, 220–222; [Google Scholar]
- 48b. Bonomo L., Dandin O., Solari E., Floriani C., Scopelliti R., Angew. Chem. Int. Ed. 1999, 38, 913–915; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 963–966; [Google Scholar]
- 48c. Floriani C., Floriani-Moro R., Adv. Organomet. Chem. 2001, 47, 167–233. [Google Scholar]
- 49. Sessler J. L., Weghorn S. J., Expanded, Contracted & Isomeric Porphyrins, Pergamon Press, Oxford, 1997. [Google Scholar]
- 50. Sessler J. L., Camiolo S., Gale P. A., Coord. Chem. Rev. 2003, 240, 17–55. [Google Scholar]
- 51.
- 51a. Gale P. A., Sessler J. L., Král V., Lynch V., J. Am. Chem. Soc. 1996, 118, 5140–5141; [Google Scholar]
- 51b. Gale P. A., Sessler J. L., Král V., Chem. Commun. 1998, 1–8. [Google Scholar]
- 52. Miyaji H., P. Anzenbacher, Jr. , Sessler J. L., Bleasdale E. R., Gale P. A., Chem. Commun. 1999, 1723–1724. [Google Scholar]
- 53.
- 53a. Gale P. A., Twyman L. J., Handlin C. I., Sessler J. L., Chem. Commun. 1999, 1851–1852; [Google Scholar]
- 53b. Camiolo S., Gale P. A., Chem. Commun. 2000, 1129–1130; [Google Scholar]
- 53c. Woods C. J., Camiolo S., Light M. E., Coles S. J., Hursthouse M. B., King M. A., Gale P. A., Essex J. W., J. Am. Chem. Soc. 2002, 124, 8644–8652; [DOI] [PubMed] [Google Scholar]
- 53d. Kim S. K., Lynch V. M., Sessler J. L., Org. Lett. 2014, 16, 6128–6131. [DOI] [PubMed] [Google Scholar]
- 54. Alešković M., Halasz I., Basarić N., Mlinarić-Majerski K., Tetrahedron 2009, 65, 2051–2058. [Google Scholar]
- 55. Medforth C. J., Smith K. M., Tetrahedron Lett. 1990, 31, 5583–5586. [Google Scholar]
- 56.
- 56a. Allen W. E., Gale P. A., Brown C. T., Lynch V. M., Sessler J. L., J. Am. Chem. Soc. 1996, 118, 12471–12472; [Google Scholar]
- 56b. Furusho Y., Aida T., Chem. Commun. 1997, 2205–2206. [Google Scholar]
- 57. Adriaenssens L., Ballester P., Chem. Soc. Rev. 2013, 42, 3261–3277. [DOI] [PubMed] [Google Scholar]
- 58.
- 58a.This Figure was generated using Mercury CSD 2.0[48b] and the data downloaded from the Cambridge Crystallographic Data Centre (CCDC): Groom C. R., Bruno I. J., Lightfoot M. P., Ward S. C., Acta Crystallogr. Sect. B 2016, 72, 171–179; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58b. Macrae C. F., Bruno I. J., Chisholm J. A., Edgington P. R., McCabe P., Pidcock E., Rodriguez-Monge L., Taylor R., van de Streek J., Wood P. A., J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar]
- 59.
- 59a. Gale P. A., Sessler J. L., Allen W. E., Tvermoes N. A., Lynch V., Chem. Commun. 1997, 665–666; [Google Scholar]
- 59b. P. Anzenbacher, Jr. , Jursíková K., Lynch V. M., Gale P. A., Sessler J. L., J. Am. Chem. Soc. 1999, 121, 11020–11021. [Google Scholar]
- 60. Escobar L., Aragay G., Ballester P., Chem. Eur. J. 2016, 22, 13682–13689. [DOI] [PubMed] [Google Scholar]
- 61.
- 61a. Sessler J. L., P. Anzenbacher, Jr. , Jursíková K., Miyaji H., Genge J. W., Tvermoes N. A., Allen W. E., Shriver J. A., Gale P. A., Král V., Pure Appl. Chem. 1998, 70, 2401–2408; [Google Scholar]
- 61b. Miyaji H., Sato W., Sessler J. L., Lynch V. M., Tetrahedron Lett. 2000, 41, 1369–1373. [Google Scholar]
- 62.
- 62a. Gale P. A., Genge J. W., Král V., McKervey M. A., Sessler J. L., Walker A., Tetrahedron Lett. 1997, 38, 8443–8444; [Google Scholar]
- 62b. Bonomo L., Solari E., Toraman G., Scopelliti R., Latronico M., Floriani C., Chem. Commun. 1999, 2413–2414; [Google Scholar]
- 62c. Sato W., Miyaji H., Sessler J. L., Tetrahedron Lett. 2000, 41, 6731–6736. [Google Scholar]
- 63. Escobar L., Arroyave F. A., Ballester P., Eur. J. Org. Chem. 2018, 1097–1106. [Google Scholar]
- 64. Gale P. A., Sessler J. L., Lynch V., Sansom P. I., Tetrahedron Lett. 1996, 37, 7881–7884. [Google Scholar]
- 65.
- 65a. Gale P. A., Coord. Chem. Rev. 2001, 213, 79–128; [Google Scholar]
- 65b. Gale P. A., P. Anzenbacher, Jr. , Sessler J. L., Coord. Chem. Rev. 2001, 222, 57–102. [Google Scholar]
- 66.
- 66a. P. Anzenbacher, Jr. , Jursíková K., Sessler J. L., J. Am. Chem. Soc. 2000, 122, 9350–9351; [Google Scholar]
- 66b. Bhatt K. D., Vyas D. J., Makwana B. A., Darjee S. M., Jain V. K., Shah H., Chin. Chem. Lett. 2016, 27, 731–737. [Google Scholar]
- 67. Král V., Sessler J. L., Shishkanova T. V., Gale P. A., Volf R., J. Am. Chem. Soc. 1999, 121, 8771–8775. [Google Scholar]
- 68.
- 68a. Gale P. A., Hursthouse M. B., Light M. E., Sessler J. L., Warriner C. N., Zimmerman R. S., Tetrahedron Lett. 2001, 42, 6759–6762; [Google Scholar]
- 68b. Yang W., Yin Z., Wang C.-H., He J., Zhu X., Cheng J.-P., Tetrahedron 2008, 64, 9244–9252. [Google Scholar]
- 69. Aydogan A., Koca A., Şener M. K., Sessler J. L., Org. Lett. 2014, 16, 3764–3767. [DOI] [PubMed] [Google Scholar]
- 70. Sessler J. L., Gale P. A., Genge J. W., Chem. Eur. J. 1998, 4, 1095–1099. [Google Scholar]
- 71.
- 71a. Cafeo G., De Rosa M., Kohnke F. H., Neri P., Soriente A., Valenti L., Tetrahedron Lett. 2008, 49, 153–155; [Google Scholar]
- 71b. Cafeo G., De Rosa M., Kohnke F. H., Soriente A., Talotta C., Valenti L., Molecules 2009, 14, 2594–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cafeo G., Kohnke F. H., Valenti L., Tetrahedron Lett. 2009, 50, 4138–4140. [Google Scholar]
- 73. Kim D. S., Sessler J. L., Chem. Soc. Rev. 2015, 44, 532–546. [DOI] [PubMed] [Google Scholar]
- 74.
- 74a. Levitskaia T. G., Marquez M., Sessler J. L., Shriver J. A., Vercouter T., Moyer B. A., Chem. Commun. 2003, 2248–2249; [DOI] [PubMed] [Google Scholar]
- 74b. Kim S. K., Sessler J. L., Acc. Chem. Res. 2014, 47, 2525–2536; [DOI] [PubMed] [Google Scholar]
- 74c. Kim S. K., Lee J., Williams N. J., Lynch V. M., Hay B. P., Moyer B. A., Sessler J. L., J. Am. Chem. Soc. 2014, 136, 15079–15085; [DOI] [PubMed] [Google Scholar]
- 74d. Kim S. K., Lynch V. M., Hay B. P., Kim J. S., Sessler J. L., Chem. Sci. 2015, 6, 1404–1413; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74e. He Q., Zhang Z., Brewster J. T., Lynch V. M., Kim S. K., Sessler J. L., J. Am. Chem. Soc. 2016, 138, 9779–9782; [DOI] [PubMed] [Google Scholar]
- 74f. Yeon Y., Leem S., Wagen C., Lynch V. M., Kim S. K., Sessler J. L., Org. Lett. 2016, 18, 4396–4399; [DOI] [PubMed] [Google Scholar]
- 74g. Ellis R. J., Reinhart B., Williams N. J., Moyer B. A., Bryantsev V. S., Chem. Commun. 2017, 53, 5610–5613. [DOI] [PubMed] [Google Scholar]
- 75. Sessler J. L., Andrievsky A., Gale P. A., Lynch V., Angew. Chem. Int. Ed. Engl. 1996, 35, 2782–2785; [Google Scholar]; Angew. Chem. 1996, 108, 2954–2957. [Google Scholar]
- 76. Gil-Ramírez G., Chas M., Ballester P., J. Am. Chem. Soc. 2010, 132, 2520–2521. [DOI] [PubMed] [Google Scholar]
- 77.
- 77a. Park J. S., Le Derf F., Bejger C. M., Lynch V. M., Sessler J. L., Nielsen K. A., Johnsen C., Jeppesen J. O., Chem. Eur. J. 2010, 16, 848–854; [DOI] [PubMed] [Google Scholar]
- 77b. Davis C. M., Lim J. M., Larsen K. R., Kim D. S., Sung Y. M., Lyons D. M., Lynch V. M., Nielsen K. A., Jeppesen J. O., Kim D., Park J. S., Sessler J. L., J. Am. Chem. Soc. 2014, 136, 10410–10417. [DOI] [PubMed] [Google Scholar]
- 78.
- 78a. Cafeo G., Carbotti G., Cuzzola A., Fabbi M., Ferrini S., Kohnke F. H., Papanikolaou G., Plutino M. R., Rosano C., White A. J. P., J. Am. Chem. Soc. 2013, 135, 2544–2551; [DOI] [PubMed] [Google Scholar]
- 78b. He Q., Kelliher M., Bähring S., Lynch V. M., Sessler J. L., J. Am. Chem. Soc. 2017, 139, 7140–7143; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78c. Ko S.-K., Kim S. K., Share A., Lynch V. M., Park J., Namkung W., Rossom W. V., Busschaert N., Gale P. A., Sessler J. L., Shin I., Nat. Chem. 2014, 6, 885–892. [DOI] [PubMed] [Google Scholar]
- 79. Lappano R., Rosano C., Pisano A., Santolla M. F., De Fracesco E. M., De Marco P., Dolce V., Ponassi M., Felli L., Cafeo G., Kohnke F. H., Abonante S., Maggiolini M., Dis. Models Mech. 2015, 8, 1237–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bernátková M., Dvořáková H., Andrioletti B., Král V., Bouř P., J. Phys. Chem. A 2005, 109, 5518–5526. [DOI] [PubMed] [Google Scholar]
- 81.
- 81a. Král V., Sessler J. L., Zimmerman R. S., Seidel D., Lynch V., Andrioletti B., Angew. Chem. Int. Ed. 2000, 39, 1055–1058; [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 1097–1100; [Google Scholar]
- 81b. Bucher C., Seidel D., Lynch V., Král V., Sessler J. L., Org. Lett. 2000, 2, 3103–3106. [DOI] [PubMed] [Google Scholar]
- 82. Buchler J. W., Puppe L., Liebigs Ann. Chem. 1970, 740, 142–163. [Google Scholar]
- 83. Bischoff I., Feng X. D., Senge M. O., Tetrahedron 2001, 57, 5573–5583. [Google Scholar]
- 84. Dehaen W., Top. Heterocycl. Chem. 2010, 24, 75–102. [Google Scholar]
- 85. Bucher C., Zimmerman R. S., Lynch V., Král V., Sessler J. L., J. Am. Chem. Soc. 2001, 123, 2099–2100. [DOI] [PubMed] [Google Scholar]
- 86.
- 86a. Sessler J. L., Zimmerman R. S., Bucher C., Král V., Andrioletti B., Pure Appl. Chem. 2001, 73, 1041–1057; [Google Scholar]
- 86b. Finnigan E. M., Giordani S., Senge M. O., McCabe T., J. Phys. Chem. A 2010, 114, 2464–2470; [DOI] [PubMed] [Google Scholar]
- 86c. Bernátková M., Andrioletti B., Král V., Rose E., Vaissermann J., J. Org. Chem. 2004, 69, 8140–8143; [DOI] [PubMed] [Google Scholar]
- 86d. Buchler J. W., Lay K. L., Lee Y. J., Scheidt W. R., Angew. Chem. Int. Ed. Engl. 1982, 21, 432–432; [Google Scholar]; Angew. Chem. 1982, 94, 456–457. [Google Scholar]
- 87. Ema T., Senge M. O., Nelson N. Y., Ogoshi H., Smith K. M., Angew. Chem. Int. Ed. Engl. 1994, 33, 1879–1881; [Google Scholar]; Angew. Chem. 1994, 106, 1951–1953. [Google Scholar]
- 88. Senge M. O. in The Porphyrin Handbook, Vol. 1 (Eds.: K. M. Kadish, K. M. Smith, R. Guilard), Academic Press, New York, 2000, pp. 239–347. [Google Scholar]
- 89. Stone A., Fleischer E. B., J. Am. Chem. Soc. 1968, 90, 2735–2748. [Google Scholar]
- 90.
- 90a. Senge M. O., Kalisch W. W., Z. Naturforsch. B 1999, 54, 943–959; [Google Scholar]
- 90b. Senge M. O., Z. Naturforsch. B 2000, 55, 336–344. [Google Scholar]
- 91. Senge M. O., Forsyth T. P., Nguyen L. T., Smith K. M., Angew. Chem. Int. Ed. Engl. 1995, 33, 2485–2487; [Google Scholar]; Angew. Chem. 1994, 106, 2554–2557. [Google Scholar]
- 92.
- 92a. Lavallee D. K., The Chemistry and Biochemistry of N-Substituted Porphyrins, VCH, Weinheim, 1987; [Google Scholar]
- 92b. Senge M. O., Kalisch W. W., Runge S., Eur. J. Org. Chem. 1997, 1345–1352; [Google Scholar]
- 92c.unpublished results.
- 93. Webb M. J., Bampos N., Chem. Sci. 2012, 3, 2351–2366. [Google Scholar]
- 94. Rosa A., Ricciardi G., Baerends E. J., Romeo A., Scolaro L. M., J. Phys. Chem. A 2003, 107, 11468–11482. [Google Scholar]
- 95. Sheinin V. B., Ratkova E. L., Mamardashvili N. Z., J. Porphyrins Phthalocyanines 2008, 12, 1211–1219. [Google Scholar]
- 96. Kruk M. M., Starukhin A. S., Mamardashvili N. Z., Mamardashvili G. M., Ivanova Y. B., Maltseva O. V., J. Porphyrins Phthalocyanines 2009, 13, 1148–1158. [Google Scholar]
- 97.
- 97a. Yoshimoto S., Sawaguchi T., J. Am. Chem. Soc. 2008, 130, 15944–15949; [DOI] [PubMed] [Google Scholar]
- 97b. Arai Y., Segawa H., Chem. Commun. 2010, 46, 4279–4281; [DOI] [PubMed] [Google Scholar]
- 97c. Gradova M. A., Kuryakov V. N., Lobanov A. V., Macroheterocycles 2015, 8, 244–251. [Google Scholar]
- 98. Honda T., Nakanishi T., Ohkubo K., Kojima T., Fukuzumi S., J. Am. Chem. Soc. 2010, 132, 10155–10163. [DOI] [PubMed] [Google Scholar]
- 99. Arai Y., Segawa H., J. Phys. Chem. B 2011, 115, 7773–7780. [DOI] [PubMed] [Google Scholar]
- 100. Nakanishi T., Kojima T., Ohkubo K., Hasobe T., Nakayama K.-i., Fukuzumi S., Chem. Mater. 2008, 20, 7492–7500. [Google Scholar]
- 101. Frühbeißer S., Mariani G., Gröhn F., Polymers 2016, 8, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zawadzka M., Wang J., Blau W. J., Senge M. O., J. Porphyrins Phthalocyanines 2013, 17, 1129–1133. [Google Scholar]
- 103. Franco R., Ma J.-G., Lu Y., Ferreira G. C., Shelnutt J. A., Biochemistry 2000, 39, 2517–2529. [DOI] [PubMed] [Google Scholar]
- 104.
- 104a. De Luca G., Romeo A., Scolaro L. M., Ricciardi G., Rosa A., Inorg. Chem. 2007, 46, 5979–5988; [DOI] [PubMed] [Google Scholar]
- 104b. Thyagarajan S., Leiding T., Årsköld S. P., Cheprakov A. V., Vinogradov S. A., Inorg. Chem. 2010, 49, 9909–9920; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104c. Aronoff S., J. Phys. Chem. 1958, 62, 428–431; [Google Scholar]
- 104d. Rudine A. B., DelFatti B. D., Wamser C. C., J. Org. Chem. 2013, 78, 6040–6049; [DOI] [PubMed] [Google Scholar]
- 104e. Neuberger A., Scott F. R. S., Scott J. J., Proc. R. Soc. London, A 1952, 213, 307–326; [Google Scholar]
- 104f. Ogoshi H., Watanabe E., Yoshida Z., Tetrahedron 1973, 29, 3241–3245. [Google Scholar]
- 105. Hirayama N., Takenaka A., Sasada Y., J. Chem. Soc. Chem. Commun. 1974, 330–331. [Google Scholar]
- 106. Honda T., Kojima T., Fukuzumi S., Chem. Commun. 2009, 4994–4996. [DOI] [PubMed] [Google Scholar]
- 107. Suzuki W., Kotani H., Ishizuka T., Ohkubo K., Shiota Y., Yoshizawa K., Fukuzumi S., Kojima T., Chem. Eur. J. 2017, 23, 4669–4679. [DOI] [PubMed] [Google Scholar]
- 108. Suzuki W., Kotani H., Ishizuka T., Shiota Y., Yoshizawa K., Kojima T., Chem. Commun. 2017, 53, 6359–6362. [DOI] [PubMed] [Google Scholar]
- 109. Almarsson Ö., Blaskó A., Bruice T. C., Tetrahedron 1993, 49, 10239–10252. [Google Scholar]
- 110.
- 110a. Al-Karadaghi S., Franco R., Hansson M., Shelnutt J. A., Isaya G., Ferreira G. C., Trends Biochem. Sci. 2006, 31, 135–142; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110b. Medlock A., Swartz L., Dailey T. A., Dailey H. A., Lanzilotta W. N., Proc. Natl. Acad. Sci. USA 2007, 104, 1789–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.
- 111a. Dailey H. A. in Biosynthesis of Heme and Chlorophylls (Ed.: H. A. Dailey), McGraw-Hill, New York, 1990, pp. 123–163; [Google Scholar]
- 111b. Walker C. J., Weinstein J. D., Plant Physiol. 1991, 95, 1189–1196; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111c. Karlberg T., Hansson M. D., Yengo R. K., Johansson R., Thorvaldsen H. O., Ferreira G. C., Hansson M., Al-Karadaghi S., J. Mol. Biol. 2008, 378, 1074–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cochran A. G., Schultz P. G., Science 1990, 249, 781–783. [DOI] [PubMed] [Google Scholar]
- 113.
- 113a. Karger G. A., Reid J. D., Hunter C. N., Biochemistry 2001, 40, 9291–9299; [DOI] [PubMed] [Google Scholar]
- 113b. Lu Y., Sousa A., Franco R., Mangravita A., Ferreira G. C., Moura I., Shelnutt J. A., Biochemistry 2002, 41, 8253–8262; [DOI] [PubMed] [Google Scholar]
- 113c. Shi Z., Franco R., Haddad R., Shelnutt J. A., Ferreira G. C., Biochemistry 2006, 45, 2904–2912. [DOI] [PubMed] [Google Scholar]
- 114. Sigfridsson E., Ryde U., J. Biol. Inorg. Chem. 2003, 8, 273–282. [DOI] [PubMed] [Google Scholar]
- 115. Ravikanth M., Chandrashekar T. K., Struct. Bonding 1995, 82, 105–188. [Google Scholar]
- 116. Senge M. O., J. Photochem. Photobiol. B 1992, 16, 3–36. [Google Scholar]
- 117.
- 117a. Hamor M. J., Hamor T. A., Hoard J. L., J. Am. Chem. Soc. 1964, 86, 1938–1942; [Google Scholar]
- 117b. Fleischer E. B., J. Am. Chem. Soc. 1963, 85, 1353–1354; [Google Scholar]
- 117c. Fleischer E. B., Miller C. K., Webb L. E., J. Am. Chem. Soc. 1964, 86, 2342–2347. [Google Scholar]
- 118.
- 118a. Evans B., Smith K. M., Fuhrhop J.-H., Tetrahedron Lett. 1977, 18, 443–446; [Google Scholar]
- 118b. Medforth C. J., Berber M. D., Smith K. M., Shelnutt J. A., Tetrahedron Lett. 1990, 31, 3719–3722; [Google Scholar]
- 118c. Senge M. O., Acc. Chem. Res. 2005, 38, 733–743. [DOI] [PubMed] [Google Scholar]
- 119. Medforth C. J., Senge M. O., Smith K. M., Sparks L. D., Shelnutt J. A., J. Am. Chem. Soc. 1992, 114, 9859–9869. [Google Scholar]
- 120.
- 120a. Barkigia K. M., Berber M. D., Fajer J., Medforth C. J., Renner M. W., Smith K. M., J. Am. Chem. Soc. 1990, 112, 8851–8857; [Google Scholar]
- 120b. Regev A., Galili T., Medforth C. J., Smith K. M., Barkigia K. M., Fajer J., Levanon H., J. Phys. Chem. 1994, 98, 2520–2526. [Google Scholar]
- 121. Yamamoto Y., Yamamoto A., − S. y. Furuta, M. Horie, M. Kodama, W. Sato, K.−y. Akiba, S. Tsuzuki, T. Uchimaru, D. Hashizume, F. Iwasaki, [DOI] [PubMed] [Google Scholar]; J. Am. Chem. Soc. 2005, 127, 14540–14541. [DOI] [PubMed] [Google Scholar]
- 122. Parusel A. B. J., Wondimagegn T., Ghosh A., J. Am. Chem. Soc. 2000, 122, 6371–6374. [Google Scholar]
- 123. Dolphin D., J. Heterocycl. Chem. 1970, 7, 275–283. [Google Scholar]
- 124.
- 124a. Takeda J., Ohya T., Sato M., Inorg. Chem. 1992, 31, 2877–2880; [Google Scholar]
- 124b. Bhyrappa P., Nethaji M., Krishnan V., Chem. Lett. 1993, 22, 869–872. [Google Scholar]
- 125.
- 125a. Kadish K. M., Caemelbecke E. V., D′Souza F., Medforth C. J., Smith K. M., Tabard A., Guilard R., Inorg. Chem. 1995, 34, 2984–2989; [Google Scholar]
- 125b. Ochsenbein P., Ayougou K., Mandon D., Fischer J., Weiss R., Austin R. N., Jayaraj K., Gold A., Terner J., Fajer J., Angew. Chem. Int. Ed. Engl. 1994, 33, 348–350; [Google Scholar]; Angew. Chem. 1994, 106, 355–357. [Google Scholar]
- 126.
- 126a. Gentemann S., Medforth C. J., Forsyth T. P., Nurco D. J., Smith K. M., Fajer J., Holten D., J. Am. Chem. Soc. 1994, 116, 7363–7368; [Google Scholar]
- 126b. Gentemann S., Medforth C. J., Ema T., Nelson N. Y., Smith K. M., J, Fajer , Holten D., Chem. Phys. Lett. 1995, 245, 441–447; [Google Scholar]
- 126c. Drain C. M., Kirmaier C., Medforth C. J., Nurco D. J., Smith K. M., Holten D., J. Phys. Chem. 1996, 100, 11984–11993; [Google Scholar]
- 126d. Gentemann S., Nelson N. Y., Jaquinod L., Nurco D. J., Leung S. H., Medforth C. J., Smith K. M., Fajer J., Holten D., J. Phys. Chem. B 1997, 101, 1247–1254. [Google Scholar]
- 127. Renner M. W., Barkigia K. M., Zhang Y., Medforth C. J., Smith K. M., Fajer J., J. Am. Chem. Soc. 1994, 116, 8582–8592. [Google Scholar]
- 128. Tagliatesta P., Li J., Autret M., Caemelbecke E. V., Villard A., D′Souza F., Kadish K. M., Inorg. Chem. 1996, 35, 5570–5576. [DOI] [PubMed] [Google Scholar]
- 129. Nurco D. J., Medforth C. J., Forsyth T. P., Olmstead M. M., Smith K. M., J. Am. Chem. Soc. 1996, 118, 10918–10919. [Google Scholar]
- 130. Fajer J., J. Pept. Res. 2000, 4, 382–385. [Google Scholar]
- 131. Barkigia K. M., Renner M. W., Furenlid L. R., Medforth C. J., Smith K. M., Fajer J., J. Am. Chem. Soc. 1993, 115, 3627–3635. [Google Scholar]
- 132. Senge M. O., Kalisch W. W., Inorg. Chem. 1997, 36, 6103–6116. [DOI] [PubMed] [Google Scholar]
- 133. Sparks L. D., Medforth C. J., Park M.-S., Chamberlain J. R., Ondrias M. R., Senge M. O., Smith K. M., Shelnutt J. A., J. Am. Chem. Soc. 1993, 115, 581–592. [Google Scholar]
- 134. Barkigia K. M., Nurco D. J., Renner M. W., Melamed D., Smith K. M., Fajer J., J. Phys. Chem. B 1998, 102, 322–326. [Google Scholar]
- 135.A putative triclinic modification, which upon closer inspection appears to be a core monoacid with a N−H-bonded ethoxide: Senge M. O., Z. Naturforsch. B 1999, 54, 821–824. [Google Scholar]
- 136. Kielmann M., Flanagan K. J., Norvaiša K., Intrieri D., Senge M. O., J. Org. Chem. 2017, 82, 5122–5134. [DOI] [PubMed] [Google Scholar]
- 137. Roucan M., Kielmann M., Connon S. J., Bernhard S. S. R., Senge M. O., Chem. Commun. 2018, 54, 26–29. [DOI] [PubMed] [Google Scholar]
- 138. Kielmann M., Prior C., Senge M. O., New J. Chem. 2018, 42, 7529–7550. [Google Scholar]
- 139. Paolesse R., Nardis S., Monti D., Stefanelli M., Di Natale C., Chem. Rev. 2017, 117, 2517–2583. [DOI] [PubMed] [Google Scholar]
- 140. Zhang Q., Zheng X., Kuang G., Wang W., Zhu L., Pang R., Shi X., Shang X., Huang X., Liu P. N., Lin N., J. Phys. Chem. Lett. 2017, 8, 1241–1247. [DOI] [PubMed] [Google Scholar]
- 141. Furusho Y., Kimura T., Mizuno Y., Aida T., J. Am. Chem. Soc. 1997, 119, 5267–5268. [Google Scholar]
- 142.
- 142a. Askim J. R., Mahmoudi M., Suslick K. S., Chem. Soc. Rev. 2013, 42, 8649–8682; [DOI] [PubMed] [Google Scholar]
- 142b. Wang L., Li H., Deng J., Cao D., Curr. Org. Chem. 2013, 17, 3078–3091; [Google Scholar]
- 142c. Ishihara S., Labuta J., Rossom W. V., Ishikawa D., Minami K., Hill J. P., Ariga K., Phys. Chem. Chem. Phys. 2014, 16, 9713–9746; [DOI] [PubMed] [Google Scholar]
- 142d. Lee H., Hong K.-I., Jang W.-D., Coord. Chem. Rev. 2018, 354, 46–73. [Google Scholar]
- 143. Venkatramaiah N., Pereira C. F., Mendes R. F., Almeida Paz F. A., Tomé J. P. C., Anal. Chem. 2015, 87, 4515–4522. [DOI] [PubMed] [Google Scholar]
- 144. Rana A., Panda P. K., RSC Adv. 2012, 2, 12164–12168. [Google Scholar]
- 145. Ema T., Ura N., Eguchi K., Ise Y., Sakai T., Chem. Commun. 2011, 47, 6090–6092. [DOI] [PubMed] [Google Scholar]
- 146. Yang J., Wang Z., Hu K., Li Y., Feng J., Shi J., Gu J., ACS Appl. Mater. Interfaces 2015, 7, 11956–11964. [DOI] [PubMed] [Google Scholar]
- 147. Neurosciences—From Molecule to Behavior: a university textbook (Eds.: C. G. Galizia, P.-M. Lledo), Springer, Heidelberg, 2013. [Google Scholar]
- 148.
- 148a. Persaud K., Dodd G., Nature 1982, 299, 352–355; [DOI] [PubMed] [Google Scholar]
- 148b. Röck F., Barsan N., Weimar U., Chem. Rev. 2008, 108, 705–725; [DOI] [PubMed] [Google Scholar]
- 148c. Suslick K., MRS Bull. 2004, 29, 720–725; [DOI] [PubMed] [Google Scholar]
- 148d. Vlasov Y., Legin A., Rudnitskaya A., Di Natale C., D′Amico A., Pure Appl. Chem. 2005, 77, 1965–1983; [Google Scholar]
- 148e. Tahara Y., Toko K., IEEE Sens. J. 2013, 13, 3001–3011. [Google Scholar]
- 149.
- 149a. Catini A., Kumar R., Capuano R., Martinelli E., Paolesse R., Di Natale C., Sensors 2016, 16, 1640; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149b. Di Natale C., Paolesse R., Macagnano A., Mantini A., Mari P., D′Amico A., Sens. Actuators B 2000, 68, 319–323; [Google Scholar]
- 149c. Di Natale C., Salimbeni D., Paolesse R., Macagnano A., D′Amico A., Sens. Actuators B 2000, 65, 220–226; [Google Scholar]
- 149d. D′Amico A., Di Natale C., Paolesse R., Macagnano A., Mantini A., Sens. Actuators B 2000, 65, 209–215; [Google Scholar]
- 149e. Di Natale C., Paolesse R., Macagnano A., Troitsky V. I., Berzina T. S., D′Amico A., Anal. Chim. Acta 1999, 384, 249–259; [Google Scholar]
- 149f. Di Natale C., Paolesse R., Macagnano A., Mantini A., Goletti C., D′Amico A., Sens. Actuators B 1998, 52, 162–168. [Google Scholar]
- 150.
- 150a. Leopold J. H., Bos L. D. J., Sterk P. J., Schultz M. J., Fens N., Horvath I., Bikov A., Montuschi P., Di Natale C., Yates D. H., Abu-Hanna A., J. Breath Res. 2015, 9, 046002; [DOI] [PubMed] [Google Scholar]
- 150b. Di Natale C., Macagnano A., Davide F., D′Amico A., Paolesse R., Boschi T., Faccio M., Ferri G., Sens. Actuators B 1997, 44, 521–526. [Google Scholar]
- 151.
- 151a. Rakow N. A., Suslick K. S., Nature 2000, 406, 710–713; [DOI] [PubMed] [Google Scholar]
- 151b. Rakow N. A., Suslick K. S. in Artificial Chemical Sensing: Olfaction and the Electronic Nose (Eds.: J. R. Stetter, W. R. Penrose), The Electrochemical Society, Pennington, 2001, pp. 8–13; [Google Scholar]
- 151c. Suslick K. S., Rakow N. A., Sen A., Tetrahedron 2004, 60, 11133–11138. [Google Scholar]
- 152.
- 152a. Askim J. R., Li Z., LaGasse M. K., Rankin J. M., Suslick K. S., Chem. Sci. 2016, 7, 199–206; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152b. Li Z., Bassett W. P., Askim J. R., Suslick K. S., Chem. Commun. 2015, 51, 15312–15315. [DOI] [PubMed] [Google Scholar]
- 153. Zhang Y., Askim J. R., Zhong W., Orlean P., Suslick K. S., Analyst 2014, 139, 1922–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Lim S. H., Feng L., Kemling J. W., Musto C. J., Suslick K. S., Nat. Chem. 2009, 1, 562–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Li Z., Suslick K. S., ACS Sens. 2016, 1, 1330–1335. [Google Scholar]
- 156. Janzen M. C., Ponder J. B., Bailey D. P., Ingison C. K., Suslick K. S., Anal. Chem. 2006, 78, 3591–3600. [DOI] [PubMed] [Google Scholar]
- 157.
- 157a. Zilberman Y., Chen Y., Sonkusale S. R., Sens. Actuators B 2014, 202, 976–983; [Google Scholar]
- 157b. Mensing J. P., Wisitsoraat A., Tuantranont A., Kerdcharoen T., Sens. Actuators B 2013, 176, 428–436; [Google Scholar]
- 157c. Yang W., Xu J., Mao Y., Yang Y., Jiang Y., Synth. React. Inorg. Met.-Org. Chem. 2016, 46, 735–740. [Google Scholar]
- 158. Brittle S. A., Richardson T. H., Dunbar A. D. F., Turega S. M., Hunter C. A., J. Mater. Chem. 2011, 21, 4882–4887. [Google Scholar]
- 159. Carturan S., Tonezzer M., Quaranta A., Maggioni G., Buffa M., Milan R., Sens. Actuators B 2009, 137, 281–290. [Google Scholar]
- 160.
- 160a. Roales J., Pedrosa J. M., Guillén M. G., Lopes-Costa T., Castillero P., Barranco A., González-Elipe A. R., Sensors 2015, 15, 11118–11132; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160b. Evyapan M., Dunbar A. D. F., Sens. Actuators B 2015, 206, 74–83; [Google Scholar]
- 160c. Çaycı D., Stanciu S. G., Çapan İ., Erdoğan M., Güner B., Hristu R., Stanciu G. A., Sens. Actuators B 2011, 158, 62–68; [Google Scholar]
- 160d. Smith V. C., Batty S. V., Richardson T., Foster K. A., Johnstone R. A. W., Sobral A. J. F. N., Gonsalves A. M. D. R., Thin Solid Films 1996, 284, 911–914; [Google Scholar]
- 160e. Nakagawa K., Kumon K., Tsutsumi C., Tabuchi K., Kitagawa T., Sadaoka Y., Sens. Actuators B 2000, 65, 138–140; [Google Scholar]
- 160f. Dunbar A. D. F., Richardson T. H., Hutchinson J., Hunter C. A., Sens. Actuators B 2008, 128, 468–481; [Google Scholar]
- 160g. Topal S. Z., Ongun M. Z., Onal E., Ertekin K., Hirel C., Dyes Pigm. 2017, 144, 102–109. [Google Scholar]
- 161. Senge M. O., Bischoff I., Eur. J. Org. Chem. 2001, 1735–1751. [DOI] [PubMed] [Google Scholar]
- 162. Thomassen I. K., McCormick L. J., Ghosh A., Cryst. Growth Des. 2018, 18, 4257–4259. [Google Scholar]